

Summary
Atopic dermatitis (AD) is a chronic itch and inflammatory disorder of the skin that affects one in ten people. Patients suffering from severe AD eventually progress to develop asthma and allergic rhinitis, in a process known as the “atopic march.” Signaling between epithelial cells and innate immune cells via the cytokine Thymic Stromal Lymphopoietin (TSLP) is thought to drive AD and the atopic march. Here we report that epithelial cells directly communicate to cutaneous sensory neurons via TSLP to promote itch. We identify the ORAI1/NFAT calcium signaling pathway as an essential regulator of TSLP release from keratinocytes, the primary epithelial cells of the skin. TSLP then acts directly on a subset of TRPA1-positive sensory neurons to trigger robust itch behaviors. Our results support a new model whereby calcium-dependent TSLP release by keratinocytes activates both primary afferent neurons and immune cells to promote inflammatory responses in the skin and airways.
Introduction
Atopic dermatitis (AD) is a chronic itch and inflammatory disorder of the skin that affects one in ten people. AD is primarily characterized by intolerable and incurable itch. Up to 70% of AD patients go on to develop asthma in a process known as the “atopic march” (He and Geha, 2010; Locksley, 2010; Spergel and Paller, 2003; Ziegler et al., 2013). Numerous studies suggest that the cytokine Thymic Stromal Lymphopoietin (TSLP) acts as a master switch that triggers both the initiation and maintenance of AD and the atopic march (Moniaga et al., 2013; Ziegler et al., 2013). TSLP is highly expressed in human cutaneous epithelial cells in AD, and bronchial epithelial cells in asthma (Jariwala et al., 2011). Over-expression of TSLP in keratinocytes, the most prevalent cell type in the skin, triggers robust itch-evoked scratching, the development of an AD-like skin phenotype and ultimately asthma-like lung inflammation in mice (Li et al., 2005; Ying et al., 2005; Ziegler et al., 2013). However, the mechanisms by which TSLP triggers itch and AD remain enigmatic.
Itch is mediated by primary afferent somatosensory neurons that have cell bodies in the dorsal root ganglia (DRG) that innervate the skin and are activated by endogenous pruritogens to drive itch behaviors (Ikoma et al., 2006; McCoy et al., 2012; Ross, 2011). Hallmarks of AD skin include robust itch sensations, increased neuronal activity and hyper-innervation (Ikoma et al., 2003; Tobin et al., 1992; Tominaga et al., 2009). While many studies have shown that epithelial cell-derived TSLP activates T cells, dendritic cells and mast cells (Ziegler et al., 2013), the role of sensory neurons in this pathway has not been studied. How does TSLP lead to sensory neuron activation to promote itch?
In vitro studies suggest that keratinocytes may directly communicate with sensory neurons via neuromodulators (Ikoma et al., 2006). Indeed, many of the factors that keratinocytes secrete act on both immune cells and primary afferent sensory neurons (Andoh et al., 2001; Fitzsimons et al., 2001; Kanda et al., 2005; Ziegler et al., 2013). Thus, TSLP may evoke itch behaviors directly, by activating sensory neurons, indirectly, by activating immune cells that secrete inflammatory mediators that target sensory neurons, or both. While TSLP's action on immune cells is well characterized, its effects on sensory neurons, and the contribution of sensory neurons to TSLP-evoked atopic disease, have not been studied. Furthermore, the mechanisms regulating TSLP release by keratinocytes are unknown.
The GPCR Protease-Activated Receptor 2 (PAR2) plays a key role in keratinocyte TSLP production. Studies have shown a correlation between PAR2 activity and TSLP expression in the skin of AD patients and in mouse models of atopic disease (Briot et al., 2009; Briot et al., 2010; Hovnanian, 2013). In addition, PAR2 activation triggers robust TSLP expression in keratinocytes (Kouzaki et al., 2009; Moniaga et al., 2013). While there is a strong correlation between PAR2 activity and TSLP levels in the skin, virtually nothing is known about the molecular mechanisms by which PAR2 leads to TSLP expression.
Here we sought to elucidate the mechanisms that regulate TSLP secretion and that promote TSLP-evoked itch. Our findings show that keratinocyte-derived TSLP activates sensory neurons directly to evoke itch behaviors. We define a new subset of sensory neurons that require both functional TSLP receptors and the ion channel, TRPA1, to promote TSLP-evoked itch behaviors, and we identify the ORAI1/NFAT signaling pathway as a key regulator of PAR2-mediated TSLP secretion by epithelial cells.
Results
TSLP evokes robust itch behaviors in mice
To identify proteins that mediate itch transduction in somatosensory neurons, we looked for biomarkers of AD (Lee and Yu, 2011) in the mouse DRG transcriptome (Gerhold et al., 2013). We were surprised to find expression of the TSLP Receptor (TSLPR) in mouse sensory ganglia. While studies have shown that TSLP acts on various immune cells, TSLP signaling in the nervous system has not been reported. TSLPR is a heterodimer, composed of the IL7 receptor alpha (IL7Rα) chain and a TSLP-specific receptor chain (TSLPR; also Crlf2; (Pandey et al., 2000). Consistent with the presence of TSLPRs in sensory neurons, we detected both TSLPR and IL7Rα transcripts in mouse and human DRG using RT-PCR (Figure 1A).
Figure 1. TSLP triggers robust itch behaviors in mice by activating sensory neurons.
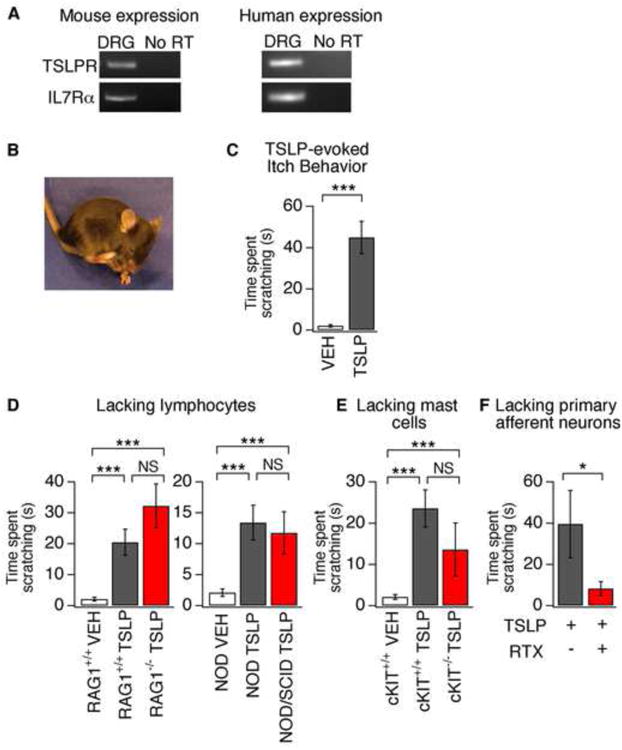
(A) PCR analysis of TSLPR and IL7Rα in mouse (left) and human (right) dorsal root ganglia (DRG). No product was amplified from the “no RT” control. (B) Image of itch-evoked scratching following intradermal injection of TSLP (2.5 μg/20 μl) into the cheek. (C) Quantification of scratching following TSLP injection in the cheek. TSLP (black) induced robust scratching compared to vehicle (white). n≥18 per group. (D) Itch behavior in RAG+/+, RAG-/-, NOD, and NOD/SCID mice following vehicle (PBS) or TSLP cheek injection. n≥8 per group. (E) Itch behavior in cKIT+/+ and cKIT-/- mice following vehicle (PBS) or TSLP injection. n≥8 per group. (F) TSLP-evoked scratching following neuronal ablation by RTX (red) versus control (black). n≥6 per group. *P<0.05; **P<0.01; ***P<0.001. Error bars represent s.e.m.
Somatosensory neurons mediate itch, touch and pain. Thus, we asked if TSLP injection triggers itch and/or pain behaviors by using a mouse cheek model of itch, which permits easy distinction between these behaviors (Shimada and LaMotte, 2008). Injection of TSLP into the cheek of wild type mice evoked robust scratching that was not observed following vehicle injection (Figure 1B-C). Wiping was never observed, indicating that TSLP triggers itch, rather than pain (Shimada and LaMotte, 2008). Intradermal injection of TSLP has been previously shown to evoke inflammation of the skin and lung over the course of hours or days (Jessup et al., 2008). However, we observed robust itch behaviors within 5 minutes of TSLP injection (latency to scratch = 4.1 ± 0.3 min).
While immune cells play a key role in long-term TSLP-evoked inflammation, whether immune cells are required for acute TSLP-triggered itch behaviors is unknown. The current model posits that TSLP acts on various immune cells to promote TH2 cell differentiation and inflammation. We thus compared TSLP-evoked itch behaviors of wild type mice to mouse strains lacking either T and B cells (RAG1-/-, NOD SCID) or mast cells (Kit(W-sh), Figure 1D-E). TSLP triggered robust itch behaviors in all strains, with no significant differences between transgenic and congenic wild type littermates. Together, these data indicate that acute TSLP-evoked itch does not specifically require lymphocytes or mast cells, nor does it require the cytokines or other products produced when these cells are activated, and suggest that TSLP may act directly on sensory neurons.
Previous studies have shown that intradermal injection of the TRPV1 agonist, resiniferatoxin (RTX), results in ablation of primary afferent sensory neurons that express TRPV1, or TRPV1 and TRPA1, and consequently eliminates pain and itch behaviors (Imamachi et al., 2009; Mitchell et al., 2010). TSLP-evoked scratching was significantly decreased in RTX-treated mice as compared to control mice (Figure 1F). These findings show for the first time that the AD cytokine, TSLP, induces itch via sensory neurons.
TSLP directly activates an uncharacterized subset of sensory neurons
We next asked whether TSLPRs are expressed in sensory neurons. DRG neurons are a heterogeneous population of cells, including a subset of small-diameter, peripherin-positive neurons that transmit itch and pain signals to the CNS, and release inflammatory mediators in the skin and other target organs (Basbaum et al., 2009). We thus examined the prevalence of TSLPR-positive neurons and co-localization with known neuronal markers. In situ hybridization revealed that TSLPR and IL7Rα were expressed in a subset of small diameter DRG neurons (Figure 2A). Using antibodies against TSLPR, we observed TSLPR protein expression in 5.9% of cells in DRG sections (Figure 2B). Co-staining of TSLPR and peripherin, a marker of small-diameter DRG neurons, demonstrated that all TSLPR-positive neurons are also peripherin-positive, with an average diameter of 18.1±0.6μm (Figure 2B). Overall, the characteristics of TSLPR-positive neurons match those of sensory neurons that mediate itch and/or pain (McCoy et al., 2013).
Figure 2. TSLP receptor components are expressed in sensory neurons.
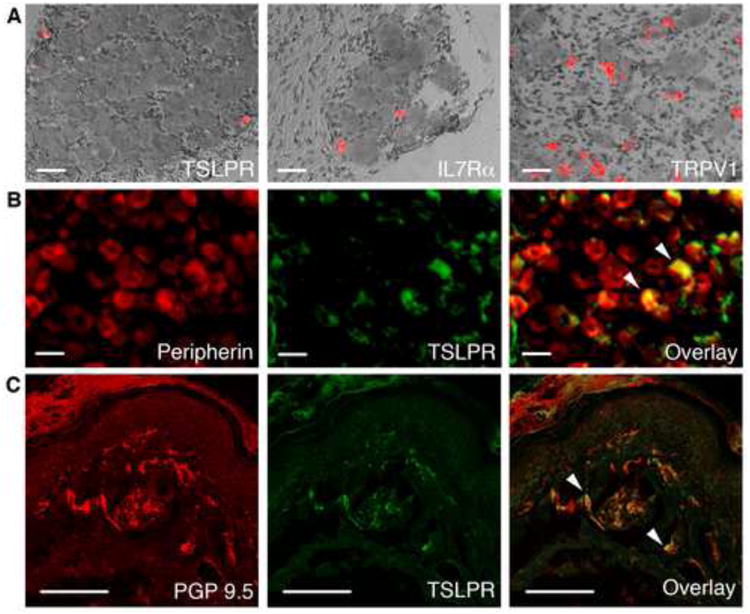
(A) DIC overlay images of in situ hybridization with cDNA probes detecting TSLPR, IL7Rα and TRPV1 in mouse DRG. Scale bar = 400μm. (B) Immunostaining of DRG sections with antibodies against peripherin and TSLPR in DRG sections. White arrows (right) mark peripherin- and TSLPR-positive neurons. Scale bar = 400μm. n≥4 mice/condition. (C) Immunostaining of PGP 9.5 and TSLPR in glabrous hind paw skin. The white arrows (right) mark PGP 9.5- and TSLPR-positive neurons. Scale bar = 200μm. n≥3 mice per condition.
If TSLPRs mediate somatosensory transduction, they should localize to primary afferent nerve terminals in the skin. We thus performed immunohistochemistry with antibodies against TSLPR and the pan-neuronal fiber marker PGP9.5 on mouse skin (Figure 2C). We observed TSLPR staining in 9% of PGP9.5-positive free nerve endings in the skin (Figure 2C). These data show that TSLPRs are localized to sensory neuronal endings that innervate the skin in close apposition to keratinocytes in the epidermis. Taken together, these data demonstrate that the TSLPR subunits are expressed in a subset of sensory neurons that innervate the skin and mediate itch and/or pain transduction.
To test whether TSLPR is functional in sensory neurons, we used ratiometric Ca2+ imaging (Figures 3A-B). We found that 4.1 ± 0.6% of DRG neurons showed robust increases in intracellular Ca2+ following TSLP application (Figure 3E); this is similar to the percentage of neurons that respond to other endogenous pruritogens, like BAM8-22 (Liu et al., 2009; Wilson et al., 2011). Previous studies have shown that small diameter sensory neurons transduce itch and/or pain signals via the ion channels TRPA1 and TRPV1 (Basbaum et al., 2009; Ross, 2011). Indeed, subsequent exposure to the TRPA1 agonist, allyl isothiocyanate (AITC), or the TRPV1 agonist, capsaicin (CAP), further increased Ca2+ levels in all TSLP-positive cells (Figures 3A-B). Similarly, TSLP triggered action potential firing in a subset of CAP-sensitive neurons (Figure 3C). These data suggest that TSLP activates a subset of TRPV1- and TRPA1-positive sensory neurons. The itch compounds histamine, chloroquine (CQ) and BAM8-22 have been shown to activate 5-20% of sensory neurons (Ikoma et al., 2006; Imamachi et al., 2009; Liu et al., 2009; Wilson et al., 2011) that express TRPA1 and/or TRPV1. TSLP appears to activate an undescribed subset of itch neurons, as most TSLP-positive neurons were insensitive to other itch compounds (Figure 3A,B,D).
Figure 3. TSLP directly activates a subset of sensory neurons.
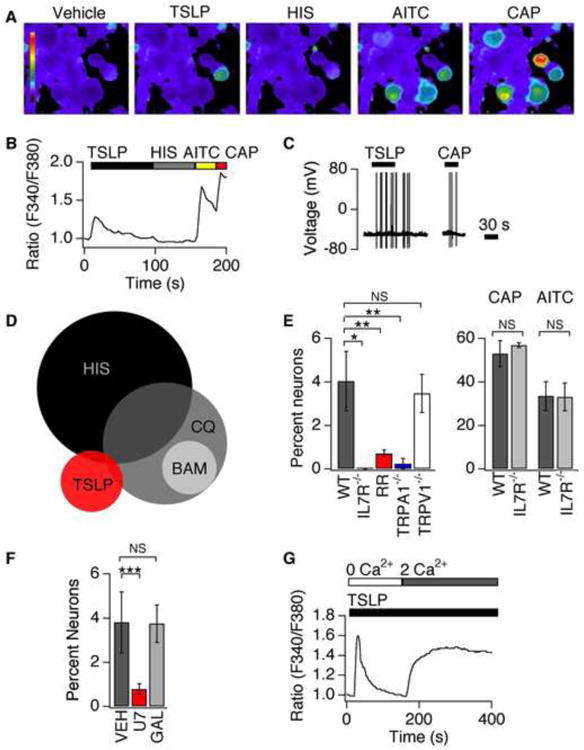
(A) Representative images of Fura-2 loaded DRG neurons treated with vehicle, TSLP (2 ng/mL), histamine (HIS, 1mM), AITC (200μM) and capsaicin (CAP, 1μM). (B) Representative trace shows a neuron that responds to TSLP, AITC and CAP, but not HIS. (C) Current-clamp recording showing TSLP- and CAP-evoked action potential firing in a DRG neuron. n≥60 cells. (D) A small percentage of the TSLP-sensitive population overlaps with the population of histamine- (HIS, 6%) or chloroquine-sensitive neurons (CQ, 6%), but not the BAM8-22 population (BAM, 0%). (E) Left: Prevalence of TSLP sensitivity in wild-type neurons (black), IL7Rα-deficient (grey) neurons, neurons treated with 20μM ruthenium red (RR; red), TRPA1-deficient neurons (blue) and TRPV1-deficient neurons (white). Right: prevalence of AITC and CAP sensitivity in wild-type (black) and IL7Rα-deficient (grey) neurons n≥1000 cells. (F) Prevalence of TSLP sensitivity in neurons pre-treated with vehicle (black), a PLC blocker, U73122 (red) and the Gβγ blocker, gallein (grey) n≥600 cells. (G) Representative response to TSLP in the absence (0mM Ca2+) and presence (2mM Ca2+) of extracellular Ca2+ n≥200 cells. *P<0.05; **P<0.01; ***P<0.001. Error bars represent s.e.m.
TSLPR and TRPA1 mediate TSLP-evoked neuronal activation
To ask whether TSLPRs mediate TSLP-evoked neuronal activation, we examined TSLP-evoked Ca2+ signals in neurons isolated from IL7Rα-deficient mice. TSLP-, but not AITC- or CAP-evoked Ca2+ signaling, was abolished in IL7α-deficient neurons (Figure 3E). These results are consistent with previous studies in immune cells showing that functional IL7Rα is required for TSLP signaling (Pandey et al., 2000). Here we show that functional TSLPRs are required for TSLP-evoked neuronal activation.
TRPV1 and TRPA1 channels are required for acute itch signaling and behavior (Ross, 2011). We thus asked whether these channels are required for TSLP-evoked neuronal activation. TRPV1 and TRPA1 inhibition by the nonselective inhibitor, ruthenium red, significantly decreased neuronal sensitivity to TSLP (Figure 3E). We also compared neurons isolated from TRPA1- and TRPV1-deficient mice to those from wild type littermates. TSLP-evoked Ca2+ signals were significantly attenuated in TRPA1-deficient neurons, but not TRPV1-deficient neurons (Figure 3E). Our results show that TRPA1 channels mediate TSLP-evoked neuronal excitability.
We next examined the mechanisms by which TSLPR activation promotes TRPA1 activity. Two signaling pathways have linked itch receptors to TRPA1 activation: Phospholipase C (PLC) signaling couples MrgprC11 to TRPA1; and, Gβγ signaling couples MrgprA3 to TRPA1 (Wilson et al., 2011). Treatment of cells with the PLC inhibitor, U73122, significantly reduced the prevalence of TSLP-sensitive neurons (Fig. 3F). In contrast, gallein, a Gβγ inhibitor, had no effect on TSLP-evoked Ca2+ signals (Fig. 3F). Consistent with TSLP activation of the PLC pathway, TSLP triggers both release of Ca2+ from intracellular stores, and subsequent Ca2+ influx in sensory neurons (Figure 3G). Overall, these experiments suggest that TSLPR and TRPA1 communicate via PLC signaling.
TSLPR and TRPA1 mediate TSLP-evoked itch
To test whether TSLP and TRPA1 receptors are required for TSLP-evoked itch behaviors, we used the cheek model of itch. TSLP-evoked scratching was significantly attenuated in IL7Rα-deficient mice, supporting a role for TSLPRs in TSLP itch signaling (Figure 4A). These mice were not generally deficient in itch behaviors, as CQ-evoked scratching, which occurs via MrgprA3 (Liu et al., 2009), was normal (Figure 4B). These data demonstrate that TSLP targets TSLPRs to trigger itch behaviors in vivo.
Figure 4. TSLP induces robust TSLPR- and TRPA1-dependent itch behaviors.
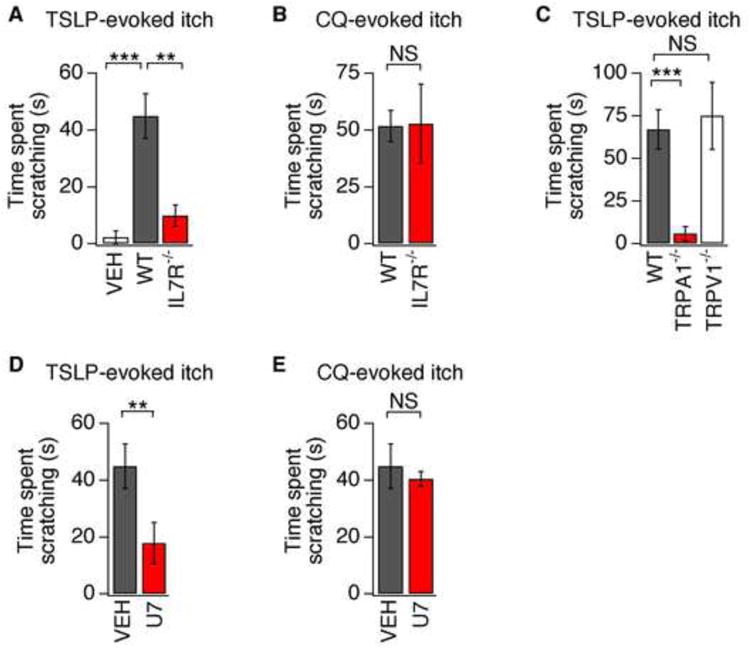
(A) Itch behaviors following intradermal cheek injection of vehicle (10μL PBS, white) or TSLP (2.5μg/10 μL) into wild type (WT; black) or IL7Rα-deficient (red) mice. (B) Scratching in WT (black) and IL7Rα-deficient (red) mice following chloroquine (CQ) injection in the cheek. (C) Scratching in WT (black), TRPA1-deficient (red) and TRPV1-deficient (white) mice following TSLP injection (2.5 μg/10 μL). (D) Attenuation of TSLP-evoked scratching by 30 min preinjection with the PLC blocker, U73122 (U7) compared to vehicle (VEH). (E) CQ-evoked scratching in mice preinjected with U73122 or vehicle. The time spent scratching was quantified for 20 min after injection. n≥7 mice/condition. **P<0.01; ***P<0.001. Error bars represent s.e.m.
We next asked whether TSLP-evoked itch behaviors require TRP channels. TSLP-evoked scratching was abolished in TRPA1-deficient mice, but normal in TRPV1-deficient mice (Figure 4D). These experiments show that both functional TSLPRs and TRPA1 channels are required for TSLP-evoked itch. PLC signaling is also required for the functional coupling between TSLPR and TRPA1 in vivo, as TSLP-evoked scratching was significantly attenuated by intradermal injection of U73122. Such treatment selectively silenced TSLP-evoked behaviors, as these mice displayed normal CQ-evoked scratching, which is PLC-independent (Wilson et al., 2011). Overall, these data demonstrate a new role for TSLP as a pruritogen and a robust activator of sensory neurons, and suggest that these neurons may contribute to the initiation of TSLP-evoked inflammatory responses in the skin in AD, and airways in asthma.
Keratinocyte release of TSLP is Ca2+-dependent
Our data establish a new cellular target for TSLP, supporting a model whereby both immune cells and sensory neurons are activated by keratinocyte-derived TSLP to drive itch and AD. What are the upstream mechanisms that govern the expression and release of TSLP by keratinocytes? Protease signaling via PAR2 plays a key role in TSLP production and AD. PAR2 activity, and levels of the endogenous PAR2 agonist, tryptase, are increased in the skin of AD patients (Steinhoff et al., 2003). Consistent with a previous study (Ui et al., 2006), injection of tryptase induced robust itch behaviors in mice (Figure 5A). Tryptase-evoked itch was significantly attenuated in both PAR2- and IL7Rα-deficient mice (Figure 5A), consistent with a pathway where PAR2 signaling promotes the release of TSLP from keratinocytes, which then acts on TSLPR-positive neurons to drive itch behaviors. We next sought to determine the signaling pathways that control PAR2-induced TSLP expression in keratinocytes.
Figure 5. PAR2 activation promotes itch behaviors and Ca2+-dependent release of TSLP.
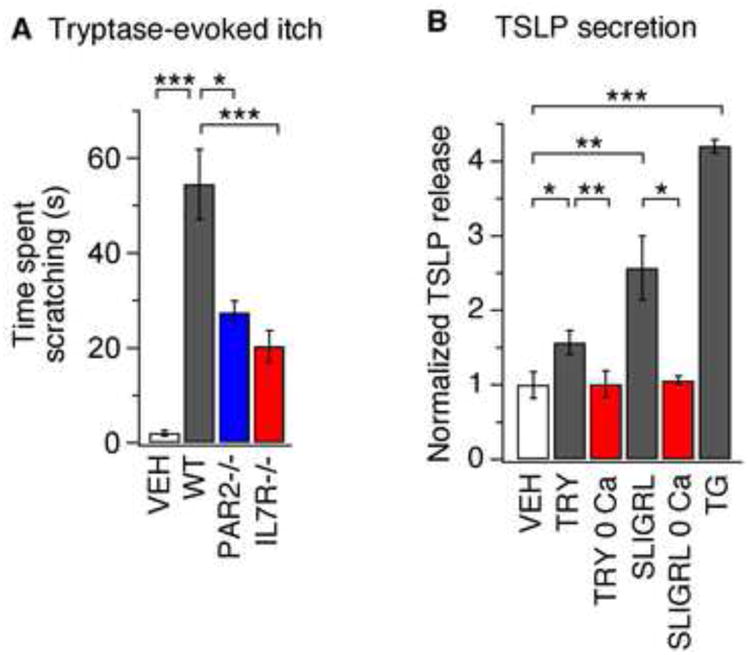
(A) Itch-evoked scratching following injection of tryptase into the cheek (100 pg/20 μL) of wild type (WT; black), PAR2-deficient (blue) or IL7Rα-deficient mice (red), or PBS (white, 20μL) injection into WT mice, n≥8 mice per condition. The time spent scratching was quantified for 1 h after injection. (B) TSLP secretion evoked by 24 h treatment with vehicle (VEH), tryptase (TRY, 100nM), tryptase in the absence of extracellular Ca2+ (TRY 0Ca), SLIGRL (100μM), SLIGRL in the absence of extracellular Ca2+ (SLIGRL 0Ca), or TG (1μM). n≥4 replicates/condition *P<0.05; **P<0.01;, ***P<0.001. Error bars represent s.e.m.
Studies on keratinocytes have shown that the endogenous PAR2 agonist, tryptase, and the widely used PAR2 ligand mimetic, Ser-Leu-Ile-Gly-Arg-Leu (SLIGRL), elicits Ca2+ influx (Schechter et al., 1998; Zhu et al., 2009) and triggers the Ca2+-dependent release of inflammatory mediators (Halfter et al., 2005; Santulli et al., 1995; Schechter et al., 1998). For example, SLIGRL triggers a rise in intracellular Ca2+ in keratinocytes (Zhu et al., 2009) and also promotes TSLP expression (Moniaga et al., 2013). We thus asked if PAR2-evoked TSLP expression is Ca2+-dependent. ELISA measurements revealed that treatment of keratinocytes with tryptase or SLIGRL, but not vehicle, triggered the robust secretion of TSLP (Figure 5B). These data show that PAR2 stimulation of keratinocytes triggers TSLP release.
TSLP secretion was highly dependent on Ca2+ First, TSLP secretion was not observed in keratinocytes treated with tryptase or SLIGRL in the absence of external Ca2+ (Figure 5B). In addition, treatment with the drug thapsigargin (TG), which promotes depletion of intracellular Ca2+ stores and subsequent Ca2+ influx, caused a significant increase in TSLP secretion (Figure 5B). These data demonstrate that Ca2+ is required and sufficient to drive TSLP secretion.
A recent study has shown that some PAR2 agonists, including SLIGRL, also activate the sensory neuron-specific itch receptor, MrgprC11 (MrgprX1 in human, (Liu et al., 2011). However, this result does not impact our in vitro studies for several reasons. First, keratinocytes do not express MrgprX1 (Supplementary Figure 1A). Second, keratinocytes are insensitive to the MrgprX1-specific ligand, BAM8-22 (Supplementary Figure 1B). Third, tryptase-evoked itch is dependent on PAR2 (Figure 5A). Finally, tryptase does not activate MrgprC11 in mice (Supplementary Figure 1C-D). Overall, our findings support a model where tryptase- and SLIGRL treatment of keratinocytes promotes PAR2-evoked Ca2+ signaling and subsequent secretion of TSLP.
ORAI1 and STIM1 are required for PAR2-evoked Ca2+ influx
We next used ratiometric Ca2+ imaging to dissect the mechanisms underlying PAR2-evoked Ca2+ signals. Consistent with previous studies, tryptase and SLIGRL evoked a rise in intracellular Ca2+ in keratinocytes (Figure 6A-C; Zhu et al., 2009). In some cells, PAR2 signals via PLC (Dai et al., 2007), and PLC activation leads to Ca2+-release from IP3-dependent stores and influx via the store-operated Ca2+ entry (SOCE) pathway. Indeed, PAR2 activation in keratinocytes induced both Ca2+ release from intracellular stores and Ca2+ influx, consistent with activation of SOCE (Figure 6A-B).
Figure 6. ORAI1 and STIM1 are required for PAR2- and TG-evoked Ca2+ influx.
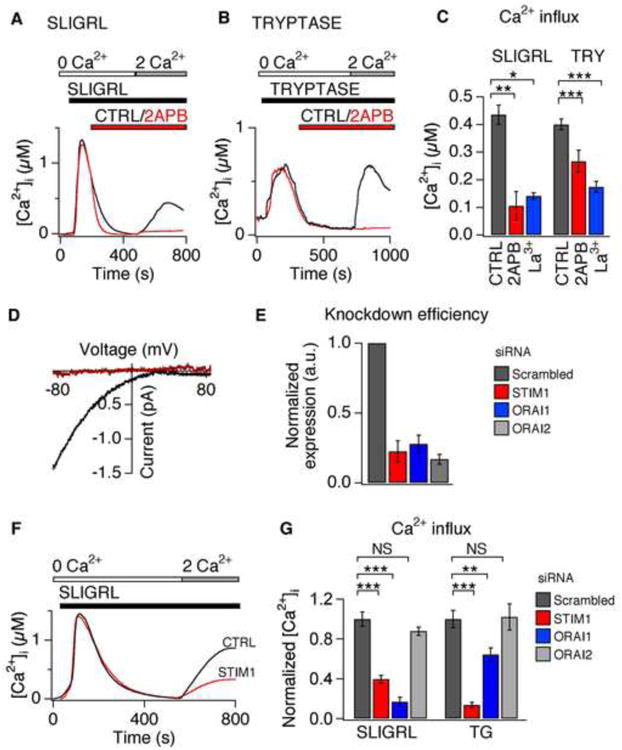
(A) Representative response to SLIGRL (100μM) following pretreatment with vehicle (black) or 2-aminoethoxydiphenyl borate (50 μM 2-APB; red). (B) Representative response to tryptase (100nM) following pretreatment with vehicle (black) or 2-APB (red). (C) Average steady state Ca2+ level following SLIGRL- or tryptase (TRY)-evoked Ca2+ influx (2 mM Ca2+), in the presence of 2-APB (red), lanthanum (50nM La3+, blue), or vehicle (CTRL, black). n≥1000 cells. (D) Representative current-voltage trace in the presence of SLIGRL (100μM) in perforated-patch, whole-cell voltage-clamp recordings. Representative baseline subtracted currents before (red) and during application of SLIGRL (black). n≥3 cells/condition. (E) siRNA-induced silencing of STIM1 (red), ORAI1 (blue), and ORAI2 (grey) mRNA in keratinocytes. Expression was normalized to scrambled-siRNA control (black). n≥1000 cells. (F) Representative traces of SLIGRL-evoked (100μM) Ca2+ signals following treatment with siRNA targeting STIM1 (red) or scrambled control (CTRL, black). (G) Average steady state Ca2+ concentration after treatment with SLIGRL (100μM) or TG (1μM) in cells treated with scrambled siRNA (black), STIM1 (red), ORAI1 (blue), or ORAI2 (grey) siRNA. n≥500 cells. *P<0.05; **P<0.01, ***P<0.001. Error bars represent s.e.m.
What are the molecules mediating PAR2-evoked SOCE in keratinocytes? Both ORAI and TRPC channels have been implicated in SOCE (Cahalan, 2009; Ramsey et al., 2006). We next asked whether PAR2 activates SOCE via ORAI or TRPC channels, which can be distinguished by their distinct pharmacological profiles (DeHaven et al., 2008; Lis et al., 2007; Zhang et al., 2008). The drugs 2-Aminoethoxydiphenyl borate (2-APB) and lanthanum (La3+) inhibit ORAI1 and ORAI2 channels, but not ORAI3 or TRPC channels (DeHaven et al., 2008; Lis et al., 2007; Zhang et al., 2008). Tryptase and SLIGRL-evoked Ca2+ influx was significantly attenuated by treatment with 2-APB or La3+. These data show that tryptase and SLIGRL activate the same SOCE pathway and support a role for ORAI channels in PAR2-evoked SOCE (Figure 6C).
ORAI and TRPC channels can also be distinguished by their distinct biophysical characteristics: ORAI1 and ORAI2 are Ca2+-selective channels that are inwardly-rectifying, while TRPC channels are outwardly-rectifying, non-selective channels (Cahalan, 2009; Owsianik et al., 2006; Yeromin et al., 2006). Thus, we measured SLIGRL-evoked currents using perforated-patch, voltage-clamp recordings on keratinocytes. Treatment with SLIGRL triggered an ORAI1/2-like current; the currents were dependent on extracellular Ca2+, displayed an inwardly-rectifying current-voltage relationship, and displayed no measurable reversal potentials below +80 mV (Figure 6D). These results implicate ORAI1 and/or ORAI2 in PAR2-evoked SOCE.
qPCR demonstrated that keratinocytes express ORAI1, ORAI2 and the ORAI regulator, Stromal Interaction Molecule 1 (STIM1). We thus examined the role of ORAI1, ORAI2, and STIM1 in SOCE using siRNA-mediated knockdown. Depletion of ORAI1 transcripts by 71% or STIM1 transcripts by 84% significantly diminished Ca2+ entry in response to SLIGRL as compared to scrambled control siRNA (Figure 6E-G). ORAI1 and STIM1 knockdown also significantly attenuated tryptase-evoked Ca2+ signals (not shown). In contrast, depletion of ORAI2 transcripts by 86% had no effect on SLIGRL-evoked SOCE (Figure 6E, 6G). These data demonstrate that ORAI1 and STIM1 are required for PAR2-evoked SOCE in human keratinocytes. ORAI1 and STIM1 knockdown also attenuated TG-evoked SOCE (Figure 6G), suggesting that ORAI1 is the primary store-operated Ca2+ pathway in keratinocytes.
PAR2-activation induces Ca2+-dependent NFAT translocation and TSLP secretion
In immune cells, ORAI1 signaling activates NFAT, which triggers cytokine expression and secretion (Feske et al., 2006; Gwack et al., 2007). The ORAI1/NFAT pathway may play a similar role in keratinocytes, promoting the expression and secretion of TSLP. Consistent with a regulatory role for NFAT in TSLP expression, two NFAT binding motifs (GGAAAATN) (Rao et al., 1997; Zhu et al., 2009) are present in the 5′-upstream regulatory region of the human TSLP gene. These findings imply that PAR2 may trigger NFAT-dependent expression and release of TSLP; however, the evidence is merely correlative. To directly test this hypothesis, we measured PAR2-dependent NFAT translocation and TSLP expression and release in keratinocytes.
Following a rise in Ca2+, NFAT is dephosphorylated by the Ca2+-dependent phosphatase calcineurin and translocates from the cytosol to the nucleus to promote transcription of target genes (Rao et al., 1997). Immunostaining demonstrated that treatment of keratinocytes with SLIGRL for 30 minutes induced robust NFAT translocation to the nucleus (Figures 7A). This translocation was attenuated by blocking ORAI channels with 2-APB, or by inhibiting NFAT activity with cyclosporine A (CsA), an inhibitor of calcineurin (Figure 7A); similar results were observed using live cell imaging of a human keratinocyte cell line, HaCat, that expressed NFAT-GFP (Figure 7B). These results show that PAR2 activation induces Ca2+-dependent NFAT translocation, which may lead to NFAT-dependent changes in gene expression. In support of this model, PAR2-evoked SOCE robustly increased expression of TSLP transcripts in keratinocytes (Figure 7C).
Figure 7. PAR2 activation promotes Ca2+-dependent NFAT translocation and TSLP secretion.
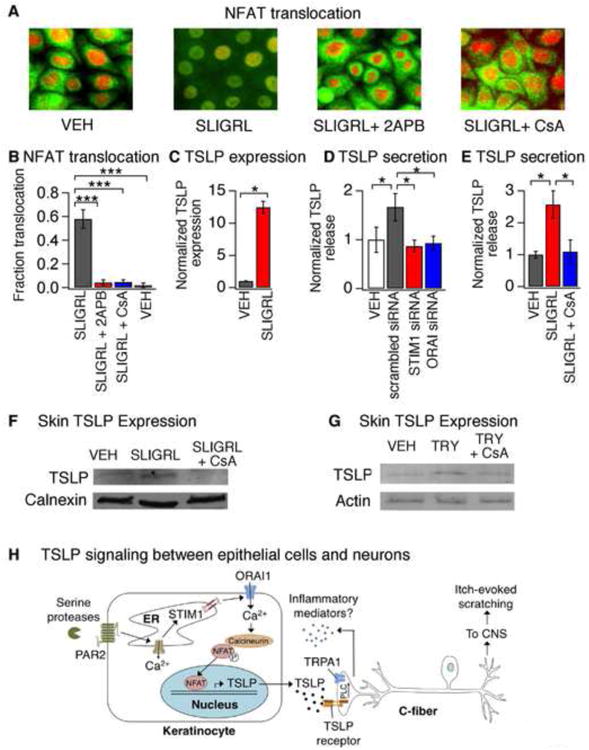
(A) Representative images displaying cytosolic and nuclear localization of NFAT (green) and DAPI (red) in keratinocytes after a 30 min incubation with vehicle (VEH), SLIGRL (100μM), SLIGRL + 2APB (50μM) or SLIGRL + CsA (1μM). Pretreatment with 2APB or CsA prevented SLIGRL-induced NFAT nuclear translocation. n≥300 cells. (B) Fraction of HaCaT keratinocytes displaying nuclear localization of NFAT-GFP following treatment with SLIGRL (100μM; black), SLIGRL and 2APB (50 μM; red), SLIGRL + CsA (1μM; blue) or vehicle (VEH; white). n≥1000 cells. (C) TSLP expression in human keratinocytes following a 3h treatment with vehicle (VEH, black) or SLIGRL (100μM, red). n≥3. (D) SLIGRL-evoked TSLP release in cells treated with scrambled (black), STIM1 (red) or ORAI1 siRNA (blue). Secretion was normalized to vehicle-treated cells (white). n≥3. (E) TSLP release in response to treatment with vehicle (VEH, black), SLIGRL (100 μM, red) or SLIGRL + CsA (1μM, blue). (F) Western blot of skin lysates from mice following intradermal injection with vehicle (VEH), SLIGRL, or SLIGRL+CsA. Samples were probed with antibodies against TSLP and calnexin (loading control). n≥3 mice. (G) Western blot of skin lysates isolated from mice following intradermal injection with vehicle (VEH), tryptase (TRY; 100pg/20μL), or tryptase+CsA (TRY + CsA). Samples were probed with antibodies against TSLP, and actin (loading control). n≥3 mice. *P<0.05; **P<0.01, ***P<0.001. Error bars represent s.e.m. (H) Schematic diagram depicting the ORAI1 signaling pathway in keratinocytes that links PAR2 to TSLP secretion and activation of itch neurons. Activation of PAR2 triggers release of Ca2+ from the ER and activation of STIM1, which opens ORAI1 channels to promote Ca2+ influx. Ca2+ activates the phosphatase calcineurin, which dephosphorylates NFAT and causes nuclear translocation, thus inducing transcription of TSLP. Secreted TSLP depolarizes a subset of C-fibers to evoke itch, in a TSLPR- and TRPA1-dependent manner. Activation of TRPA1-expressing sensory neurons can then lead to release of neuropeptides in the skin in a process known as neurogenic inflammation.
We next addressed whether ORAI1/NFAT signaling mediates PAR2-evoked TSLP release. We found that siRNA-mediated knockdown of ORAI1 or STIM1 significantly attenuated SLIGRL-evoked TSLP release by keratinocytes, suggesting that ORAI1 is required for PAR2-evoked TSLP secretion (Figure 7D). Likewise, inhibition of NFAT-mediated transcription with CsA also attenuated TSLP release (Figure 7E), but had no effect on SOCE-evoked Ca2+ signals (not shown). In addition to cutaneous epithelial cells, airway epithelial cells of patients with allergic rhinitis, AD and asthma also display high TSLP expression (Ziegler et al., 2013). Previous studies have shown that TG induces ORAI1-dependent Ca2+ signals in human airway epithelial cells (Gusarova et al., 2011). Interestingly, we found that, like keratinocytes, SOCE triggers robust TSLP expression in human airway epithelial cells, which can be blocked by CsA (not shown). These data identify ORAI1-dependent NFAT activation as a regulator of TSLP expression and release in both cutaneous and airway epithelial cells.
We next tested the hypothesis that NFAT promotes TSLP expression in vivo. Mice were treated with SLIGRL, SLIGRL and CSA, or vehicle via intradermal injection into the back. TSLP protein levels in treated skin were measured three hours after injection. Co-injection of CsA significantly attenuated SLIGRL-evoked TSLP protein expression in skin (Figure 7F). Similar results were also observed with the endogenous PAR2 agonist, tryptase (Figure 7G), demonstrating that PAR2 triggers TSLP expression via the Ca2+-calmodulin/NFAT pathway in vivo.
Discussion
TSLP plays a key role in the triad of atopic diseases: asthma, allergic rhinitis and atopic dermatitis (Ziegler et al., 2013). Recent studies have also implicated TSLP in a number of disorders, including cancer, gastrointestinal diseases, and autoimmunity (Ziegler et al., 2013). As such, there is much interest in understanding the mechanisms of TSLP expression and downstream effects of TSLP secretion. Here we present molecular, cellular and behavioral data showing that ORAI1/NFAT signaling regulates TSLP release by keratinocytes, and that TRPA1 is required for TSLP-evoked activation of sensory neurons and subsequent itch behaviors. Our data support a new model whereby TSLP released from keratinocytes acts directly on sensory neurons to trigger robust itch-evoked scratching (Figure 7H).
Sensory neurons mediate TSLP-evoked itch
Studies on the role of TSLP in promoting atopic disease have focused solely on its effects on immune cells. A variety of immune cells are activated by TSLP, including dendritic cells, T cells, B cells, natural killer cells, mast cells, basophils and eosinophils, which together promote allergic inflammation (Ziegler et al., 2013). The inflammatory cytokines produced by these immune cells can activate sensory neurons (Cevikbas et al., 2007). TSLP expression in keratinocytes leads to robust scratching in mice, which was previously assumed to occur solely downstream of immune cell cytokine release (Bogiatzi et al., 2012; Yoo et al., 2005).
The current model is that sensory neurons are activated downstream of TSLP-activated immune cells to induce itch. Our data support the direct activation of sensory neurons by TSLP. First, we show that mast cell release of histamine, or other pruritogens, is not required for TSLP-evoked itch behaviors. In addition, histamine-dependent itch requires TRPV1 (Imamachi et al., 2009), and our data show that TRPV1-deficient mice display normal TSLP-evoked itch behaviors. Finally, we show that acute TSLP-evoked itch does not require lymphocytes. These results were surprising given the well-known role of immune cells in TSLP-evoked atopic disease. However, until now, studies have focused on the long-term, rather than the acute effects of TSLP. These data suggest that the acute versus chronic phases of TSLP-evoked inflammation may be mediated by distinct mechanisms. In addition, because activation of primary afferent neurons triggers the release of inflammatory agents that promote immune cell chemoattraction and activation (e.g., substance P; Basbaum et al., 2009), neuron-to-immune cell communication may also play a key role in the development of AD. Thus far, all published studies have focused on global knockouts of TSLPR. Future studies using tissue specific TSLPR knockout mice are required to determine the relative contributions of sensory neurons and immune cells to both the acute and chronic phases of AD.
TRPA1 is required for TSLP-evoked itch
TSLP activates a subset of sensory neurons that express TSLPRs and the irritant receptor TRPA1. TRPA1-positive sensory neurons are required for the transmission of itch and pain stimuli to the CNS (Basbaum et al., 2009; Ross, 2011). Recent studies have shown that TRPA1 is also required for dry skin- and allergen-evoked chronic itch (Liu et al., 2013; Wilson et al., 2013), but the endogenous signaling molecules that promote TRPA1 activation in these itch models are unknown. We now show that the endogenous AD cytokine, TSLP, leads to TRPA1 activation, downstream of TSLPR. Inhibition of PLC significantly attenuates such coupling both in vitro and in vivo. Despite the extensive literature on TSLP in immune cells, little is known about the signaling pathways activated downstream of TSLPR. The JAK/STAT pathway has been implicated in TSLP signaling but thus far, neither TSLPR nor IL7R have been linked to PLC (Ziegler et al., 2013).
TRPA1 afferents are not restricted to the skin, but also densely innervate the airways and gastrointestinal tract (Bautista et al., 2013). Indeed, TRPA1 activation promotes lung inflammation in mouse models of airway inflammation and asthma and triggers inflammation in mouse models of inflammatory bowel disease (Bautista et al., 2013). Interestingly, we found that like keratinocytes, Ca2+ signaling through ORAI1 triggers robust TSLP expression in human airway epithelial cells (data not shown). Thus, crosstalk between sensory neurons and epithelial cells via TSLP and TRPA1 may not be restricted to the skin, but may also occur in the airways and gut. The “atopic march” has been largely attributed to the actions of epithelial and immune cells (Holgate, 2007). Future studies using tissue specific TSLPR-deficient animals are required to resolve the contributions of neuronal and immune cell TSLP signaling to atopic disease. Nonetheless, our findings highlight a potential new role for TRPA1 and sensory neurons in promoting the atopic march.
ORAI1/NFAT regulates TSLP release in keratinocytes
ORAI1 was first identified as the channel that mediates store-operated Ca2+ influx required for NFAT-dependent cytokine expression during immune cell activation; loss of function mutations in ORAI1 and STIM1 lead to severe combined immunodeficiencies in patients (Feske, 2010; Feske et al., 2006; Prakriya et al., 2006; Yeromin et al., 2006). Our work shows that in addition to lymphocytes, epithelial cells also utilize ORAI1-mediated Ca2+ influx to regulate cytokine expression and release, suggesting that ORAI1 plays a more general role in the pathogenesis of inflammatory disease. Thus, ORAI1 may represent a new therapeutic target for atopic disease.
A role for the ORAI1/NFAT pathway in AD is consistent with a number of disparate clinical findings. First, SNPs in the ORAI1 gene have been linked to susceptibility to atopic disease in humans (Chang et al., 2012) but the role of ORAI1 in AD had not been studied. Second, NFAT displays an abnormally high degree of nuclear localization in the keratinocytes of chronic itch patients (Al-Daraji et al., 2002), but the consequences of NFAT activation on AD was unknown. Finally, CsA, an inhibitor of NFAT-mediated transcription, is a potent immunosuppressant drug and is often prescribed for itchy inflammatory skin diseases, such as psoriasis and AD (Madan and Griffiths, 2007). While its effects have been mainly attributed to immune cell inhibition, our work suggests that the effectiveness of CsA in treating chronic itch may, in part, be due to its effects on keratinocyte-mediated TSLP release.
Experimental Procedures
Cell Culture
Primary human epidermal keratinocytes (PromoCell) and HaCaT cells were cultured in PromoCell Keratinocyte Medium 2 and DMEM, respectively. siRNA directed against ORAI1, ORAI2, and STIM1 (Qiagen; 100ng) were transfected using HiPerFect (Qiagen). HaCaT cells were transiently transfected with Lipofectamine 2000 (Invitrogen) using 1μg HA-NFAT1(1-460)-GFP plasmid (Addgene 11107). DRG neurons were isolated from P18-30 mice and cultured as previously described (Wilson et al., 2011). All media and cell culture supplements were purchased from the UCSF Cell Culture Facility.
Ca2+ imaging
Ca2+ imaging was carried out as previously described (Wilson et al., 2011). Physiological Ringer solution: 140mM NaCl, 5mM KCl, 10mM HEPES, 2mM CaCl2, 2mM MgCl2, 10mM D-(+)-glucose, pH 7.4 with NaOH. Images were collected and analyzed using MetaFluor (Molecular Devices). [Ca2+]i was determined from background-corrected F340/F380 ratio images using the relation [Ca2+]I = K*(R−Rmin)/(Rmax−R) (Almers 1985), with the following parameters measured in keratinocytes: Rmin=0.3; Rmax=2.2; and K*=3μM. Cells were classified as responders if [Ca2+]i increased 15% above baseline.
Electrophysiology
Recordings were collected at 5 kHz and filtered at 2 kHz using an Axopatch 200B and PClamp software. Electrode resistances were 2-6 MΩ. Perforated patch internal solution: 140mM CsCl, 5mM EGTA,10mM HEPES, pH 7.4 with CsOH, 0.24 mg ml−1 Amphotericin B (Rae et al., 1991). Stimulation protocol: 10ms step to -80 mV, 150ms ramp from -80mV to +80mV. Current clamp internal solution: 140mM KCl, 5mM EGTA and 10mM HEPES (pH 7.4 with KOH). Series resistance of all cells were <30 MΩ and liquid junction potentials were < 5mV (no correction).
RT-PCR
RNA was extracted using RNeasy (Qiagen) and reverse transcription was performed using Superscript III. RT-PCR was carried out using SYBR Green (Invitrogen) on a StepOnePlus ABI machine. Threshold cycles for each transcript (Bogiatzi et al.) were normalized to GAPDH (ΔCt). Calibrations and normalizations used the 2-ΔΔCt method where ΔΔCt = [(Ct (target gene) - Ct (reference gene)] - [Ct (calibrator) - Ct (reference gene)]; GAPDH=reference gene; scrambled siRNA=calibrator. Experiments were performed in triplicate.
Histology
Histology was carried out as previously described (Gerhold, 2013). Antibodies: rabbit anti-PGP9.5 and rabbit anti-peripherin (Millipore) 1:1000; goat anti-TSLPR and mouse anti-NFATc1 (Santa Cruz Biotechnology) 1:100. IL7Rα and TSLPR probes (Panomics) were used for in situ hybridization following the Quantigene protocol (Panomics).
Protein detection
TSLP protein levels were measured using the DuoSet ELISA kit (R&D Systems) on media collected 24h after stimulation. TSLP release was normalized to vehicle. For western blots, 50μg of cleared tissue lysate was resolved by SDS-PAGE, transferred to nitrocellulose membranes and probed with Anti-TSLP (1:250, Genetex), Anti-Calnexin (1:2,000, Abcam) and Anti-Actin (1:2,000).
Mice and Behavior
Mice (20-35g) were housed in 12h light-dark cycle at 21°C. Behavioral measurements were performed as previously described (Wilson et al., 2011). Compounds injected: 2.5μg TSLP, 200μg CQ, 100pg tryptase dissolved in PBS, or RTX 1μg/mL in 0.05% ascorbic acid and 7% Tween 80 (two days prior to pruritogen injection). For AITC behavior, 5μL 10% AITC in mineral oil was applied to the right hind paw. Behavioral scoring was performed while blind to treatment and genotype. All experiments were performed under the policies and recommendations of the International Association for the Study of Pain and approved by the University of California, Berkeley Animal Care and Use Committee.
Data analysis
Data are shown as mean ± s.e.m. Statistical significance was evaluated using a oneway ANOVA followed by a Tukey-Kramer post hoc test or unpaired two-tailed Student's t-test for comparing difference between two samples. *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
Highlights.
Epithelial cells communicate to sensory neurons via TSLP to promote itch
ORAI1/NFAT calcium signaling regulates release of TSLP from keratinocytes
TSLP is a robust pruritogen that promotes itch-evoked scratching
TSLP-evoked itch behaviors require TRPA1 ion channels that promote inflammation
Acknowledgments
We are grateful to Drs. Hariharan and Heald and lab members for critical comments on the manuscript. We thank K.Gerhold, J. Meeuwsen, T. Morita, M. Newton and M. Tsunozaki for behavior data analyses. This work was supported by NIH grants AR059385 and DOD007123A (DMB) and NSF Fellowships (SRW & LT). The authors declare no competing financial interests. We thank the Neurobiology course at the Marine Biological Laboratory for pilot experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Daraji WI, Grant KR, Ryan K, Saxton A, Reynolds NJ. Localization of calcineurin/NFAT in human skin and psoriasis and inhibition of calcineurin/NFAT activation in human keratinocytes by cyclosporin A. J Invest Dermatol. 2002;118:779–788. doi: 10.1046/j.1523-1747.2002.01709.x. [DOI] [PubMed] [Google Scholar]
- Almers N. The Ca signal from fura-2 loaded mast cells depends strongly on the method of dye-loading. FEBS Letters. 1985;192:13–18. doi: 10.1016/0014-5793(85)80033-8. [DOI] [PubMed] [Google Scholar]
- Andoh T, Katsube N, Maruyama M, Kuraishi Y. Involvement of leukotriene B(4) in substance P-induced itch-associated response in mice. J Invest Dermatol. 2001;117:1621–1626. doi: 10.1046/j.0022-202x.2001.01585.x. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Pellegrino M, Tsunozaki M. TRPA1: A gatekeeper for inflammation. Annu Rev Physiol. 2013;75:181–200. doi: 10.1146/annurev-physiol-030212-183811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogiatzi SI, Guillot-Delost M, Cappuccio A, Bichet JC, Chouchane-Mlik O, Donnadieu MH, Barillot E, Hupe P, Chlichlia K, Efremidou EI, et al. Multiple-checkpoint inhibition of thymic stromal lymphopoietin-induced TH2 response by TH17-related cytokines. J Allergy Clin Immunol. 2012;130:233–240 e235. doi: 10.1016/j.jaci.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, Dubus P, Hovnanian A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206:1135–1147. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briot A, Lacroix M, Robin A, Steinhoff M, Deraison C, Hovnanian A. Par2 inactivation inhibits early production of TSLP, but not cutaneous inflammation, in Netherton syndrome adult mouse model. J Invest Dermatol. 2010;130:2736–2742. doi: 10.1038/jid.2010.233. [DOI] [PubMed] [Google Scholar]
- Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevikbas F, Steinhoff A, Homey B, Steinhoff M. Neuroimmune interactions in allergic skin diseases. Curr Opin Allergy Clin Immunol. 2007;7:365–373. doi: 10.1097/ACI.0b013e3282a644d2. [DOI] [PubMed] [Google Scholar]
- Chang WC, Lee CH, Hirota T, Wang LF, Doi S, Miyatake A, Enomoto T, Tomita K, Sakashita M, Yamada T, et al. ORAI1 genetic polymorphisms associated with the susceptibility of atopic dermatitis in Japanese and Taiwanese populations. PLoS One. 2012;7:e29387. doi: 10.1371/journal.pone.0029387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H, Noguchi K. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest. 2007;117:1979–1987. doi: 10.1172/JCI30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW., Jr Complex actions of 2-aminoethyldiphenyl borate on store-operated Ca2+ entry. J Biol Chem. 2008;283:19265–19273. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S. CRAC channelopathies. Pflugers Arch. 2010;460:417–435. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Fitzsimons C, Engel N, Duran H, Policastro L, Cricco G, Martin G, Molinari B, Rivera E. Histamine production in mouse epidermal keratinocytes is regulated during cellular differentiation. Inflamm Res. 2001;50 Suppl 2:S100–101. doi: 10.1007/PL00022378. [DOI] [PubMed] [Google Scholar]
- Gerhold KA, Pellegrino M, Tsunozaki M, Morita T, Leitch DB, Tsuruda PR, Brem RB, Catania KC, Bautista DM. The star-nosed mole reveals clues to the molecular basis of mammalian touch. PLoS One. 2013;8:e55001. doi: 10.1371/journal.pone.0055001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M, Sznajder JI. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol Cell Biol. 2011;31:3546–3556. doi: 10.1128/MCB.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y, Feske S, Srikanth S, Hogan PG, Rao A. Signalling to transcription: store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium. 2007;42:145–156. doi: 10.1016/j.ceca.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Halfter UM, Derbyshire ZE, Vaillancourt RR. Interferon-gamma-dependent tyrosine phosphorylation of MEKK4 via Pyk2 is regulated by annexin II and SHP2 in keratinocytes. Biochem J. 2005;388:17–28. doi: 10.1042/BJ20041236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Geha RS. Thymic stromal lymphopoietin. Ann N Y Acad Sci. 2010;1183:13–24. doi: 10.1111/j.1749-6632.2009.05128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgate ST. The epithelium takes centre stage in asthma and atopic dermatitis. Trends Immunol. 2007;28:248–251. doi: 10.1016/j.it.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289–300. doi: 10.1007/s00441-013-1558-1. [DOI] [PubMed] [Google Scholar]
- Ikoma A, Rukwied R, Stander S, Steinhoff M, Miyachi Y, Schmelz M. Neuronal sensitization for histamine-induced itch in lesional skin of patients with atopic dermatitis. Arch Dermatol. 2003;139:1455–1458. doi: 10.1001/archderm.139.11.1455. [DOI] [PubMed] [Google Scholar]
- Ikoma A, Steinhoff M, Stander S, Yosipovitch G, Schmelz M. The neurobiology of itch. Nat Rev Neurosci. 2006;7:535–547. doi: 10.1038/nrn1950. [DOI] [PubMed] [Google Scholar]
- Imamachi N, Park GH, Lee H, Anderson DJ, Simon MI, Basbaum AI, Han SK. TRPV1-expressing primary afferents generate behavioral responses to pruritogens via multiple mechanisms. Proc Natl Acad Sci U S A. 2009;106:11330–11335. doi: 10.1073/pnas.0905605106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariwala SP, Abrams E, Benson A, Fodeman J, Zheng T. The role of thymic stromal lymphopoietin in the immunopathogenesis of atopic dermatitis. Clin Exp Allergy. 2011;41:1515–1520. doi: 10.1111/j.1365-2222.2011.03797.x. [DOI] [PubMed] [Google Scholar]
- Jessup HK, Brewer AW, Omori M, Rickel EA, Budelsky AL, Yoon BR, Ziegler SF, Comeau MR. Intradermal administration of thymic stromal lymphopoietin induces a T cell- and eosinophil-dependent systemic Th2 inflammatory response. J Immunol. 2008;181:4311–4319. doi: 10.4049/jimmunol.181.6.4311. [DOI] [PubMed] [Google Scholar]
- Kanda N, Koike S, Watanabe S. Prostaglandin E2 enhances neurotrophin-4 production via EP3 receptor in human keratinocytes. J Pharmacol Exp Ther. 2005;315:796–804. doi: 10.1124/jpet.105.091645. [DOI] [PubMed] [Google Scholar]
- Kouzaki H, O'Grady SM, Lawrence CB, Kita H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J Immunol. 2009;183:1427–1434. doi: 10.4049/jimmunol.0900904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Yu HS. Biomarkers for itch and disease severity in atopic dermatitis. Curr Probl Dermatol. 2011;41:136–148. doi: 10.1159/000323307. [DOI] [PubMed] [Google Scholar]
- Li M, Messaddeq N, Teletin M, Pasquali JL, Metzger D, Chambon P. Retinoid X receptor ablation in adult mouse keratinocytes generates an atopic dermatitis triggered by thymic stromal lymphopoietin. Proc Natl Acad Sci U S A. 2005;102:14795–14800. doi: 10.1073/pnas.0507385102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, Penner R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Escalera J, Balakrishna S, Fan L, Caceres AI, Robinson E, Sui A, McKay MC, McAlexander MA, Herrick CA, et al. TRPA1 controls inflammation and pruritogen responses in allergic contact dermatitis. FASEB J. 2013 doi: 10.1096/fj.13-229948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng HJ, Geng Y, et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell. 2009;139:1353–1365. doi: 10.1016/j.cell.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Weng HJ, Patel KN, Tang Z, Bai H, Steinhoff M, Dong X. The distinct roles of two GPCRs, MrgprC11 and PAR2, in itch and hyperalgesia. Sci Signal. 2011;4:ra45. doi: 10.1126/scisignal.2001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locksley RM. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Griffiths CE. Systemic ciclosporin and tacrolimus in dermatology. Dermatol Ther. 2007;20:239–250. doi: 10.1111/j.1529-8019.2007.00137.x. [DOI] [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Street SE, Pribisko AL, Zheng J, Zylka MJ. Peptidergic CGRPalpha Primary Sensory Neurons Encode Heat and Itch and Tonically Suppress Sensitivity to Cold. Neuron. 2013;78:138–151. doi: 10.1016/j.neuron.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Zylka MJ. CGRPalpha-expressing sensory neurons respond to stimuli that evoke sensations of pain and itch. PLoS One. 2012;7:e36355. doi: 10.1371/journal.pone.0036355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K, Bates BD, Keller JM, Lopez M, Scholl L, Navarro J, Madian N, Haspel G, Nemenov MI, Iadarola MJ. Ablation of rat TRPV1-expressing Adelta/C-fibers with resiniferatoxin: analysis of withdrawal behaviors, recovery of function and molecular correlates. Mol Pain. 2010;6:94. doi: 10.1186/1744-8069-6-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniaga CS, Jeong SK, Egawa G, Nakajima S, Hara-Chikuma M, Jeon JE, Lee SH, Hibino T, Miyachi Y, Kabashima K. Protease activity enhances production of thymic stromal lymphopoietin and basophil accumulation in flaky tail mice. Am J Pathol. 2013;182:841–851. doi: 10.1016/j.ajpath.2012.11.039. [DOI] [PubMed] [Google Scholar]
- Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol. 2006;68:685–717. doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, Ziegler SF, Leonard WJ, Lodish HF. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol. 2000;1:59–64. doi: 10.1038/76923. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Ross SE. Pain and itch: insights into the neural circuits of aversive somatosensation in health and disease. Curr Opin Neurobiol. 2011;21:880–887. doi: 10.1016/j.conb.2011.10.012. [DOI] [PubMed] [Google Scholar]
- Santulli RJ, Derian CK, Darrow AL, Tomko KA, Eckardt AJ, Seiberg M, Scarborough RM, Andrade-Gordon P. Evidence for the presence of a protease-activated receptor distinct from the thrombin receptor in human keratinocytes. Proc Natl Acad Sci U S A. 1995;92:9151–9155. doi: 10.1073/pnas.92.20.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter NM, Brass LF, Lavker RM, Jensen PJ. Reaction of mast cell proteases tryptase and chymase with protease activated receptors (PARs) on keratinocytes and fibroblasts. J Cell Physiol. 1998;176:365–373. doi: 10.1002/(SICI)1097-4652(199808)176:2<365::AID-JCP15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Shimada SG, LaMotte RH. Behavioral differentiation between itch and pain in mouse. Pain. 2008;139:681–687. doi: 10.1016/j.pain.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. 2003;112:S118–127. doi: 10.1016/j.jaci.2003.09.033. [DOI] [PubMed] [Google Scholar]
- Tobin D, Nabarro G, Baart de la Faille H, van Vloten WA, van der Putte SC, Schuurman HJ. Increased number of immunoreactive nerve fibers in atopic dermatitis. J Allergy Clin Immunol. 1992;90:613–622. doi: 10.1016/0091-6749(92)90134-n. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Tengara S, Kamo A, Ogawa H, Takamori K. Psoralen-ultraviolet A therapy alters epidermal Sema3A and NGF levels and modulates epidermal innervation in atopic dermatitis. J Dermatol Sci. 2009;55:40–46. doi: 10.1016/j.jdermsci.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Ui H, Andoh T, Lee JB, Nojima H, Kuraishi Y. Potent pruritogenic action of tryptase mediated by PAR-2 receptor and its involvement in anti-pruritic effect of nafamostat mesilate in mice. Eur J Pharmacol. 2006;530:172–178. doi: 10.1016/j.ejphar.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, Bautista DM. TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor-mediated itch. Nat Neurosci. 2011;14:595–602. doi: 10.1038/nn.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SR, Nelson AM, Batia L, Morita T, Estandian D, Owens DM, Lumpkin EA, Bautista DM. The Ion Channel TRPA1 Is Required for Chronic Itch. J Neurosci. 2013;33:9283–9294. doi: 10.1523/JNEUROSCI.5318-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee TH, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, Comeau MR, Campbell DJ, Ziegler SF. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. 2005;202:541–549. doi: 10.1084/jem.20041503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Kozak JA, Jiang W, Yeromin AV, Chen J, Yu Y, Penna A, Shen W, Chi V, Cahalan MD. Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem. 2008;283:17662–17671. doi: 10.1074/jbc.M801536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wang XR, Peng C, Xu JG, Liu YX, Wu L, Zhu QG, Liu JY, Li FQ, Pan YH, et al. Induction of leukotriene B(4) and prostaglandin E(2) release from keratinocytes by protease-activated receptor-2-activating peptide in ICR mice. Int Immunopharmacol. 2009;9:1332–1336. doi: 10.1016/j.intimp.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H. The Biology of Thymic Stromal Lymphopoietin (TSLP) Adv Pharmacol. 2013;66:129–155. doi: 10.1016/B978-0-12-404717-4.00004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.