

Abstract
Estrogen receptor α (ERα) interacts with DNA directly or indirectly via other transcription factors, referred to as “tethering.” Evidence for tethering is based on in vitro studies and a widely used “KIKO” mouse model containing mutations that prevent direct estrogen response element DNA- binding. KIKO mice are infertile, due in part to the inability of estradiol (E2) to induce uterine epithelial proliferation. To elucidate the molecular events that prevent KIKO uterine growth, regulation of the pro-proliferative E2 target gene Klf4 and of Klf15, a progesterone (P4) target gene that opposes the pro-proliferative activity of KLF4, was evaluated. Klf4 induction was impaired in KIKO uteri; however, Klf15 was induced by E2 rather than by P4. Whole uterine chromatin immunoprecipitation-sequencing revealed enrichment of KIKO ERα binding to hormone response elements (HREs) motifs. KIKO binding to HRE motifs was verified using reporter gene and DNA-binding assays. Because the KIKO ERα has HRE DNA-binding activity, we evaluated the “EAAE” ERα, which has more severe DNA-binding domain mutations, and demonstrated a lack of estrogen response element or HRE reporter gene induction or DNA-binding. The EAAE mouse has an ERα null–like phenotype, with impaired uterine growth and transcriptional activity. Our findings demonstrate that the KIKO mouse model, which has been used by numerous investigators, cannot be used to establish biological functions for ERα tethering, because KIKO ERα effectively stimulates transcription using HRE motifs. The EAAE-ERα DNA-binding domain mutant mouse demonstrates that ERα DNA-binding is crucial for biological and transcriptional processes in reproductive tissues and that ERα tethering may not contribute to estrogen responsiveness in vivo.
The estrogen receptor α (ERα) controls transcriptional rates of target genes in cells by directly interacting with the estrogen response element (ERE) DNA motifs. A second “tethering” mechanism has been described using in vitro studies, in which ERα interacts with other DNA motif binding transcription factors, such as FOS/JUN dimers on AP1 sites (1). To elucidate biological processes mediated by direct ERE vs indirectly tethered responses, mutations were introduced at amino acids 207 and 208 in the second “knuckle” of the first zinc finger of the mouse ERα, a region referred to as the proximal box (P-box) (Figure 1A), because these residues govern DNA sequence selectivity (2, 3). When the mutations were knocked in to the mouse ERα locus (4), females heterozygous for the ERα DNA-binding domain (DBD) mutation (KIWT) were infertile, developed abnormally enlarged uteri, and were anovulatory (4). To overcome these issues and produce a mouse model with the DBD-mutated ERα as its only functional ERα allele, KIWT males were bred to females heterozygous for the ERα-null allele (WTKO) (5). The resulting compound heterozygous (KIKO) females were infertile, and although initial findings suggested an ability to mount uterine proliferative responses to estrogen (5), later studies indicated that KIKO uteri are refractory to estrogen response in terms of weight increase and epithelial cell proliferation (6, 7). Further evaluation of the KIKO uterine response revealed lack of estradiol (E2)-mediated induction of IGF-1 (Igf1) (7), a growth factor essential for uterine growth (8). A second ERα DBD mutant mouse model (EAAE), which has 4 amino acid mutations that flank the P-box region (amino acids 201, 210, 214, and 215) (Figure 1A), also serves as an in vivo model to discern ERE vs tethered responses (9). This second model differed from the KIKO model in that EAAE mice displayed more of the phenotypes observed in ERα-null mouse models and lacked female liver transcriptional responses (9). In addition, females heterozygous for the EAAE ERα mutations are fertile (9).
Figure 1.
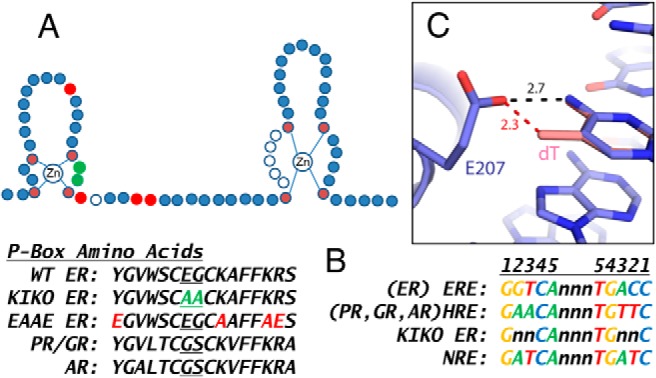
Zinc finger domain, DNA motifs, and structural evaluation of the role of E207 in ERE binding selectivity. A, Schematic diagram showing the structure of 2 Zn+2 fingers of nuclear receptor DBD. Each finger contains 4 cysteine residues (shown in dark red), which coordinately bind a Zn+2 ion. The first finger contains 3 amino acids that determine specificity for the DNA motif binding (indicated by 2 circles colored green and 1 open circle, the proximal [P] box). The 2 P-box residues colored green were mutated to alanine in the KIKO ERα. The amino acids mutated in the EAAE ERα are colored red. The second finger contains a region involved in ERα dimerization (distal [D] box, open circles). The P-box amino acid residues for ERα (residues 201–216 in the mouse), GR, AR, and PR are shown. The amino acid substitutions used in the KIKO mouse (highlighted in green) and in the EAAE mouse (highlighted in red) are shown. B, Consensus ERE and HRE motif DNA sequences, the preferred motif of KIKO ERα, and NRE, a motif demonstrated to be nonresponsive to nuclear receptors, with palindromic arms numbered, are shown. C, Role of E207 in steric exclusion. Crystal structure of the ERα DBD bound to DNA (blue) (Protein Data Bank code 1HCQ). Modeling of the HRE consensus dT (pink) at position 2 onto the ERE DNA, places the dT C13 methyl group 2.3 Å from E207 of ERα, generating a significant steric clash. In addition, ER-ERE E207 forms a hydrogen bond with the G/C at position 2 of the ERE. Mutation of E207 to A disrupts formation of this hydrogen bond, reducing affinity for the ERE, and also relieves the steric clash with HRE, reducing the selectivity against the HRE.
The activities of ERα and progesterone receptor (PR) in uterine tissue are interrelated. Proper uterine function requires intricate regulation of responses to fluctuation in estrogen and progesterone hormone levels, both temporally and in terms of uterine cell type-specific events (10). These effects are balanced, with the PR-mediated-P4 function tempering the pro-proliferative activity of the ERα-mediated E2 response. Two Kruppel-like factors (KLFs) have been implicated in E2 and P4 modulation of uterine proliferation; KLF4 is increased by E2 and promotes DNA replication, whereas KLF15 is increased by P4 and inhibits growth via regulation of minichromosome maintenance 2 (Mcm2) (11). We hypothesized that tethered ERα signaling mediated by the DBD mutant was insufficient to regulate uterine biological functions. Therefore, we evaluated the regulation of Klf4 and Klf15 to determine whether their mis-regulation might contribute to the inability of E2 to induce uterine growth. Our study has revealed not only an inability of KIKO ERα to induce uterine Klf4 but also an unexpected E2 induction of the normally P4-responsive Klf15 by KIKO ERα.
Recently, we described the uterine ERα “cistrome,” showing sites of interaction between ERα and chromatin in mouse uterine tissue, by using chromatin immunoprecipitation sequencing (ChIP-seq) (12). Similarly, Rubel et al (13) have described the uterine PR cistrome. Therefore, to evaluate the mechanisms that might underlie the E2 induction of Klf15 by KIKO ERα, we examined the KIKO ERα cistrome to determine sites of interaction of KIKO ER with uterine chromatin, expecting to determine tethered sites but have instead revealed altered DNA motif interaction of the KIKO ERα.
Materials and Methods
Animals and tissue samples
All procedures with animals were performed under an animal study protocol approved by the National Institute of Environmental Health Sciences Animal Care and Use Committee in accordance with policies detailed in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult female wild-type (WT) (C57B/6), KIWT (B6;129P2-Esr1<tm1Lja>), EAAE (B6;129P2-Esr1<tm2.1Gsc>, WTKO (B6.129-Esr1<tm4.2Ksk>), and KIKO (B6;129-Esr1<tm4.2Ksk>Esr1<tm1Lja>) mice were produced in our breeding colony at Charles River Laboratories as described previously (6) and shipped to National Institute of Environmental Health Sciences. For the KIWT uterine enlargement study, tissue from 7- to 18-week-old female mice was collected and weighed. For hormone response studies, adult female mice were ovariectomized and then were rested for 10 to 14 days to allow endogenous ovarian hormones to clear. WTKO female mice were used as controls for the KIKO mice because they express ERα from 1 allele, as do the KIKO mice, and are called “WT” in the text to simplify the descriptions. Animals were treated as indicated, and uterine tissue was collected and snap-frozen in liquid nitrogen for RNA, protein, or chromatin isolation. RNA isolation and RT-PCR, microarray, and ChIP-PCR were performed as described previously, including most primer sequences (6, 12, 14) with the addition of those listed in Supplemental Table 1. Microarray data were deposited at Gene Expression Omnibus (GSE56423). For ChIP-PCR, a primer set was selected in the aquaporin 5 (Aqp5) transcript region with no ER enrichment as a negative control. For Western blotting, whole-tissue homogenates were made as described previously (7), and 10 μg of protein was loaded on each lane of a 10% NuPAGE gel (Invitrogen). Proteins were transferred to nitrocellulose using iBlot (Invitrogen) and probed for ERα (1:1000, sc-542; Santa Cruz Biotechnology) and β-tubulin (1:5000, sc-9104; Santa Cruz Biotechnology) in 5% Blotto (Santa Cruz Biotechnology) in Tris-buffered saline-Tween 20 buffer and detected using infrared fluorescence–labeled (800 nm) antirabbit antibody (1:20 000; LI-COR) and scanned and quantified using an Odyssey Fc instrument and Image Studio software (LI-COR).
ChIP-seq
For ERα ChIP-seq, E2 (250 ng in 100 μL of saline; Research Plus) or 100 μL of saline was injected intraperitoneally in each of 5 mice/treatment group. One hour after injection, mice were killed. Then uterine tissue was collected, snap-frozen in liquid nitrogen, pooled, and shipped to Active Motif for FactorPath ChIP-seq. Each dataset was obtained from a single pool of uterine tissue (n = 5 mice). Details of the ChIP-seq using anti-ERα antibody (sc-542; Santa Cruz Biotechnology) and alignment to the reference genome were as described previously (12). Mapped reads were deduplicated using MarkDuplicates.jar from the Picard tools package (v1.62; http://picard.sourceforge.net). The Partek Genomics Suite (v6.11.1101; Partek, Inc) was used to make peak calls for ERα with default parameters, followed by further filtering to exclude peaks with a Mann-Whitney P value of >.01. Partek Genomics Suite was also used for empirical estimation of the average fragment size per ChIP-seq library. To enable comparison to PR, the PR ChIP-seq dataset (13, GEO GSE 34927) was reanalyzed using the same deduplication, Partek peak calling, and peak filtering steps described above. Characteristics of the ER and PR ChIP-seq data are summarized in Supplemental Table 2. Data were deposited in Gene Expression Omnibus (WT ER GSE36455, KIKO ER GSE56466).
Motif enrichment analysis
Sequences of peaks unique to WT or KIKO ERα or peaks in common were normalized to 300 nucleotides, evaluated with the GADEM tool (15) for motifs, and then compared for relative enrichment.
Reporter assays
Candidate motifs identified in ChIP-seq peaks were synthesized as oligonucleotides (Sigma Genosys) (sequences in Supplemental Table 1), annealed, phosphorylated with T4 polynucleotide kinase (New England Biolabs) and then inserted into the EcoRV site of the pGL4.23 luciferase (luc) vector (Promega), which is engineered for minimal basal activity, to test candidate response elements. Clones were sequenced to verify the presence of inserted response elements, and clones containing 2 copies of Fkbp5 hormone response element (HRE), Fkbp5 nonresponsive element (NRE), Indian hedgehog (Ihh) HRE, or Epstein-Barr virus nuclear antigen (EBNA) or 3 copies of Igf1 ERE or Igf1 NRE were used in experiments. NRE was made by changing the nucleotides in the Fkbp5 HRE and Igf1 ERE motifs (Supplemental Table 1 and Figure 1B) to match an NRE motif with a motif sequence intermediate between ERE and HRE, which was previously shown to lack activity through ER or glucocorticoid receptor (GR) (16, 17). EAAE ERα was made by amplifying the SmaI fragment of mouse ERα cDNA in pGEM-3Zf with site-directed mutagenesis primers (Supplemental Table 1) to recreate the originally described point mutations (Figure 1A) (9). The NotI-XbaI fragment of mouse ERα containing the EAAE mutations was used to replace the same fragment of the WT mouse ERα cDNA in pCDNA3. The pAP1-luc plasmid was purchased from Stratagene. HeLa cells were transfected as described previously (18) with 0.1 μg of reporter constructs and 0.1 μg of pCDNA3 expression vectors containing WT, KIKO, or EAAE mouse ERα or human PRB, and 0.1 μg of pRL-TK control vector. After hormone treatment (0.1% ethanol [vehicle], 10 nM E2, 100 nM P4 [Sigma-Aldrich], or 100 nM ICI 182,780 [Tocris Bioscience]), cell lysates were assayed for luc as described previously (18) and normalized to a Renilla luc vector transfection control. Values were plotted relative to pCDNA3 expression vector plus empty PGL4.23 luc vector transfection with vehicle treatment, except for the AP1-luc assay, which was plotted relative to empty pCDNA3 plus AP1-luc with vehicle treatment.
DNA-binding assay
Ovariectomized WT, KIKO, or EAAE mice were injected intraperitoneally with 250 ng of E2 in saline vehicle, and uterine tissue was collected and snap-frozen in liquid nitrogen 1 hour after injections. Pools of 3 to 5 frozen uteri were pulverized, and nuclear extracts (NEs) were prepared (N-PER Kit; Thermo Scientific), as described in the manufacturer's protocol by homogenization in 200 μL of CERI buffer. The ERα level in each NE was evaluated using Western blotting, as described above so that the amounts of ERα protein used in the binding assays of WT, KIKO, or EAAE samples were the same. NEs were used with the NoShift DNA-binding assay kit according to the protocol provided (Millipore Corp). Biotinylated ERE or HRE probes (Sigma Genosys) (sequences in Supplemental Table 1) were mixed with 2 μL of NEs in the presence of 150 nM E2 and P4 and allowed to bind for 60 minutes on ice. Binding reactions were transferred to avidin-coated wells and incubated at 37°C for 1 hour. Bound ER-biotinylated DNA complexes were detected with anti ERα antibody (1:300) followed by horseradish peroxidase-labeled antirabbit IgG (Cell Signaling Technology), developed with tetramethylbenzidine substrate, and stopped with 1 N HCl. Wells were scanned to read A450 (SpectraMax Plus 384; Molecular Devices). Specificity of binding was evaluated by including 10- to 20-fold excess unbiotinylated ERE or HRE DNA in the binding step to inhibit complex formation. In addition, excess unbiotinylated NRE, made by altering ERE or HRE, or EBNA, a motif for a different type of transcription factor, was added to some binding reactions because these should not interfere with complex formation.
Statistical analyses
For RT-PCR, luc reporter activity assays, and ChIP-PCR, data (means ± SEM) were analyzed using the statistical tools in GraphPad Prism 6. Treated groups within each genotype were compared with the vehicle group using 2-way ANOVA; further details are found in the figure legends.
Results
E2 induces DNA synthesis inhibitor Klf15 in KIKO uterus
Recently, 2 KLFs were implicated in E2 and P4 modulation of uterine proliferation; KLF4 is increased by E2 and promotes DNA replication, whereas KLF15 is increased by P4 and inhibits growth via regulation of Mcm2, which is important for the initiation of DNA synthesis (11). Because uterine epithelial proliferation is absent in the KIKO uterus, we reasoned that tethered ERα signaling might be insufficient to establish proper regulation of these KLFs; therefore, we evaluated their transcripts. E2 robustly increased the Klf4 transcript in WT uteri, but the increase was minimal in KIKO uteri (Figure 2A). As was previously reported in WT mice (11), the Klf15 transcript was increased by P4; however, KIKO uteri exhibit E2-mediated induction of Klf15 (Figure 2A), consistent with the lack of a KIKO uterine proliferative response.
Figure 2.

Altered hormonal regulation of uterine KLf4 and Klf15. A, RT-PCR of uterine RNA after treatment of WT or KIKO mice for 6 hours with vehicle (V), E2, or P4. Statistical analyses included 2-way ANOVA, multiple comparisons of means to results for vehicle treatment, and Bonferroni multiple test correction. ***, P < .001; ****, P < .0001. B, ChIP-seq datasets near Klf15 displayed in University of California Santa Cruz (UCSC) Genome Browser showing WT and KIKO ERα (blue) and PR (red) ChIP-seq tracks from mice treated for 1 hour with vehicle, E2, or P4 and input tracks (blue). The arrow shows the HRE motif containing the peak, and the motif sequence that was inserted in pGL4.23 plasmid and tested in the in vitro DNA-binding assay is shown. The HRE motif is indicated by bold text with consensus-matching nucleotides underlined. C, ChIP-PCR for enrichment of ERα at HRE in the Klf15 gene from WT and KIKO uterine samples 1 hour after vehicle or E2 injection. Statistical analyses included 2-way ANOVA, multiple comparisons of means to results for vehicle treatment, and Bonferroni multiple test correction. **, P < .01; ****, P < .0001.
Enriched transcription factor motifs within ERα ChIP-Seq peak DNA sequences suggests KIKO DNA-binding mutant binds HRE motifs
To evaluate the mechanism by which the DBD mutant KIKO ERα regulates this normally P4-responsive transcript, KIKO ERα binding to uterine chromatin was evaluated by ChIP-seq and compared with our previously reported datasets from WT uteri. A total of 5605 (30%) of 18 990 KIKO E2 peaks overlapped with WT E2 peaks (Table 1 and Figure 3A). These findings are consistent with the unique E2-induced transcriptional responses of the KIKO uterus (6). To discover transcription factors involved in tethering the KIKO ERα to uterine chromatin, WT and KIKO ERα ChIP-seq datasets were evaluated for DNA-binding protein motif enrichment using the GADEM tool (15). For this analysis, the WT and KIKO 1-hour E2 peaks were divided into three categories: those unique to the WT set (15 163 WT selective, DNA-binding), those unique to the KIKO set (13 385 KIKO selective), and those common between WT and KIKO (5605 overlapping, presumably tethered ER binding; referred to as WT+KIKO overlap hereafter). WT selective, KIKO selective, or WT+KIKO overlap sequences were then evaluated for transcription factor–binding motifs and motif enrichment relative to one another. As anticipated, GGTCA/ERE motifs were significantly enriched in WT selective peaks in comparison to the WT+KIKO overlap (Supplemental Table 3). Similarly, motifs for numerous known mediators of tethered ERα interaction, including Sp1 and AP2, were enriched in WT+KIKO overlap vs WT selective peaks (Supplemental Table 4). The enriched motifs represent potential tethering mediators used in vivo by ERα in uterine tissue. Remarkably, analysis of motifs enriched at sites of KIKO ERα binding revealed that androgen receptor (AR), PR, and GR motifs (androgen response element, progesterone response element, and glucocorticoid response element) dominate those enriched in the KIKO selective vs WT+KIKO overlapping peaks (Table 2). The DNA motifs for these 3 receptors share homology and are often called HREs. To evaluate the possibility that KIKO ERα was enriched at PR-binding sites, mouse uterine ChIP-seq profiles from WT PR (13), KIKO ERα, and WT ERα were compared (Figure 3A and Table 1). A total of 2095 (17%) WT PR peaks colocalize with WT ERα peaks, whereas 4051 (32%) WT PR peaks overlap with KIKO ERα binding peaks (Table 1 and Figure 3A), indicating more overlap between WT PR and KIKO ERα than between WT PR and WT ERα. Based on these observations, we hypothesized that the KIKO ERα DBD mutation altered DNA motif specificity. Supporting this hypothesis, the HRE binding motif computed from the PR binding peaks is similar to one computed from the KIKO selective peaks (Figure 3B). To explore whether KIKO ERα induces the Klf15 transcript via an HRE motif, the ChIP-seq data were examined and revealed PR and KIKO ERα enrichment in the second intron that contains a consensus HRE (Figure 2B). ChIP-PCR confirmed E2-dependent enrichment of KIKO ERα at this HRE (Figure 2C).
Table 1.
Comparison of WT ERα, KIKO ERα. and PR ChIP-seq called peaks
| Sample “X” | No. of Peaks in Sample X | No of Peaks in Sample Y (%) |
|||||
|---|---|---|---|---|---|---|---|
| 1-h V WT ERα | 1-h E2 WT ERα | 1-h V KIKO ERα | 1-h E2 KIKO ERα | 1-h V WT PR | 1-h P4 WT PR | ||
| 1-h V WT ERα | 5496 | − | 3992 (72.6) | 1125 (20.5) | 2596 (47.2) | 1169 (21.3) | 1133 (20.6) |
| 1-h E2 WT ERα | 20792 | 3968 (19.1) | − | 1352 (6.5) | 5629 (27.1) | 1549 (7.4) | 2090 (10.1) |
| 1-h V KIKO ERα | 2908 | 1124 (38.7) | 1360 (46.8) | − | 2037 (70) | 723 (24.9) | 838 (28.8) |
| 1-h E2 KIKO ERα | 18990 | 2580 (13.6) | 5605 (29.5) | 2025 (10.7) | − | 1938 (10.2) | 4021 (21.2) |
| 1-h V WT PR | 3541 | 1169 (33.0) | 1558 (44.0) | 720 (20.3) | 1947 (55.0) | − | 2133 (60.2) |
| 1-h P4 WT PR | 12590 | 1136 (9.0) | 2095 (16.6) | 835 (6.6) | 4051 (32.2) | 2132 (16.9) | − |
| 1-h E2 WT ERα selective | 15163 | − | − | − | − | − | 586 (4.0) |
| 1-h E2 KIKOERα selective | 13385 | − | − | − | − | − | 2468 (18.0) |
| 1-h E2 WT ERα +1-h E2 KIKO ERα | 5605 | − | − | − | − | − | 1388 (25.0) |
Comparing WT ERα, KIKO ERα, and WT PR ChIP-seq peaks reveals KIKO ERα selective enriched sites and overlap with PR enriched regions. Peaks (% of total peaks) from different samples that share one or more genomic bases in common are considered overlapping peaks. Of the peaks called in sample X, how many (what % of X) overlap with a peak called in sample Y? Note that the reciprocal comparisons should be very similar, but perhaps not identical because 1 peak from sample X might overlap with 2 peaks in sample Y, which is counted as 1 for X vs Y, but as 2 for Y vs X. Most overlapping peaks share the majority of their genomic range. One-hour E2 WT or KIKO ERα selective peaks are those that do not overlap with each other (total peaks minus overlapping peaks). Comparisons in bold type are discussed in the text.
Figure 3.

KIKO ERα selective computed motif is HRE-like, and in vitro assays demonstrate KIKO binding to the Fkbp5 HRE-motif. A, Called peaks in each dataset (1-hour E2 WT ERα [blue]: 20 792 peaks, 1-hour E2 KIKO ERα [yellow]: 18 990 peaks, and 1-hour P4 WT PR [red]: 12 590 peaks) were compared and are considered overlapping if the coordinate ranges of the peak calls share one or more genomic positions (Table 1). Most overlapping peaks share most their genomic range. Numbers indicate numbers of peaks in each region of the Venn diagram. B, Progesterone response element (PRE) motifs computed from KIKO ERα selective peaks or WT PR ChIP-seq peaks. A total of 12 590 WT P4 PR peaks were scanned with GADEM analysis seeded with the TRANSFAC PRE motif model (WT PR: 12 590 peaks scanned, 4577 PRE sites [37.8% peaks]; KIKO ERα: 13 385 peaks scanned; 4000 PRE sites [30% of peaks]). C, RT-PCR. Analysis and statistical analysis were performed as described for Figure 2A. D, ChIP-seq datasets near Fkbp5 displayed in the UCSC Genome Browser as described for Figure 2B. E, Luciferase reporter gene activity. Fkbp5 HRE-luc with empty pCDNA3, WT, KIKO, or EAAE ERα or PR, with no hormone (vehicle [V]), E2 (10 nM), or P4 (100 nM). Values were calculated relative to those for vehicle treatment of empty expression plasmid and empty reporter plasmid transfected cells. Fold change relative to vehicle treatment is indicated above the bars. Statistical analysis included 2-way ANOVA, multiple comparisons of means to results for vehicle treatment, and Bonferroni multiple test correction. ***, P < .001; **** P < .0001. F, ChIP-PCR. ChIP-PCR analysis and statistical analysis were performed as described for Figure 2C. G, In vitro DNA-binding assay. Biotinylated Fkbp5 HRE binding with nuclear protein extracts from WT, KIKO or EAAE uteri. ERα-DNA complexes were detected as described in Materials and Methods. Nonbiotinylated (unlabeled) DNA (positive control, Fkbp5 HRE; negative control, Fkbp5 NRE) (sequences in Supplemental Table 1) was added to binding reactions at 10× higher levels than the biotinylated probe to compete for ERα binding and demonstrate specificity. Probe, NE sample contained no nuclear extract.
Table 2.
HRE Motifs (ARE, GRE, and PRE) Dominate Enriched Motifs in 1-Hour E2 KIKO ERα Selective vs 1-Hour E2 WT ERα + 1-Hour E2 KIKO ERα Overlapping Peaks
| Model Name | Enrichment P Value |
|---|---|
| AR_01 | 1.34E–105 |
| AR_02 | 1.35E–100 |
| AR_04 | 1.23E–95 |
| AR_03 | 3.21E–67 |
| GR_Q6 | 3.47E–55 |
| PR_02 | 1.52E–52 |
| GR_01 | 1.10E–39 |
| PR_01 | 2.73E–33 |
| AR_Q2 | 2.37E–23 |
| GR_Q6_01 | 5.15E–16 |
| PR_Q2 | 9.96E–10 |
| GRE_C | 8.67E–09 |
| AREB6_02 | 2.14E–08 |
| HSF1_01 | 7.39E–08 |
| SOX4_01 | 1.13E–07 |
| SOX9_B1 | 1.86E–07 |
| DELTAEF1_01 | 6.20E–07 |
| AREB6_01 | 9.95E–07 |
Abbreviations: ARE, androgen response element; GRE, glucocorticoid response element; PRE, progesterone response element.
In vitro HRE reporter and DNA-binding assays demonstrate interaction between KIKO ERα and HRE motifs
To further test our hypothesis that the KIKO ERα can bind HRE motifs, we developed in vitro assays by evaluating typically P4-responsive transcripts that are induced by E2 in KIKO uteri. FK506 binding protein 5 (Fkbp5) is a large-molecular-weight immunophilin and is a well-known target of PR or GR (19). RT-PCR analysis of uterine RNA shows that Fkbp5 is significantly increased by P4 in the WT uterus (Figure 3C). However, in the KIKO uterus, the transcript is robustly increased by E2. Examination of the WT and KIKO ERα and PR ChIP-seq profiles near the Fkbp5 transcript revealed a region located in an intron with E2-dependent WT ERα recruitment, P4-independent PR binding, and E2-dependent KIKO ERα-binding (Figure 3D). An HRE motif was identified in the sequence within this peak (Supplemental Table 1) and inserted into a pGL4.23 luc reporter vector to evaluate the ability of WT, ERα, both KIKO and EAAE ERα (DBD mutants) and PR to bind to the HRE and mediate hormone-dependent transcription of the luc reporter gene. We reasoned that an ability of KIKO ERα to bind HRE motifs would be reflected in an estrogen-dependent increase in luc activity. WT ERα, KIKO ERα, and PR all induced hormone-dependent luc activity, whereas EAAE ERα was inactive (Figure 3E). ERα ChIP-PCR revealed E2-dependent recruitment of KIKO but not WT ERα to this site in uterine tissue (Figure 3F). An in vitro DNA-binding assay revealed KIKO ERα but not WT or EAAE ERα interaction with this Fkbp5 HRE (Figure 3G).
Ihh is a member of the hedgehog signaling family, and its proper regulation is essential to establish pregnancy (10). Ihh is increased in WT uteri after P4 injection (Figure 4A). This transcript is increased by P4 or E2 in the KIKO uterus. Peaks of hormone-dependent PR or KIKO ERα binding enrichment are seen 5′ of Ihh, in an intron of Nhej1, a region that was previously described as a potential mediator of PR induction of Ihh (20) (Supplemental Figure 1A). Some WT ERα recruitment also occurs in this region. An HRE motif was identified in the sequences in the peak (Supplemental Table 1). This motif mediated E2-dependent KIKO ERα luc activity or P4-dependent PR activity (Figure 4B), reflecting interaction with the HRE sequence. WT and EAAE ERα were unable to induce the Ihh HRE luc reporter. ChIP-PCR confirmed recruitment of KIKO ERα to this site in uterine tissue (Figure 4C). An in vitro DNA-binding assay revealed KIKO ERα but not WT or EAAE ERα interaction with this Ihh HRE (Figure 4D).
Figure 4.

In vitro assays demonstrate KIKO binding to the Ihh HRE motif. A, RT-PCR. Analysis and statistical analysis were performed as described for Figure 2A. B, Luciferase reporter gene activity. Ihh HRE-luc analysis and statistical analyses were performed as described for Figure 3E. C, ChIP-PCR. ChIP-PCR analysis and statistical analyses were performed as described for Figure 2C. D, In vitro DNA-binding assay. Biotinylated Ihh HRE binding to WT, KIKO, or EAAE ERα-DNA and statistical analysis were performed as described for Figure 3G. Nonbiotinylated (unlabeled) DNA positive control, Ihh HRE; negative control, Fkbp5 NRE.
Our previous analysis demonstrated that the Igf1 transcript is increased by E2 in the WT uterus but not in the KIKO uterus (Figure 5A) (7). In addition, using ChIP-seq we have identified 5 sites of ERα enrichment 50 kb 5′ of the Igf1 transcription start site (12). Some KIKO ERα enrichment occurs at 2 of the 5 sites in this region (Supplemental Figure 1B). To demonstrate the validity of our in vitro assay analysis strategy, we tested an ERE motif that was identified in one of the sites that lacked KIKO ERα enrichment (Supplemental Table 1). WT ERα robustly induced this ERE-luc reporter, but KIKO ERα induced minimal activity, and EAAE ERα was inactive (Figure 5B). ChIP-PCR revealed selective recruitment of WT ERα to this site in uterine tissue (Figure 5C). Although KIKO ERα showed little Igf1 ERE-luc reporter activity, the in vitro DNA-binding assay revealed both WT ERα and KIKO ERα interaction with this ERE (Figure 5D), whereas EAAE ERα lacked interaction. KIKO ERα binding to the ERE is also apparent in the increased basal ERE-luc activity compared with that for EBNA motif-luc (compare Figure 5B and Supplemental Figure 2), a motif that does not bind KIKO ERα, as including 10-fold excess unlabeled EBNA motif DNA does not prevent KIKO ERα binding to any of the biotinylated probes (Supplemental Figure 3).
Figure 5.

In vitro assays demonstrate WT binding to the Igf1 ERE motif. A, RT-PCR. Analysis and statistical analyses were performed as described for Figure 2A. B, Luciferase reporter gene activity. Igf1 ERE-luc analysis and statistical analyses were performed as described for Figure 3E. C, ChIP-PCR. ChIP-PCR analysis and statistical analyses were performed as described for Figure 2C. D, In vitro DNA-binding assay. Biotinylated Igf1 ERE-binding to WT, KIKO, or EAAE ERα-DNA and statistical analysis were performed as described for Figure 3G. Nonbiotinylated (unlabeled) DNA positive control, Igf1 ERE; negative control, Igf1 NRE.
Phenotypic differences between KIKO and EAAE DNA-binding-deficient mouse models
The EAAE ERα DBD mutant mouse has phenotypes similar to those of ERα-null animals (9). Although KIKO uteri are similar in size to WT uteri, the EAAE uteri are hypoplastic, similar to ERα-null uteri (9) (Figure 6A); however, neither DBD model exhibits uterine epithelial proliferation (Figure 6A). Female mice that are heterozygous for the KIKO allele (WTKI) develop enlarged uterine tissue after puberty (Supplemental Table 5) and are infertile (4); however, females heterozygous for the EAAE ERα mutation are fertile (9), indicating a difference in the biological functions mediated by these two DBD mutant estrogen receptors. Unlike the KIKO ERα, the EAAE ERα completely lacks binding to both ERE and HRE motifs (Figures 3G, 4D, and 5D) and is unable to induce ERE-luc or HRE-luc reporter constructs (Figures 3E, 4B, and 5B and Supplemental Figure 2). EAAE ERα induces an AP1-luc reporter comparably with WT or KIKO ERα (Figure 6B). To examine potentially tethered functions mediated by EAAE ERα in uterine tissue, we evaluated genes regulated by E2 after 2 hours by microarray and RT-PCR (Figure 6C and Supplemental Figure 4A). Although the EAAE ERα protein is expressed in the tissue (Supplemental Figure 4B), most of the transcriptional responses are absent (Figure 6C and Supplemental Figure 4C), including both induced and repressed transcripts. This result demonstrates that ERα DNA-binding is required for both up- and down-regulation of transcription in the uterus. Of the 3163 probes shown clustered in Figure 6C, only 53 probes representing 44 genes (1.6%) were E2 regulated in the EAAE uteri (>2-fold change, false discovery rate < 0.05) (Supplemental Figure 4C). Of the 53 differentially expressed EAAE transcripts, 68% were increased, 32% were decreased, and, overall, only 13 of the 53 probes were regulated in the same direction in WT and EAAE uteri, whereas 29 probes were regulated in opposite directions, and 13 were not differentially expressed in the WT. Significantly, transcripts that were previously observed as E2 responsive in the KIKO uterus (6) are not responsive in the EAAE uterus (Supplemental Figure 4, A and D). Overall, these in vitro and in vivo findings suggest that ERα tethering in uterine tissue is not sufficient to mediate hormonal responsiveness including most transcriptional responses, because the EAAE mouse model lacks uterine estrogen responses.
Figure 6.

Evaluation of DBD mutant ERα activities in vivo and in vitro. A, Uterine cross sections from ovariectomized WT, KIKO, EAAE, or ERα-null mice that were treated for 24 hours with E2. Sections were evaluated for the proliferative marker, Ki67 (brown). Scale bar corresponds to 0.1 mm. B, WT, KIKO, and EAAE ERα mediate AP1-luc responses similarly. WT, KIKO, and EAAE ERα with AP1-luc. Fold changes relative to vehicle (V) treatment are indicated above each bar. E2, 10 nM; ICI 182,780 (ICI), 100 nM. Statistical analysis included 2-way ANOVA, multiple comparisons of means vs results for vehicle treatment, and Bonferroni multiple test correction. ***, P < .001; ****, P < .0001. C, Microarray profile. A hierarchical cluster of normalized ratios (E2 2h/V) of WT and EAAE uterine RNA is shown. Uterine RNA from WT or EAAE ovariectomized mice collected 2 hours after vehicle (V) or E2 injection (n = 3 each condition) was compared by microarray. The cluster was created by combining replicates and building ratios (E2 treated/vehicle treated) and filtering for probes that met the following criteria: absolute fold change of > 2 in at least 1 genotype, probe intensity of >100 in at least 1 condition, and false discovery rate < 0.05. This resulted in 3162 probes. Red indicates induced transcripts; green signifies repression.
Discussion
Altered DNA-binding specificity rather than DNA-binding deficiency
Nuclear receptors can be categorized according to their preferences for DNA motif sequence binding. ER, vitamin D receptor and thyroid receptor prefer motifs with GGTCA arms, whereas AR, GR, and PR prefer GAACA (21, 22). Analysis of the sequences in KIKO ERα selective peaks revealed a preference for motifs with the GAACA sequence, normally associated with AR, PR, or GR binding. The mutations in KIKO ERα (E207A and G208A) are located at the base of the first zinc finger (Figure 1A). Glutamate and glycine (EG) are found in this P-box region of all other nuclear receptors that bind the GGTCA motif, including thyroid receptor and vitamin D receptor (2, 22). The amino acids in the “knuckle” of the first zinc finger in the P-box of nuclear receptors that bind to the GAACA motif (GR, PR, and AR) are glycine and serine (GS) (22). Examination of crystal structures of DBDs of ERα, PR, and GR bound to their cognate DNA motifs (ERE, PRE, and GRE) as a basis to predict the effect of the amino acid substitutions in the KIKO ERα suggests that E207 may contribute to the specificity of ERα for the ERE over HRE sequences by steric exclusion of the dT:dA at position 2 nucleotide present in the HRE (Figure 1C). Modeling of a dT:dA at ERE position 2 places the dT C7 methyl group 2.3 Å from the side chain of E207, generating a significant clash (Figure 1C). Removal of the side chain for E207, as in the KIKO ERα E207A mutation, would remove this steric clash with HRE or NRE motifs. Note that the preferred motif for KIKO ERα that we determined in this study indicates relaxed preference at position 2 of the motif (Figures 1B and 3B). In contrast, when the structure of the EAAE mutant ERα is examined, an H bond normally formed between K210 of WT ERα and G/C at position 1 in the ERE is lost; thus, interaction with ERE is not favored. Moreover, interaction between the EAAE ERα and GAACA HRE motifs is excluded, because this mutant retains E207, maintaining the steric conflict with A:T at position 2. Therefore, these modeling observations suggest that the KIKO ERα interaction with HRE is possible, but that interaction between EAAE ERα and HRE is not. In concordance with these observations, mutation of the human ERα K210 (equivalent to mouse K214) decreases ERE binding to a degree similar to that of mutating human ERα E203A (equivalent to mouse E207), but does not lead to interaction with A:T at position 2 (23). In addition, a single mutation of human ERα E203 to A remained permissive for interaction with ERE (23), consistent with the KIKO ERα ERE binding observed in the DNA-binding assay (Figures 5D and Supplemental Figure 3). Similar to what we have reported using the KIKO mouse uterus (6), transcriptional profiling of E2-treated cells with ERα P-box mutations that “switch” the EG to GS revealed regulation of transcripts that were unique to the DBD mutant ERα (24), and the ERα ChIP-seq revealed ERα-binding sites that were unique to the DBD mutant ERα and enriched for HRE motifs (24).
Recently, a role for tethering through AP1 has been proposed in an assisted loading mechanism whereby activation of GR leads to increased chromatin accessibility for ERα at a small fraction of ERα-binding sites in mammary epithelial cells (25). The KIKO DBD mutant ERα was used to demonstrate that ERE-binding activity was not needed for the assisted loading (25). The sites that mediate the assisted loading were shown to be enriched for the HRE motif (25); thus, the HRE binding of the KIKO DBD mutant ERα that we demonstrate in our study may be directing the binding. AP1 binding is enriched and required at these assisted loading regions (25), but its role in the mechanism might need to be further clarified.
KIKO ERα DNA-binding motif specificity
Our in vitro DNA-binding analysis (Figures 3G, 4D, and 5D and Supplemental Figure 3) indicated that the KIKO ERα bound both ERE and HRE sequences, and, in addition, the negative control unlabeled competitor (NRE; Supplemental Table 1 and Figure 1B) was effective in competing for interaction with KIKO ERα. The NRE, with a sequence intermediate between ERE and HRE (Figure 1B), was used based on work indicating that this motif did not bind ERα (17) or mediate ER or GR transcription (16). Therefore, the ability of the NRE to inhibit KIKO ERα binding to the ERE or HRE suggests that the DNA motif preference of KIKO ERα is quite relaxed and includes sequences that are typically unable to bind steroid receptors. Specificity was demonstrated using the unrelated EBNA motif, which did not bind KIKO ERα (Supplemental Figure 3). In the reporter assay study (Figures 3E, 4B, and 5B and Supplemental Figure 2), KIKO ERα was active via HRE-luc and NRE-luc but not ERE-luc or EBNA-luc reporters. However, KIKO ERα increased basal activity of the Igf1 ERE-luc reporter (Figure 5B), suggesting that some interaction is occurring, but that E2 is unable to induce transcription via the KIKO ERα on the ERE. Overall, by comparing the motifs used to test binding to KIKO ERα and its ability to induce luc reporter activity, the common nucleotides indicate a “consensus” motif of GnnCAnnnTGnnC (Figure 1B and Supplemental Table 6).
WT ERα showed a strict preference for ERE in the in vitro DNA-binding assay (Figure 5D and Supplemental Table 6); nonetheless, WT ERα mediated E2-dependent activity via the Fkbp5 HRE-luc (Figure 3E) and the Fkbp5 NRE-luc (Supplemental Figure 2) but not the Ihh HRE-luc (Figure 4B). The activity may be driven by DNA sequences flanking the Fkbp5 HRE and NRE, because comparable WT ERα transcriptional activity is seen with Fkbp5 HRE-luc (Figure 3B), and Fkbp5 NRE-luc (Supplemental Figure 2), but none occurs via Igf1 NRE-luc (Supplemental Figure 2). Although there were indications in these in vitro assays for a WT ERα interaction with Fkbp5 HRE-luc, this interaction was not apparent in the conditions found in the uterine tissue, because WT ERα was not enriched by ChIP-PCR (Figure 3F). The HRE motif tested in the Fkbp5 construct was previously identified and similarly demonstrated by reporter assay and gel shift to interact with PR and GR (19).
Potential biological significance
AR, GR, and PR are all present in the mouse uterus, but the biological role of PR is most closely linked to that of ERα, allowing responses to cycling ovarian hormones, E2 and P4. ERα and PR exhibit a well-characterized balance in activities in terms of cycle stage (periovulatory vs early pregnancy) and uterine tissue compartments (stroma vs epithelia). PR is restricted to the epithelia until the preovulatory increase in E2 leads to decreased epithelial and increased stromal expression of PR. ERα is found in all cell types of the uterine tissue, and proper levels and patterns of PR help to maintain an appropriate estrogen response (10). We speculate that rather than a model for tethered-selective ERα signaling, the KIKO mouse exhibits estrogen regulation of target genes normally controlled by PR and inappropriate induction of PR target genes in the stromal compartment. Perhaps this underlies the inability of E2 to cause KIKO uterine proliferation (6, 7), because PR normally inhibits epithelial proliferation to allow embryo attachment and implantation (26).
As an example, Ihh, which has been shown to be involved in the PR-mediated inhibition of epithelial proliferation necessary for embryo implantation (27), becomes E2 inducible in the KIKO uterus. The uterine enlargement reported for the KIWT heterozygotes (Supplemental Table 5 and Ref. 4) is similar to observed uterine overgrowth in a model with constitutive activation of the IHH transducer, smoothened (SmoM2+) (28). We hypothesize that estrogen might be increasing IHH via KIKO ERα HRE interaction in a setting that maintains ERα ERE-mediated responses through the WT allele. Consistent with our hypothesis, KIWT uterine enlargement develops with the onset of puberty (Supplemental Table 5). When estrogen elevates Ihh/SMO activation in the KIKO uterus, ERα signaling via ERE is impaired. Conversely, when estrogen elevates Ihh in KIWT uterine tissue, resulting in SMO activation, in a milieu where WT ERα signaling is intact, enlargement develops. This difference emphasizes an important role for ERE-mediated responses in the overgrowth. Similarly, normally P4, but not E2, increases epithelial KLF15, which then inhibits expression of Mcm2, thereby preventing epithelial proliferation (11). However, KIKO ERα binds a chromatin site within the Klf15 transcript that contains a consensus HRE, and Klf15 is increased by E2 (Figure 2), which may prevent the KIKO uterine proliferation.
More severe mutation of the DBD was used in the EAAE ERα and leads to an ERα null-like female reproductive tract phenotype and lack of estrogen induced uterine transcriptional regulation (9) (Figure 6A and Supplemental Figure 4). This indicates that ERα deficient in DNA-binding is not sufficient to mediate biological or transcriptional responses in uterine tissue as was reported for the liver of the EAAE mice (9), which lacked transcriptional responses as well. The implications of this finding in the larger context indicate a need to reexamine ER tethering in vivo. This mechanism was initially proposed to account for the effects of ERα on AP1-, SP-, or nuclear-κB-mediated responses in studies conducted in vitro with reporter plasmids that lack the complexity of chromatin that ERα and these other transcription factors normally encounter. Clearly, AP1 activity is affected by EAAE ERα in vitro (Figure 6B); however, in uterine tissues, ERα-ERE binding might enable interactions between AP1 and ERα by altering chromatin structure. Thus, discovering whether our findings indicate that ERα tethering to non-ERE motifs requires ERα-ERE binding to permit the necessary ERα interactions with transcription factors or whether tethering occurs in vivo but lacks physiological significance will require further study. Ultimately, future ERα ChIP-seq analysis of EAAE tissues will indicate how loss of DNA-binding impacts ERα interaction with chromatin. Alternatively, the in vitro effects of ERα on AP1-luc reporter activity could reflect an artifact such as sequestering of transcription factors or displacement of AP1.
Some E2 responses, such as vascular dilation, are initiated from membrane-localized ERα (29), and because membrane localization results from palmitoylation of ERα C451, the EAAE ERα should retain these responses if they are independent from DNA-binding. When C451 was mutated to prevent palmitoylation, vasodilation was disrupted, whereas uterine proliferative and transcriptional responses were largely unaffected, indicating that ER membrane signaling is not the primary mechanism in operation in the uterus but greatly affects the vasculature (29). Thus, future studies will be performed to evaluate endpoints in EAAE mice to explore whether DNA-binding affects membrane ERα-mediated responses.
Our unexpected findings have demonstrated a defect in a model used by numerous laboratories in efforts to dissect effects mediated by direct or indirect interactions between ERα and target genes. We have established that the KIKO model introduces an aberrant mode of signaling to the tissues that has complicated interpretation of findings regarding mechanistic details of ERα signaling in reproductive, neuroendocrine, metabolic, skeletal, breast cancer and circadian studies (Table 3). Our previous studies and those of other investigators should be revisited to consider the novel activity we have uncovered here. Future work will require utilization of a truly HRE and ERE DNA-binding-deficient model, such as the EAAE mouse, to indicate the biological importance of tethered ERα signaling in physiological systems.
Table 3.
Systems Studied Using the KIKO Mouse Model
Additional material
Supplementary data supplied by authors.
Acknowledgments
We appreciate Dr Yukitomo Arao for his advice in cloning the EAAE mutation. We thank Dr Grace Kissling for statistical analysis of KIWT uterine weights, and Drs Heather Franco and Harriet Kinyamu for critical reading of our study. We thank members of the National Institute of Environmental Health Sciences (NIEHS) histology and microarray cores for their expertise. We appreciate the surgical expertise and husbandry provided by the NIEHS Comparative Medicine Branch.
This work was supported in part by the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health (Projects Z0170065 [to K.S.K.] and Z0101765 [to L.L.]) and National Institutes of Health (Grant 20 R01HD042311 [to F.J.D.]). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ChIP-seq
- chromatin immunoprecipitation sequencing
- DBD
- DNA-binding domain
- E2
- estrogen
- EBNA
- Epstein-Barr virus nuclear antigen
- ER
- estrogen receptor
- ERE
- estrogen response element
- GR
- glucocorticoid receptor
- HRE
- hormone response element
- Ihh
- Indian hedgehog
- NE
- nuclear extract
- NRE
- nonresponsive element
- KIKO
- heterozygous
- KLF
- Kruppel-like factor
- KIWT
- DBD mutation
- P4
- progesterone
- luc
- luciferase
- Mcm2
- minichromosome maintenance 2
- P-box
- proximal box
- PR
- progesterone receptor
- WT
- wild-type
- WTKO
- ERα-null allele.
References
- 1. McDevitt MA, Glidewell-Kenney C, Jimenez MA, et al. New insights into the classical and non-classical actions of estrogen: evidence from estrogen receptor knock-out and knock-in mice. Mol Cell Endocrinol. 2008;290:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Danielsen M, Hinck L, Ringold GM. Two amino acids within the knuckle of the first zinc finger specify DNA response element activation by the glucocorticoid receptor. Cell. 1989;57:1131–1138. [DOI] [PubMed] [Google Scholar]
- 3. Mader S, Kumar V, de Verneuil H, Chambon P. Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid-responsive element. Nature. 1989;338:271–274. [DOI] [PubMed] [Google Scholar]
- 4. Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL. An estrogen receptor (ER) α deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol. 2002;16:2188–2201. [DOI] [PubMed] [Google Scholar]
- 5. O'Brien JE, Peterson TJ, Tong MH, et al. Estrogen-induced proliferation of uterine epithelial cells is independent of estrogen receptor α binding to classical estrogen response elements. J Biol Chem. 2006;281:26683–26692. [DOI] [PubMed] [Google Scholar]
- 6. Hewitt SC, O'Brien JE, Jameson JL, Kissling GE, Korach KS. Selective disruption of ERα DNA-binding activity alters uterine responsiveness to estradiol. Mol Endocrinol. 2009;23:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem. 2010;285:2676–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu L, Pollard JW. Estradiol-17β regulates mouse uterine epithelial cell proliferation through insulin-like growth factor 1 signaling. Proc Natl Acad Sci USA. 2007;104:15847–15851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahlbory-Dieker DL, Stride BD, Leder G, et al. DNA binding by estrogen receptor-alpha is essential for the transcriptional response to estrogen in the liver and the uterus. Mol Endocrinol. 2009;23:1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wetendorf M, DeMayo FJ. The progesterone receptor regulates implantation, decidualization, and glandular development via a complex paracrine signaling network. Mol Cell Endocrinol. 2012;357:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ray S, Pollard JW. KLF15 negatively regulates estrogen-induced epithelial cell proliferation by inhibition of DNA replication licensing. Proc Natl Acad Sci USA. 2012;109:E1334–E1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hewitt SC, Li L, Grimm SA, et al. Research resource: whole-genome estrogen receptor α binding in mouse uterine tissue revealed by ChIP-Seq. Mol Endocrinol. 2012;26:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rubel CA, Lanz RB, Kommagani R, Franco HL, Lydon JP, Demayo FJ. Research resource: genome-wide profiling of progesterone receptor binding in the mouse uterus. Mol Endocrinol. 2012;26:1428–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J. 2010;24:4660–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li LP. GADEM: a genetic algorithm guided formation of spaced dyads coupled with an EM algorithm for motif discovery. J Comput Biol. 2009;16:317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klock G, Strahle U, Schutz G. Oestrogen and glucocorticoid responsive elements are closely related but distinct. Nature. 1987;329:734–736. [DOI] [PubMed] [Google Scholar]
- 17. Curtis SW, Korach KS. Uterine estrogen receptor interaction with estrogen-responsive DNA sequences in vitro: effects of ligand binding on receptor-DNA complexes. Mol-Endocrinol. 1990;4:276–286. [DOI] [PubMed] [Google Scholar]
- 18. Winuthayanon W, Piyachaturawat P, Suksamrarn A, et al. Diarylheptanoid phytoestrogens isolated from the medicinal plant Curcuma comosa: biologic actions in vitro and in vivo indicate estrogen receptor-dependent mechanisms. Environ Health Perspect. 2009;117:1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hubler TR, Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones. 2004;9:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Franco HL, Rubel CA, Large MJ, et al. Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 2012;26:1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khorasanizadeh S, Rastinejad F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem Sci. 2001;26:384–390. [DOI] [PubMed] [Google Scholar]
- 22. Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486. [DOI] [PubMed] [Google Scholar]
- 23. Nguyen D, Bail M, Pesant G, et al. Rational design of an estrogen receptor mutant with altered DNA-binding specificity. Nucl Acids Res. 2007;35:3465–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stender JD, Kim K, Charn TH, et al. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010;30:3943–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miranda TB, Voss TC, Sung MH, et al. Reprogramming the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res. 2013;73:5130–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Conneely OM, Mulac-Jericevic B, DeMayo F, Lydon JP, O'Malley BW. Reproductive functions of progesterone receptors. Recent Progr Horm Res 2002;57:339–355. [DOI] [PubMed] [Google Scholar]
- 27. Franco HL, Lee KY, Broaddus RR, et al. Ablation of Indian hedgehog in the murine uterus results in decreased cell cycle progression, aberrant epidermal growth factor signaling, and increased estrogen signaling. Biol Reprod. 2010;82:783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franco HL, Lee KY, Rubel CA, et al. Constitutive activation of smoothened leads to female infertility and altered uterine differentiation in the mouse. Biol Reprod. 2010;82:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA. 2014;111:E283–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yao L, Cooke PS, Meling DD, Shanks RD, Jameson JL, Sherwood OD. The effect of relaxin on cell proliferation in mouse cervix requires estrogen receptor α binding to estrogen response elements in stromal cells. Endocrinology. 2010;151:2811–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McDevitt MA, Glidewell-Kenney C, Weiss J, Chambon P, Jameson JL, Levine JE. Estrogen response element-independent estrogen receptor (ER)-α signaling does not rescue sexual behavior but restores normal testosterone secretion in male ERα knockout mice. Endocrinology. 2007;148:5288–5294. [DOI] [PubMed] [Google Scholar]
- 32. Glidewell-Kenney C, Weiss J, Lee EJ, et al. ERE-independent ERα target genes differentially expressed in human breast tumors. Mol Cell Endocrinol. 2005;245:53–59. [DOI] [PubMed] [Google Scholar]
- 33. Glidewell-Kenney C, Weiss J, Hurley LA, Levine JE, Jameson JL. Estrogen receptor α signaling pathways differentially regulate gonadotropin subunit gene expression and serum follicle-stimulating hormone in the female mouse. Endocrinology. 2008;149:4168–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao Z, Park C, McDevitt MA, et al. p21-Activated kinase mediates rapid estradiol-negative feedback actions in the reproductive axis. Proc Natl Acad Sci USA. 2009;106:7221–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gottsch ML, Navarro VM, Zhao Z, et al. Regulation of Kiss1 and dynorphin gene expression in the murine brain by classical and nonclassical estrogen receptor pathways. J Neurosci. 2009;29:9390–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Christian CA, Glidewell-Kenney C, Jameson JL, Moenter SM. Classical estrogen receptor α signaling mediates negative and positive feedback on gonadotropin-releasing hormone neuron firing. Endocrinology. 2008;149:5328–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Glidewell-Kenney C, Hurley LA, Pfaff L, Weiss J, Levine JE, Jameson JL. Nonclassical estrogen receptor α signaling mediates negative feedback in the female mouse reproductive axis. Proc Natl Acad Sci USA. 2007;104:8173–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park CJ, Zhao Z, Glidewell-Kenney C, et al. Genetic rescue of nonclassical ERα signaling normalizes energy balance in obese ERα-null mutant mice. J Clin Invest. 2011;121:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong WP, Tiano JP, Liu S, et al. Extranuclear estrogen receptor-alpha stimulates NeuroD1 binding to the insulin promoter and favors insulin synthesis. Proc Natl Acad Sci USA. 2010;107:13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu S, Le May C, Wong WP, et al. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes. 2009;58:2292–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mamounis KJ, Yang JA, Yasrebi A, Roepke TA. Estrogen response element-independent signaling partially restores post-ovariectomy body weight gain but is not sufficient for 17beta-estradiol's control of energy homeostasis. Steroids. 2014;81:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Syed FA, Modder UI, Fraser DG, et al. Skeletal effects of estrogen are mediated by opposing actions of classical and nonclassical estrogen receptor pathways. J Bone Miner Res. 2005;20:1992–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Modder UI, Rudnik V, Liu G, Khosla S, Monroe DG. A DNA binding mutation in estrogen receptor-α leads to suppression of Wnt signaling via beta-catenin destabilization in osteoblasts. J Cell Biochem. 2012;113:2248–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Syed FA, Fraser DG, Spelsberg TC, et al. Effects of loss of classical estrogen response element signaling on bone in male mice. Endocrinology. 2007;148:1902–1910. [DOI] [PubMed] [Google Scholar]
- 45. Syed FA, Fraser DG, Monroe DG, Khosla S. Distinct effects of loss of classical estrogen receptor signaling versus complete deletion of estrogen receptor alpha on bone. Bone. 2011;49:208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martin-Millan M, Almeida M, Ambrogini E, et al. The estrogen receptor-α in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol. 2010;24:323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Almeida M, Martin-Millan M, Ambrogini E, et al. Estrogens attenuate oxidative stress and the differentiation and apoptosis of osteoblasts by DNA-binding-independent actions of the ERα. J Bone Miner Res. 2010;25:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kousteni S, Almeida M, Han L, Bellido T, Jilka RL, Manolagas SC. Induction of osteoblast differentiation by selective activation of kinase-mediated actions of the estrogen receptor. Mol Cell Biol. 2007;27:1516–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chokalingam K, Roforth MM, Nicks KM, et al. Examination of ERα signaling pathways in bone of mutant mouse models reveals the importance of ERE-dependent signaling. Endocrinology. 2012;153:5325–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blattner MS, Mahoney MM. Circadian parameters are altered in two strains of mice with transgenic modifications of estrogen receptor subtype 1. Genes Brain Behav. 2012;11:828–836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.