

Abstract
We developed a homogeneous phenotypic fluorescence endpoint assay for cytotoxic T lymphocyte lytic granule exocytosis. This flow cytometric assay measures binding of an antibody to a luminal epitope of a lysosomal membrane protein (LAMP-1) that is exposed by exocytosis to the extracellular solution. Washing to remove unbound antibody is not required. Confirming the assay’s ability to detect novel active compounds, we screened at a concentration of 50 μM a synthetic diversity library of 91 compounds in a 96-well plate format, identifying 17 compounds that blocked by 90% or more. The actions of six structurally related tetracyano-hexahydroisoindole compounds that inhibited by ~90% at a concentration of 10 μM were investigated further. Four reduced elevations in intracellular Ca2+; it is likely that depolarization of the cells’ membrane potential underlies the effect for at least two of the compounds. Another compound was found to be a potent inhibitor of the activation of the MAP kinase ERK. Finally, we transferred the assay to a 384-well format and screened the Prestwick Compound Library using high-throughput flow cytometry. Our results indicate that our assay will likely be a useful means of screening libraries for novel compounds with important biological activities.
Keywords: Cytotoxic T lymphocytes, exocytosis, flow cytometry, high-throughput screen, phenotypic assay
Introduction
Cell-based assays offer some significant advantages for high throughput screens.1, 2 Entire pathways can be interrogated, even if the molecular basis of a given process is incompletely understood, and the active compounds obtained are guaranteed to be effective in a cellular context, which is not always the case for in vitro biochemical assays. Fluorescently-labeled antibodies are powerful tools that have found widespread use in cell biology3, but their use in cell-based phenotypic screens, particularly those conducted at higher throughput, has been limited. The basic problem is that, for a plate-based assay in the absence of a wash step to remove unbound antibody, antibody binding doesn’t result in an easily detectable signal. An antibody may redistribute quite dramatically, but this does not result in a change in the total amount of fluorescence. Polarization methods are of no help, because the rotational lifetime of antibodies far exceeds the lifetime of the excited fluorophore. Imaging plate readers can be used to visualize antibody binding, but signal dynamic range will be small, because the total amount of antibody present in the solution contributes a large background signal.4 While background fluorescence due to excess antibody could be eliminated by incorporating wash steps into the assay, these add significant complexity and cost to HTS assays, and so are typically avoided.
Flow cytometry offers a powerful method to overcome the problem of background antibody fluorescence, since only a small volume of solution accompanies cells as they pass through the interrogating lasers4, as first noted by Sklar and Finney5 and subsequently reviewed6. High-throughput flow cytometers, consisting of a sampling probe attachment coupled to a modified cytometer, can read a 384-well plate in ~15 minutes.7 More recently, the ability of flow cytometers to analyze signals due to antibody binding without washing was exploited by Kim et al. to study T cell receptor-stimulated integrin activation8 and by Chigaev et. al9 to screen for allosteric integrin regulators.
For a number of years, we10–13 and others14–16 have used binding of antibodies raised against an intraluminal domain of lysosome associated membrane protein 1 (LAMP-1) as a tool to measure lytic granule exocytosis by cytotoxic T lymphocytes (CTLs) and natural killer cells (NKs). CTLs are a subset of lymphocytes critical to the immune response to intracellular pathogens, and which play a key role in transplant rejection and autoimmunity, while NK cells are components of the innate immune system. Both cell types kill their targets using a similar mechanism that involves release of cell-killing agents from lytic granules that are secretory lysosomes. The intraluminal domain of LAMP-1 is exposed to the extracellular solution when the CTL’s lytic granules, which are secretory lysosomes, fuse with the plasma membrane during exocytosis. Thus, antibodies present in the extracellular solution can bind and serve as a quantitative measure of granule exocytosis. We suspected that it might be possible to exploit flow cytometry and fluorescent antibody binding in a homogeneous phenotypic endpoint assay to identify inhibitors of lytic granule exocytosis, which could represent novel immunosuppressants. Here, we provide evidence of the validity of the approach and have used it to screen two libraries, one at medium throughput in 96 well plates and the second at high throughput in 384 well plates.
Materials and Methods
Cells, solutions, chemicals and reagents
Tall-104 human leukemic CTLs were from the American Type Culture Collection (Manassas, VA). They were grown in Iscove’s medium (Sigma Aldrich, St. Louis, MO) supplemented with 10% FBS (Hyclone, Logan, UT), 2% L-glutamine (Gemini, West Sacramento, CA) 4% Pen-Strep (Gemini, West Sacramento, CA), and 100 IU IL-2/ml (NCl–Frederick, Frederick, Maryland). Cells were maintained at a density of 0.5 – 1×106 per ml in 10% CO2 at 37°C. Phorbol 12-myristate 13 acetate (PMA) was from Alexis Biochemicals (San Diego, CA). Thapsigargin (TG) was from AdipoGen (San Diego, CA). Paraformaldehyde (PFA) was from Electron Microscopy Sciences (Hatfield, PA) and was diluted to 4% in PBS. Fura-2/AM was from Invitrogen (Grand Island, NY) and was made to 1 mM in DMSO. Bis-(1,3-dibutylbarbituric acid) trimethine oxonol (bis oxonol) was from Molecular Probes (Grand Island, NY). A 10mM stock solution was made in DMSO. FACs buffer consisted of PBS containing 0.01% NaN3 (Fisher Scientific, Waltham, MA) and 5% FBS. Anti-LAMP antibody (clone H43A) was from BD (Franklin Lakes, NJ). It was conjugated to Alexafluor 647 using a kit (A20173) from Molecular Probes/Invitrogen (Grand Island, NY), and the stock concentration after conjugation was adjusted to 0.1 mg/ml by adding FACS buffer. The Prestwick Library, a broad spectrum collection of compounds that includes numerous off-patent, marketed drugs with known safety and bioavailability in humans, was from Prestwick Chemical (Illkirch, France).
Normal Ringer’s was made with 145 mM NaCl, 4.5 mM KCl, 1mM MgCl2, 2mM CaCl2 (all from Fisher Scientific, Waltham, MA), 5mM HEPES (Sigma Aldrich, St. Louis, MO) and 10mM glucose (Fisher Scientific, Waltham, MA). The pH was adjusted to 7.4 with NaOH. K Ringer contained 160 mM KCl, no NaCl and was adjusted to pH 7.4 with KOH (Fisher Scientific, Fairlawn, NJ).
Flow Cytometry
Flow cytometry (except for the HTS screen of the Prestwick Library) was performed on a Becton-Dickinson FACSCalibur (Franklin Lakes, NJ) at the University of Connecticut Biotechnology Center’s Flow Cytometry and Confocal Microscopy Center. For 96 well experiments, approximately 5000 events were collected per sample. Data were analyzed using Flow Jo software (TreeStar, Ashland, OR).
Medium- throughput LAMP assay and plate uniformity testing
1 μl of compounds (5 mM stock) or DMSO was dispensed into wells of 96 well Corning Costar tissue culture treated plates. 100 μl of TALL-104 cells that had been washed and resuspended in Normal Ringers at 2–3×106 cells/ml were dispensed into the wells, mixed and incubated for 30 minutes at room temperature. During this time, microcentrifuge tubes for the anti-LAMP antibody-containing solutions were blocked with 5%BSA in PBS for 30 minutes. 10 μl of the appropriate anti-LAMP antibody-containing solutions was then added and the plates were thoroughly mixed and were incubated for 50 minutes with rotation in the dark. Well contents were fixed by adding 100 μl of PBS + 2% paraformaldehyde and were then transferred into Biorad Titertube microtubes (Biorad, Hercules CA) that were then placed into standard flow tubes. Stimulation solution was made of 5 μl of 1 mM TG, 5 μl of 50 μM PMA, 10 μl of LAMP Ab and 380 μl Normal Ringer’s. The non-stimulating solution was made of 10 μl LAMP Ab and 390 μl Normal Ringer’s. For plate uniformity testing, the medium stimulation solution was made of 5 ul 1 mM TG, 10 μl of anti-LAMP Ab and 385 μl of Normal Ringer’s. The final anti-LAMP antibody concentration in these solutions was 2.5 ng/ml. All antibody-containing solutions were made in the blocked tubes (described above). All addition and mixing steps were done using multichannel pipetters with automated dispense/mix functions.
Fura2 measurements
TALL-104 cells were loaded with 1 uM Fura2-AM in Iscove’s medium at room temperature for 25 minutes, washed by centrifugation to remove excess dye, and resuspended in Ringer’s at 2.5×106 cells/ml. 100 μl of the cell suspension was incubated in 96-well plates with compounds or DMSO for 10 minutes, then stimulated by adding 2 μl of 100 μM TG. Fluorescence excited at 340 and 380 nm was measured at > 510 nm emission using a BioTek Synergy HT plate reader (BioTek, Winooski, VT), for 30 minutes after TG stimulation. The 340/380 ratio was calculated after subtracting from each wavelength a blank, measured from wells containing Ringer’s and compounds. Ratios from DMSO-treated cells washed and stimulated in Ca2+-free Ringer’s were also measured, allowing the inhibitory effect of compounds on Fura-2 ratios to be calculated as (Eq. (1)):
Bisoxonol measurements
To test the effect of compounds on membrane potential, 2.5 ×105 cells per condition were incubated with compounds or DMSO in 250 ul of Ringer’s for 15 min at room temperature. Bisoxonol was added from the stock solution to a final concentration of 100 nM, and samples were incubated for an additional 15 minutes before being analyzed via flow cytometry. Samples treated with compounds were read in an interleaved fashion with DMSO-treated cells suspended in Ringer’s or K+ Ringer’s so as to account for a tendency of bisoxonol signals to decline with time (see Figure 3). We do not know why the decline occurs. The effect of compounds was calculated as (Eq. (2)):
Figure 3. Assessing the mechanism of action of tetracyano-hexahydroisoindole compounds.

A) Representative Fura-2 ratio data for the six tetracyano-hexahydroisoindole compounds that inhibited lytic granule exocytosis by ~90% when tested at 10 μM. TG was added as indicated to trigger Ca2+ influx. For some traces, some symbols have been omitted for clarity. The lines connecting the symbols are drawn through those missing points. B) Representative bisoxonol data from one experiment of three. The geometric mean of bisoxonol fluorescence (Fl-1) is plotted for compounds (filled circles), Normal Ringer’s (open circles) and K+ Ringer’s (open squares). C) Cells treated as indicated were fixed, permeabilized and then stained with an anti-phospho ERK antibody followed by a CY5-labeled secondary antibody.
Both F values had the fluorescence measured in Normal Ringer’s in the absence of compound subtracted from them prior to the calculation.
BLT esterase assays
BLT esterase assays17 were performed and analyzed as described previously10.
Trypan blue exclusion assay of cell viability
Trypan blue solution (0.4%, Sigma Aldrich, St. Louis, MO) was mixed 1:1 with a sample of control (DMSO) or compound-treated cells 50 minutes after stimulation with 1 μM TG and 50 nM PMA. After 5–10 minutes, the fraction of blue-stained cells was assessed by counting on a hemocytometer. 125–200 total cells were counted per condition.
High throughput screening of the Prestwick Compound Library
Plates were prepared as follows. 10 μl of medium was added to columns 1–22 of Greiner Bio-One polypropylene 384-well microtiter plates (Greiner Bio-One North America, Monroe, NC), followed by the addition of 0.2 μl of test compounds at a concentration of 1 mM in DMSO to columns 3–22. Column 23 and 24 were left blank for analysis purposes. Negative and positive controls samples (columns 1 and 2) received 0.2 μL of DMSO. 1.3×105 cells in a volume of 3 μl were added to columns 1–22 and the plates were mixed by vortexing then incubated for 30 minutes at 37°C. This protocol resulted in a final compound concentration of 15.2 μM with a DMSO concentration of ~ 1.5%. Following incubation with compounds, column 1 (unstimulated control wells) received 2 μl of medium containing anti-LAMP antibody (final dilution 1:500), while wells in columns 2–22 received 2 μl of stimulation solution containing 1 mM thapsigargin, 50 μM PMA, and a 1/40 dilution of labeled anti-LAMP antibody. This addition resulted in wells that contain 1 uM TG, 50 nM PMA and ~1/500 anti-lamp. Plates were incubated with rotation for 120 minutes. Fluorescence was assessed using a HyperCyt® platform comprised of a dual laser CyAn flow cytometer (Beckman-Coulter, Fort Collins, CO) and a Gilson autosampler (Gilson, Middleton, WI) controlled using HyperSip software (Intellicyt, Albuquerque, NM). Forward and side scatter gates were used to identify viable cells. Fluorescence of alexafuor 647-conjugated anti-LMP1 antibody was excited at 635 nm and detected with a 665/20 optical bandpass filter. Samples were acquired for a total duration of 1 second, resulting in the delivery of >1000 cells/well for analysis. Using this setting, which aspirated 1–2 μl of sample, it took ~12 minutes to read a 384-well plate. Mean channel fluorescence (MCF) of individual wells was acquired and saved as a single file that was parsed into individual well data and analyzed using Intellicyte Hyperview software Version 2.5.1.
Results
LAMP externalization can be detected reliably in a homogeneous format
Stimulating TALL-104 cells with thapsigargin (TG, a drug that indirectly triggers the opening of plasma membrane Ca2+ channels) together with phorbol myristate acetate (PMA, a synthetic analog of diacylglycerol that activates PKC and ERK) in the presence of alexafluor 647-conjugated anti-LAMP-1 antibody causes a large increase in membrane-associated antibody fluorescence10. We tested whether signals due to anti-LAMP antibody binding could be measured in flow cytometry without washing (Figure 1A). For these experiments, cells were incubated in Ringer’s medium with or without TG + PMA in the presence of anti-LAMP1 in a total volume of 200 μl, then were either washed with FACS buffer and resuspended in 200 μl of PBS or left unwashed. We found that, although there was a slight increase in the signal intensity of the unstimulated unwashed cells compared to those that were washed (geometric means of anti-LAMP 1 fluorescence 23.9 vs. 4.5, respectively), the intensity of the stimulated unwashed sample was increased as well compared to the washed cells (geometric means of anti-LAMP 1 fluorescence 430 vs. 294) and so there was no diminution of the signal (Figure 1A). We determined that the assay can tolerate DMSO concentrations of up to 2.5% before its quality is significantly degraded (data not shown). We optimized the assay with respect to antibody concentration and cell number to obtain a reliable and robust signal while using as little antibody as possible (data not shown). We then conducted plate uniformity tests using unstimulated, partially-stimulated (treated with TG only), and fully-stimulated (treated with both TG and PMA) cells laid out in wells in an interleaved pattern. Three plates were tested on different days. To assess the suitability of the assay for screening, we computed the Z′ value.18 All three plates had Z′ > 0.6, and the average Z′ for the three plates was 0.71. Data from one representative plate is shown (Fig. 1B.) A Z′> 0.5 is considered as indicating acceptable assay performance.18
Figure 1. Detecting exocytosis via anti-LAMP antibody binding without washing.

A) Histograms of anti-LAMP antibody fluorescence for cells that were washed (top) or not washed (bottom). Unstimulated cells are represented by a dashed line, while stimulated cells are denoted by a solid line. Note the shift in the fluorescence of the unwashed unstimulated cells compared to the washed unstimulated cells. B) Plot of the geometric mean of anti-LAMP antibody fluorescence from a test conducted in a 96 well plate.
Probing a synthetic diversity set at medium throughput
We next tested, at a compound concentration of 50 μM, a small synthetic diversity library comprised of 91 compounds containing 16 distinct chemical scaffolds19 (Figure 2). We chose this high concentration of compound for the initial screen because it was used in the previous work on this library19, and we wanted to be able to compare our results to the results of that study. Cells in Normal Ringer’s were pre-incubated with compounds or DMSO for 30 minutes before stimulation with TG and PMA. A representative test of the library is shown in Figure 2. Figure 2A shows flow data from a negative control well, a positive control well, and a well containing compound 9b, while Figure 2B shows the overall plate data. Each point in Figure 2B represents the geometric mean of LAMP antibody fluorescence from an individual well. This initial library screen was performed twice; a scatter plot of the data from the two runs demonstrates excellent reproducibility (Fig. 2C). Seventeen compounds reduced anti-LAMP1 binding by 90% or more, a cutoff chosen due to the high initial test concentration. None of the hits decreased the proportion of cells in the live cell forward scatter vs. side scatter gate, suggesting that they were not toxic at the concentration tested (data not shown). Interestingly, the majority of the active compounds possessed one of three scaffolds. Those three scaffolds are represented by compounds 9a, 10a and 13e. The scaffold exemplified by compound 9a was present in the largest number of active compounds, a series of tetracyano-hexahydroisoindoles, and we decided to investigate the effects of these compounds further (Figure 3 and Table 1). As preincubation times of 10 minutes were found sufficient to produce maximal effects (data not shown), this time period was used for subsequent experiments.
Figure 2. Screening a 91-compound library in plate format.

A) Histograms of anti-LAMP antibody fluorescence from a negative control well, a positive control well, and a well treated with compound 9b. Cells were gated on forward and side scatter so as to exclude dead cells; for all conditions, ~40% of cells were in the live cell gate. B) Plot of the geometric mean of anti-LAMP antibody fluorescence from one screen of the synthetic 91 compound library. Data have been rearranged from the original plate format so that compounds containing each of the 16 different scaffolds are displayed in a contiguous manner. Positive control (filled squares) and negative control (filled circles) wells are shown first, and dashed lines separate compounds containing different scaffolds. Structures of three compounds possessing scaffolds present in four or more inhibitory compounds are shown above the plot. C) Plot of the geometric mean of anti-LAMP antibody fluorescence from one screen of the 91 compound library vs. that from a second screen, demonstrating reproducibility.
Table 1.
Analysis of the effects of tetracyano-hexahydroisoindole compounds
| Compound1 | Structure | Block of LAMP response2 | Block of BLT esterase2 | IC50/Hill Coeff3 | Block of Fura-2 ratio2 | Increase in Bisoxonol fluorescence2 |
|---|---|---|---|---|---|---|
| 9a |

|
91 +/− 7 | 19 +/−10 | 4.5/1.9 | 22 +/− 4 | 14 +/− 1 |
| 9b |
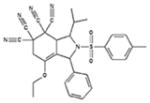
|
99 +/− 1 | 94 +/− 6 | 0.8/4.9 | 70 +/− 3 | 39 +/− 3 |
| 9c |
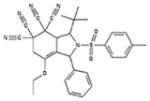
|
91 +/− 4 | 98 +/− 10 | 0.5/2.1 | 60 +/− 5 | 53 +/− 14 |
| 9d |

|
87 +/− 10 | 82 +/− 10 | 1.7/2.2 | 72 +/− 5 | 33 +/− 2 |
| 9e |

|
100 +/− 0 | 103 +/− 8 | 0.8/4.1 | 71 +/− 1 | 72 +/− 14 |
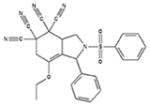
| 9f |
|
100 +/− 0 | 87 +/− 20 | 1.0/3.2 | 11+/− 7 | 25 +/− 2 |
Compound designations are from Wang et al. 2010.
Percent inhibition measured at 10 μM from at least three separate determinations. Data are mean +/− SD.
Hill number and inhibitory constant (in μM) obtained from fits of the Hill equation to three dose response curves for inhibition of LAMP antibody binding.
Although all 7 tetracyano-hexahydroisoindole compounds in the library inhibited exocytosis by >90% when tested at 50 μM, when retested at 10 μM, compound 9g only blocked by ~20%. The other 6 compounds blocked by > 85%. We performed dose-response measurements on those 6 compounds. They blocked with IC50 values ranging from 0.5 – ~5 μM, and Hill numbers for all were ~2 or more (Table 1). Additionally, all except 9a blocked release of granzyme B by >80% as assessed using BLT esterase assays17, confirming their exocytosis-inhibiting activity (Table 1). None of the compounds appeared to be cytotoxic at 10 μM, as assessed by measuring trypan blue dye exclusion (Supplementary Table 1).
Calcium influx, likely occurring through CRAC channels20, is critical for lytic granule exocytosis and is relatively easy to assess in a fluorescent plate reader. As an initial step in investigating the mechanism of action of these compounds we tested whether they affected Ca2+ signaling. We loaded cells with Fura-221 and examined effects of compounds at 10 μM on TG-stimulated fluorescence ratio changes reflecting Ca2+ influx (Figure 3A and Table 1). We found that compounds 9b, 9c, 9d and 9e caused a 60–70% reduction in the magnitude of the Fura-2 fluorescence signal, while 9a and 9f had much smaller effects. 9f also consistently produced an elevation in the resting signal and increased the rate at which the maximum ratio following stimulation was attained.
In principal, the effect of the compounds that reduced Fura-2 ratios could be explained by a direct inhibition of Ca2+ influx, by a depolarization of the cells’ membrane potential (Vm), which would reduce the driving force for Ca2+ entry (see22 for a discussion of this), or by a combination of both effects. To distinguish between these possibilities, we used the membrane-permeant dye bisoxonol23 to assess relative changes in Vm in control and compound-treated cells, detecting fluorescence using flow cytometry (Figure 3B and Table 1). Bisoxonol is an anionic dye that distributes across the membrane in a Nernstian fashion, accumulating in cells when they are depolarized, resulting in increased fluorescence. We compared compound-induced fluorescence changes to increases in fluorescence caused by bathing cells in Ringer’s solution that contained 160 mM K+ (high [K+]o solution, which depolarizes Vm). We found that compounds caused an increase in bisoxonol fluorescence ranging from ~15% of the high-[K+]o stimulated value for 9a to ~70% of the high-[K+]o stimulated value for 9e (Table 1). Among the compounds that significantly decreased Fura-2 ratio changes (9b, 9c, 9d and 9e), bisoxonol fluorescence increases ranged from 30% to 70% of the high-[K+]o value. Thus, compounds that inhibit Ca2+ signaling depolarize cells to varying degrees.
Since compounds 9a and 9f failed to affect Ca2+ signals, we next tested their effect on activation of ERK, a process known to be required for granule exocytosis that can conveniently be monitored with phosphospecific antibodies via flow cytometry. We fixed and permeabilized cells, and then stained them with an antibody that detects phosphorylated (i.e. active) ERK24, followed by a Cy5-labeled secondary Ab (Fig 3C). We found that compound 9f but not compound 9a inhibited the increase in fluorescence that occurs upon stimulation; in three experiments, 9f inhibited ERK activation as assessed using antibody staining by 87 +/−14%, while 9a was without significant inhibitory effect. These results suggest that 9f inhibits the action of upstream kinases that are involved in phosphorylating and thus activating ERK, but 9a does not. As the activation of PKC is known to participate in activating ERK when cells are stimulated with PMA12, we tested whether 9f inhibits PKC activity. We stained fixed and permeabilized cells with an antibody that detects proteins that are phosphorylated on the consensus PKC substrate site, an approach we have used before25. The stimulation-dependent increase in staining with this antibody in cells treated with compound 9f was not significantly different than in DMSO-treated control cells (data not shown), suggesting that the effects of 9f lie downstream of PKC activation.
Probing the Prestwick Compound Library with a high throughput version of the LAMP assay
Our final step in confirming the ability of flow cytometry to enable a homogeneous fluorescent antibody-based high throughput assay of exocytosis was to use the method to screen the Prestwick Compound Library (PCL) in a 384-well plate format using the HyperCyt high throughput flow cytometry platform (Figure 4). Figure 4A shows representative raw flow cytometry data from one plate’s set of negative and positive control wells. There is a small number of more highly-responding cells in the negative control population in early wells compared to later wells in this sample. This was not typical, and, although we do not know the cause, the effect is small and does not affect the results enough to degrade assay performance. On the left is a scatter plot of the well data, separated by bubbles as the sampling probe was moved, and on the right are corresponding histograms of the fluorescence values. Representative data from one entire plate are shown in two different ways in Figure 4B and 4C. Figure 4B is a plot of the geometric mean of individual well data. Values from compound-containing wells are represented by open circles, while values from positive and negative control wells are represented by filled squares and triangles, respectively. Figure 4C shows a heat map of the data, in which the lowest geometric mean value is denoted by black, while the highest is represented by white.
Figure 4. Screening the Prestwick Compound Library in high-throughput format.

A) Left, sample data from the high-throughput cytometer system showing the acquisition of 16 negative and 16 positive control wells. Data from different wells are separated by air bubbles, allowing them to be discriminated off-line. Right, representative histograms of anti-LAMP antibody fluorescence from the experiment shown on the left. B) Plot of the geometric mean of anti-LAMP fluorescence from one run of Plate 1 of the PCL. Negative (closed triangles) and positive (closed squares) control data are indicated, and compound-containg test wells are represented by open circles. C) Heat map of the data shown in (B). The lookup table is set such that the lowest mean fluorescence intensity is represented by black, while the highest is represented by white. D) Plot of the number of hits (solid symbols, left axis) and the Jaqqard similarity index (open triangles) for two runs of the entire PCL analyzed at inhibitory thresholds of 40, 50 and 60 %.
The PCL was screened twice. In both cases, the overall Z′ for the inlier control wells in the 4 plates was > 0.5, indicating suitable assay performance. To assess assay reproducibility, we analyzed hits from the two runs at three different thresholds for inhibition: 40%, 50% and 60% (Figure 4C and Supplementary Table 1). The two library screens yielded similar numbers of hits at all three thresholds, and there was significant overlap between the hits that were identified in the two runs. As a means to assess how well the two runs agreed, we computed the Jaccard Similarity Index, defined as (Eq. 3):
This serves as a measure of the fraction of hits that were identified in both runs. Increasing the threshold from 40% to 60% decreased the number of hits three-fold (from ~45 in each of the runs to ~15) and increased J (Run1, Run2) from ~0.3 to ~0.6. This result indicates that a higher stringency likely results in more reliable identification of active compounds. Among the hits observed in both runs at a threshold of 60% were amodiaquin, an antimalarial compound related to chloroquine26, and auranofin, a gold-containing compound27. Interestingly, both compounds have been used to treat rheumatoid arthritis, consistent with immunosuppressive effects. We confirmed the inhibitory effects of amodiaquin on granule exocytosis and performed preliminary dose-response measurements in complete cell culture medium that indicate an IC50 < 1 μM (data not shown). Another very potent inhibitory compound we identified was 1,4- chrysenequinone, a component of petroleum fuel exhaust fumes28, which in preliminary confirmatory experiments inhibited LAMP externalization with an IC50 in cell culture medium of < 1 μM (data not shown). The mechanism of action of these compounds on lytic granule exocytosis remains to be determined. A full listing of compounds that were identified as hits in the screens of the PCL is provided in Supplemental Table 1.
Discussion
Our results demonstrate that binding of fluorescently-labeled anti-LAMP1 antibodies can serve as the basis of a homogeneous high-throughput screen of exocytosis when flow cytometry is used to detect the signal. Flow cytometry enables this by restricting the volume of solution that is interrogated so that background antibody fluorescence is negligible. We note that confocal plate readers might also serve the same purpose, exploiting the confocal effect to minimize background fluorescence from antibody in solution. Supporting this idea, in preliminary no-wash experiments that used coverslip-bottomed 96-well plates and were conducted under conditions identical to those of Figure 1A, we were able to obtain Z′ values > 0.5 for replicates of 5 unstimulated and 5 stimulated samples (data not shown). Since confocal-capable plate readers such as Qiagen’s Opera and Molecular Devices’ ImageXpress Ultra are becoming more readily available, and may be available to researchers who do not have access to high-throughput flow cytometry, this may be a worthwhile strategy to pursue. However, we encountered several technical issues that would have to be overcome to implement such an assay. A reproducible focal plane would be needed to achieve a suitable Z′, and a second stain would have to be used label membranes for analysis. A confocal approach would undoubtedly be more difficult to implement in high-throughput format than flow cytometry, in which focal plane is not an issue and quantification of signals is simple.
Our screen of the 91-compound synthetic library19 confirms that our assay can identify molecules with interesting biological activities. Six of the seven tetracyano-hexahydroisoindole compounds blocked exocytosis with relatively high affinity, with estimated IC50 values ranging from 0.5–5 μM. All but one of the compounds, 9a, also almost completely blocked granzyme release in BLT-esterase assays. The effect of 9a seems unlikely to be either a case of a false positive in the LAMP assay or of a false negative in the BLT-esterase assay. We confirmed that adding 9a together with the anti-LAMP antibody to cells that had been previously stimulated to undergo exocytosis in the absence of the antibody did not cause a reduction in signal intensity compared to cells that were not treated with 9a, demonstrating that 9a does not quench alexafluor 647 fluorescence or inhibit antibody binding to exposed LAMP (data not shown). We also determined that 9a does not react with the detection reagents to produce a color change and mimic exocytosis in the BLT esterase assay (data not shown). Assuming the effect of 9a is a real one and not an artifact, what kind of biological action could result in a block of antibody binding but not release of granzyme B? One possibility is a block in the dilation of a fusion pore that forms when the vesicle and plasma membrane coalesce. If 9a were to arrest the fusion pore at a stage in which it was wide enough to allow granzyme B (39 KDa) to diffuse out but not wide enough to allow the anti-LAMP mAB (150 KDa) to diffuse in, it would produce a discrepancy between the LAMP and BLT assays. Recent work has demonstrated two modes of fusion pore behavior in exocytosis by NK cells, one of which leads to restricted diffusion of vesicular cargo.29
Analysis of the effects of the tetracyano-hexahydroisoindole compounds leads to the following four conclusions about their structure activity relations and plausible mechanisms of action. First, inhibition of [Ca2+]i elevation appears to require bulky substituents at the 3 position, since 9a and 9f, which lack a substituent, do not significantly affect Fura-2 ratios. Second, for some compounds that reduce [Ca2+]i, such as 9c and 9e, effects on Vm may be sufficient to account for the entire effect on [Ca2+]i, since mimicking the depolarization they produce (by elevating [K+]o) is sufficient to cause a 60–70% reduction in Fura-2 ratio (data not shown), comparable to the reduction that 9c and 9e cause. For other compounds, such as 9b and 9d, however, effects on Vm may not entirely account for the effects on [Ca2+]i, since mimicking the depolarization they produce reduced Fura-2 ratios by < 60–70% (data not shown). Thus, Ca2+ influx may also be inhibited directly by compounds 9b and 9d. The simplest possible mechanism by which compounds could depolarize Vm is by blocking K+ channels22, although it is also possible that they could inhibit Na+-K+ ATPases, resulting in a change in the equilibrium potential for K+. We note that both K+ channels and Ca2+ channels in T cells are targets of interest as immunosuppressants. Third, compound 9f did not block [Ca2+]i increases but did block ERK activation, likely by inhibiting the ability of upstream MAP kinase kinases to phosphorylate ERK. The other compounds that do block [Ca2+]i increases may also have this effect; our unpublished observations suggest that reducing [Ca2+]i levels by ~70% (by decreasing extracellular [Ca2+]) only blocks exocytosis by ~50%, which is less than the block we observed. This is consistent with that idea that effects in addition to block of [Ca2+]i increases, possibly block of ERK activation, may contribute to the inhibition of exocytosis by 9b, 9c, 9d and 9e.
Block by all of the tetracyano-hexahydroisoindole compounds was characterized by dose response curves with Hill numbers > 1 (Table 1), which is usually interpreted as indicating a cooperative mechanism of inhibition. There has been discussion in the literature that Hill numbers > 1 can reflect non-specific modes of inhibition such as compound aggregation, but this does not necessarily apply to cell-based assays 30. In the case of the compounds that block [Ca2+]i increases, the effect is likely due to a non-linear dependence of exocytosis on [Ca2+]. Our unpublished observations regarding the dependence of TG-stimulated lytic granule on [Ca2+]i reveal cooperativity (Shreyassee Kumbhar, Alexander J. Meeske and Adam Zweifach), as expected for a process that ultimately depends on Ca2+ binding to calmodulin to activate calcineurin 31. Thus, even a simple competitive inhibitor of Ca2+ influx would be expected to generate an apparently cooperative block of exocytosis. We speculate that lytic granule exocytosis also has an underlying nonlinear dependence on ERK activity, which would explain why compound 9F also has a Hill slope >1.
Our screen of the PCL confirmed that our method is suitable for use in a true HTS campaign. Note that we do not know whether the miniaturized version of the assay and the 96-well version have the same sensitivity. Testing this would require that we examine a set of compounds with both versions of the assay, which we have not done. In fact, we have preliminary evidence that the two versions of the assay may not behave similarly for some compounds (data not shown). We suspect that this is because the miniaturized assay was performed in serum-containing medium to prevent cells from clogging the sampling tubing. Binding to proteins in serum likely alters the free concentration of some compounds.
We envision at least three potential applications for our assay of lytic granule exocytosis. First, it will serve as a means of identifying novel immunosuppressants. There is a great deal of similarity between the signaling underlying lytic granule exocytosis and the signals promoting cytokine production by helper T cells32, so compounds active in a screen of CTLs could represent novel broadly immunosuppressive compounds. Helper T cell activation requires hours, so screening CTL granule exocytosis is much more convenient from an assay standpoint. The need for novel immunosuppressants is well known.33 Alternatively, inhibitory compounds could, depending upon their mechanism of action, represent CTL-specific immunosuppressants, which might enable a much more finely-tuned kind of immunosuppression than is currently feasible. Determining whether compounds affect CTLs only, or CTLs and other immune cells would require examining the effects of hits on other cell types so as to assess specificity. Second, compounds identified in a screen of lytic granule exocytosis may also be useful in a forward chemical genetics campaign to identify novel pathways and molecules that are involved in lytic granule exocytosis. Such an approach would involve identifying compounds that do not work via any of the currently-known pathways that control lytic granule exocytosis and then seeking to identify their molecular targets. This could most likely be done via affinity chromatography using optimized versions of lead compounds.34 We plan in the near future to screen the NIH’s Molecular Libraries Small Molecule Repository with these goals in mind. A third use of the assay might be to screen diversity libraries, whether comprised of natural or synthetic products, as a means of broadly probing chemical space so as to identify compounds with biological activity. Because lytic granule exocytosis involves multiple signaling pathways and cellular processes, we expect it to yield more hits than a screen of a single cellular function, which is the way diversity libraries are typically screened. Consistent with this, our hit rate when we screened the 91-compound synthetic library was almost 6-fold higher than when the same library was screened at the same concentration in an assay of cell migration.19 We note that the three compounds active in that study were also active in our assay, although the mechanisms underlying the compounds’ antimigratory activity is unclear.
We suspect that the basic strategy underlying our assay could be used to create assays for a variety of other cellular functions. Inhibition of exocytosis by other cell types could be monitored, provided a suitable vesicular protein was identified. This protein would have to possess a luminal domain for which an antibody was available. The same basic strategy could also be used to screen for agents that alter the expression of any surface marker for which an antibody is available, provided that the fold change in expression is large and variability small enough to yield a sufficiently robust assay. High throughput flow cytometry would be the modality of choice for such endeavors.
Supplementary Material
Acknowledgments
We would like to thank Dr. Arun Pores-Fernando and Michelle Y. D. Ranaghan for preliminary experiments, Shreyasee Kumbhar and Alexander J. Meeske for unpublished observations, Ziyan Zhao for helpful comments on the manuscript, and Yi Chiao Fan for the supply of the synthetic diversity library of 91 compounds. Supported by NIH grants R21 NS066462 to AZ, U54 MH084690 to LAS and MKH and R01 GM071779 and P41 GM081282 to OK.
References
- 1.Johnston PA, Johnston PA. Cellular platforms for HTS: three case studies. Drug Discov Today. 2002;7:353–363. doi: 10.1016/s1359-6446(01)02140-7. [DOI] [PubMed] [Google Scholar]
- 2.An WF, Tolliday N. Cell-based assays for high-throughput screening. Mol Biotechnol. 2010;45:180–186. doi: 10.1007/s12033-010-9251-z. [DOI] [PubMed] [Google Scholar]
- 3.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 4.Black CB, Duensing TD, Trinkle LS, Dunlay RT. Cell-based screening using high-throughput flow cytometry. Assay Drug Dev Technol. 2011;9:13–20. doi: 10.1089/adt.2010.0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sklar LA, Finney DA. Analysis of ligand-receptor interactions with the fluorescence activated cell sorter. Cytometry. 1982;3:161–165. doi: 10.1002/cyto.990030304. [DOI] [PubMed] [Google Scholar]
- 6.Nolan J, Chambers JD, Sklar LA. Flow Cytometric Analysis of Ligand-Receptor Interactions. Cytometry Approaches to Cellular Analysis. 1998:19–46. [Google Scholar]
- 7.Kuckuck FW, Edwards BS, Sklar LA. High throughput flow cytometry. Cytometry. 2001;44:83–90. [PubMed] [Google Scholar]
- 8.Kim K, Wang L, Hwang I. A novel flow cytometric high throughput assay for a systematic study on molecular mechanisms underlying T cell receptor-mediated integrin activation. PLoS One. 2009;4:e6044. doi: 10.1371/journal.pone.0006044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chigaev A, Wu Y, Williams DB, Smagley Y, Sklar LA. Discovery of very late antigen-4 (VLA-4, alpha4beta1 integrin) allosteric antagonists. J Biol Chem. 2011;286:5455–5463. doi: 10.1074/jbc.M110.162636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pores-Fernando AT, Bauer RA, Wurth GA, Zweifach A. Exocytic responses of single leukaemic human cytotoxic T lymphocytes stimulated by agents that bypass the T cell receptor. J Physiol. 2005;567:891–903. doi: 10.1113/jphysiol.2005.089565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pores-Fernando AT, Gaur S, Doyon MY, Zweifach A. Calcineurin-dependent lytic granule exocytosis in NK-92 natural killer cells. Cell Immunol. 2008 doi: 10.1016/j.cellimm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pores-Fernando AT, Gaur S, Grybko MJ, Zweifach A. ERK activation is only one role of PKC in TCR-independent cytotoxic T cell granule exocytosis. Biochem Biophys Res Commun. 2008;371:630–634. doi: 10.1016/j.bbrc.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pores-Fernando AT, Gaur S, Doyon MY, Zweifach A. Calcineurin-dependent lytic granule exocytosis in NK-92 natural killer cells. Cell Immunol. 2009;254:105–109. doi: 10.1016/j.cellimm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 16.Betts MR, Koup RA. Detection of T-cell degranulation: CD107a and b. Methods Cell Biol. 2004;75:497–512. doi: 10.1016/s0091-679x(04)75020-7. [DOI] [PubMed] [Google Scholar]
- 17.Takayama H, Trenn G, Sitkovsky MV. A novel cytotoxic T lymphocyte activation assay: optimized conditions for antigen receptor triggered granule enzyme secretion. J Immunol Meth. 1987;104:183–190. doi: 10.1016/0022-1759(87)90502-3. [DOI] [PubMed] [Google Scholar]
- 18.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Castellano S, Kinderman SS, Argueta CE, Beshir AB, Fenteany G, Kwon O. Diversity Through a Branched Reaction Pathway: Generation of Multicyclic Scaffolds and Identification of Antimigratory Agents. Chemistry. 2010 doi: 10.1002/chem.201002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maul-Pavicic A, Chiang SC, Rensing-Ehl A, Jessen B, Fauriat C, Wood SM, Sjoqvist S, Hufnagel M, Schulze I, Bass T, Schamel WW, Fuchs S, Pircher H, McCarl CA, Mikoshiba K, Schwarz K, Feske S, Bryceson YT, Ehl S. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A. 2011;108:3324–3329. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 22.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Ann Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 23.Tatham PER, Delves PJ. Flow cytometric detection of membrane potential changes in murine lymphocytes induced by concanavalin A. Biochemical Journal. 1984;221:137–146. doi: 10.1042/bj2210137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chow S, Patel H, Hedley DW. Measurement of MAP kinase activation by flow cytometry using phospho-specific antibodies to MEK and ERK: potential for pharmacodynamic monitoring of signal transduction inhibitors. Cytometry. 2001;46:72–78. doi: 10.1002/cyto.1067. [DOI] [PubMed] [Google Scholar]
- 25.Pores-Fernando AT, Ranaghan MY, Zweifach A. No specific subcellular localization of protein kinase C is required for cytotoxic T cell granule exocytosis. J Biol Chem. 2009;284:25107–25115. doi: 10.1074/jbc.M109.011866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pomery H, Warren C, Mills D, Clark GM. The effect of amodiaquin (camoquin) on the course of rheumatoid arthritis. Arthritis Rheum. 1959;2:396–402. doi: 10.1002/1529-0131(195910)2:5<396::aid-art1780020504>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 27.Lorber A, Simon TM, Leeb J, Peter A, Wilcox SA. Effect of chrysotherapy on parameters of immune response. J Rheumatol Suppl. 1979;5:82–90. [PubMed] [Google Scholar]
- 28.Misaki K, Kawami H, Tanaka T, Handa H, Nakamura M, Matsui S, Matsuda T. Aryl hydrocarbon receptor ligand activity of polycyclic aromatic ketones and polycyclic aromatic quinones. Environ Toxicol Chem. 2007;26:1370–1379. doi: 10.1897/06-465r.1. [DOI] [PubMed] [Google Scholar]
- 29.Liu D, Martina JA, Wu XS, Hammer JAr, Long EO. Two modes of lytic granule fusion during degranulation by natural killer cells. Immunol Cell Biol. 2011;89:728–738. doi: 10.1038/icb.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shoichet BK. Interpreting steep dose-response curves in early inhibitor discovery. Journal of medicinal chemistry. 2006;49:7274–7277. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 31.Grybko MJ, Bartnik JP, Wurth GA, Pores-Fernando AT, Zweifach A. Calcineurin activation is only one calcium-dependent step in cytotoxic T lymphocyte granule exocytosis. J Biol Chem. 2007;282:18009–18017. doi: 10.1074/jbc.M702222200. [DOI] [PubMed] [Google Scholar]
- 32.Pores-Fernando AT, Zweifach A. Calcium influx and signaling in cytotoxic T-lymphocyte lytic granule exocytosis. Immunol Rev. 2009;231:160–173. doi: 10.1111/j.1600-065X.2009.00809.x. [DOI] [PubMed] [Google Scholar]
- 33.Woodcock J. Coping with rejection: immunosuppressants and organ transplantation. Drug Discov Today. 2005;10:11–13. doi: 10.1016/S1359-6446(04)03323-9. [DOI] [PubMed] [Google Scholar]
- 34.Ong SE, Schenone M, Margolin AA, Li X, Do K, Doud MK, Mani DR, Kuai L, Wang X, Wood JL, Tolliday NJ, Koehler AN, Marcaurelle LA, Golub TR, Gould RJ, Schreiber SL, Carr SA. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc Natl Acad Sci U S A. 2009;106:4617–4622. doi: 10.1073/pnas.0900191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.