

Abstract
Hypoxia induces angiogenesis and glycolysis for cell growth and survival, and also leads to growth arrest and apoptosis. HIF-1α, a basic helix–loop–helix PAS transcription factor, acts as a master regulator of oxygen homeostasis by upregulating various genes under low oxygen tension. Although genetic studies have indicated the requirement of HIF-1α for hypoxia-induced growth arrest and activation of p21cip1, a key cyclin-dependent kinase inhibitor controlling cell cycle checkpoint, the mechanism underlying p21cip1 activation has been elusive. Here we demonstrate that HIF-1α, even in the absence of hypoxic signal, induces cell cycle arrest by functionally counteracting Myc, thereby derepressing p21cip1. The HIF-1α antagonism is mediated by displacing Myc binding from p21cip1 promoter. Neither HIF-1α transcriptional activity nor its DNA binding is essential for cell cycle arrest, indicating a divergent role for HIF-1α. In keeping with its antagonism of Myc, HIF-1α also downregulates Myc-activated genes such as hTERT and BRCA1. Hence, we propose that Myc is an integral part of a novel HIF-1α pathway, which regulates a distinct group of Myc target genes in response to hypoxia.
Keywords: cell cycle, HIF-1α, hypoxia, Myc, p21cip1
Introduction
Under low oxygen tension (hypoxia), mammals elicit an array of cellular processes, in an attempt to maintain ATP production by improving oxygen delivery and enhancing glycolysis. It appears that HIF-1α, a basic helix–loop–helix (bHLH) transcription factor of the PAS family (Wang et al, 1995), is responsible for upregulating most, if not all, hypoxia-inducible genes (Semenza, 1999; Wenger, 2002). HIF-1α is constitutively transcribed and translated (Huang and Bunn, 2003), but is rapidly degraded in normoxia by the ubiquitin-proteasomal pathway (Salceda and Caro, 1997; Huang et al, 1998; Kallio et al, 1999). Polyubiquitination of HIF-1α is mediated by the von Hippel-Lindau tumor suppressor (Maxwell et al, 1999; Cockman et al, 2000; Kamura et al, 2000; Ohh et al, 2000; Tanimoto et al, 2000), a part of an E3 ubiquitin ligase, that binds to the oxygen-dependent degradation domain (ODD) (Huang et al, 1998; Huang and Bunn, 2003) of HIF-1α. Thus, removal of the ODD renders HIF-1α stable in normoxia, enabling transcriptional activation of its target genes and induction of hypervasculature in vivo (Huang et al, 1998; Elson et al, 2001). The VHL binding is controlled by hydroxylation of two proline residues in HIF-1α: Pro-402 (Masson et al, 2001) and Pro-564 (Ivan et al, 2001; Jaakkola et al, 2001). These prolines are modified in normoxia by the HIF prolyl-4-hydroxylases that require oxygen, iron, and 2-oxoglutarate as cofactors (Bruick and McKnight, 2001; Epstein et al, 2001). Therefore, depletion of oxygen or iron chelation inhibits the enzymatic activity, resulting in stabilization of HIF-1α, followed by heterodimerization with ARNT and recruitment of the transcription co-activator p300/CBP (Arany et al, 1996; Kallio et al, 1998; Kung et al, 2000) and SRC-1 (Carrero et al, 2000). Consequently, HIF-1 heterodimer binds to the hypoxia-responsive element (HRE) in the promoter, upregulating its target genes including vascular endothelial growth factor (VEGF), glucose transporters, and glycolytic enzymes for cell growth and survival (Semenza, 1999).
On the other hand, hypoxia/HIF-1α has also been suggested to inhibit cell growth (Carmeliet et al, 1998; Gardner et al, 2001; Green et al, 2001; Goda et al, 2003) and to promote apoptosis (Carmeliet et al, 1998; Bruick, 2000). Although the results from Hif1α null cells indicate that HIF-1α is required for hypoxia-induced upregulation of p21cip1, a key cyclin-dependent kinase inhibitor that controls G1 checkpoint, the role of HIF-1α in cell cycle has been controversial (Carmeliet et al, 1998; Gardner et al, 2001; Goda et al, 2003). Furthermore, it remains puzzling how HIF-1α activates p21cip1 because no HIF-1α-bound HRE has been identified in the promoter. To gain insights into the mechanism underlying hypoxia-induced cell growth arrest, we took advantage of an ODD-deficient HIF-1α (ΔODD) that is stable and functional in normoxia (Huang et al, 1998), and demonstrated that HIF-1α, even in the absence of hypoxic signal, induces cell cycle arrest by antagonizing Myc activity, resulting in derepression of p21cip1 (Herold et al, 2002; Seoane et al, 2002). Remarkably, neither HIF-1α transcriptional activity nor its DNA binding is required; instead, HIF-1α does so via its N-terminal region that interacts with Myc. Thus, our findings signalize a novel HIF-1α–Myc pathway regulating a new array of hypoxia-responsive genes that are distinct from those activated through HIF-1α transcriptional activation.
Results
HIF-1α induces cell cycle arrest by activating p21cip1
To demonstrate that it is HIF-1α, rather than other hypoxia-associated effects, that induces cell cycle arrest, we tested the effects of HIF-1α in normoxia by taking advantage of an ODD-deficient HIF-1α (ΔODD). Infection of HCT116 cells with three multiplicities of infection (moi) of adenoviruses expressing the HIF-1α variant (Ad-ΔODD) gave rise to pronounced changes in cell cycle (Figure 1A, top panels); there was an ∼7-fold decrease of cells in the S-phase and a concomitant increase in the G1-phase. In addition, the G2-phase was moderately increased. By contrast, adenoviruses expressing green fluorescent protein (Ad-GFP) produced no obvious effects. The GFP expression was confirmed with fluorescence microscopy and Western analysis (data not shown). As expected, a stable full-length HIF-1α, resulting from double mutations of Pro-402 and Pro-564, also induced G1 arrest (Supplementary Figure). In agreement with the effect of adenoviral infections, treatment with 1% oxygen resulted in similar changes in cell cycle (Figure 1B, left panel). Therefore, these results provide evidence that a stable HIF-1α is sufficient for cell cycle arrest in normoxia.
Figure 1.
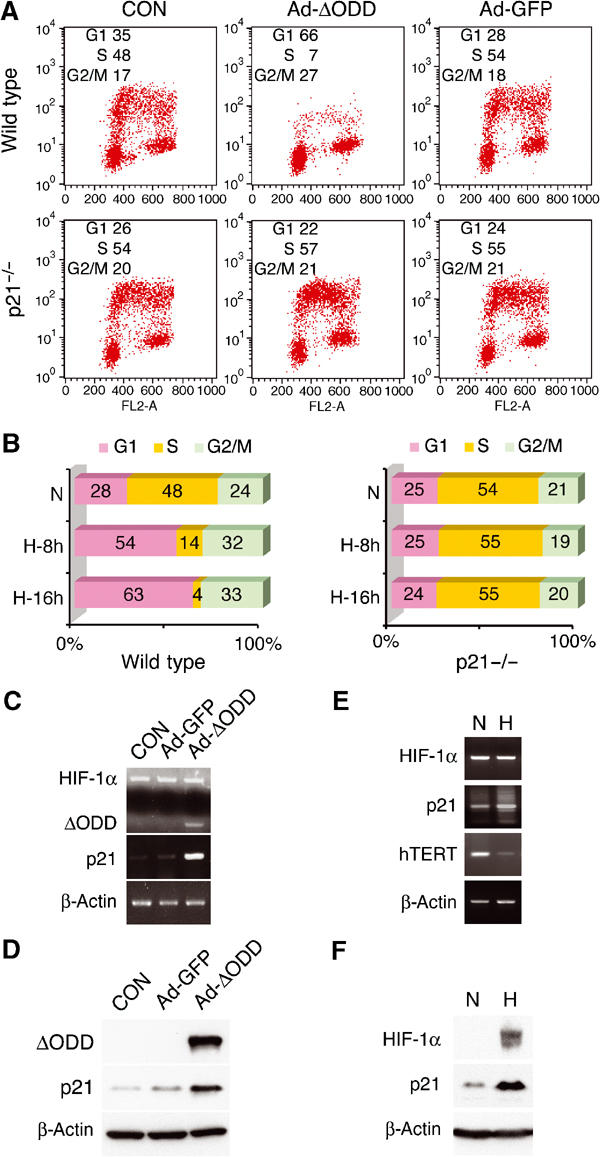
HIF-1α expression is sufficient to induce cell cycle arrest by activating p21cip1. (A) HCT116 wild-type and p21−/− variant were analyzed for BrdU incorporation after adenoviral infection, as indicated. The original data are presented, together with the percentage of each phase in the inlet. CON, no infection. (B) HCT116 wild-type and p21−/− cells were subjected to hypoxia (H) for 8 or 16 h prior to cell cycle analysis. The percentage of each phase is shown in bar graphs. N, normoxia. (C, D) Adenovirus-infected cells were assayed for mRNA (C) and protein (D) levels of specified genes by RT–PCR and Western blot, respectively. Both endogenous HIF-1α and ΔODD mRNA levels were determined in the same PCR reactions, and β-actin levels served as loading controls. (E, F) Hypoxic induction of p21cip1 expression was examined at mRNA (E) and protein levels (F). hTERT mRNA levels were also included as a control (see below).
To test whether HIF-1α-induced cell cycle arrest requires p21cip1, we determined the effect of HIF-1α in p21cip1 null HCT116 cells (Bunz et al, 1998). As a result of the genetic alteration, these cells exhibited an increased S-phase along with a decreased G1-phase (Figure 1A, bottom panels). However, neither Ad-ΔODD infection nor hypoxic treatment (Figure 1B, right panel) altered the cell cycle profile, confirming the requirement of p21cip1 for HIF-1α-mediated cell cycle arrest (Carmeliet et al, 1998; Goda et al, 2003). In keeping with this notion, Ad-ΔODD infection of wild-type HCT116 cells enhanced p21cip1 expression at both mRNA and protein levels (Figure 1C and D). Similar results were obtained when cells were treated with hypoxia (Figure 1E and F).
p21cip1 upregulation is independent of HIF-1α transcriptional activity and its DNA binding
To elucidate the mechanism underlying HIF-1α-mediated p21cip1 expression, we determined the involvement of HIF-1α C-terminal activation domain, which is responsible for the transcriptional activity of ΔODD by binding to p300/CBP. Unlike wild-type HIF-1α, ΔODD constitutively activated HIF-1-mediated reporter gene in transient transfection (Figure 2A). We then inactivated ΔODD transcriptional activity via simultaneous mutations (L795V, C800S, L818S, and L822V) that disrupt p300/CBP binding (Gu et al, 2001; Dames et al, 2002; Freedman et al, 2002). As expected, this ΔODD LCLL mutant was unable to activate the HIF-1 reporter (Figure 2A). In contrast, substitution of ΔODD Cys-800 with valine (C800V) further stimulated transcriptional activity in reference to ΔODD (Gu et al, 2001). Likewise, adenoviral infection with the ΔODD LCLL mutant failed to induce the expression of VEGF (Figure 2B) and Glut-1 (data not shown), another HIF-1 target gene. Therefore, these results confirm that the ΔODD LCLL mutant is transcriptionally inactive.
Figure 2.
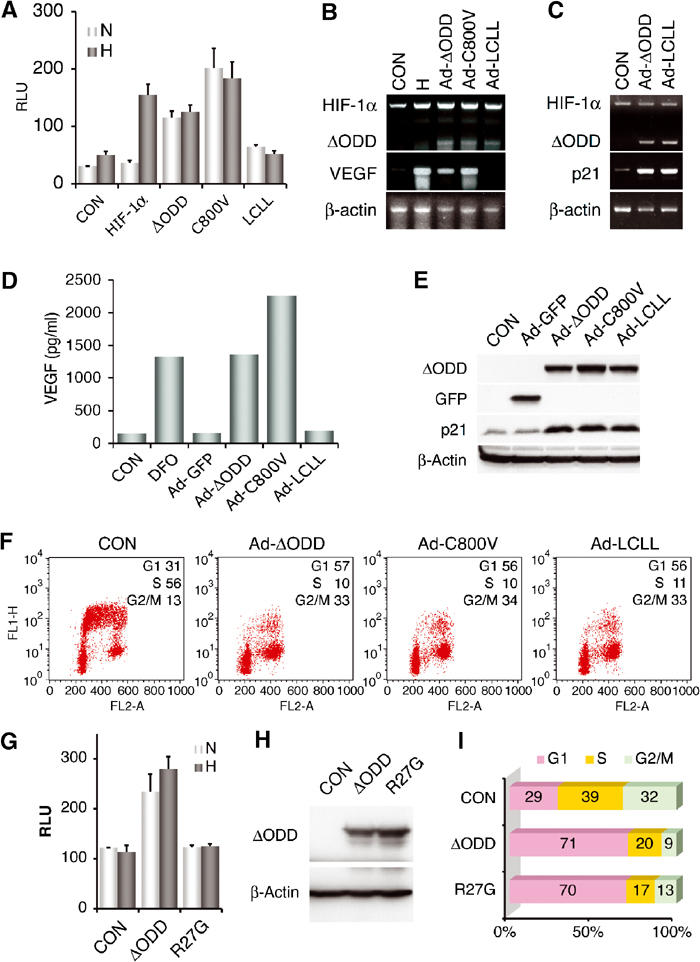
HIF-1α induction of p21cip1 and G1 arrest is independent of its DNA-binding and transcriptional activity. (A) Transcriptional activity of HIF-1α, ΔODD, and ΔODD plus C800V or LCLL mutations was determined in Cos-7 cells in a HIF-1-mediated reporter system (Huang et al, 1996). Relative luciferase units (RLU) are presented with means plus standard errors from three independent experiments. (B, D) HCT116 cells were assayed for VEGF expression by RT–PCR (B) or VEGF secretion by ELISA (D) after adenoviral infection as indicated. DFO, desferrioxamine. (C, E) ΔODD LCLL mutant was analyzed for its effects on p21cip1 expression at mRNA (C) and protein (E) levels. (F) Effect of the ΔODD LCLL mutant on cell cycle profile was determined as above, with the percentage of each phase indicated in the inlet. (G–I) ΔODD and its R27G mutant were assayed in Cos-7 cells for HIF-1-mediated reporter activity (G) and Western analysis of protein levels (H), and in HCT116 cells for induction of cell cycle arrest (I). CON, transfected with pcDNA3.
Next, we tested the effects of ΔODD LCLL on cell cycle progression. Surprisingly, cells infected with Ad-LCLL also manifested slowed growth, similar to those infected with Ad-ΔODD, or treated with desferrioxamine, a hypoxia mimetic (data not shown). Whereas Ad-LCLL infection failed to increase VEGF secretion (Figure 2D), cell cycle arrest was observed in the infected HCT116 cells (Figure 2F) and normal human fibroblasts (data not shown). Furthermore, both p21cip1 mRNA and protein levels were elevated upon Ad-LCLL infection (Figure 2C and E), arguing against the requirement of HIF-1α transcriptional activity for p21cip1 activation.
This surprising finding suggested that HIF-1α activates p21cip1 in a way that is distinct from the activation of classic hypoxia-inducible genes such as VEGF and glycolytic enzyme genes. To test this hypothesis, we investigated whether HIF-1α DNA binding is required for growth arrest. To this end, the basic region required for HIF-1α DNA binding was mutated by replacing Arg-27 with glycine. Despite its equivalent level of expression to that of ΔODD, the ΔODD R27G mutant failed to activate the HIF-1 reporter (Figure 2G and H), indicating the loss of DNA-binding ability. However, the mutant was still able to induce G1 arrest (Figure 2I). Thus, we conclude that neither HIF-1α transcriptional activity nor its DNA binding is required for p21cip1 activation.
HIF-1α functionally antagonizes Myc
As p21cip1 is transcriptionally repressed by Myc (Herold et al, 2002; Seoane et al, 2002; Gartel and Shchors, 2003), we hypothesized that HIF-1α-mediated p21cip1 activation is a result of HIF-1α counteraction of Myc repression. To this end, a Myc-activated hTERT promoter (Horikawa et al, 2002) was tested for the HIF-1α antagonism. Whereas transient Myc expression resulted in a moderate increase (Eisenman, 2001; Levens, 2002) in hTERT reporter activity, co-expression of ΔODD significantly inhibited the induction (Figure 3A). Likewise, Ad-ΔODD infection also markedly downregulated the expression of Myc-activated genes including hTERT and BRCA1 (Figure 3B). Real-time PCR confirmed a seven-fold reduction in hTERT expression (data not shown). In addition, infection with the transcriptionally inactive mutant Ad-LCLL also inhibited the Myc-activated genes (Supplementary Figure). It is noteworthy that neither Ad-ΔODD nor hypoxia (data not shown) altered c-myc mRNA and protein levels, supporting the notion that HIF-1α functionally antagonizes Myc.
Figure 3.
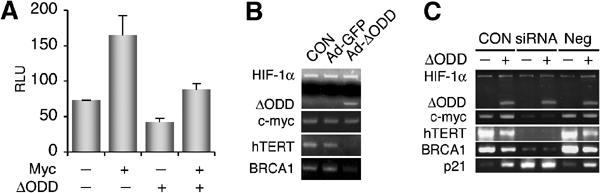
HIF-1α counteracts Myc activity. (A) Effect of ΔODD on Myc transcriptional activity was examined with an hTERT luciferase reporter in HCT116 cells. The graph represents three independent experiments in duplicate. (B) Myc target genes, as indicated, were analyzed by RT–PCR after adenoviral infection. c-myc mRNA levels were also determined. (C) After transfection with c-myc siRNA (siRNA), HCT116 cells were infected with Ad-ΔODD and analyzed with RT–PCR for Myc target gene expression, as indicated. CON, no siRNA. Neg, negative control siRNA.
To confirm the requirement of Myc for HIF-1α effects on p21cip1, hTERT, and BRCA1, we employed c-myc small interfering RNA (siRNA) that effectively abrogated c-myc expression (Figure 3C). Consequently, hTERT and BRCA1 expression was inhibited, whereas p21cip1 expression was upregulated, confirming an essential role of Myc in regulating these genes. More importantly, c-myc siRNA abrogated the HIF-1α antagonism, as evidenced by the lack of further changes in the expression of p21cip1, hTERT, and BRCA1. These results confirm an indispensable role of Myc in HIF-1α counteraction.
Hypoxia-induced HIF-1α dictates Myc target gene expression
To gain insights into the underlying mechanism of the HIF-1α antagonism, we determined HIF-1α and Myc binding to the p21cip1 promoter. The results from chromatin-immunoprecipitation assays showed that in normoxia Myc, but not HIF-1α, bound the proximal region of the promoter (Figure 4A, top panel), consistent with a previous report (Seoane et al, 2002). In addition, both Miz-1 and Sp1 also interacted with this region (Gartel and Shchors, 2003). Upon Ad-ΔODD infection or hypoxia (data not shown), HIF-1α occupied the same region of the p21cip1 promoter, whereas Myc binding was significantly weakened, suggesting that HIF-1α has a higher affinity than Myc for the p21cip1 promoter. However, when the upstream p53-binding region of the promoter was analyzed, only p53 binding was detected (bottom panel), arguing for the specificity of Myc and HIF-1α binding. Therefore, these results indicate that HIF-1α activation of p21cip1 is mediated by the displacement of Myc from the p21cip1 promoter.
Figure 4.
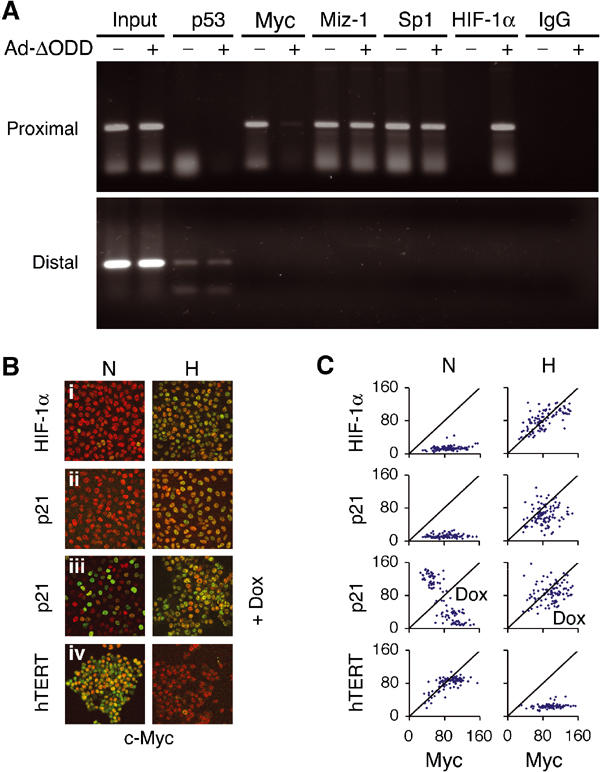
HIF-1α displaces Myc binding from p21cip1 promoter and overrides Myc target gene expression. (A) After Ad-ΔODD infection of HCT116 cells, chromatin immunoprecipitation assays were performed with indicated antibodies at the top. Both the proximal and distal regions of p21cip1 promoter were analyzed. Input, genomic DNA prior to immunoprecipitations. (B) Normoxic (N) and hypoxic (H) HCT116 cells were stained by immunofluorescence with anti-Myc antibody (in red) and antibodies against HIF-1α, p21cip1, or hTERT (in green). Only merged images are presented. The cells in panel iii were treated with doxorubicin (Dox). (C) Myc expression levels from 100 individual cells in (B) were plotted in dots along the x-axis against those of HIF-1α, p21, or hTERT in the y-axis. Cells expressing both proteins are depicted in dots along the diagonal lines.
To substantiate the biological relevance of HIF-1α effects on Myc target gene, we evaluated the hypoxic effects on Myc target gene expression. Immunofluorescence staining showed that vast majority of the cells co-expressed Myc with p21cip1 and HIF-1α under hypoxia (Figure 4B and C). Although treatment with DNA-damaging agents such as doxorubicin rendered cells expressing Myc or p21cip1, but not both (Seoane et al, 2002), additional hypoxic treatment converted the cells to co-expression of the two, indicating an authoritative role of hypoxia in Myc target gene expression. Consistently, hypoxia abolished normoxic co-expression of hTERT and Myc. Similar results were obtained with Ad-ΔODD infection (data not shown). The immunofluorescence staining of Myc and p21cip1 was validated by knocking down their expression with siRNAs; both Myc and p21cip1 siRNAs ablated the staining of the corresponding genes and altered the expression patterns (Supplementary Figure). Thus, these results support that hypoxia-induced HIF-1α dictates Myc target gene expression.
HIF-1α interacts with Myc in vivo
To ascertain the biochemical aspect of the HIF-1α antagonism, we asked whether HIF-1α displaces Myc through protein–protein interactions. To this end, both ΔODD and Myc were transiently co-expressed in normoxic cells for co-immunoprecipitation assays. Figure 5A showed that Myc was weakly detected in an anti-hemagglutinin (HA) immunoprecipitation targeting ΔODD. Conversely, ΔODD was present in the anti-Myc immunocomplexes (Figure 5B), suggesting in vivo complex formation between HIF-1α and Myc. The complex specificity was confirmed further by antibody omissions and isotype-matched normal immunoglobulins (Figure 5C). To provide the physiological relevance of this finding, we examined endogenous HIF-1α and Myc interaction in hypoxic cells. Again, Myc was weakly detected in an anti-HIF-1α co-immunoprecipitation (Figure 5D). Therefore, we conclude that hypoxia-induced HIF-1α weakly interacts with Myc in vivo.
Figure 5.
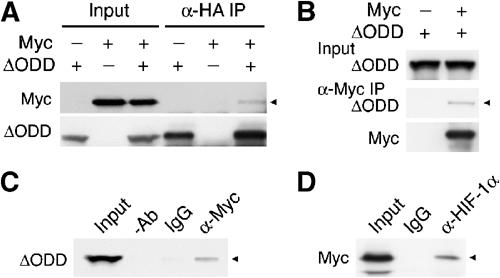
HIF-1α forms a weak complex with Myc in vivo. (A, B) ΔODD and Myc, as indicated, were co-expressed in 293 cells. Co-immunoprecipitations with antibodies against HA (α-HA IP) or Myc (α-Myc IP) were performed, followed by Western analyses detecting co-immunoprecipitates (marked with arrow heads), as well as immunoprecipitates. Input, whole-cell extracts subjected to direct Western analysis. (C) Omission of antibody (−Ab) and use of normal immunoglobulin (IgG) were included as controls for an anti-Myc co-immunoprecipitation. (D) The interaction of endogenous HIF-1α and Myc in HCT116 cells was shown in an anti-HIF-1α co-immunoprecipitation, followed by anti-Myc Western blotting.
The N-terminal HIF-1α is sufficient for cell cycle arrest
In search of the minimum requirement for HIF-1α to form a complex with Myc, we tested a set of HIF-1α mutants with C-terminal deletions (Figure 6A). Whereas HIF-1α mutants (amino acids 1–400 and 1–329) remained interactive with Myc (Figure 6B), further removal of the PAS-B domain from the C terminus abolished the interaction (Figure 6C), suggesting that Myc interaction requires the N-terminal HIF-1α containing bHLH and PAS domains (Semenza, 1999). To validate the functional relevance of the N-terminal HIF-1α interaction with Myc, we tested the effects of the N-terminal HIF-1α fragments on cell cycle checkpoint. In agreement with the above results, HIF-1α 1–400 not only stimulated p21cip1 reporter expression (Figure 6D) but also induced G1 arrest (Figure 6E). In contrast, HIF-1α 1–167 showed no effect. To confirm the dispensable role of HIF-1α DNA binding in cell cycle arrest, we again mutated the basic region with R27G mutation. As expected, a similar G1 arrest was detected with this mutant (Supplementary Figure). Therefore, we conclude that the N-terminal HIF-1α is sufficient for functionally counteracting Myc, resulting in derepression of p21cip1.
Figure 6.
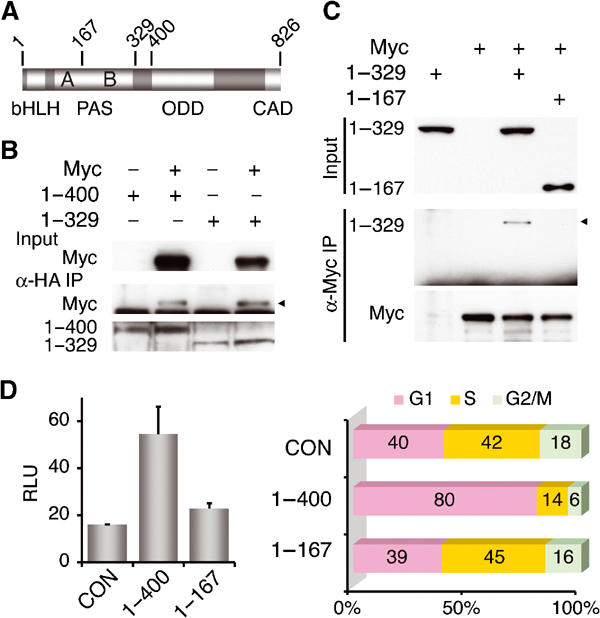
The N-terminal HIF-1α interacts with Myc and induces cell cycle arrest. (A) A schematic representation of HIF-1α. Structural domains are indicated at the bottom (Huang and Bunn, 2003), and the relevant codon numbers at the top. (B, C) HIF-1α mutants (amino acids 1–400, 1–329, and 1–167) were transfected into 293 cells, and analyzed by co-immunoprecipitation assays to determine the minimum requirement for Myc binding. (D, E) The N-terminal HIF-1α (1–400 and 1–167) was examined for its ability to activate p21cip1 promoter in Cos-7 cells (D), and to alter cell cycle in HCT116 cells (E). CON, transfected with pcDNA3.
Discussion
In this study, not only did we provide evidence that HIF-1α, even in the absence of hypoxic signal, is sufficient to induce cell cycle arrest by upregulating p21cip1 expression, but more significantly we unravelled the mechanism by which HIF-1α activates p21cip1. In particular, we demonstrated that HIF-1α counteracts Myc activity by displacing Myc binding from the p21cip1 promoter. Remarkably, it is the N-terminal HIF-1α comprising bHLH and PAS domains that functionally antagonizes Myc, a novel mechanism distinct from HIF-1α C-terminal transactivation of the classic hypoxia-inducible genes (e.g. VEGF, Glut-1). Therefore, HIF-1α employs at least two mechanisms for regulating gene expression: in addition to its C-terminal transactivation of genes possessing HREs, HIF-1α, via its N-terminal counteraction of Myc activity, independently overrides the expression of Myc target genes lacking the canonical HRE. Interestingly, HIF-1α splice variants devoid of the C-terminal transactivation domains have been reported (Chun et al, 2001, 2002), although their role in counteracting Myc activity remains to be determined. The HIF-1α antagonism of Myc is not limited to p21cip1 activation, because HIF-1α also downregulates the expression of hTERT and BRCA1, known targets of Myc (Eisenman, 2001; Levens, 2002). In addition, HIF-1α has also been implicated in hypoxic induction of cyclin-dependent kinase inhibitor p27Kip1 (Goda et al, 2003), another Myc target (Obaya et al, 2002) that might be regulated by the same mechanism. It should be stressed that none of these genes possesses a canonical HRE in the promoter. Thus, HIF-1α counteraction of Myc may serve as a general mechanism underlying up- or downregulation of Myc target genes in response to hypoxia (Figure 7).
Figure 7.
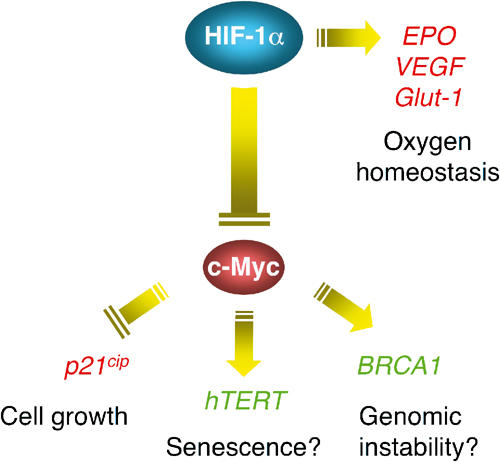
An HIF-1α–Myc pathway regulating hypoxia-responsive genes lacking the canonical HRE. Apart from activating the classic hypoxia-inducible genes such as erythropoietin (EPO), VEGF, and Glut-1, HIF-1α functionally counteracts Myc, thereby overriding Myc target gene expression. The upregulated genes are in red, and the downregulated in green.
Our finding that neither HIF-1α DNA binding nor its transcriptional activity is required for p21cip1 activation and cell cycle arrest does not support the possibility that HIF-1α directly binds to the promoter of p21cip1, which contains ACGTG sequence (Salnikow et al, 2000), hTERT, and BRCA1, thereby transcriptionally activating or inactivating gene expression. Furthermore, this finding also argues against the hypothesis that the HIF-1α antagonism of Myc results directly from HIF-1α transcriptional activation of target genes that in turn repress Myc activity, because it is highly improbable that an HIF-1α mutant lacking DNA-binding and transcriptional activity is able to transactivate Myc repressor genes. Although direct HIF-1α DNA binding to the promoter cannot be completely ruled out, the overall evidence favors the HIF-1α–Myc interaction as a primary mode of action in altering the Myc target gene expression under hypoxia.
We showed that the HIF-1α antagonism is mediated by Myc displacement, resulting in derepression of p21cip1 promoter. In fact, not only hypoxia, but a stable HIF-1α in normoxia as well, is able to displace Myc binding, suggesting an authoritative role of HIF-1α in Myc target gene expression. This notion is consistent with and supported by another line of evidence that hypoxia overrides Myc target gene expression, as shown in Figure 4B and C. HIF-1α displacement of Myc is also in good agreement with the observation of weak HIF-1α–Myc interaction in vivo, presumably resulting from the transient protein–protein interactions during competition, or the residual Myc binding after competition. The biological significance of this weak interaction, however, is underscored by the finding that the N-terminal HIF-1α not only binds Myc, but also activates p21cip1 promoter and thereby induces cell cycle arrest (Figure 6), arguing for the functional relevance of the interaction.
It is noteworthy that, despite its constitutive binding to the distal region of p21cip1 promoter (Figure 4A), p53 is not absolutely required for hypoxia-induced p21cip1 activation (unpublished data), in agreement with a previous report (Goda et al, 2003). This finding is supported by the observation that hypoxia-induced p53 is transcriptionally incompetent for activating downstream target genes (Koumenis et al, 2001). Therefore, it is likely that p53 has a limited, yet nonessential role in facilitating hypoxic activation of p21cip1 promoter.
Myc is known to play a pivotal role in a transcription factor network that regulates cellular proliferation, growth, differentiation, and apoptosis (Dang, 1999). It has been reported recently that Myc is essential for vasculogenesis and angiogenesis during development and tumor progression (Baudino et al, 2002), apparently in disagreement with the theory of HIF-1α counteraction of Myc. Furthermore, Myc has also been shown to activate glucose transporter 1 and glycolytic genes (Osthus et al, 2000), transcriptional targets of HIF-1α as well. These seemingly contradictory observations can be reconciled, however, by the interpretation that Myc is only responsible for normoxic induction of these genes, whereas in hypoxia HIF-1α seizes control of gene expression as a result of its counteraction of Myc. Therefore, both HIF-1α and Myc may function coherently to enhance glycolysis in tumor tissues. A crucial experiment to test this hypothesis would be to examine the requirement of Myc for increased glycolysis in hypoxia. Another possible explanation is that Myc downregulates the expression of HIF-1α E3 ubiquitin ligase, the von Hippel–Lindau protein (O'Connell et al, 2003), thereby giving rise to a more stable HIF-1α that accounts for glycolysis. Given the complexity of tumor biology, how HIF-1α and Myc contribute to glycolysis and angiogenesis needs in-depth investigation.
Although HIF-1α-induced cell cycle arrest may seem paradoxical to its documented role in tumor growth, recent reports from well-controlled studies support our hypothesis that HIF-1α inhibits cell proliferation. HIF-1α−/− tumors unexpectedly grew faster (Carmeliet et al, 1998) and became more invasive than its HIF-1α+/+ counterpart, when oxygen supply was adequate (Blouw et al, 2003). Furthermore, HIF-α stabilization failed to promote, but rather decreased, tumor growth (Mack et al, 2003). Interestingly, a stable HIF-2α (Kondo et al, 2002), but not HIF-1α (Maranchie et al, 2002), overrides the tumor-suppressing activity of the von Hippel–Lindau protein, suggesting differential roles for HIF-1α and HIF-2α in tumor growth. Undoubtedly, further investigation is warranted for a better understanding of the divergent roles of HIF-1α and HIF-2α in tumorigenesis.
Materials and methods
Cell culture and hypoxic treatment
HCT116p53+/+ and HCT116p21−/− (gifts from B Vogelstein) were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum. Normal human fibroblasts (MRC5, GM07532), 293, and Cos-7 cells were grown in DMEM medium supplemented with 10% bovine calf serum. Hypoxic treatment was carried out in an incubator (NAPCO) with 1% O2, 5% CO2, and 94% N2, or in the presence of 200 μM desferrioxamine as indicated.
Adenoviruses
Recombinant adenoviruses expressing ODD-deleted HIF-1α, or its variant C800V and LCLL were created with the AdEasy adenoviral vector system (Stratagene). Adenoviral infection was carried out in 5% fetal bovine serum media for 1 h at 37°C. In general, 3 moi was used for tumor cell lines and 10 moi for nontransformed cells. Cells were harvested after 8 h infection.
Cell cycle analysis
Cells were labeled with 10 μM bromodeoxyuridine (BrdU) for 30 min, and fixed in 70% ethanol for 1 h. Subsequently, cells were treated with 2 N hydrochloric acid plus 0.5% Triton X-100. After wash, cells were stained with anti-BrdU antibody (BD Biosciences) and propidium iodide, and analyzed by FlowJo (Treestar). Figures are representatives of at least three independent and reproducible experiments.
Cell cycle profile was also determined from cells transfected with 1 μg of indicated p(HA)HIF1α mutant constructs plus 0.1 μg pEGFP-F (Clontech). The GFP-positive population was analyzed with propidium iodide staining (Nagashima et al, 2001).
Transfection
Reporter assays were performed essentially as described previously with 0.25 μg reporter and 0.1 μg pEYFP-Nuc for normalization (Huang et al, 2002). hTERT promoter activity was assayed with 0.1 μg pMyc (Horikawa et al, 2002) and 0.2 μg p(HA)HIF1α(401Δ603) (Huang et al, 1998). HIF-1 reporter assays were performed with pEpoE-luc containing a 3′ enhancer of erythropoietin (Huang et al, 1996) co-transfected with 0.1 μg pARNT and p(HA)HIF1α, p(HA)HIF1α(401Δ603), or its variants. p21cip1 reporter (pWWP-luc, a gift from B Vogelstein) was assayed with 0.1 μg pHIF1α(1–400) and 0.1 μg pMyc.
The interaction between Myc and HIF-1α was determined by transfecting cells in six-well plates with 0.6 μg Myc and 1 μg p(HA)HIF1α(401Δ603), p(HA)HIF1α(1-400), p(HA)HIF1α(1-329), or p(HA)HIF1α(1-167) (Huang et al, 1998), followed by co-immunoprecipitations.
Western blot and co-immunoprecipitation
Western blot was conducted as described previously (Huang et al, 1998). Co-immunoprecipitation was carried out in NETN buffer (Gu et al, 2001), followed by Western blot to identify precipitates and co-precipitates. One-fifth of the whole-cell extracts were subject to direct Western blot for quantification of the input amounts. Monoclonal antibodies against HIF-1α and p21cip1 were purchased from BD Biosciences, and anti-HA antibody from Roche. Polyclonal anti-Myc antibody was from Upstate.
For detection of endogenous HIF-1α–Myc complex, HCT116 cells grown in two 10 cm plates were treated with 12.5 μM Cbz-LLL in hypoxia for 4 h. Subsequently, cells were lysed in a lysis buffer containing 25 mM Tris, 300 mM NaCl, 1% Triton X-100, and protease inhibitor Complete (Roche). The lysates were pre-cleared by incubation with protein A agarose beads for 1 h, and divided into two: one for immunoprecipitation with polyclonal anti-HIF-1α antibody (Santa Cruz) and the other with normal rabbit IgG. A monoclonal anti-Myc antibody (Oncogene) was used for Western blot.
Reverse transcription–polymerase chain reaction (RT–PCR)
Total RNA was extracted with Trizol (Invitrogen) according to the manufacturer's instructions. One-step RT–PCR was carried out using AccessQuick RT–PCR kit (Promega) with 0.5 μg total RNA. Primers for HIF-1α flank the ODD so that both endogenous and ODD-deleted HIF-1α can be detected simultaneously.
Chromatin immunoprecipitation assays
Following 16-h treatment with hypoxia or adenoviral infection, HCT116 cells were crosslinked with formaldehyde as described previously (Boyd et al, 1998). Chromatin immunoprecipitations were performed by using the Chromatin Immunoprecipitation Assay Kit (Upstate) following the manufacturer's instructions. Antibodies used include rabbit polyclonal anti-Myc (Upstate), mouse monoclonal anti-p53 (BD Biosciences), goat polyclonal anti-Miz-1, mouse monoclonal anti-Sp1, rabbit polyclonal anti-HIF-1α, and normal rabbit IgG (Santa Cruz Biotechnology). The PCR primers spanning the distal region of the p21cip1 promoter (−2312 to −2131) are 5′-CAGGCTGTGGCTCTGATTGG-3′ (forward) and 5′-TTCAGAGTAACAGGCTAAGG-3′ (reverse), and those covering the proximal region (−194 to +88), 5′-ACCGGCTGGCCTGCTGGAACT- 3′ (forward) and 5′-TCTGCCGCCGCTCTCTCACCT (reverse).
Enzyme-linked immunoabsorbant assay (ELISA)
VEGF levels in the cell culture medium were determined with a human VEGF ELISA kit (American Research Products). The VEGF level was quantified by comparing its optical density to the standard curve in accordance with the manufacturer's protocol.
Immunofluorescence staining and confocal microscopy
HCT116 cells were maintained in normoxia or incubated in hypoxia for 8 h. Some were pretreated with 0.34 μM doxorubicin. Cells were then fixed in 4% paraformaldehyde in PBS for 15 min, and permeabilized with 0.1% Triton X-100 in PBS for another 15 min. Subsequently, cells were incubated with 5% chicken serum. Primary antibodies used at 1:100 dilutions include rabbit anti-Myc (Upstate), mouse ant-Myc (Oncogene), mouse anti-p21 (BD Biosciences), rabbit anti-HIF-1α (Santa Cruz), and goat anti-hTERT (Santa Cruz). Secondary antibodies against mouse, rabbit or goat were conjugated with FITC, Texas red (Molecular Probes) or AMCA (Jackson Immuno Research). Confocal laser scanning was carried out using a Zweiss LSM 510 microscope.
The area and mean pixel intensities of Myc, HIF-1α, p21cip1, and hTERT were quantified, respectively, from 100 individual cells and analyzed with Quantitative analysis software (Zweiss). Myc expression levels in each cell were plotted in x-axis in dots, and the HIF-1α, p21cip1, or hTERT expression levels in the corresponding cells were in y-axis to demonstrate the co-expression of these proteins in single cells. Cells co-expressing both proteins are distributed along the diagonal line.
RNA interference
Silencer c-myc siRNA, negative control siRNA, and Silencer siRNA transfection kit were purchased from Ambion, and used according to the manufacturer's instructions. At 36 h after transfection with siRNA, cells were infected with Ad-ΔODD.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We thank Xin W Wang for the helpful suggestions on the cell cycle study, Bert Vogelstein for reagents, and Curt C Harris for critical reading of the manuscript. We are grateful to the Gene Expression Laboratory of the SAIC Frederick Inc. for adenovirus productions, and Susan Garfield and Remy Pedeux for their advice on immunofluorescence staining and confocal microscopy.
References
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM (1996) An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA 93: 12969–12973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL (2002) c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16: 2530–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, Johnson RS, Bergers G (2003) The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 4: 133–146 [DOI] [PubMed] [Google Scholar]
- Boyd KE, Wells J, Gutman J, Bartley SM, Farnham PJ (1998) c-Myc target gene specificity is determined by a post-DNA binding mechanism. Proc Natl Acad Sci USA 95: 13887–13892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK (2000) Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA 97: 9082–9087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294: 1337–1340 [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshet E (1998) Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394: 485–490 [DOI] [PubMed] [Google Scholar]
- Carrero P, Okamoto K, Coumailleau P, O'Brien S, Tanaka H, Poellinger L (2000) Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol 20: 402–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun YS, Choi E, Kim TY, Kim MS, Park JW (2002) A dominant-negative isoform lacking exons 11 and 12 of the human hypoxia-inducible factor-1alpha gene. Biochem J 362: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun YS, Choi E, Yeo EJ, Lee JH, Kim MS, Park JW (2001) A new HIF-1 alpha variant induced by zinc ion suppresses HIF-1-mediated hypoxic responses. J Cell Sci 114: 4051–4061 [DOI] [PubMed] [Google Scholar]
- Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH (2000) Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel–Lindau tumor suppressor protein. J Biol Chem 275: 25733–25741 [DOI] [PubMed] [Google Scholar]
- Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE (2002) Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA 99: 5271–5276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (1999) c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman RN (2001) Deconstructing myc. Genes Dev 15: 2023–2030 [DOI] [PubMed] [Google Scholar]
- Elson DA, Thurston G, Huang LE, Ginzinger DG, McDonald DM, Johnson RS, Arbeit JM (2001) Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev 15: 2520–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107: 43–54 [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ (2002) Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA 99: 5367–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV (2001) Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem 276: 7919–7926 [DOI] [PubMed] [Google Scholar]
- Gartel AL, Shchors K (2003) Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp Cell Res 283: 17–21 [DOI] [PubMed] [Google Scholar]
- Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS (2003) Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23: 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SL, Freiberg RA, Giaccia AJ (2001) p21(Cip1) and p27(Kip1) regulate cell cycle reentry after hypoxic stress but are not necessary for hypoxia-induced arrest. Mol Cell Biol 21: 1196–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Milligan J, Huang LE (2001) Molecular mechanism of hypoxia-inducible factor 1alpha-p300 interaction. A leucine-rich interface regulated by a single cysteine. J Biol Chem 276: 3550–3554 [DOI] [PubMed] [Google Scholar]
- Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, Syvaoja J, Saluz HP, Haenel F, Eilers M (2002) Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell 10: 509–521 [DOI] [PubMed] [Google Scholar]
- Horikawa I, Cable PL, Mazur SJ, Appella E, Afshari CA, Barrett JC (2002) Downstream E-box-mediated regulation of the human telomerase reverse transcriptase (hTERT) gene transcription: evidence for an endogenous mechanism of transcriptional repression. Mol Biol Cell 13: 2585–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Arany Z, Livingston DM, Bunn HF (1996) Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem 271: 32253–32259 [DOI] [PubMed] [Google Scholar]
- Huang LE, Bunn HF (2003) Hypoxia-inducible factor and its biomedical relevance. J Biol Chem 278: 19575–19578 [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF (1998) Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA 95: 7987–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Pete EA, Schau M, Milligan J, Gu J (2002) Leu-574 of HIF-1alpha is essential for the von Hippel–Lindau (VHL)-mediated degradation pathway. J Biol Chem 277: 41750–41755 [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468 [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292: 468–472 [DOI] [PubMed] [Google Scholar]
- Kallio PJ, Okamoto K, O'Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L (1998) Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J 17: 6573–6586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio PJ, Wilson WJ, O'Brien S, Makino Y, Poellinger L (1999) Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. J Biol Chem 274: 6519–6525 [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW (2000) Activation of HIF1alpha ubiquitination by a reconstituted von Hippel–Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci USA 97: 10430–10435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG (2002) Inhibition of HIF is necessary for tumor suppression by the von Hippel–Lindau protein. Cancer Cell 1: 237–246 [DOI] [PubMed] [Google Scholar]
- Koumenis C, Alarcon R, Hammond E, Sutphin P, Hoffman W, Murphy M, Derr J, Taya Y, Lowe SW, Kastan M, Giaccia A (2001) Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol Cell Biol 21: 1297–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AL, Wang S, Klco JM, Kaelin WG, Livingston DM (2000) Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med 6: 1335–1340 [DOI] [PubMed] [Google Scholar]
- Levens D (2002) Disentangling the MYC web. Proc Natl Acad Sci USA 99: 5757–5759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack FA, Rathmell WK, Arsham AM, Gnarra J, Keith B, Simon MC (2003) Loss of pVHL is sufficient to cause HIF dysregulation in primary cells but does not promote tumor growth. Cancer Cell 3: 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD (2002) The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 1: 247–255 [DOI] [PubMed] [Google Scholar]
- Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J 20: 5197–5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399: 271–275 [DOI] [PubMed] [Google Scholar]
- Nagashima M, Shiseki M, Miura K, Hagiwara K, Linke SP, Pedeux R, Wang XW, Yokota J, Riabowol K, Harris CC (2001) DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc Natl Acad Sci USA 98: 9671–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, Sedivy JM (2003) A large scale genetic analysis of c-Myc-regulated gene expression patterns. J Biol Chem 278: 12563–12573 [DOI] [PubMed] [Google Scholar]
- Obaya AJ, Kotenko I, Cole MD, Sedivy JM (2002) The proto-oncogene c-myc acts through the cyclin-dependent kinase (Cdk) inhibitor p27(Kip1) to facilitate the activation of Cdk4/6 and early G(1) phase progression. J Biol Chem 277: 31263–31269 [DOI] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel–Lindau protein. Nat Cell Biol 2: 423–427 [DOI] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275: 21797–21800 [DOI] [PubMed] [Google Scholar]
- Salceda S, Caro J (1997) Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 272: 22642–22647 [DOI] [PubMed] [Google Scholar]
- Salnikow K, Costa M, Figg WD, Blagosklonny MV (2000) Hyperinducibility of hypoxia-responsive genes without p53/p21-dependent checkpoint in aggressive prostate cancer. Cancer Res 60: 5630–5634 [PubMed] [Google Scholar]
- Semenza GL (1999) Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol 15: 551–578 [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Massague J (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419: 729–734 [DOI] [PubMed] [Google Scholar]
- Tanimoto K, Makino Y, Pereira T, Poellinger L (2000) Mechanism of regulation of the hypoxia-inducible factor-1alpha by the von Hippel–Lindau tumor suppressor protein. EMBO J 19: 4298–4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix–loop–helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92: 5510–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger RH (2002) Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J 16: 1151–1162 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4