

Abstract
In the mammalian central nervous system, the postsynaptic small-conductance Ca2+-dependent K+ (SK) channel has been shown to reduce postsynaptic depolarization and limit Ca2+ influx through N-methyl-d-aspartate receptors. To examine further the role of the postsynaptic SK channel in synaptic transmission, we studied its action at the Drosophila larval neuromuscular junction (NMJ). Repetitive synaptic stimulation produced an increase in postsynaptic membrane conductance leading to depression of excitatory postsynaptic potential amplitude and hyperpolarization of the resting membrane potential (RMP). This reduction in synaptic excitation was due to the postsynaptic Drosophila SK (dSK) channel; synaptic depression, increased membrane conductance and RMP hyperpolarization were reduced in dSK mutants or after expressing a Ca2+ buffer in the muscle. Ca2+ entering at the postsynaptic membrane was sufficient to activate dSK channels based upon studies in which the muscle membrane was voltage clamped to prevent opening voltage-dependent Ca2+ channels. Increasing external Ca2+ produced an increase in resting membrane conductance and RMP that was not seen in dSK mutants or after adding the glutamate-receptor blocker philanthotoxin. Thus it appeared that dSK channels were also activated by spontaneous transmitter release and played a role in setting membrane conductance and RMP. In mammals, dephosphorylation by protein phosphatase 2A (PP2A) increased the Ca2+ sensitivity of the SK channel; PP2A appeared to increase the sensitivity of the dSK channel since PP2A inhibitors reduced activation of the dSK channel by evoked synaptic activity or increased external Ca2+. It is proposed that spontaneous and evoked transmitter release activate the postsynaptic dSK channel to limit synaptic excitation and stabilize synapses.
Keywords: postsynaptic Ca2+, SK channel; synaptic strength
the regulation of synaptic strength is crucial to nervous system function and a number of mechanisms act in parallel to produce short- and long-term changes in synapses. The presynaptic intracellular free Ca2+ concentration ([Ca2+]i) regulates synaptic strength at a broad range of synapses through the production of facilitation, augmentation and posttetanic potentiation (Zucker and Regehr 2002). Postsynaptic [Ca2+]i also regulates synaptic strength by triggering both long-term potentiation (LTP) and long-term depression at central synapses (Malenka and Bear 2004); however, there is much less evidence for a role of postsynaptic Ca2+ at peripheral synapses. Although small-conductance Ca2+-dependent K+ (SK) channels were initially noted for their contribution to the after-hyperpolarization and control of action potential firing frequencies, it appears that Ca2+ activation of postsynaptic SK channels can regulate synaptic strength and plasticity (Faber and Sah 2007). Synaptic activation of postsynaptic SK channels in pyramidal neurons of the rat lateral amygdala and CA1 neurons in mouse hippocampus led to a reduction in excitatory postsynaptic potential (EPSP) amplitude (Faber et al. 2005; Ngo-Anh et al. 2005). Here, the SK channels were closely coupled to the N-methyl-d-aspartate (NMDA) receptor and specifically acted to limit Ca2+ influx through NMDA receptors by reducing postsynaptic depolarization.
It remained unclear whether postsynaptic SK channels played a more general role in regulating synaptic excitation. To examine this, we studied the effect of the postsynaptic Drosophila SK channel (dSK) in regulating synaptic excitation at the larval neuromuscular junction (NMJ). The Drosophila NMJ has become a popular model system for the study of synapses, and these identified synapses are particularly good to study the regulation of synaptic strength (Keshishian et al. 1996). Here synaptic strength must be precisely regulated since synaptic depolarization grades muscle fiber contraction, and very few fibers are used to produce movement. The larval muscle fibers have non-NMDA glutamate receptors that admit Ca2+, and the muscle also appeared to contain a dSK channel (Abou Tayoun et al. 2011; Chang et al. 1994). In addition, we had observed that experimental increases in postsynaptic [Ca2+]i produced reduced synaptic excitation due to activation of a Ca2+-dependent K+ conductance (gKCa). In the current experiments, we studied whether transmitter release activated the postsynaptic dSK channel. We found that Ca2+ entry at the postsynaptic membrane during spontaneous and evoked transmitter release activated the dSK channel to hyperpolarize the membrane and reduce EPSP amplitude. We propose that the postsynaptic SK channel acts generally as a control mechanism to limit synaptic excitation and stabilize synapses.
MATERIALS AND METHODS
Experiments were performed on muscle fiber 6 in segments 3 and 4 of Drosophila wandering third-instar larvae. The following Drosophila stocks were used: wild type, Canton-S (CS); dSK−, which contains a deletion in the Drosophila SK gene (Abou Tayoun et al. 2011); slo1 (Bloomington stock 4587), which eliminates gCF in larval muscle; P(GawB)how24B (Bloomington stock 1767), expresses GAL4 in all embryonic and larval somatic muscles; and UAS-dSKDNmyc, which contains a dominant-negative dSK subunit (Abou Tayoun et al. 2011). P(GawB)how24B and UAS-dSKDNmyc were crossed to express the dominant-negative dSK subunit in only muscle fibers (Brand and Perrimon 1993). After an incision through the dorsal body wall, the larvae were pinned out in a physiology chamber, and the internal organs were removed to expose the body-wall muscles. In our initial studies, the preparation was bathed in HL3 saline (Stewart et al. 1994) containing 1 mM Ca2+, and in later studies, we used HL3.1 saline (Feng et al. 2004) with 0 or 1.5 mM Ca2+.
Electrophysiology.
To evoke synaptic responses, the cut end of the segmental nerve was stimulated with a suction electrode connected to a S11 stimulator (Grass-Telefactor, West Warwick, RI). Both axons were stimulated to record the compound EPSPs or excitatory postsynaptic currents (EPSCs) (referred to as simply EPSPs or EPSCs). EPSPs or EPSCs were recorded using sharp microelectrodes (20–30 MΩ filled with 3 M KCl) connected to Axoclamp 2A or GeneClamp 500 (Molecular Devices, Sunnyvale, CA). Data were acquired (sampling rate 5–10 kHz) and analyzed using a Digidata 1440A digitizer (Molecular Devices) and pCLAMP 10.3 software (Molecular Devices). For voltage clamping, a grounded shield was placed between the electrodes to reduce capacitive coupling, and the holding potential was set at −60 mV. Input conductance (Gin) was measured in current clamp with a single electrode by passing 5 nA of hyperpolarizing current; the bridge was balanced or the electrode resistance was digitally subtracted. During voltage clamp, Gin was measured with −20 mV, 0.4-s voltage steps. For all experiments, the initial EPSP amplitudes represent the mean of 10 responses evoked at 0.1 Hz. To inhibit protein phosphatase 2A (PP2A), we added 100 nM calyculin A (EMD Millipore Chemicals, Billerica, MA) or okadaic acid (Sigma-Aldrich, St. Louis, MO) to the saline.
Data analysis.
SigmaPlot 12.3 (SPSS, Plover, WI) was used for data transformation and statistical analysis. The mean values are presented as mean ± SE, and the n values represent the number of larvae, unless otherwise noted.
Western blot.
For Western blots, larval brain and muscle proteins were separated by SDS-PAGE and electroblotted to nitrocellulose membranes (Laemmli 1970; Towbin et al. 1979). Briefly, brains were isolated from third-instar larvae and homogenized in loading buffer containing reducing agent (NuPAGE kit, Invitrogen). For muscle tissue samples, the brains were first removed from dissected larvae, and then the muscle fibers were scraped off from the underlying body wall. The rest of the procedure was as previously described (Abou Tayoun et al. 2011; Lnenicka et al. 2006).
RESULTS
During repetitive synaptic activity, there was an increase in Gin and resting membrane potential (RMP) due to an increase in postsynaptic [Ca2+]i.
We found that increasing the muscle fiber [Ca2+]i either by blocking the plasma membrane Ca2+ ATPase or direct Ca2+ injection resulted in membrane hyperpolarization and a reduction in EPSP amplitude. This was due to activation of a gKCa and was consistent with previous studies showing a fast, inactivating gKCa (gCF) and a slow gKCa (gCS) in larval muscle fibers (Gho and Mallart 1986; Salkoff 1983; Singh and Wu 1989; Wu et al. 1983).
We determined whether synaptic activity produced an increase in the RMP and membrane conductance (Gm) that was consistent with activation of gKCa. Both motor axons innervating muscle fiber 6 were stimulated at 20 Hz for 60 s, and we measured Gin and RMP before and after stimulation. We used HL3 with reduced Ca2+ (1 mM), since 20-Hz stimulation in this saline produced minimal contraction and allowed stable intracellular recordings. During stimulation, there was a decrease in EPSP amplitude and hyperpolarization of the RMP, which combined to reduce synaptic excitation (Fig. 1A, left). In addition, there was an increase in Gin as a result of 20-Hz stimulation (Fig. 1A, left). We used the EPSP peak potential as a measure of synaptic excitation, since this reflected both the decrease in EPSP amplitude and RMP hyperpolarization. The combined data (Fig. 1B) showed that the final EPSP peak (−56.7 ± 1.9 mV) was significantly more negative than the initial one (−24.7 ± 0.9 mV). This resulted from depression of EPSP amplitude by ∼22 mV and hyperpolarization of the RMP by ∼10 mV (Fig. 1C). The increase in Gin during the train was 0.81 ± 0.11 μS (Fig. 1D); this would have produced about one-third of the decrease in EPSP amplitude seen during 20-Hz stimulation (see below). Surprisingly, there was a small increase in Gin that persisted for almost 10 min after the train, which is likely after the postsynaptic [Ca2+]i had returned to baseline.
Fig. 1.

The effect of repetitive synaptic activity on excitatory postsynaptic potential (EPSP) amplitude, resting membrane potential (RMP) and input conductance (Gin) in control larvae and larvae expressing parvalbumin (24B/PV) in the muscle. A: for muscle fiber 6, EPSPs recorded during 20-Hz stimulation for 60 s (every other EPSP is shown) and current (I) injection before (Initial) and after (Final) stimulation are shown. Calibration for I injection: Canton-S (CS): voltage (V) = 15 mV, 100 ms; 24B/PV: V = 10 mV, 100 ms. I amplitude was 5 nA for both. B: combined data showed that the EPSP peak became more negative during 20-Hz stimulation for CS larvae (n = 55). 24B/PV larvae (n = 8) showed no significant change in the EPSP peak during 20-Hz stimulation. C: the initial and final values measured at the beginning and end of 20-Hz stimulation. For control larvae (CS and UAS-PV, n = 5), the final EPSP amplitude was significantly smaller than the initial one, and the final RMP was significantly greater than the initial RMP. For 24B/PV larvae, the final EPSP amplitude and RMP were not significantly different from the initial ones. D: for control larvae, there was a significant increase in Gin at the end of stimulation (final Gin was measured 1 s after the last EPSP). For 24B/PV larvae, there was also a significant increase in Gin, but it was smaller than that seen in the controls. For CS, Gin was followed for 20 min after stimulation, and a significant increase was still seen 10 min poststimulation. Values are means ± SE. For all statistical tests, comparisons were made to the initial value. *P < 0.05, **P < 0.01,***P < 0.001, paired t-test.
We examined whether the increase in Gin and RMP was due to an increase in postsynaptic [Ca2+]i by expressing the Ca2+ buffer, parvalbumin (PV) in the muscle. Flies with UAS-PV transgenes on both the second and third chromosomes (Harrisingh et al. 2007) were crossed to P(GawB)how24B flies to produce 24B/PV larvae expressing PV in the muscles. The UAS-PV transgene contained a myc epitope tag, and expression of PV in the muscle was confirmed using an antibody to myc (data not shown). During 20-Hz stimulation, PV expression appeared to eliminate the change in the EPSP peak and Gin that was seen in CS larvae (Fig. 1A, right). For 24B/PV larvae, the combined data showed no significant change in the EPSP peak, EPSP amplitude or RMP during 20-Hz stimulation (Fig. 1, B and C). The UAS-PV larvae also served as a control and showed a reduction in EPSP amplitude and an increase in RMP and Gin that was similar to CS larvae. The 24B/PV larvae showed a significant increase in Gin, but the increase was significantly smaller (P < 0.05, ANOVA, Dunn test) than seen for either of the controls (Fig. 1D).
PV expression appears to have resulted in a reduction in transmitter release since the basal EPSP was smaller, and this was not due to a change in resting Gin. The reduction in basal transmitter release would result in less postsynaptic Ca2+ influx, and this could contribute to the reduced change in RMP and Gin in 24B/PV larvae. (However, there may have also been less depression of transmitter release during 20-Hz stimulation, which would have the opposite effect.) To examine this further, we selected CS larvae with the smallest initial EPSPs (not significantly different from the 24B/PV larvae; P > 0.05, t-test) and compared them to the 24B/PV larvae. These CS larvae (n = 5) had a significantly greater increase in RMP (−9.0 ± 3.1 mV) and Gin (0.34 ± 0.11 μS) than 24B/PV larvae (2.3 ± 2.7 mV; P < 0.05 and 0.04 ± 0.01 μS; P < .001, t-test); this argues that the postsynaptic Ca2+ buffering prevented the increase in Gin and RMP.
Repetitive synaptic stimulation activated dSK channels to reduce synaptic excitation.
We assumed that the increase in Gin resulted from gCS, since gCF shows rapid inactivation. It was proposed that gCS in larval muscle was produced by the Drosophila homologue of the mammalian SK (dSK) channel (Abou Tayoun et al. 2011). Thus we examined the effects of repetitive synaptic activity on dSK− larvae with a deletion in the gene for the dSK channel or P(GawB)how24B/UAS-dSKDNmyc larvae (24B/dSKDN) in which the dSK dominant-negative subunit was expressed in the muscle. We used Western blots to confirm that the dSK protein isoform seen in muscle was missing in dSK−. Using an antibody generated against the predicted protein isoforms (Abou Tayoun et al. 2011), our Western blots showed a single band in larval brain and muscle, which was absent in the mutant (Fig. 2A). Also, we confirmed expression of dSKDN in the muscle using an antibody to myc (data not shown).
Fig. 2.

The effect of synaptic activity on EPSPs, RMP and Gin in mutants lacking functional Drosophila small-conductance Ca2+-dependent K+ (dSK) channels. The CS data from Fig. 1 have been added for comparison. A: Western blot of CS larval brain (B) and muscle (M) tissue (left) and dSK− larval brain and muscle tissue (right) probed with an antibody specific to the dSK channel. The band seen in CS is missing in dSK−. B: for a dSK− larvae, the EPSPs during 20-Hz stimulation (every other EPSP) and I injection before (Initial) and at the end (Final) of stimulation are shown. There was no change in the EPSP peak or amplitude and no increase in Gin. Calibration for I injection: V = 20 mV, 400 ms; I = 5 nA, 400 ms. C: combined data show that, during 20-Hz stimulation, there was no significant change in the EPSP peak for dSK− (n = 7), 24B/dSKDN (dSKDN) (n = 5) and dSKDN 1.5 Ca (n = 6) larvae. This contrasts with the results from CS larvae taken from Fig. 1. All experiments were performed in HL3 (1.0 mM Ca2+), except for dSKDN 1.5 Ca, which used HL3.1 with 1.5 mM Ca2+. D: combined data for dSK−, dSKDN, and dSKDN 1.5 Ca2+ larvae showed that there was no significant change in EPSP amplitude, RMP or Gin as a result of 20-Hz stimulation. This differed from the previous results obtained from CS larvae. Values are means ± SE. ***P < 0.001, paired t-test.
We found that during 20-Hz stimulation of dSK− larvae the EPSP peak potential remained constant, and there was no increase in Gin (Fig. 2B). The combined data clearly showed that, for both dSK− and 24B/dSKDN, there was no significant change in the EPSP peak during 20-Hz stimulation (Fig. 2C). Furthermore, the combined results showed that eliminating a functional dSK channel prevented the changes in EPSP amplitude, RMP and Gin that were previously seen in CS larvae (Fig. 2D). Similar to the PV-expressing larvae, it appeared that dSK− and 24B/dSKDN had reduced basal transmitter release since the EPSP was smaller, especially for 24B/dSKDN. To increase transmitter release for 24B/dSKDN, we repeated the synaptic stimulation in HL3.1 (1.5 mM Ca2+); HL3.1 has a lower Mg2+ concentration (4 mM) than HL3 (20 mM) and produces greater presynaptic Ca2+ influx than HL3 (Desai and Lnenicka 2011). Under these conditions, we still did not see an effect of repetitive stimulation on EPSP amplitude, RMP or Gin (Fig. 2, C and D), providing strong evidence that synaptic activity activates dSK.
The synaptic activation of the dSK conductance (gSK), and not gCF, was supported by repeating this experiment using slo1 larvae, which lack gCF in their muscle fibers (Singh and Wu 1989). The experiments with slo1 larvae gave results similar to those using CS larvae. During 20-Hz stimulation, the EPSP peak became more negative (−19.3 ± 3.2 mV to −49.6 ± 2.0 mV), EPSP amplitude was reduced (30.0 ± 3.6 mV to 11.1 ± 5.1 mV), the RMP hyperpolarized (−49.3 ± 0.8 mV to −63.6 ± 2.4 mV) and Gin increased (0.34 ± 0.08 μS to 0.80 ± 0.07 μS, all P < 0.05, paired t-tests, n = 4).
Ca2+ entry at the postsynaptic membrane was sufficient to activate the dSK channel.
During synaptic activity, the dSK channel could be activated by Ca2+ entering through glutamate-activated channels at the postsynaptic membrane (Desai and Lnenicka 2011; Guerrero et al. 2005) and/or voltage-dependent Ca2+ channels, which are presumably distributed throughout the muscle membrane (Chang et al. 1994; Wu et al. 1983). To determine whether Ca2+ entry at the postsynaptic membrane was sufficient, we stimulated the synapses at 20 Hz for 60 s while voltage clamping the muscle fiber at −60 mV to prevent the voltage-dependent Ca2+ channels from opening (Ren et al. 1998). We used the same saline as in the previous current-clamp experiments (HL3, 1 mM Ca2+). During synaptic stimulation, the leakage current was measured during a step hyperpolarization every 10 s to determine Gin (Fig. 3A). We found that Gin continually increased during stimulation, and the increase reached 3.0 ± 0.3 μS at the end of stimulation (Fig. 3B). This increase in Gin was greater than that seen during 20-Hz stimulation in current clamp (Fig. 1), presumably because there was a greater synaptic current during voltage clamp (see below) and greater Ca2+ influx. During the voltage-clamp, Ca2+ influx should have been restricted to the postsynaptic membrane; therefore, Ca2+ entry at the postsynaptic membrane was sufficient to increase Gin. We also stimulated the nerve for 60 s at lower frequencies, 5 and 10 Hz, and measured the increase in Gin at the end of the stimulation. The results show that these lower stimulation frequencies produced a smaller increase in Gin, and the relationship between stimulation frequency and gSK appeared to be nonlinear (Fig. 3B).
Fig. 3.
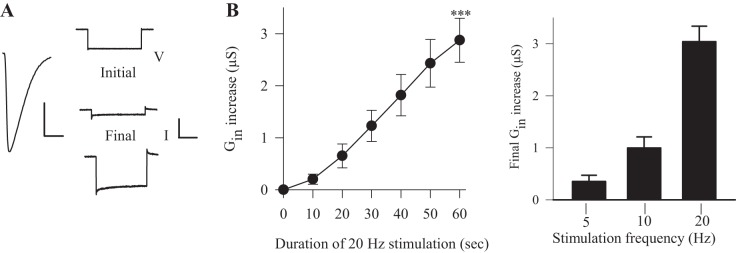
Synaptic activity produced an increase in Gin even when the muscle fiber was voltage clamped. A, left: the muscle membrane was voltage clamped, and the EPSC was observed during 20-Hz stimulation for 60 s. Right: representative traces of the membrane currents produced by a −20-mV voltage step before (Initial) and at the end of stimulation (Final). Calibration: V = 20 mV, 100 ms; I = 20 nA, 100 ms. B, left: Gin gradually increases during synaptic stimulation at 20 Hz, such that Gin at the end of stimulation was significantly greater than at the beginning (n = 8). ***P < 0.001, paired t-test. Right: the increase in Gin produced by the 60-s trains was greater as stimulation frequency was increased: 5 Hz (n = 3) and 10 Hz (n = 6). Values are means ± SE.
The dSK channel influenced resting Gm and the RMP; it was apparently activated by spontaneous transmitter release.
Previous studies have reported that increases in external Ca2+ produced hyperpolarization of the RMP (Jan and Jan 1976; Krans et al. 2010); it was proposed that this resulted from greater membrane-to-electrode sealing and a reduction in the electrode shunt conductance (Jan and Jan 1976). Alternatively, the greater RMP seen in high external Ca2+ could result from greater activation of dSK channels. To distinguish between these possibilities, we altered Ca2+ in HL3.1 saline and measured the RMP and Gin (Fig. 4A). Measurements were made from muscle fiber 6 in segments 3 and 4 from one side of the animal, and then external Ca2+ was changed, and we remeasured the original fibers and also made measurements from the contralateral fibers. In the end, we combined the values from the two sides since they were not significantly different; i.e., the prior electrode penetration of the muscle fiber did not affect a subsequent measurement of RMP and Gin. We either increased external Ca2+ from nominally 0 to 1.5 mM and or reduced it from 1.5 mM to 0, and, since both experiments gave similar values, these data were also combined. For CS larvae, these within-animal comparisons clearly showed that physiological Ca2+ (1.5 mM) gave a higher Gin and greater RMP than 0 Ca2+ (Fig. 4, A and B); this is consistent with activation of gSK. A decrease in the electrode shunt conductance in 1.5 mM Ca2+ would have given a lower Gin, and this was not seen.
Fig. 4.
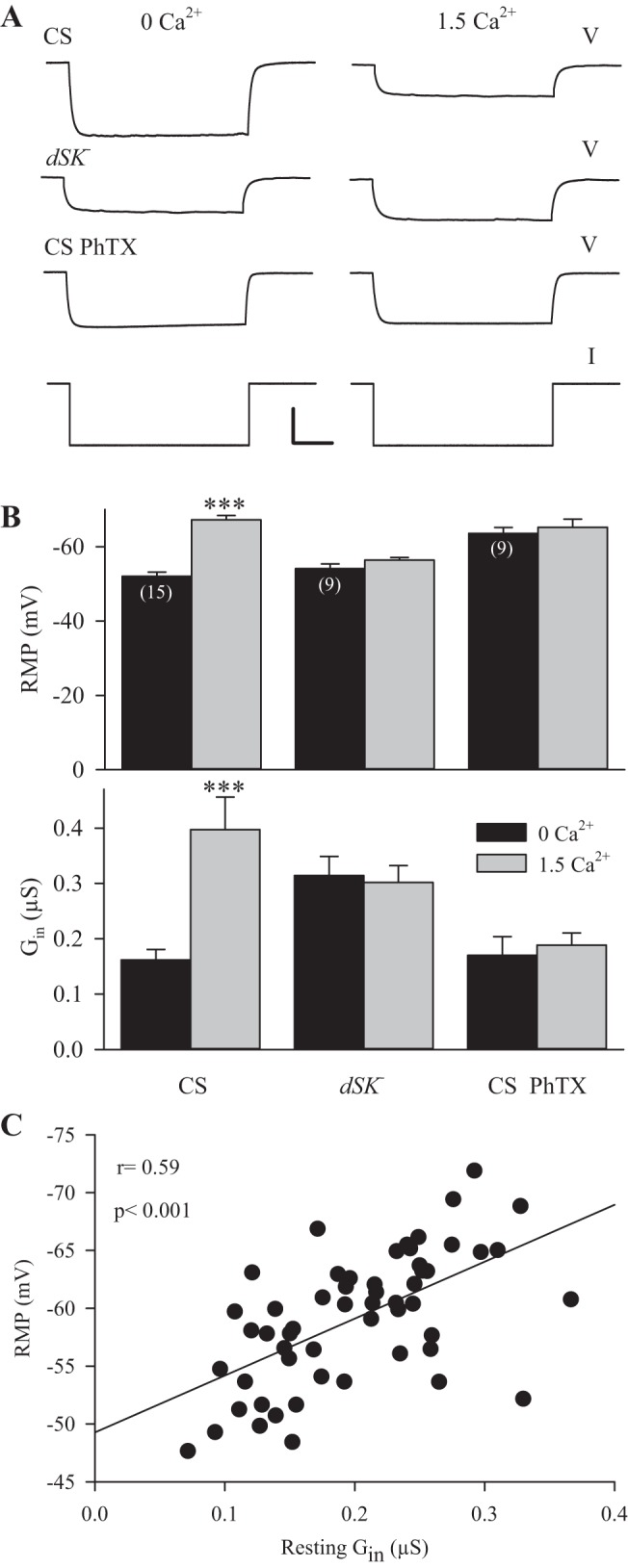
Resting Gin and RMP were influenced by dSK channels and spontaneous transmitter release. The effect of external Ca2+ on Gin and RMP was examined for muscle fiber 6 in segments 3 and 4. A: representative measurements of Gin in single fibers when the external Ca2+ was changed from 0 to 1.5 mM. V traces for wild-type larvae (CS), dSK− larvae and CS larvae after adding philanthotoxin (CS PhTX) are shown. The I trace was the same for all. Calibration: V = 15 mV, 200 ms; I = 3 nA. B: combined data showed the change in Gin and RMP when external Ca2+ was increased from 0 to 1.5 mM. For each animal, multiple muscle fibers were sampled, and the average Gin and RMP values were determined. For CS larvae, there was a significant increase in Gin and RMP as Ca2+ increased; however, dSK− larvae and CS PhTX larvae did not show a significant change in Gin or RMP. Values are means ± SE; n are listed in parentheses. ***P < 0.001, paired t-test. C: measurements of resting Gin and RMP from the CS larvae in Fig. 1. For individual fibers, there was a significant correlation between Gin and RMP.
To determine whether the effects of altering external Ca2+ were dependent on dSK channels, we repeated the experiments in dSK− larvae. Both the Gin and RMP in dSK− larvae were insensitive to changes in external Ca2+ (Fig. 4, A and B). This confirmed that external Ca2+ influences the resting Gin and RMP by acting on dSK channels.
It appeared that the leakage conductance (gL) for dSK− larvae was greater than that seen in CS larvae, since Gin in 0 Ca2+ was significantly greater for the dSK− fibers (0.31 ± 0.03 μS) compared with the CS fibers (0.16 ± 0.02 μS; P < 0.001; t-test). Note we assumed that in 0 Ca2+ gSK was not active so that Gin results entirely from gL. Thus it appears that in dSK− larvae there was an increase in gL, presumably to compensate for the lack of dSK channels.
What was responsible for activation of dSK channels at rest? One possibility is that the postsynaptic Ca2+ transients produced by spontaneous transmitter release activated these channels (Desai and Lnenicka 2011). To test this, we blocked glutamate receptors with 4 μM philanthotoxin (PhTX) and again increased external Ca2+ from 0 to 1.5 mM (Fig. 4, A and B). Note we found that 4 μM PhTX produced a 65% decrease in miniature EPSP (minEPSP) amplitude, which was similar to previously reported values (Frank et al. 2006). PhTX application blocked the increase in Gin and RMP normally seen when Ca2+ was increased to 1.5 mM. Thus activation of glutamate receptors is necessary for the effects of external Ca2+, and presumably Ca2+ enters through glutamate receptors during spontaneous transmitter release to activate the dSK channels.
Although PhTX blocked the increase in RMP when Ca2+ was increased, it appeared to have an independent effect on the RMP, since the RMP measured in 0 Ca2+ in CS larvae (Fig. 4B) was greater in the presence of PhTX (−63.6 ± 1.6 mV) than in the absence of PhTX (−52.1 ± 1.1 mV; P < 0.001; t-test). To confirm these results, we performed within-animal comparisons in 0 Ca2+ before and after adding PhTX. Adding PhTX produced a significant increase in the RMP from −54.0 ± 1.2 mV to −63.8 ± 2.1 mV (n = 7; P < 0.001, paired t-test) and a small decrease in Gin, which was not significant (0.16 ± 0.02 μS to 0.14 ± 0.02 μS; P > 0.1, paired t-test). This confirmed that PhTX produced about a 10-mV hyperpolarization of the RMP that was apparently independent of a significant change in Gin. We further examined the effect of PhTX by adding it to muscle fibers bathed in 1.5 mM Ca2+. Adding PhTX resulted in a significant reduction in Gin (0.33 ± 0.09 μS to 0.18 ± 0.04 μS, n = 11; P < 0.001, paired t-test) as expected, since it would reduce activation of the dSK channels. There was no significant change in the RMP (−65.7 ± 1.3 mV to −64.7 ± 1.1 mV; P > 0.1, paired t-test), which is consistent with PhTX having dual opposing effects on RMP. One possible explanation for the independent effect of PhTX on the RMP is that the glutamate receptors are activated at rest, possibly due to nonvesicular glutamate release, as has been proposed (Featherstone et al. 2002). In this scenario, the resting activation of glutamate receptors depolarizes the membrane by activating a synaptic conductance (Gsyn), but the density of open glutamate receptors would be too low to produce Ca2+ signals large enough to activate dSK channels. Given a reversal potential of −1 mV (Jan and Jan 1976), the 10-mV hyperpolarization could occur with very little change in Gin. In fact, if the small decrease in Gin seen when adding PhTX to 0 Ca2+ saline resulted from a decrease in Gsyn, then the new RMP would be calculated to be −64.4 mV, which is very close to that seen experimentally (−63.8 mV).
We examined whether the normal variability in the RMP resulted from variability in the activation of gSK. If this is the case, there should be a positive correlation between Gin and the RMP; i.e., greater activation of gSK should result in a larger Gin and RMP. We examined Gin and the RMP for the data in Fig. 1 (before 20-Hz stimulation) where we had the largest number of experiments performed in the same saline. In these experiments, there was approximately a fourfold range of resting Gin values, and the RMP had a range of about a 20 mV. We found a significant correlation between Gin and the RMP (Fig. 4C). We performed the same analysis on dSK mutants using the data from Fig. 2 and found no significant correlation between Gin and the RMP (r = 0.13, n = 32, P > 0.10). Thus the dSK channel plays a role in setting the RMP, and normal variability in gSK contributes to variability in the RMP.
Activation of the dSK channel is reduced by phosphatase 2A inhibitors.
In mammals, the Ca2+ sensitivity of the SK channel is increased by dephosphorylation by PP2A; it appears that an increase in [Ca2+]i favors dephosphorylation and increased Ca2+ sensitivity (Allen et al. 2007). Activation of the dSK channels by increased [Ca2+]i, seen here, could involve the action of PP2A. To examine this, we repeated experiments involving 20-Hz stimulation (Fig. 1) and increasing external Ca2+ (Fig. 4) in the presence of PP2A inhibitors. We utilized two phosphatase inhibitors: a specific PP2A inhibitor (calyculin A), and a general phosphatase inhibitor (okadaic acid). These inhibitors have been used to block PP2A function in Drosophila Schneider cells, and we used 100 nM concentrations for both inhibitors (Banreti et al. 2012; Itagaki et al. 2004; Sathyanarayanan et al. 2004). We repeated the 20-Hz stimulation experiments in the presence of the inhibitors, which were added 10 min prior to stimulation, and compared the results to the control (CS) values from Fig. 1. Overall the changes in EPSP amplitude, RMP and Gin were less in the presence of the inhibitors than in the control (Fig. 5A). All of the changes seen after the addition of calyculin A were significantly less than for the control, and for okadaic acid only the change in RMP was not significantly different from the control.
Fig. 5.
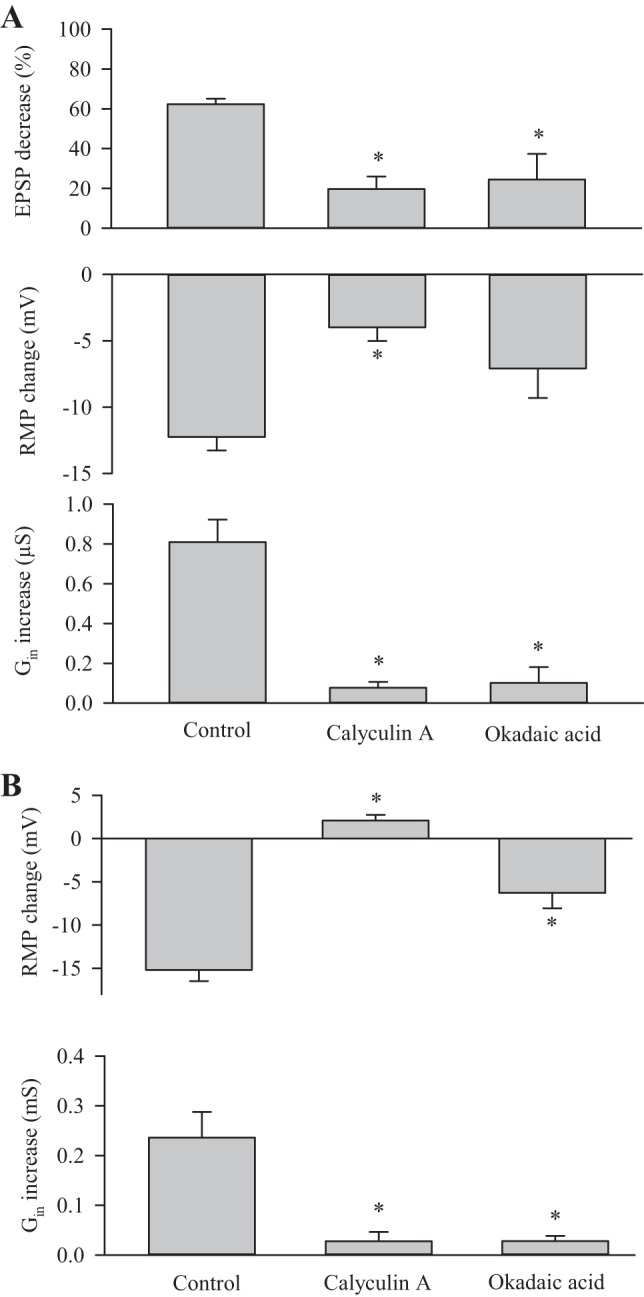
Inhibition of protein phosphatase 2A (PP2A) prevents activation of the dSK channel. A: combined data show the changes in EPSP amplitude, RMP, and Gin resulting from 20-Hz synaptic stimulation for control (CS) data from Fig. 1 and in the presence of PP2A inhibitors. In general, the inhibitors calyculin A (n = 8) and okadaic acid (n = 5) reduced the changes in EPSP amplitude, RMP and Gin produced by 20-Hz stimulation. All experiments were performed in HL3 with 1 mM Ca2+. B: combined data show the change in Gin and RMP when external Ca2+ was increased from 0 to 1.5 mM. For each animal, multiple muscle fibers were sampled, and the average Gin and RMP values were calculated to determine the change. The change in Gin and RMP was significantly less for the PP2A inhibitors than for the control (CS) data from Fig. 4. Values for the inhibitors were compared with the control. Values are means ± SE. *P < 0.05, ANOVA, Dunn test.
The EPSP amplitude after applying calyculin A was 30.3 ± 1.7 mV, which was significantly smaller than controls (34.5 ± 0.7 mV; P < 0.05, t-test). If the smaller EPSP was due to a decrease in transmitter release, then there would less activation of glutamate receptors, and this could result in less activation of the dSK channel. To rule out this possibility, we repeated the calyculin A experiments in HL3.1 (1.5 mM Ca2+) to increase transmitter release as in Fig. 2. Under these conditions, the EPSP amplitude was larger (37.1 ± 2.1, n = 8), and the EPSP decrease (33.8 5.3%), RMP change (−2.1 ± 1.7 mV) and Gin increase (0.05 0.01 uS) remained significantly less (P < 0.01 for all, t-test) compared with controls. Thus we conclude that calyculin A is acting postsynaptically to produce its effect.
To examine further the role of PP2A, we repeated the Ca2+ exchange experiments described in Fig. 4 in the presence of the inhibitors and compared the results to the previous control (CS) data. The inhibitor was added to HL3.1 (0 Ca2+) 10 min before measuring RMP and Gin, and the saline was replaced with HL3.1 (1.5 mM Ca2+) also containing the inhibitor. For both inhibitors, the increase in RMP and Gin was significantly less than for controls (Fig. 5B). These results suggest that an increase in postsynaptic [Ca2+]i produces an increase in the Ca2+ sensitivity of the dSK channel involving PP2A.
Modeling the role of gCS in regulating synaptic strength.
To examine the effect of gSK on EPSPs, we modeled the larval NMJ. Muscle fiber 6 has been found to be virtually isopotential (Jan and Jan 1976) and can be treated as a sphere rather than a cable; the specific Gm was estimated by the Gin × surface area (SA) of the muscle fiber. Measurements of muscle fiber 6 (n = 99 fibers) in segments 3 and 4 gave a mean dorsal SA of 54,804 ± 472 μm2, a perimeter of 1,297 ± 5 μm and a thickness of 19.2 ± 0.1 μm. Assuming a curved edge with a radius of 9.6 μm, the total apparent SA would be 122,187 μm2. Measurements of the specific Gm and time constant gave a specific membrane capacitance of 3.9 μF/cm2. These measurements will overestimate the membrane capacitance and specific Gm due to the contribution of the t-tubules; however, these apparent values can be used in calculating the electrical response of the membrane to synaptic currents (Gage 1976).
We used virtual cell software (Schaff et al. 1997; Slepchenko et al. 2003) to model the generation of the EPSP in muscle fiber 6. The time course of the Gsyn was simulated using an alpha function (Johnston and Wu 1995), and a value was chosen for alpha that best replicated the waveform of the EPSCs (Fig. 3). Our model included two parallel conductances: the gL in series with the leakage potential and gSK in series with the K+ equilibrium potential (EK). (This virtual cell model, epsp_gcs, is available in the public domain at http://www.vcell.org/ under the shared username gregL.) Values for gL and leakage potential were based on Gin and RMP in the absence of gSK, which should be given by measurements made in 0 external [Ca2+]; these values from Fig. 4B were 0.16 ± 0.01 μS and −53.4 ± 0.8 mV, respectively. EK was calculated from the Nernst equation to be −79.0 mV, assuming that the internal K+ concentration was 115 mM, as measured in larval blowfly muscle (Dawson and Djamgoz 1988). One obtains a similar value for EK (−77.5 mV) when it is calculated based on the change in RMP and Gin that we observed when increasing external [Ca2+] from 0 to 1.5 mM.
We modeled the effect of increasing gSK up to 2 μS on synaptic excitation; this covered much of the range in gSK we observed when stimulating the synapse (Fig. 1). We used a peak Gsyn of 250 nS, 500 nS or 1,000 nS, since this spanned most of the values obtained from the compound EPSCs (Fig. 3); typically, the single EPSCs would fall between 250 nS and 500 nS. Of course, these measurements were made in HL3 with reduced Ca2+, and the Gsyn would be larger if measured in HL3.1 with 1.5 mM Ca2+. Increasing gSK clearly reduced synaptic excitation by hyperpolarizing the RMP and reducing EPSP amplitude (Fig. 6A). Small increases in gSK produced a large hyperpolarization of the RMP (Fig. 6B); this effect was seen at the resting gSK, particularly in HL3.1, consistent with the experimental results. The EPSP amplitude did not decrease during small increases in gCS, since the synaptic current (Isyn) increased; e.g., the simulation predicted that the peak Isyn would increase from 36 nA to 50 nA (Gsyn = 1,000 nS) for an increase in gSK from 0 to 0.5 μS. [Note that the peak Isyn values were larger (59 nA) when the membrane potential was clamped −60 mV.] As gSK exceeded 0.5 μS, further increases in gSK produced less RMP hyperpolarization and a greater decrease in EPSP amplitude (Fig. 6, B and C).
Fig. 6.
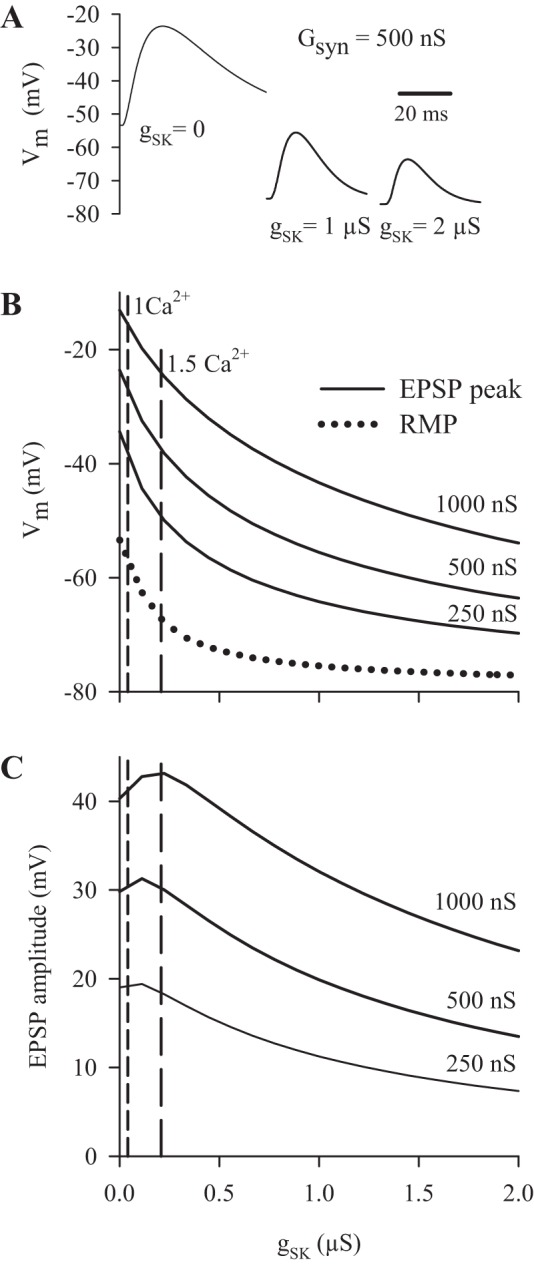
Modeling the effect of changes in slow Ca2+-dependent K+ conductance on the EPSP in muscle fiber 6. A: simulated EPSPs produced by a peak synaptic conductance (Gsyn) of 500 nS in the absence of dSK conductance (gSK) (0) and as the gSK for muscle fiber 6 was increased to 1 μS and 2 μS. B: the change in EPSP peak and RMP as gSK was increased from 0 to 2 μS. For the EPSP peak, plots are shown for a Gsyn of 250 nS, 500 nS and 1,000 nS. The dashed vertical lines show the mean gSK at rest when measured in HL3 with 1 mM Ca2+ (1 Ca2+) and HL3.1 containing 1.5 mM Ca2+ (1.5 Ca2+). C: the EPSP amplitudes during an increase in gSK for all three values of Gsyn. Vm, membrane potential.
The results of Fig. 1 provided evidence that an increase in gSK contributed to synaptic depression during 20-Hz stimulation. The modeling supported this conclusion. For example, during 20-Hz stimulation, the gSK increased from its resting level (0.04 μS) to 0.85 μS, and, according to Fig. 6C, this should reduce EPSP amplitude by about one-third. Alternatively, we used simulations to determine the amplitude of the final EPSP, assuming gSK remained constant during stimulation. At the end of 20-Hz stimulation, the EPSP amplitude was 12.7 mV, and, based upon the final gSK (0.85 μS), the simulation gave a final Gsyn of 260 nS. If gSK did not increase during stimulation, then the final GSyn would give a 20.2-mV EPSP. Thus activation of gSK contributed to synaptic depression by reducing the final EPSP by 37%.
DISCUSSION
Ca2+ entering at the postsynaptic membrane activates postsynaptic dSK channels to reduce synaptic excitation.
Stimulation of the neuromuscular synapses at 20 Hz resulted in an increase in resting Gin, and a decrease in synaptic excitation due to membrane hyperpolarization and a decrease in EPSP amplitude. Our simulations predicted that the increase in resting Gin during stimulation accounted for about one-third of the depression of EPSP amplitude due to shunting of the synaptic current. Synaptic depression is generally held to be due to a reduction in transmitter release (Zucker and Regehr 2002); thus the remaining decrease in EPSP amplitude may have resulted from a decline in transmitter release. Nonetheless, we provide evidence for a change in postsynaptic conductance contributing to synaptic depression. The increase in Gin and RMP was consistent with activation of a gKCa; this was further supported by the reduced increase in Gin and RMP seen after expressing PV in the muscle.
Larval muscle fibers contain two types of Ca2+-dependent K+ channels producing gCF and gCS. gCF was found to result from the slowpoke channel, a BK-type channel showing voltage- and Ca2+-dependent activation and voltage-dependent inactivation (Elkins et al. 1986; Komatsu et al. 1990; Salkoff et al. 2006), and the dSK channel was proposed to produce gCS (Abou Tayoun et al. 2011). Our Western blots showed a single dSK isoform in larval brain and muscle; a Western blot of adult fly brains showed two closely spaced bands, which could have represented two dSK isoforms or a single isoform along with its posttranslationally altered form (Abou Tayoun et al. 2011). SK channels are not voltage-gated, and calmodulin acts as their Ca2+ sensor, resulting in a relatively high Ca2+ sensitivity (Xia et al. 1998). Repetitive synaptic stimulation activated the postsynaptic dSK channels, since the increase in Gin and RMP was eliminated in dSK− or 24B/dSKDN larvae, but not in slo1 larva. As expected, there was reduced synaptic depression in dSK mutants and PV-expressing larvae; however, it was surprising that synaptic depression was completely eliminated. This suggests that there was also less depression of transmitter release in these larvae.
We assume that the dSK channel is activated during evoked transmitter release in vivo, since the 20-Hz stimulation applied here likely falls within the physiological range of activity. The Ib synapses on muscle fiber 6 fire on average 20–30 Hz, and the Is terminals fire less than 10 Hz in dissected larvae (Chouhan et al. 2010, 2012); however, these firing frequencies may be greater in vivo since the waves of contraction are slow in dissected larvae, probably due to lack of appropriate sensory input (Song et al. 2007). Also, since we used HL3 with low Ca2+ for our stimulation procedure, the postsynaptic Ca2+ influx was less than would occur under physiological conditions.
Ca2+ entering at the postsynaptic membrane was sufficient to activate dSK channels, since evoked transmitter release produced an increase in Gin even when the muscle was voltage clamped at −60 mV. Under these conditions, we expect that Ca2+ entry was limited to glutamate receptors, since voltage-dependent Ca2+ channels in larval muscle open at membrane potentials more positive than −30 mV (Ren et al. 1998). However, we cannot rule out the possibility that there was a voltage drop across the subsynaptic reticulum (SSR), such that voltage-dependent Ca2+ channels near the glutamate receptors remained unclamped. In this case, both the glutamate receptors and nearby Ca2+ channels could have been the source of Ca2+ that activated the dSK channel. In fact, dSK channels in larval muscle can be activated by Ca2+ entering through voltage-dependent Ca2+ channels since depolarization of the muscle in the absence of synaptic activity produced an increase in gCS (Gho and Mallart 1986). Additional evidence that the glutamate receptor can act as the Ca2+ source comes from the activation of the dSK channel by spontaneous transmitter release; here, it seems unlikely that the SSR resistance would be great enough for quantal currents to open voltage-dependent Ca2+ channels. It appears that dSK channels exist at the postsynaptic membrane near the glutamate receptors, and this is consistent with findings in mammals, where the SK channel has been shown to be closely coupled to postsynaptic nicotinic acetylcholine receptors and NMDA receptors (Ngo-Anh et al. 2005; Oliver et al. 2000).
The postsynaptic dSK channels were activated by spontaneous transmitter release.
The dSK channels were activated at rest and were responsible for the previously reported dependence of the RMP on external Ca2+ (Jan and Jan 1976; Krans et al. 2010). This was based upon our finding that the increase in Gin and RMP typically seen when increasing external Ca2+ from 0 to 1.5 mM was not seen in dSK− larvae. In fact, it appeared that activation of the dSK channel is responsible for much of the normal variability in the RMP, since resting Gin and the RMP were positively correlated in wild-type larvae, but not in dSK− larvae. Spontaneous transmitter release apparently activates dSK channels to set the resting Gin and RMP, since blocking the glutamate receptors prevented the rise in Gin and RMP produced by increasing external Ca2+. This is a novel function for spontaneous transmitter release, but not entirely unexpected, since spontaneous transmitter release produced postsynaptic Ca2+ transients similar in amplitude to evoked release (Desai and Lnenicka 2011).
The spontaneous Ca2+ transients must have produced an increase in resting Gin that outlasted the Ca2+ signal, since the frequency of spontaneous events in this muscle is usually about 1 Hz, and the spontaneous Ca2+ transients had a decay time constant of ∼50 ms (Desai and Lnenicka 2011). The increase in Gin produced by repetitive synaptic stimulation also appeared to outlast the increase in [Ca2+]i, since it persisted for about 10 min after stimulation. We previously found that after 5 s of 10-Hz stimulation, postsynaptic [Ca2+]i decayed with a time constant of about 100 ms, and this decay was largely due to the plasma membrane Ca2+ ATPase, which appeared to be highly enriched in the SSR (Desai and Lnenicka 2011). The SSR may limit Ca2+ diffusion to enhance the amplitude of postsynaptic Ca2+ transients; however, it seems likely that it also reduces their duration by providing efficient postsynaptic Ca2+ extrusion. Thus it seems improbable that postsynaptic [Ca2+]i remained elevated for 10 min after the stimulation train.
We found that there was an increase in gL in dSK− larvae. This is consistent with previous studies showing covariation of ion conductances (MacLean et al. 2003). In particular, reduced activation of Ca2+-dependent K+ currents in Drosophila cultured neurons resulted in a compensatory upregulation of the transient K+ current, apparently due to increased expression of transient K+ current channels (Peng and Guo 2007). Although the channels responsible for gL have not been characterized in larval muscle, they may include ORK1 which was identified as a K+ leak channel in Drosophila and appears to be expressed in adult muscle (Goldstein et al. 1996); it is a member of the two-pore domain K+ channels identified in mammals (Enyedi and Czirjak 2010). The nonselective cation channel NALCN produces the Na+ leak conductance in mammalian neurons (Lu et al. 2007) and its ortholog na is expressed in Drosophila neurons (Nash et al. 2002), raising the possibility that it could also be responsible for the Na+ leak conductance in larval muscle.
The dSK channel is modulated by PP2A.
In mammals, CK2 and PP2A are constitutively bound to the SK channel and regulate the phosphorylation state of calmodulin, which determines the Ca2+ sensitivity and deactivation rate of the SK channel. Dephosphorylation of calmodulin by PP2A can reduce EC50 to 0.3 μM Ca2+ and phosphorylation of the SK channel by CK2 can increase EC50 to 2.0 μM (Allen et al. 2007; Bildl et al. 2004). An increase in [Ca2+]i should shift the balance toward dephosphorylation and produce an increase in Ca2+ sensitivity, since CK2 cannot phosphorylate calmodulin when the channel is open (Allen et al. 2007). Our results showed that the dSK channel is also regulated by PP2A, since inhibiting PP2A reduced the increase in Gin and RMP produced by repetitive stimulation or increasing external Ca2+. During repetitive stimulation, the increase in postsynaptic [Ca2+]i may have increased the Ca2+ sensitivity of the dSK channel. Similarly, it may be that the Ca2+ transients produced by spontaneous transmitter release produced an increase in Ca2+ sensitivity so that the dSK channel is partially activated by resting [Ca2+]i. If the Ca2+-dependent increase in Ca2+ sensitivity persisted, this could explain the lasting increase in Gin seen after the end of 20-Hz stimulation and after spontaneous Ca2+ transients.
The dSK channel acts to regulate synaptic excitation.
Our results demonstrate that Ca2+ entering at the postsynaptic membrane during transmitter release provides negative feedback on synaptic excitation. This could rapidly stabilize the synapse and dampen the effects of changes in impulse activity, transmitter release or postsynaptic sensitivity. The effect of gSK on synaptic excitation was modeled using a Gsyn of 500 nS. The resting gSK in 1 mM (HL3) and 1.5 mM external Ca2+ (HL3.1) would make the EPSP peak about 3 mV and 14 mV more negative, respectively, mainly due to hyperpolarization of the RMP. An increase in gSK from 0 to 1 μS (approximately the value seen after 20 Hz stimulation) made the EPSP peak 22 mV more negative due to both RMP hyperpolarization and a decrease in EPSP amplitude. Activation of the dSK channel by evoked transmitter release is consistent with this negative-feedback mechanism, but what is the function of dSK channel activation by spontaneous transmitter release? minEPSP frequency is elevated after repetitive synaptic activity, and this could produce a persistent reduction in synaptic excitation. Also, since greater minEPSP frequency and amplitude would predict a larger EPSP, spontaneous transmitter release could set gSK to oppose strong synaptic excitation.
This action of the postsynaptic dSK channel is similar to that found at mammalian central nervous system synapses, where the SK channel counteracts synaptic changes: experiments blocking SK channels with apamin, or overexpressing them, showed that activation of SK channels increased the threshold for the induction of LTP and impaired learning (Behnisch and Reymann 1998; Faber et al. 2005; Hammond et al. 2006; Ngo-Anh et al. 2005; Stackman et al. 2002). At these synapses, it was proposed that SK channels act specifically to restrict Ca2+ entry through NMDA receptors limiting LTP and learning and memory (Faber and Sah 2007). Our findings show that dSK channels play a more general role in regulating synaptic excitation.
The dSK channel could act in concert with forms of homeostatic synaptic plasticity; these have involved compensatory changes in transmitter release at the NMJ, and most operate over long time scales (DeRosa and Govind 1978; DiAntonio et al. 1999; Lnenicka and Mellon 1983; Petersen et al. 1997). A failure of the dSK channel to maintain appropriate synaptic excitation could result in compensatory changes in transmitter release; this is supported by the apparent decrease in transmitter release seen when dSK channel activity was reduced in dSK mutants or by expressing PV in the muscle. This is consistent with previous findings that expressing additional K+ channels in Drosophila larval muscle resulted in a homeostatic increase in transmitter release (Paradis et al. 2001).
At mammalian central synapses, the SK channel reduced the amplitude of the EPSC; single EPSPs and EPSCs were larger after adding the SK-channel blocker apamin (Faber 2005, 2010; Ngo-Anh et al. 2005). Thus the SK channel was rapidly activated during synaptic transmission so that the resultant EPSC was a composite of the inward current through the postsynaptic receptor and outward current through the SK channel. We have not directly examined whether the dSK channel influences the amplitude of the EPSC; this is made difficult since apamin does not block dSK channels. We have evidence that dSK currents reduced the duration of the EPSC, but further experiments are required to characterize fully the contribution of the dSK current to the EPSC.
GRANTS
The Virtual Cell is supported by National Institute of General Medical Sciences Grant P41 GM-103313 from the National Center for Research Resources. Research supported by National Science Foundation Grant IOS1051605 (G. A. Lnenicka).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.M.G. and G.A.L. conception and design of research; D.M.G. and S.D. performed experiments; D.M.G. analyzed data; D.M.G. and G.A.L. interpreted results of experiments; D.M.G. prepared figures; D.M.G. edited and revised manuscript; G.A.L. drafted manuscript; G.A.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Patrick Dolph for fly lines and antibodies, Dr. Richard Ribchester for helpful discussions, and Dr. Harold Atwood for comments on an earlier version of the manuscript.
REFERENCES
- Abou Tayoun AN, Li X, Chu B, Hardie RC, Juusola M, Dolph PJ. The Drosophila SK channel (dSK) contributes to photoreceptor performance by mediating sensitivity control at the first visual network. J Neurosci 31: 13897–13910, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci 27: 2369–2376, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banreti A, Lukacsovich T, Csikos G, Erdelyi M, Sass M. PP2A regulates autophagy in two alternative ways in Drosophila. Autophagy 8: 623–636, 2012 [DOI] [PubMed] [Google Scholar]
- Behnisch T, Reymann KG. Inhibition of apamin-sensitive calcium dependent potassium channels facilitate the induction of long-term potentiation in the CA1 region of rat hippocampus in vitro. Neurosci Lett 253: 91–94, 1998 [DOI] [PubMed] [Google Scholar]
- Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is coassembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron 43: 847–858, 2004 [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415, 1993 [DOI] [PubMed] [Google Scholar]
- Chang H, Ciani S, Kidokoro Y. Ion permeation properties of the glutamate receptor channel in cultured embryonic Drosophila myotubes. J Physiol 476: 1–16, 1994 [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Ivannikov MV, Lu Z, Sugimori M, Llinas RR, Macleod GT. Cytosolic calcium coordinates mitochondrial energy metabolism with presynaptic activity. J Neurosci 32: 1233–1243, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Zhang J, Zinsmaier KE, Macleod GT. Presynaptic mitochondria in functionally different motor neurons exhibit similar affinities for Ca2+ but exert little influence as Ca2+ buffers at nerve firing rates in situ. J Neurosci 30: 1869–1881, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson J, Djamgoz MB. Quantitative analysis of resting membrane electrogenesis in insect (diptera) skeletal muscle. I. Intracellular K+, Na+ and Cl− activities, measured using liquid ion-exchanger and neutral ion-carrier microelectrodes. J Exp Biol 136: 417–432, 1988 [DOI] [PubMed] [Google Scholar]
- DeRosa RA, Govind CK. Transmitter output increases in an identifiable lobster motoneurone with growth of its muscle fibres. Nature 273: 676–678, 1978 [DOI] [PubMed] [Google Scholar]
- Desai SA, Lnenicka GA. Characterization of postsynaptic Ca2+ signals at the Drosophila larval NMJ. J Neurophysiol 106: 710–721, 2011 [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, Goodman CS. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci 19: 3023–3032, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins T, Ganetzky B, Wu CF. A Drosophila mutation that eliminates a calcium-dependent potassium current. Proc Natl Acad Sci U S A 83: 8415–8419, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90: 559–605, 2010 [DOI] [PubMed] [Google Scholar]
- Faber ES. Functional interplay between NMDA receptors, SK channels and voltage-gated Ca2+ channels regulates synaptic excitability in the medial prefrontal cortex. J Physiol 588: 1281–1292, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci 8: 635–641, 2005 [DOI] [PubMed] [Google Scholar]
- Faber ES, Sah P. Functions of SK channels in central neurons. Clin Exp Pharmacol Physiol 34: 1077–1083, 2007 [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Rushton E, Broadie K. Developmental regulation of glutamate receptor field size by nonvesicular glutamate release. Nat Neurosci 5: 141–146, 2002 [DOI] [PubMed] [Google Scholar]
- Feng Y, Ueda A, Wu CF. A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. J Neurogenet 18: 377–402, 2004 [DOI] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52: 663–677, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage PW. Generation of end-plate potentials. Physiol Rev 56: 177–247, 1976 [DOI] [PubMed] [Google Scholar]
- Gho M, Mallart A. Two distinct calcium-activated potassium currents in larval muscle fibres of Drosophila melanogaster. Pflügers Arch 407: 526–533, 1986 [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Price LA, Rosenthal DN, Pausch MH. ORK1, a potassium-selective leak channel with two pore domains cloned from Drosophila melanogaster by expression in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 93: 13256–13261, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero G, Reiff DF, Agarwal G, Ball RW, Borst A, Goodman CS, Isacoff EY. Heterogeneity in synaptic transmission along a Drosophila larval motor axon. Nat Neurosci 8: 1188–1196, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci 26: 1844–1853, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrisingh MC, Wu Y, Lnenicka GA, Nitabach MN. Intracellular Ca2+ regulates free-running circadian clock oscillation in vivo. J Neurosci 27: 12489–12499, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki K, Kannan KB, Singh BB, Hauser CJ. Cytoskeletal reorganization internalizes multiple transient receptor potential channels and blocks calcium entry into human neutrophils. J Immunol 172: 601–607, 2004 [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol 262: 189–214, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Wu SM. Foundations of Cellular Neurophysiology. Cambridge, MA: MIT Press, 1995 [Google Scholar]
- Keshishian H, Broadie K, Chiba A, Bate M. The Drosophila neuromuscular junction: a model system for studying synaptic development and function. Annu Rev Neurosci 19: 545–575, 1996 [DOI] [PubMed] [Google Scholar]
- Komatsu A, Singh S, Rathe P, Wu CF. Mutational and gene dosage analysis of calcium-activated potassium channels in Drosophila: correlation of micro- and macroscopic currents. Neuron 4: 313–321, 1990 [DOI] [PubMed] [Google Scholar]
- Krans JL, Parfitt KD, Gawera KD, Rivlin PK, Hoy RR. The resting membrane potential of Drosophila melanogaster larval muscle depends strongly on external calcium concentration. J Insect Physiol 56: 304–313, 2010 [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685, 1970 [DOI] [PubMed] [Google Scholar]
- Lnenicka GA, Grizzaffi J, Lee B, Rumpal N. Ca2+ dynamics along identified synaptic terminals in Drosophila larvae. J Neurosci 26: 12283–12293, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lnenicka GA, Mellon D., Jr. Changes in electrical properties and quantal current during growth of identified muscle fibres in the crayfish. J Physiol 345: 261–284, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Su Y, Das S, Liu J, Xia J, Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell 129: 371–383, 2007 [DOI] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Activity-independent homeostasis in rhythmically active neurons. Neuron 37: 109–120, 2003 [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 44: 5–21, 2004 [DOI] [PubMed] [Google Scholar]
- Nash HA, Scott RL, Lear BC, Allada R. An unusual cation channel mediates photic control of locomotion in Drosophila. Curr Biol 12: 2152–2158, 2002 [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci 8: 642–649, 2005 [DOI] [PubMed] [Google Scholar]
- Oliver D, Klocker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron 26: 595–601, 2000 [DOI] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW. Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30: 737–749, 2001 [DOI] [PubMed] [Google Scholar]
- Peng Y, Guo A. Novel stimulus-induced calcium efflux in Drosophila mushroom bodies. Eur J Neurosci 25: 2034–2044, 2007 [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19: 1237–1248, 1997 [DOI] [PubMed] [Google Scholar]
- Ren D, Xu H, Eberl DF, Chopra M, Hall LM. A mutation affecting dihydropyridine-sensitive current levels and activation kinetics in Drosophila muscle and mammalian heart calcium channels. J Neurosci 18: 2335–2341, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salkoff L. Drosophila mutants reveal two components of fast outward current. Nature 302: 249–251, 1983 [DOI] [PubMed] [Google Scholar]
- Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the SLO family. Nat Rev Neurosci 7: 921–931, 2006 [DOI] [PubMed] [Google Scholar]
- Sathyanarayanan S, Zheng X, Xiao R, Sehgal A. Posttranslational regulation of Drosophila PERIOD protein by protein phosphatase 2A. Cell 116: 603–615, 2004 [DOI] [PubMed] [Google Scholar]
- Schaff J, Fink CC, Slepchenko B, Carson JH, Loew LM. A general computational framework for modeling cellular structure and function. Biophys J 73: 1135–1146, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Wu CF. Complete separation of four potassium currents in Drosophila. Neuron 2: 1325–1329, 1989 [DOI] [PubMed] [Google Scholar]
- Slepchenko BM, Schaff JC, Macara I, Loew LM. Quantitative cell biology with the Virtual Cell. Trends Cell Biol 13: 570–576, 2003 [DOI] [PubMed] [Google Scholar]
- Song W, Onishi M, Jan LY, Jan YN. Peripheral multidendritic sensory neurons are necessary for rhythmic locomotion behavior in Drosophila larvae. Proc Natl Acad Sci U S A 104: 5199–5204, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci 22: 10163–10171, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A 175: 179–191, 1994 [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Nat Acad Sci U S A 76: 4350–4354, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CF, Ganetzky B, Haugland FN, Liu AX. Potassium currents in Drosophila: different components affected by mutations of two genes. Science 220: 1076–1078, 1983 [DOI] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395: 503–507, 1998 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405, 2002 [DOI] [PubMed] [Google Scholar]