

Abstract
Fluorescence redistribution after photobleaching has been used to show that a cytoplasmic GFP fusion is immobile in dormant spores of Bacillus subtilis but becomes freely mobile in germinated spores in which cytoplasmic water content has increased ≈2-fold. The GFP immobility in dormant spores is not due to the high levels of dipicolinic acid in the spore cytoplasm, because GFP was also immobile in germinated cwlD spores that had excreted their dipicolinic acid but where cytoplasmic water content had only increased to a level similar to that in dormant spores of several other Bacillus species. The immobility of a normally mobile protein in dormant wild-type spores and germinated cwlD spores is consistent with the lack of metabolism and enzymatic activity in these spores and suggests that protein immobility, presumably due to low water content, is a major reason for the metabolic dormancy of spores of Bacillus species.
Spores of various Bacillus species are formed in the process of sporulation, and these spores are adapted for long-term survival because they are resistant to many environmental stresses and are metabolically dormant (1–4). However, given the proper stimulus, generally the appearance of specific nutrients in the environment, the dormant spores can return to life through the process of spore germination and then outgrowth (2). A major factor in spore dormancy and resistance is the low level of water in the spore cytoplasm or core [25–55% of the mass of the hydrated dormant spore core depending on the species (1, 3–5)]; another factor may be the high level of pyridine-2,6-dicarboxylic acid [dipicolinic acid (DPA)] (≈10% of the mass of the hydrated stage II-germinated spore core) in the spore core, likely present as a chelate with divalent cations, predominantly Ca2+ (1, 6). DPA is released in the first minutes of spore germination and is replaced with water as the spore-core water content rises slightly; these spores are said to have completed stage I of germination (2, 7). Subsequently, the large peptidoglycan cortex around the spore core is degraded, and removal of this restraint allows the spore core to expand rapidly by uptake of water and thus complete stage II of germination (2, 7). This latter event results in a level of core water (75–80% of wet weight) that is similar to that in growing cells (8). As noted above, there is neither metabolism nor enzyme action in the cytoplasm of dormant spores, although there are several enzyme–substrate pairs located in this region of the spore (3). There is also no detectable metabolism or enzyme action in the core of stage I-germinated spores that have excreted their DPA but have not degraded their cortex (7). However, in fully germinated (stage II) spores in which the cortex has been degraded and the cytoplasm is fully hydrated, enzyme action and metabolism begin rapidly (2).
One explanation that has been put forward for the lack of enzyme action in dormant and stage I-germinated spores is that the level of water in these spores is too low to allow enzyme action (3). Unfortunately, although the levels of total water in the cores of spores of a number of species are known (5), the percentage of this water that is free water (if any) is not known. However, it is certainly possible that the amount of water in the spore core is too low to allow sufficient macromolecular movement for enzyme action. Indeed, there are data that have been interpreted as indicating that (i) ions in the spore core are relatively immobile (9, 10), and (ii) the dormant spore core is in a glass-like state (11–13). In addition, the levels of DPA in the spore core are far above its solubility, and available data indicate that at least the great majority of spore DPA is not in solution (14, 15). However, there are also other data suggesting that there is some mobility of phosphorylated molecules in the spore core, although this may be only vibrational motion (14).
Despite strong circumstantial evidence that macromolecules in the spore core are likely to be immobile, there is no direct evidence for this. One way in which molecular movement within cells can be assessed directly is the technique of fluorescence redistribution after photobleaching (FRAP) (16–20). In this technique a fluorescence microscope is used to monitor fluorescence intensity before and after a high-intensity laser is used to photobleach one region of a cell. By monitoring the kinetics and the extent of movement of fluorescent molecules from the unbleached area into the bleached area, both the percentage of fluorescent molecules that are mobile and their diffusion coefficient can be calculated. This technique has been used successfully to measure the movement of a number of macromolecules in bacteria (21–25). The fluorescent molecule we chose for use was the GFP from Aequoria victoria, because this protein and its many variants have been used to measure the mobility of soluble proteins in a number of systems (26–28). GFP was expressed to high levels in the cytoplasm or core of spores of Bacillus subtilis (29), and FRAP analysis was used to determine the mobility of this GFP in dormant spores as well as in spores at both stages I and II of spore germination.
Methods
Strains and Spore and Cell Preparation.
The B. subtilis strains used in this work are all derivatives of strain 168 and were PS832, a prototrophic derivative of strain 168; PS3207, cwlD Cmr (7); CW355 (obtained from R. Losick, Harvard University, Cambridge, MA), encoding GFP fused to the first 21 codons of the sspE gene under control of the strong forespore-specific sspE-2G promoter with an adjacent K (10 μg/ml) marker (29, 30); PS3518, sspE-gfp K
(10 μg/ml) marker (29, 30); PS3518, sspE-gfp K , made by transformation of chromosomal DNA from strain CW355 into PS832 and selection for K
, made by transformation of chromosomal DNA from strain CW355 into PS832 and selection for K ; and PS3519, cwlD sspE-gfp Cmr K
; and PS3519, cwlD sspE-gfp Cmr K , made by transformation of chromosomal DNA from strain CW355 into PS3207 and selection for K
, made by transformation of chromosomal DNA from strain CW355 into PS3207 and selection for K .
.
Spores of all strains were prepared at 37°C on 2× SG medium agar plates and harvested, cleaned, and stored as described (31). All dormant spore preparations used in this work were free (≥98%) of growing cells, germinated spores, and cell debris. To prepare germinated spores, the spores at an OD600 of ≈20 in water were first heat-shocked for 30 min at 70°C and then cooled. The heat-shocked spores (1 ml) were added to 19 ml of prewarmed (37°C) 10 mM Tris⋅HCl (pH 8.8) and 10 mM l-alanine and incubated at 37°C for 60 min (PS3518 spores) or 120 min (PS3519 spores) before purification of germinated spores by density-gradient centrifugation as described (32). The spore suspension was centrifuged, suspended in 30% Nycodenz (Sigma), and layered on a 35–50% Nycodenz density gradient (≈2 ml total volume), and the tube was centrifuged for 1 h at 13,000 rpm in the small swinging bucket head of the TLS55 ultracentrifuge (Beckman–Spinco). The light-germinated spore band was removed by aspiration, diluted to ≈1.5 ml with sterile PBS [25 mM KPO4 (pH 7.4)/0.15 M NaCl], washed several times by centrifugation with PBS, and suspended in 50 μl of PBS before application to microscope slides. Growing cells of strain PS3518 were obtained by heat shocking 0.5 ml of spores at an OD600 of 10 as described above. The heat-shocked spores were germinated at 37°C in 4.5 ml of 2× YT medium (per liter: 5 g of NaCl/16 g of tryptone/10 g of yeast extract) plus 4 mM l-alanine. After ≈2 h of incubation at 37°C, the original ovoid spore had elongated to bacillary form, the culture was harvested, and the pellet fraction was suspended in 50 μl of PBS before microscopy.
Microscopy and FRAP Analysis.
Aliquots of spore or cell suspensions were applied to either agarose- or polylysine-coated microscope slides; after a coverslip was added, the edges of the coverslip were sealed with clear nail polish (Revlon). Images were collected on a Zeiss LSM510 confocal microscope by using a ×100, 1.4-numerical aperture planapocromat objective. The pinhole was opened fully such that all fluorescence from the cell was collected. Excitation used the 488-nm line from an argon laser, and the confocal scan axis was oriented such that the x direction of the scan axis was perpendicular to one axis of the cell as shown in Fig. 1A. To minimize the time for image collection, the scan area was limited to a region of interest consisting of a box slightly larger than the spore. By using the time series function in the Zeiss AIM software, five prebleach images of the spore were collected, and a region of interest consisting of a box overlying one half of the spore was photobleached rapidly by using unattenuated laser light. Immediately (4–6 msec) after photobleaching, 45 successive images were collected to monitor the redistribution after the photobleaching. In most cases images were collected with no delay between images, and the total time between successive images (collection time plus blank time before the next image was collected) ranged from 79 to 209 msec. To control for the fraction of GFP that exhibits “reversible bleaching” (33, 34), control time series were collected by using spores in which the entire spore was photobleached. To determine the effect of bleaching during monitoring, spores were monitored in an identical fashion except that no light was used during the bleach.
Fig 1.

Diagram showing photobleaching protocol and quantitative analysis of the images. (A) For collection of time series before and after photobleaching, the confocal scanning was restricted to a box slightly larger than the spore, aligned such that one axis of the spore was parallel to the y axis of the scan. After collecting prebleach images, a rectangular region covering half the spore was photobleached rapidly during one pass of the scan by using unattenuated laser light. (B) For the quantitative analysis, fluorescence intensity was averaged across the cell at each position along the x axis of the cell, yielding a plot of average fluorescence intensity along the y axis of the cell. Diffusion coefficient and mobile fraction then are calculated from the intensity versus position data obtained from successive images in the time series as described. (C) Diagrammatic representation of FRAP theory. The fluorescence image, F(r, t), is written as a convolution of the impulse–response function of the optical detection process, I(r), and the concentration of fluorescent molecules, c(r, t). The initial bleaching of approximately one-half of the concentration is shown in the dotted lines. Above the representation of the fluorescence image, F(r, t), is the corresponding sinusoidal component in Fourier space (see FRAP Theory for details).
Image processing was done by using METAMORPH software (Universal Imaging, Downingtown, PA). An average background value determined from a cell-free area in the image was first subtracted for each image in a time series, and average intensity values were calculated perpendicular to a line drawn down the long axis of the spore (see Fig. 1B). The analysis of the fluorescence redistribution was performed with the software system MLAB, constructed by Civilized Software (Bethesda). The diffusion coefficient (D) and mobile fraction (R) for GFP were calculated based on a modification of the normal-mode analysis described in ref. 18 and below.
FRAP Theory.
The FRAP analysis is based in principle on the normal-mode analysis described previously for membrane components (18). The normal-mode analysis is an appropriate method in cases where the sample geometry is not infinitely large compared with the bleached area but rather is a bounded region on the same scale as the bleached region and has a relatively symmetric geometry. The problem is first reduced to a one-dimensional problem by determining average fluorescence along one dimension of the two-dimensional image, and the fluorescence intensity related to concentration of fluorescent probe based on the convolution of the fluorescence signal in the optical system. In the one-dimensional case, the general solution for the concentration is the sum of Fourier spatial components, and the coefficient of the appropriate sinusoidal component in Fourier space will decay exponentially proportional to the diffusion coefficient D. To determine D, the Fourier transform of measured intensity is first corrected for bleaching during monitoring and for any asymmetry of the prebleach intensity.
The fluorescence image, F(r, t), can be written as a convolution of the impulse–response function of the optical detection process, I(r), and the concentration of fluorescent molecules, c(r, t),
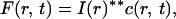 |
where ** signifies the convolution operation,
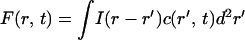 |
(see Fig. 1C). We can apply the well known relationship for the Fourier transform of a convolution, giving
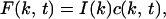 |
where the Fourier transform is written as
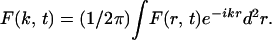 |
We can then see that F(k, t) is simply proportional to c(k, t) for any value of k.
Consider the case where c(r, t) is limited in space to the one-dimensional equivalent of a box of 2a0 in y, where we place a bleaching box to the lower half of the area (see Fig. 1C). We define a function g1(t) that is proportional to |F(k0, t)| but is corrected for bleaching during monitoring and normalized for any asymmetry of the prebleach intensity. For the normalized bleaching asymmetry at k0 we have
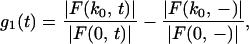 |
where the minus sign signifies the average of the prebleach Fourier transform, and k0 equals π/a0. We estimate a0 from the points halfway to the top of the prebleach intensity as a function of x. g1(t) is proportional to |F(k0, t)|, but it is corrected for bleaching during monitoring by |F(0, t)| and corrected for any slight asymmetry of the prebleach intensity by |F(k0, −)|/|F(0, −)|.
If we assume that the fluorescent molecules in question undergo simple irreversible photobleaching, and c(r, t) assumes an isotropic two-dimensional diffusion with diffusion coefficient D, we then have
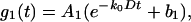 |
where the immobile component is represented by b1. We calculate the mobile fraction, R,
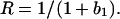 |
We know that the fluorescence emission of GFP is not as simple as assumed. Aside from irreversible photobleaching, it undergoes light-driven “on–off” blinking (33, 34). With a certain probability the molecule undergoes a transition from an excited state to a long-lived dark state, which then undergoes the reversible transition to a singlet ground state with a slow characteristic time in the range of seconds, independent of light. Only then, when the singlet state absorbs a photon, will fluorescence resume. This reversible photobleaching cannot affect molecules with diffusion times faster than this slow characteristic time. It can make the apparent diffusion times of molecules seem shorter than they actually are when they range up to the slow characteristic time and above.
Let us see what happens when we take a population of fluorophores that are immobile but undergo a certain percentage of reversible photobleaching. We bleach one half of the cell, as before, and monitor the fluorescence as it appears as a function of time. Let us assume there is no bleaching during monitoring. After the bleach of time T,
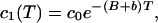 |
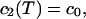 |
where B is the irreversible photobleaching, and b is the reversible photobleaching. If we see where the fluorophores have gone, we have
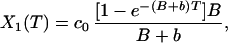 |
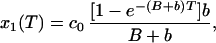 |
where X1(T) and x1(T) are the irreversible photobleached component and the reversible photobleached component, respectively. If we then see that the x1(T) reversibly decays down to 0, as it reappears as a component of c1(t), we then have
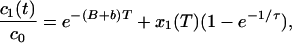 |
where τ is the reversible time constant. We can then make g1(t) proportional to c1(t) and c2(t).
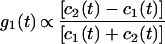 |
A plot of g1(t), as functions of b/(B + b) for (B + b)T equals 1.5, and τ = 1.6 sec, is shown in Fig. 4, which is published as supporting information on the PNAS web site, www.pnas.org.
Results and Discussion
The spores used for the FRAP analysis were all from B. subtilis strains with or without the A. victoria gfp gene fused to the first 21 codons of the B. subtilis sspE gene under the control of the sspE-2G promoter (29). This latter promoter is extremely strong and directs high levels of expression only in the forespore compartment of the sporulating cell (30). Consequently, these spores have extremely high levels of GFP in the spore core (ref. 29 and see below). However, in all other respects, these GFP-containing spores appear normal (data not shown).
Dormant spores of strains CW355 or PS3518 were subjected to FRAP analysis. Strikingly, >70% of the GFP in dormant spores was immobile over a 10-sec time course, and no difference was observed between spores of strains CW355 and PS3518. A representative experiment is shown in Fig. 2 A and B (see Movie 1, which is published as supporting information on the PNAS web site). Sixteen dormant spores (5 of strain CW355 and 11 of strain PS3518) monitored over this time scale showed little FRAP, yielding a calculated mobile fraction of 0.31 ± 0.17 with a diffusion coefficient (D) of (7.7 ± 5.2) × 10−10 cm2/sec. This diffusion coefficient is ≈3 orders of magnitude slower than expected for a soluble protein of similar size.
Fig 2.

FRAP of GFP in wild-type dormant and germinated spores of B. subtilis (strain PS3518). (A, C, and E) Fluorescence intensity averaged across the spore along a line drawn down the longest axis of the spore (y) before and after photobleaching. ○, average of five prebleach scans; ▵, first postbleach scan; □, last postbleach scan. The lower values for the last postbleach scan are the result of some photobleaching that occurs during continuous monitoring. Bleaching during monitoring is corrected for in the subsequent analysis (see Methods). The vertical bars show the regions defined as the beginning and end of the spore for the determination of the diffusion coefficient. (B, D, and F) Fits to Eq. 6 from which D and R are derived. (A and B) A representative dormant spore in which half the spore was photobleached. (C and D) The composite results from control spores and include the average of five control dormant spores in which the entire spore was photobleached and five control spores that were not photobleached but were otherwise monitored the same as photobleached spores. Both halves of each spore monitored without bleaching were averaged together and plotted as for the left half of the spore, whereas both halves of the five control spores in which the entire spore was photobleached were average together as the “right” half of the spore. These data then were fitted to Eq. 6, and the results are shown in D. (E and F) A representative germinated spore in which half the spore was photobleached. Redistribution of fluorescence was essentially complete before the first postbleach scan was obtained.
Because of the slow characteristic time of the observed recovery of fluorescence in the bleached region, we undertook control experiments to determine what fraction of the increased fluorescence intensity in the bleached region might be due to reversible photobleaching of GFP. A form of “blinking” of GFP fluorescence on the time scale of seconds is known to result from the light-induced conversion of the GFP chromophore from a fluorescent (light state) to a nonfluorescent (dark state) form, which can revert back slowly to the fluorescent form (33, 34). The prevalence of formation of the dark state and the characteristic time for conversion from dark to light states are variable depending on the exact form of GFP used and its chemical environment. To determine the extent of reversible photobleaching in our experiments, the entire dormant spore (strain PS3518) was bleached, and fluorescence intensity was monitored over time. In parallel, time series were also collected of spores that received no high-intensity bleach to also account for residual affects of bleaching during monitoring alone. The control experiments revealed that for GFP in the milieu of the dormant spore core, exposure to the bleaching pulse induced conversion of a significant percentage of the GFP molecules to a transiently dark state that subsequently become fluorescent (Fig. 2 C and D; see FRAPTheory for a complete discussion of this analysis). The time constant (1.5 sec) and recovery fraction (0.28) observed in g1(t) in control spores (Fig. 2D) are similar to the time constant (1.7 sec) and mobile fraction (0.30) calculated for the apparent redistribution observed in dormant spores when half the cell was photobleached (Fig. 2B). This indicates that essentially all of the small amount of slow apparent fluorescence redistribution of GFP observed in dormant spores is due to the conversion of bleached GFP from a dark state back to a light-emitting state without diffusion within the spore. When we analyzed dormant spores for postbleach times of ≈20 sec with a function that contained a second exponential, rather than a constant representing an immobile fraction, we obtained a time constant of >600 sec, corresponding to a D of <10−12 cm2/sec. Thus, we conclude that all of the GFP in the dormant spore is essentially immobilized on the time scale of FRAP experiments down to a D of <10−12 cm2/sec.
When GFP in the germinated spore (strain PS3518) was subjected to photobleaching, redistribution was essentially complete within the first postbleach scan for 12 of 16 germinated spores examined. The fastest possible time course for data collection using the confocal scanning mirrors (≈70 msec per scan) was insufficient to accurately determine a characteristic diffusion time for these spores examined, indicating that the diffusion coefficient of GFP in these germinated spores was >10−8 cm2/sec. An example is shown in Fig. 2 E and F (for time series, see Movie 2, which is published as supporting information on the PNAS web site). This value should be compared with D = 7.7 × 10−8 cm2/sec, previously reported for GFP diffusion in the cytoplasm of Escherichia coli (35). Four germinated spores exhibited a somewhat slower characteristic time for diffusion of GFP, and in these cases we were able to calculate a diffusion coefficient of (2–8) × 10−9 cm2/sec. It is possible that these four spores have not completed the transition into stage II of germination (see below), resulting in reduced diffusion of GFP compared with the other 12 germinated spores measured. Control experiments such as those described above for GFP in the dormant spore demonstrated that reversal from the dark to light state of GFP in germinated spores does not contribute significantly to the observed redistribution in germinated spores; conversion from the dark to light state represented only 9% of the total GFP molecules and had a time constant of 0.16 sec. Because dark- to light-state transition rates for GFP depend on the chemical environment, we speculate that the unique environment of GFP in the dormant spore is likely responsible for the large difference in dark- to light-state transitions in dormant spores compared with germinated spores. FRAP analysis of vegetative cells isolated shortly after completion of outgrowth of GFP-containing spores (strain PS3518) again showed that GFP was mobile in these cells with a characteristic time of redistribution similar to that in fully germinated spores (data not shown).
As noted above, a likely reason for the immobility of GFP in the cytoplasm of dormant spores is the low core water content. However, an alternative or at least a contributing factor could be the high core levels of DPA and divalent cations. To test the contribution of DPA to GFP immobility we used spores carrying both the gfp fusion and a cwlD mutation. This latter mutation results in a modified spore cortex lacking muramic acid lactam, the recognition determinant for the enzymes that initiate cortex hydrolysis during spore germination (2, 8, 36, 37). Consequently, although cwlD spores will initiate germination in response to nutrients and release DPA rapidly, they cannot progress past stage I of germination, and the core water content of germinated cwlD spores [≈60% of wet weight (7, 8)] is only slightly above that of dormant spores of several other Bacillus species (5). As was found with GFP in dormant wild-type spores, GFP in dormant cwlD spores was also essentially immobile (Fig. 3 A and B and Movie 3, which is published as supporting information on the PNAS web site). However, in contrast to the mobility of GFP in germinated wild-type spores, GFP was also immobile in germinated cwlD spores (Fig. 3 C and D and Movie 4, which is published as supporting information on the PNAS web site). Because DPA and associated divalent cations are not present in germinated cwlD spores, this latter finding indicates that these molecules are not critical for the immobility of GFP in the spore cytoplasm.
Fig 3.

FRAP of GFP in dormant and germinated cwlD spores of B. subtilis (strain PS3519). (A and C) Fluorescence intensity averaged across the spore along a line perpendicular to the longest axis of the cell (y) before and after photobleaching. ○, average of all five prebleach scans; ▵, first postbleach scan; □, last postbleach scan. The vertical bars show the regions defined as the beginning and end of the spore for determination of the diffusion coefficient. (B and D) Fits to Eq. 6 from which D and R are derived. (A and B) Data from a representative dormant cwlD spore in which half the spore was photobleached. (C and D) Results from a representative germinated cwlD spore in which half the spore was photobleached.
The immobility of GFP in the cytoplasm of dormant and stage I-germinated spores (i.e., germinated cwlD spores) by inference suggests that all proteins are immobile in the cytoplasm of these spores, and this is certainly consistent with the lack of enzyme action in these spores. It is possible, of course, that there is mobility of proteins in the spore core on a time scale longer than can be used in FRAP analyses and that there is also some difference in the mobility of proteins in dormant and stage I-germinated spores. Indeed, because the cores of stage I-germinated spores have a higher water content than do that of dormant spores (≈60% versus ≈40%; refs. 8 and 36), this might be expected. However, because of the interference caused by the reversible photobleaching of the GFP in the spore core, the FRAP experiments described here do not allow this discrimination.
It is clear that there is a difference of at least 4 orders of magnitude in the diffusion coefficients of the great majority of GFP in the core of dormant (D ≤ 10−12 cm2/sec) and fully germinated (D ≥ 10−8 cm2/sec) spores. If this result for GFP is also true for other proteins in the spore core, this could explain in part the stability of enzymes within the spore core, because enzymes very often are stabilized by immobilization (38). Protein immobility may also explain the lack of interaction of some enzyme–substrate pairs in the spore core, because at least one of these pairs are a protease and its protein substrate (3). However, because immobilized enzymes often exhibit significant if not full enzyme activity on small molecule substrates (39), lack of enzyme mobility seems unlikely of itself to fully explain the lack of enzyme activity in the spore core. Two other factors can be imagined as likely involved in the latter phenomenon. (i) The lack of protein mobility in dormant and stage I-germinated spores may be only one manifestation of the low core water content that may also prevent enzyme action by not providing sufficient water for enzyme function. Unfortunately there are no data available on the amount of free water in dormant spores or on the amount of water bound to core proteins. (ii) Not only proteins but also small molecules may be immobile in dormant spores. If this is the case, then enzyme action in the spore core would be precluded further by the prevention of interaction of enzymes and their small molecule substrates. Indeed, as noted above there is evidence that has been interpreted as indicating that at least ions in the dormant spore core are immobile and that the spore core is in a glass-like state (9–13). In any event, the environment within the core of the dormant and stage I-germinated spore is a very special and unique one, and the features of this environment clearly contribute to the dormancy as well as the resistance of the dormant spore.
Supplementary Material
Acknowledgments
We are grateful to R. Losick for the gift of strain CW355. This work was supported by a grant from the U.S. Army Research Office. The Center for Biomedical Imaging Technology is also supported by National Institutes of Health Grant RR13186.
Abbreviations
DPA, pyridine-2,6-dicarboxylic acid (dipicolinic acid)
FRAP, fluorescence redistribution after photobleaching
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Driks A. & Setlow, P. (1999) in Prokaryotic Development, eds. Brun, Y. V. & Shimkets, L. J. (Am. Soc. Microbiol., Washington, DC), pp. 191–218.
- 2.Paidhungat M. & Setlow, P. (2002) in Bacillus subtilis and Its Relatives: From Genes to Cells, eds. Hoch, J. A., Losick, R. & Sonenshein, A. L. (Am. Soc. Microbiol., Washington, DC), pp. 537–548.
- 3.Setlow P. (1994) J. Appl. Bacteriol. 76,Suppl., 49S-60S. [DOI] [PubMed] [Google Scholar]
- 4.Setlow P. (2000) in Bacterial Stress Responses, eds. Storz, G. & Hengge-Aronis, R. (Am. Soc. Microbiol., Washington, DC), pp. 217–230.
- 5.Gerhardt P. & Marquis, R. E. (1989) in Regulation of Prokaryotic Development, eds. Smith, I., Slepecky, R. A. & Setlow, P. (Am. Soc. Microbiol., Washington, DC), pp. 43–64.
- 6.Paidhungat M., Setlow, B., Driks, A. & Setlow, P. (2000) J. Bacteriol. 182, 5505-5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Setlow B., Melly, E. & Setlow, P. (2001) J. Bacteriol. 183, 4894-4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popham D. L., Helin, J., Costello, C. E. & Setlow, P. (1996) Proc. Natl. Acad. Sci. USA 93, 15405-15410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carstensen E. L., Marquis, R. E., Child, S. Z. & Bender, G. R. (1979) J. Bacteriol. 140, 917-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carstensen E. L., Marquis, R. E. & Gerhardt, P. (1971) J. Bacteriol. 107, 106-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ablett S., Darke, A. H., Lillford, P. J. & Martin, D. R. (1999) Int. J. Food Sci. Technol. 34, 59-69. [Google Scholar]
- 12.Leuschner R. G. & Lillford, P. J. (2003) Int. J. Food Microbiol. 80, 131-143. [DOI] [PubMed] [Google Scholar]
- 13.Sapru V. & Labuza, T. P. (1993) J. Food Sci. 58, 445-448. [Google Scholar]
- 14.Leuschner R. G. & Lillford, P. J. (2000) Microbiology 146, 49-55. [DOI] [PubMed] [Google Scholar]
- 15.Leuschner R. G. & Lillford, P. J. (2001) Int. J. Food Microbiol. 63, 35-50. [DOI] [PubMed] [Google Scholar]
- 16.Edidin M., Zagyansky, Y. & Lardner, T. J. (1976) Science 191, 466-468. [DOI] [PubMed] [Google Scholar]
- 17.Koppel D. E. (1979) Biophys. J. 28, 281-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koppel D. E. (1985) Biophys. J. 47, 337-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobson K., Derzko, Z., Wu, E. S., Hou, Y. & Poste, G. (1976) J. Supramol. Struct. 5, 565-576. [DOI] [PubMed] [Google Scholar]
- 20.Schlessinger J., Koppel, D. E., Axelrod, D., Jacobson, K., Webb, W. W. & Elson, E. L. (1976) Proc. Natl. Acad. Sci. USA 73, 2409-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brass J. M., Higgins, C. F., Foley, M., Rugman, P. A., Birmingham, J. & Garland, P. B. (1986) J. Bacteriol. 165, 787-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foley M., Brass, J. M., Birmingham, J., Cook, W. R., Garland, P. B., Higgins, C. F. & Rothfield, L. I. (1989) Mol. Microbiol. 3, 1329-1336. [DOI] [PubMed] [Google Scholar]
- 23.Sarcina M., Tobin, M. J. & Mullineaux, C. W. (2001) J. Biol. Chem. 276, 46830-46834. [DOI] [PubMed] [Google Scholar]
- 24.Schindler M., Osborn, M. J. & Koppel, D. E. (1980) Nature 285, 261-263. [DOI] [PubMed] [Google Scholar]
- 25.Stricker J., Maddox, P., Salmon, E. D. & Erikson, H. P. (2002) Proc. Natl. Acad. Sci. USA 99, 3171-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dayel M. J., Hom, E. F. & Verkman, A. S. (1999) Biophys. J. 76, 2843-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reits E. A. & Neefjes, J. J. (2001) Nat. Cell Biol. 3, E145-E157. [DOI] [PubMed] [Google Scholar]
- 28.Swaminathan R., Hoag, C. P. & Verkman, A. S. (1997) Biophys. J. 72, 1900-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webb C. D., Decatur, A., Teleman, A. & Losick, R. (1995) J. Bacteriol. 177, 5906-5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun D., Fajardo-Cavazos, P., Sussman, M. D., Tovar-Rojo, F., Cabrera-Martinez, R.-M. & Setlow, P. (1991) J. Bacteriol. 173, 7867-7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicholson W. L. & Setlow, P. (1990) in Molecular Biological Methods for Bacillus, eds. Harwood, C. R. & Cutting, S. M. (Wiley, Chichester, U.K.), pp. 391–450.
- 32.Lindsay J. A., Beaman, T. C. & Gerhardt, P. (1985) J. Bacteriol. 163, 735-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dickson R. M., Cubitt, A. B., Tsien, R. Y. & Moerner, W. E. (1997) Nature 388, 355-358. [DOI] [PubMed] [Google Scholar]
- 34.Weber W., Helms, V., McCammon, J. A. & Langhoff, P. W. (1999) Proc. Natl. Acad. Sci. USA 96, 6177-6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elowitz M. B., Surette, M. G., Wolf, P.-E., Stock, J. B. & Leibler, S. (1999) J. Bacteriol. 181, 197-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sekiguchi J., Akeo, K., Yamamoto, H., Khasanov, F. K., Alonso, J. C. & Kuroda, A. (1995) J. Bacteriol. 177, 5582-5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Atrih A., Zollner, P., Allmaier, G. & Foster, S. J. (1996) J. Bacteriol. 178, 6173-6183.. (Am. Soc. Microbiol., Washington, DC), pp. 191–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kilbanov A. (1979) Anal. Biochem. 93, 1-25. [PubMed] [Google Scholar]
- 39.Mosbach K. (1976) Methods Enzymol. 44, 3-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}