

Abstract
Humpback whales (Megaptera novaeangliae) annually undertake the longest migrations between seasonal feeding and breeding grounds of any mammal. Despite this dispersal potential, discontinuous seasonal distributions and migratory patterns suggest that humpbacks form discrete regional populations within each ocean. To better understand the worldwide population history of humpbacks, and the interplay of this species with the oceanic environment through geological time, we assembled mitochondrial DNA control region sequences representing approximately 2700 individuals (465 bp, 219 haplotypes) and eight nuclear intronic sequences representing approximately 70 individuals (3700 bp, 140 alleles) from the North Pacific, North Atlantic and Southern Hemisphere. Bayesian divergence time reconstructions date the origin of humpback mtDNA lineages to the Pleistocene (880 ka, 95% posterior intervals 550–1320 ka) and estimate radiation of current Northern Hemisphere lineages between 50 and 200 ka, indicating colonization of the northern oceans prior to the Last Glacial Maximum. Coalescent analyses reveal restricted gene flow between ocean basins, with long-term migration rates (individual migrants per generation) of less than 3.3 for mtDNA and less than 2 for nuclear genomic DNA. Genetic evidence suggests that humpbacks in the North Pacific, North Atlantic and Southern Hemisphere are on independent evolutionary trajectories, supporting taxonomic revision of M. novaeangliae to three subspecies.
Keywords: diversity, gene flow, genomic, mitochondrial, whale, cetacean
1. Introduction
The humpback whale (Megaptera novaeangliae) is an iconic and globally distributed migratory species. Within each ocean basin, humpbacks breed and calve in tropical and subtropical seas during winter, migrating to high latitudes to feed during summer. Despite an absence of geographical barriers to dispersal, populations show significant genetic structure between and within ocean basins [1–7], with the strongest restrictions to maternal gene flow across the equatorial boundary [1,8]. Observations of naturally marked and genotyped individuals suggest that maternally directed fidelity to both breeding and feeding grounds may be responsible for this population structure [9–14]. Some population structure has also been identified in the nuclear genome; a recent study using multiple nuclear introns to survey Atlantic diversity [15] found evidence of population structuring between the North Atlantic and Southern Hemisphere, but not among breeding and feeding grounds within the North Atlantic.
Although phylogenetic reconstructions of mtDNA show evidence of long-term gene flow between oceans [1], no permanent dispersal between populations in different hemispheres has been documented. Although seasonal breeding cycles are asynchronous between the hemispheres, two Southern Hemisphere breeding grounds extend north of the equator—Ecuador and Costa Rica in the Pacific [16], and Gabon and Guinea in the Atlantic [17,18]—demonstrating that inter-hemisphere movements are biologically possible. Encounters on common breeding grounds between whales at the end or start of their respective winter breeding seasons could result in male-mediated gene flow, but genetic patterns of population structure and haplotype distribution show no evidence of this to date [7].
Despite the evidence for limited gene flow between oceans, there has been no recent taxonomic investigation of humpback whales [19]. In 1946, Tomilin [20] proposed humpbacks in the two hemispheres as subspecies, on the basis of a greater measured body length in the Southern Hemisphere form. Subsequent investigation [21] found no significant variation in lengths between oceans and, along with a later review of cetacean taxonomy [22], concluded that there was insufficient evidence for subspecies. Multiple lines of evidence for genetic divergence [22] or the diagnosis of fixed character differences [23] could be a reason to revisit their status.
Unlinked nuclear DNA markers provide multiple independent lines of evidence for reconstructing evolutionary histories of population structuring within species [24]. Here, we assess the genetic evidence for humpback global population structure, using the largest global genetic dataset for this species to date, including eight nuclear loci (approx. 3700 bp in total length) from more than 70 individuals and mtDNA control regions from more than 2900 individuals inhabiting all three ocean basins. We use conventional frequency and coalescent-based population genetic approaches to (i) describe the pattern and magnitude of organismal (mtDNA) and gametic (nuclear DNA) gene flow between oceans, and (ii) estimate the time frame of radiation of extant humpback whale lineages. We consider these patterns of gene flow in the context of the criteria currently recognized as defining subspecies for cetaceans.
2. Material and methods
(a). mtDNA and nuclear datasets
Sequences spanning 465 bp of the mtDNA control region were compiled from North Pacific [5,25] (n = 396), South Pacific and southeastern Indian Ocean [13] (n = 804), southwestern Atlantic [26] (n = 48), southwestern Indian Ocean [27] (n = 1137) and western North Atlantic [25,28] (n = 348) studies. Blue (Balaenoptera musculus) and fin (Balaenoptera physalus) whales were used as outgroups in phylogenetic reconstructions (GenBank AY582748, NC_001321, NC_001601 and AY582748) following Sasaki et al. [29]. Sequences were aligned by eye in MacClade v. 4.0 [30]. Another mtDNA dataset was assembled from shorter sequences, allowing greater sample representation from North Atlantic locations [3,4] (n = 246); on alignment, these overlapped with the 465 bp worldwide dataset by 285 bp. Eight nuclear loci (ACT, CAT, CHRNA1, ESD, FGG, GBA, LAC and RHO; GenBank GQ407914–408186) were phased as described by Jackson et al. [25] for 70–80 humpback whales worldwide (figure 1). Exonic regions were excluded from analysis. ModelTest v. 3.7 [31] was used to determine the Akaike information criterion best-fitting model for all datasets, with branch lengths included as parameters [32].
Figure 1.

Worldwide distribution of humpback whale mtDNA source locations. Wintering regions (circles), feeding grounds (crosses) and migratory routes (grey circles) are shown. Double circles show nuclear DNA source locations. Double-lined boxes denote ocean–basin groupings.
(b). Mitochondrial population structure and gene flow
Nucleotide diversity (π) and haplotype diversity (h) were estimated following Nei [33] using Arlequin v. 3.1 [34]. Differentiation between oceans was estimated using FST and ϕST with 50 000 matrix permutations. To correct for multiple substitutions, ϕST was adjusted using the Kimura 2-parameter + Γ mutation model (as supported by ModelTest for the humpback-only dataset). Hierarchical AMOVA tests were conducted to measure partitioning of variance (1) between oceans, (2) among regions (breeding and feeding grounds) within oceans, and (3) within regions (figure 1). Two groupings were considered; (1) ‘5-oceans’ (North Pacific, South Pacific, North Atlantic, South Atlantic and Southern Indian Ocean) and (2) three ocean basins (‘3-oceans’: North Pacific, North Atlantic and Southern Hemisphere).
The effective population size Θ and numbers of effective female migrants per generation 2Nfmf (equivalent to Nemf) were estimated for both mtDNA datasets, partitioned by 3-oceans and 5-oceans, using the Bayesian inference program MIGRATE-N v. 3.5.1 [35,36]. Each oceanic partition was sub-sampled to generate computationally tractable datasets containing 150 and 200 animals. Sampling was stratified within each ocean basin or ocean so that roughly equal numbers of samples were randomly selected from each breeding or feeding ground. Analyses were run with four Markov chains and gamma-distributed priors on 2Nfmf (range 0–20 for 3-oceans, 0–100 for 5-oceans) and Θ (range 0–0.1). The heating scheme was set to temperatures 1.0, 1.5, 3.0 and 100 000.0. Analyses were conducted with 100 replicates, with 10 000 steps recorded every 100 generations and 50% burn-in, totalling 50 million retained parameter values.
(c). Mitochondrial phylogeny and divergence times
A phylogeny of mtDNA control region sequences was reconstructed in MrBayes v. 4.0 [37] using a HKY + Γ model of sequence evolution (as supported by ModelTest for humpback whales plus outgroups). Analysis was conducted for 25 million generations (sampling every 1000 generations), with 10% discarded as burn-in and split frequencies monitored for convergence. Posterior parameter distributions were examined using TRACER v. 1.5 [38].
To estimate within-species divergence times, a humpback-only dataset was analysed in BEAST v. 1.7.4 [39], containing 150 individuals from each of the five oceans, chosen by stratified random sub-sampling as above. As humpback whales exhibit complex sub-structure, we tested the fit of three coalescent models (expansion, logistic and constant size), and to strict and relaxed lognormal clock rates. Analyses were run for 25 million generations. Bayes factors (BFs) were calculated in TRACER to determine the best-fitting population model. We imposed a strict clock with a rate of 3.94% bp−1 Myr−1 for the humpback control region as determined by phylogenetic approaches [25]. To assess the impact of assumed substitution rate, we repeated the analysis using a higher rate of substitution (mean 14.9% bp−1 Myr−1), as estimated by Ho et al. [40] for bowhead whales using ancient DNA sequences.
(d). Neutrality and population expansion
Tajima's D [41,42] and Fu's Fs [43] were estimated to test for selection (versus population neutrality) worldwide and for haplotype clades within each ocean basin. Mismatch distributions were generated to test null hypotheses of population expansion for the Southern Ocean basin and for the three Northern Hemisphere mtDNA clades using Arlequin [34], with 1000 bootstrap replicates.
(e). Nuclear diversity, structuring and gene flow
Nuclear heterozygosity was estimated for phased alleles and sequences (π), and FST and ϕST estimates of differentiation between oceans and ocean basins were measured with 50 000 permutations in Arlequin. We used Jombart's discriminant analysis of principal components (DAPC [44]) in the R package adegenet to evaluate the level of support for different numbers of distinct genetic clusters (K = 1–20) in the absence of a priori ocean divisions. DAPC can discriminate complex patterns like hierarchical clustering or stepping stone structures—realistic possibilities, as humpbacks exhibit fidelity to multiple migratory routes between various breeding and feeding areas. Sequential K-mean clustering was applied to find the best-supported cluster size. All 21 principal components (PCs) were retained initially. We then applied the discriminant function dapc, which constructs synthetic variables in order to maximize variation between, and minimize variation within, each cluster group. After inspecting the cumulative variance retained by the PCs, the a-score was used to calculate that five PCs are sufficient to characterize population structure. Based on these discriminant functions, posterior probabilities of membership to each of the three ocean basins (North Atlantic, North Pacific and Southern Hemisphere) were calculated for each individual and within each ocean basin.
Population differentiation and partitioning of allelic variance was calculated (1) within individuals, (2) among individuals within three ocean basins and (3) among individuals between three ocean basins, using the standard AMOVA test in Arlequin [45]. For this we included only individuals for which more than 75% of intronic loci had been sequenced. Effective population size for each ocean basin and effective numbers of migrants were co-estimated using MIGRATE-N [35,36] with gamma distributed priors ranging between 0–0.1 and 0–20, respectively. Analyses were conducted for 100 replicates, with 10 000 steps recorded every 1000 generations and 50% removed as burn-in, totalling 500 million parameter values retained.
Extended nuclear DNA sequences (including intronic and exonic sequences described in [25]) were aligned with a selection of Balaenopteridae (blue, fin and Antarctic minke whales, B. bonaerensis) in MacClade [30]. Patterns of allelic divergence between humpback, blue and fin whales were calculated using statistical parsimony networks with the program TCS v. 1.21 [46].
3. Results
The mtDNA control region sequences (length 465 bp) were compiled for 2733 individuals worldwide (2979 individuals at length 285 bp). This corresponds to 1.3 Mb of data. The eight nuclear loci corresponded to 3.7 kb, yielding 140–160 alleles per locus, a total dataset size of 663 kb (table 1).
Table 1.
Basic diversity estimates of nuclear genomic and mtDNA control region sequences used in this study. CR 465 and CR 285 refer to the two mtDNA datasets, ‘introns’ shows statistics summed over all intronic loci. Numbers in parentheses represent alleles private to each ocean basin. Levels of nucleotide diversity (π) and their standard deviations (s.d.) are reported.
| ACT | CHRNA1 | GBA | CAT | LAC | RHO | ESD | FGG | introns | CR 465 | CR 285 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| no. of chromosomes (2n) | 550 | 158 | 158 | 140 | 146 | 156 | 140 | 142 | 170 | 2733 | 2979 |
| length (bp) | 474 | 306 | 118 | 422 | 334 | 122 | 661 | 1008 | 3445 | 465 | 285 |
| no. of polymorphic sites | 11 | 3 | 1 | 3 | 3 | 5 | 11 | 13 | 50 | 85 | 78 |
| no. of indels | 1 | 0 | 0 | 0 | 0 | 2 | 2 | 1 | 6 | ||
| alleles: North Atlantic | 3(0) | 2(0) | 1(0) | 2(0) | 1(0) | 10(4) | 5(0) | 4(1) | 22(21) | 41(38) | |
| alleles: North Pacific | 4(1) | 2(0) | 2(1) | 2(0) | 2(1) | 6(0) | 5(1) | 6(3) | 19(17) | 18(16) | |
| alleles: Southern Hemisphere | 7(3) | 3(0) | 1(0) | 4(2) | 3(2) | 7(0) | 7(2) | 3(0) | 181(178) | 153(148) | |
| total no. of alleles | 8 | 3 | 2 | 4 | 4 | 10 | 8 | 7 | 219 | 209 | |
| obs. heterozygosity | 0.2418 | 0.0971 | 0.0127 | 0.1762 | 0.0137 | 0.2237 | 0.1286 | 0.0607 | 0.1983 | 0.9846a | 0.9828a |
| s.d. | 0.2389 | 0.0073 | 0 | 0.2561 | 0 | 0.0790 | 0.1517 | 0.1256 | 0.1758 | 0.0006 | 0.0006 |
| π | 0.0061 | 0.0010 | 0.0001 | 0.0013 | 0.0000 | 0.0132 | 0.0023 | 0.0008 | 0.0022 | 0.0214 | 0.0416 |
| s.d. | 0.0035 | 0.0011 | 0.0006 | 0.0012 | 0.0002 | 0.0087 | 0.0015 | 0.0006 | 0.0012 | 0.0108 | 0.0208 |
aHaplotype diversity is reported for the control region dataset.
(a). Mitochondrial diversity and phylogeny
Within the 465 bp mtDNA control region dataset, we found 85 variable sites, resolving 219 haplotypes worldwide. The 285 bp dataset contained 78 polymorphic sites, resolving 209 haplotypes. In both cases, a number of sites have undergone multiple substitutions (‘hits’). At these mutational hotspots, phylogenetic signal may be diminished by the noise created by multiple mutations (including back-mutations). Nearly all haplotypes in the 465 bp dataset were private to an ocean basin, with only one haplotype shared between the North Atlantic and Southern Hemisphere, and two shared between the North Pacific and Southern Hemisphere. Control region sequences showed no fixed ‘diagnostic’ substitutions unique to each ocean basin.
Total worldwide nucleotide diversity π was 2.14% for the 465 bp dataset. Ocean basin estimates of π were similar to those obtained in previous studies [1,7]: 1.13% in the North Pacific, 1.97% in the North Atlantic and 2.48% in the Southern Hemisphere (electronic supplementary material, tables S1 and S2). The 285 bp control region dataset yielded higher global π (4.16%) because most variable sites in the 465 bp dataset fall within the 285 bp fragment. A similar pattern was observed for oceanic population size Θ, which was similar for both northern oceans at 285 bp consensus length (0.009) but lower for the North Atlantic at the 465 bp length (0.004) compared with the North Pacific (0.007), probably as a consequence of more limited geographical sampling of the 465 bp dataset.
The Bayesian majority-rule phylogeny (Ts : Tv = 43.4, α = 0.136) of the 465 bp haplotypes supported the grouping of humpback mtDNA control region sequences into four previously recognized clades (electronic supplementary material, figure S1) [1,13]. The largest clade is ‘CD’ (96% Bayesian posterior probability support, PP), which contains haplotypes from all oceans and includes a haplotype shared across the Pacific equator. The next largest (‘IJ’, 82% PP) includes haplotypes from all oceans except the north Pacific, and one haplotype shared across the Atlantic equator. The smallest clade, ‘SH’ (89% PP) includes only Southern Hemisphere haplotypes. A fourth ‘AE’ clade contains mostly North Pacific haplotypes, but fell as a basal polytomy. The 285 bp dataset expands the haplotypic diversity of the North Atlantic IJ clades (table 1) and increases support for IJ (100% PP), while providing weak support for clade CD (72% PP), and none for SH and AE, probably due to a reduction in variable sites (electronic supplementary material, figure S2). The phylogenetic interrelationships among clades are inconsistent and weakly supported. This may be due to saturation of the control region obscuring true signal, or rapid radiation of clades close to the origin of the present-day humpback mtDNA lineages.
BFs supported the constant size model over other growth models (BF > 4). Using the phylogenetically derived substitution rate (figure 2), the median root age was 880 ka (95% PI 55–132 ka). Northern Ocean clades diverged from the Southern Hemisphere subsequently, with the earliest North Pacific clade radiating 170 ka (95% PI 14–80 ka) followed by a second radiation 70 ka. The earliest North Atlantic clade radiated 87 ka (95% PI 1.200–146 ka), followed by subsequent radiation of two extant clades ca 55 and 38 ka. As the imposed molecular clock did not include any variance, however, these values provide only broad guidance rather than representing the full range of uncertainty. Using the ancient DNA-derived bowhead rate, the median root age was 232 ka (95% PI 137–346 ka) with the earliest northern ocean radiation (AE clade, North Pacific) dated to 47 ka (95% PI 21–90 ka) and the North Atlantic radiations ranging from 14 to 23 ka (data not shown).
Figure 2.

Bayesian chronogram of divergence and radiation of humpback mtDNA haplogroups (465 bp), identified as CD, AE, IJ and SH (following [1,13]). North Atlantic clades are red, North Pacific clades are blue and Southern Hemisphere clades are unshaded. Dashed lines show 95% posterior estimates of key divergence times.
(b). Neutrality and population expansion
The null hypothesis of population equilibrium was rejected by Fu's FS for the Southern Hemisphere and North Atlantic IJ haplotype clade (electronic supplementary material, table S4), suggesting that both populations expanded in the past. For the Southern Hemisphere, the SSD test for spatial (but not sudden) expansion was rejected, suggesting humpbacks may have undergone a sudden expansion in this ocean. For the IJ clade, the test for sudden expansion was rejected, so this clade may have instead undergone a spatial expansion. Alternatively, true history may have involved more complex expansion scenarios not captured by these tests. Spatial and sudden population expansions were rejected for the North Atlantic CD clade, which is estimated to diverge into the North Atlantic more recently than the IJ clade. Test statistics do not support long-term expansion of any North Pacific clades.
(c). Mitochondrial oceanic differentiation and gene flow
The hierarchical AMOVA showed strong differentiation among oceans (electronic supplementary material, table S4). Greater differentiation was found between the three ocean basins (28% and 10% of total molecular and haplotypic genetic variation, respectively) than between the five oceans (18% molecular, 6% haplotypic), suggesting that gene flow has been more restricted between inter-hemispheric oceans than across the Southern Hemisphere oceans (electronic supplementary material, table S5). Molecular differentiation was greater than haplotypic differentiation in both cases, indicating that substantial genetic divergence as well as drift has been occurring between the three ocean basins [47]. The level of overall molecular differentiation between the five oceans was similar to that between individual breeding and feeding populations (around 18% of total variation), suggesting similar genetic divergence at both spatial scales, and in both cases much lower divergence than that seen at the inter-basin scale.
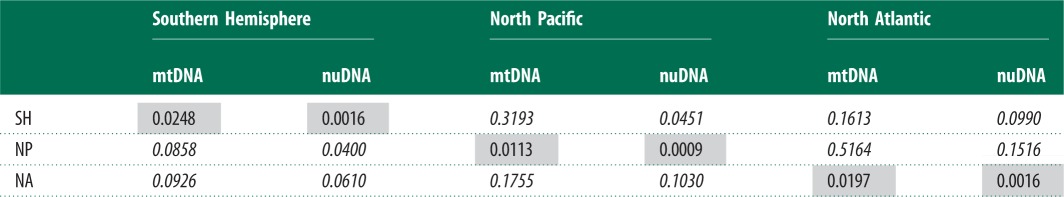
The greatest population differentiation was found between the Northern Hemisphere oceans (mtDNA 465 bp, FST = 0.18, ϕST = 0.51 for 465 bp mtDNA). Similar differentiation was found between the North Pacific and the three Southern Hemisphere oceans (all ϕST ≈ 0.35; table 2; electronic supplementary material, table S6). Nucleotide differentiation of roughly half this magnitude was estimated between the North Atlantic and the three Southern Hemisphere oceans (ϕST = 0.16–0.18). This may be because the North Pacific clade AE diverged from the Southern Hemisphere earlier than the North Atlantic clades (figure 2). Differentiation among oceans of the Southern Hemisphere was two orders of magnitude weaker (electronic supplementary material, table S6), with varying levels of differentiation between Southern Hemisphere oceans [48].
Table 2.
Inter-ocean genetic differentiation of humpback whales at nuclear loci and the 465 bp mtDNA control region. FST and ϕST measures are shown below and above the diagonal, respectively. Within-ocean diversity of each locus (π) is shown in the shaded diagonal (Tajima-Nei corrected pairwise distances for introns, Kimura 2-parameter correction for mtDNA control region sequences, α = 0.1364). Italicized values indicate values significant at p < 0.05 after Bonferroni correction.
 |
Coalescent estimates of maternal gene flow between ocean basins were low (figure 3). Gene flow (immigrants per generation) was slightly higher from the Northern Hemisphere to the Southern Hemisphere (approx. 3 Nemf), with wider confidence intervals and an upper 95% boundary up to 8.5, compared with Southern Hemisphere movements into the North, with mean values of 0.6–1.1 Nemf and an upper 95% boundary of less than 2.8 (table 3). The 5-ocean analysis showed a similar pattern of restricted gene flow across the equator, but gene flow between the Southern Hemisphere oceans (electronic supplementary material, table S6) was so high that Nemf values were truncated by the maximum upper boundary of the prior, indicating little restriction to migration flow.
Figure 3.

Posterior distributions of migration rates (Nem) between ocean basins from MIGRATE. MtDNA posteriors in black (465 bp solid, 285 bp dashed) represent mtDNA gene flow Nemf. NuDNA posteriors (red) represent nuclear gene flow (Nemf+m). Prior distribution, dashed grey.
Table 3.
MIGRATE Nem coalescent estimates of gene flow between ocean basins. nuDNA (4Nemf+m) is divided by 4 for comparability with mtDNA. PP refers to posterior probabilities. Southern hemisphere, SH; North Atlantic, NA; North Pacific, NP.
| SH |
NA |
NP |
|||||
|---|---|---|---|---|---|---|---|
| mean | 95% PP | mean | 95% PP | mean | 95% PP | ||
| 465 bp | from SH | 0.87 | 0.00–2.08 | 0.69 | 0.00–1.85 | ||
| from NA | 3.26 | 0.00–8.15 | 0.40 | 0.00–1.25 | |||
| from NP | 2.98 | 0.00–8.28 | 0.26 | 0.00–0.85 | |||
| 285 bp | from SH | 1.07 | 0.00–2.71 | 0.72 | 0.00–1.81 | ||
| from NA | 3.87 | 0.00–9.20 | 0.33 | 0.00–1.03 | |||
| from NP | 3.27 | 0.04–7.81 | 0.31 | 0.00–0.99 | |||
| nuDNA | from SH | 0.82 | 0.00–2.08 | 1.51 | 0.00–3.59 | ||
| from NA | 1.04 | 0.00–2.54 | 0.58 | 0.00–1.60 | |||
| from NP | 1.65 | 0.00–3.84 | 0.92 | 0.00–2.40 | |||
(d). Nuclear diversity and networks
The numbers of alleles at each intron ranged from 2 (GBA) to 10 (RHO). A total of 50 variable sites were identified across the entire dataset, of which six were insertions/deletions. Heterozygosity varied from 0.01 (LAC) to 0.22 (RHO). Within-ocean nucleotide diversity (π) ranged 30-fold across nuclear loci, from 0.00% (LAC) to 1.32% (RHO). Average worldwide genomic π was 0.22%. Within ocean basins, mean π diversity was 0.09% in the North Pacific, 0.16% in the North Atlantic and 0.12% in the Southern Hemisphere (table 1).
Most common alleles were found in similar frequencies among the three oceans (electronic supplementary material, figure S3). A number of region-specific (‘private’) alleles were found in the Southern Hemisphere, but there were no fixed or diagnostic differences. One highly divergent actin allele [49] is widely distributed and relatively common. This allele is equidistant between fin and humpback whale clades (average distance to these is 0.012–0.013), while average distance within the humpback clade is 0.006. The allele could represent an ‘ancestral’ balaenopterid lineage that originated prior to the evolutionary radiation of humpbacks and which might be under selection, considering the absence of closely related alleles. Alternatively, the allele could be divergent due to past genetic introgression from other balaenopterids (e.g. by hybridization). CAT and ESD were strongly differentiated from nearest neighbours (eight and 19 mutation steps, respectively, from humpback whales). For other loci, the distance to outgroups was less than or equal to the maximum distance between alleles within humpbacks. Divergences among balaenopterids are therefore low for most loci, reflecting a slow mutation rate [25] and possibly also inter-species introgression (e.g. [50]).
(e). Nuclear oceanic differentiation
A weaker pattern of oceanic differentiation was seen in the nuclear dataset, compared with mtDNA (electronic supplementary material, tables S8 and S9), with overall FST = 0.12 between the three ocean basins. When the Southern Hemisphere was partitioned into southeastern Indian Ocean and South Pacific regions, this reduced to FST = 0.06. Levels of differentiation between Northern Hemisphere oceans (combined nuclear FST = 0.15) were similar in magnitude to those obtained for mtDNA, while differentiation between the two northern oceans and Southern Hemisphere was much weaker (combined nuclear differentiation from North Pacific and North Atlantic was FST = 0.05 and 0.09, respectively; table 2). No significant FST or ϕST differentiation between the southeastern Indian and South Pacific oceans was detected by any nuclear loci.
DAPC analyses yielded six to nine clusters as a good fit to the dataset. In each repeat analysis over K = 6–9, a Southern Hemisphere-only cluster and predominantly North Atlantic cluster were stably recovered. Probabilities of membership within each ocean were all over 70% when five PCs were used (electronic supplementary material, figures S4 and S5).
Coalescent estimates of Θ across loci (electronic supplementary material, table S10) revealed a pattern of lower diversity in the Northern Hemisphere and higher diversity in the Southern Hemisphere. Coalescent migration rates between ocean basins (figure 3) were slightly lower than those obtained from mtDNA, but in a very similar magnitude range, with upper percentiles less than four migrants per generation. Similarly to mtDNA, gene flow into the Southern Hemisphere was greater than gene flow into the North Atlantic, but unlike mtDNA there was fairly symmetrical nuclear gene flow estimated between the North Pacific and Southern Hemisphere.
4. Discussion
(a). Genetic diversity: cultural maintenance?
Our diversity metrics reveal higher mtDNA nucleotide genetic diversity in humpbacks than other baleen whales [51–55], with comparable levels only found in the southern right whale [56]. High nucleotide diversity may reflect large ancestral population sizes [54] or may be driven by strong population structuring and restricted gene flow between populations. For humpback whales and southern right whales [56], mtDNA haplotype frequencies show marked differences between breeding/calving grounds. Photo-identification and genetic evidence suggests that this is driven by maternal fidelity to natal breeding grounds (e.g. [12]), so this behaviour may be the major driver influencing high global mtDNA diversity levels. By contrast, nuclear genetic diversity of humpbacks by ocean basin is lower than comparable estimates in Antarctic minke whales [15], but similar to levels estimated for grey whales [55], suggesting that humpbacks and grey whales may have had smaller past population sizes or a greater loss of diversity as a consequence of population bottlenecks due to whaling.
(b). Gene flow of baleen whales across the equator
Strongly significant mtDNA differentiation of humpbacks in each of the world's ocean basins indicates that extensive genetic drift and mutational divergence has occurred between populations. However, much more divergence has occurred between inter-hemispheric ocean basins, suggesting that each basin is isolated by equatorial barriers to movement. Our inter-population divergence levels are consistent with previous analyses finding significant differentiation and sometimes also divergence between breeding populations [13,27], but here we demonstrate that humpback divergence between ocean basins is an order of magnitude greater, strong enough even to drive population differentiation in slowly evolving nuclear intronic genes. Recent worldwide analyses of fin whale mitogenomes also showed strong population divergence between the North Pacific, North Atlantic and Southern Ocean, suggesting similar restrictions to trans-equatorial gene flow for fin whales [57]. Levels of mutational divergence (ϕST) between the North Pacific and North Atlantic humpbacks are equivalent to divergence between right whale species inhabiting the Southern Hemisphere and North Atlantic ocean basins [52]. However, in contrast with right whales, no diagnostic mtDNA or nuclear sites have been identified between the two Northern Hemisphere oceans in this study for humpback whales.
Two types of gene flow have been measured in this study—gametic (from nuclear DNA) and female-mediated organismal (from mtDNA). If gene flow occurs as a result of whales from the two hemispheres mating at the extreme edge of their wintering seasons, such exchange would only be detected using nuclear genes as females remain in their natal hemispheres. Estimates of gene flow from the Southern to the Northern Hemisphere are similar for both nuclear DNA and mtDNA (less than 1.6 migrants per generation), suggesting that both organismal and gametic exchange is infrequent and is not sex-biased (e.g. R ∼ 1 [58]). The low migration rates reported here could be a consequence of regular, low-level genetic exchange or no regular exchange but occasional pulses of migrants over time, possibly as a consequence of unusual oceanographic or environmental conditions. Surprisingly, female-mediated gene flow is slightly higher than biparental gene flow across the Atlantic equator. Females crossing the equator are more likely to produce offspring than males (as males compete for mates), so this may be the driving mechanism. This suggests migration across the equator may have been more influential in determining Atlantic gene flow than mating on common wintering grounds (which would not be reflected in mtDNA gene flow).
In all cases, southward migration across the equator was higher, though not significantly so, than northward migration. Oceanic shifts in temperature shifted upwelling centres during periods of glacial expansion, which may have reduced available habitat in many Northern Hemisphere areas (e.g. [59,60]). This reduction may have led to southward shifts in humpback distribution on feeding grounds, and possibly also breeding grounds, increasing the chance of southward gene flow.
(c). Oceanic subspecies?
Reeves et al. [22] recommended that the ranking of subspecies be used to ‘embrace groups of organisms that appear to have been on independent evolutionary trajectories (with minor continuing gene flow), as demonstrated by morphological evidence or at least one line of appropriate genetic evidence’ (p. 7). We consider that oceanic populations of humpback whales meet these criteria. Two lines of genetic evidence support an independent evolutionary trajectory for humpback whales in the three ocean basins: (1) differentiation and divergence of mtDNA, reflecting low organismal gene flow; and (2) differentiation of multiple nuclear DNA loci, reflecting reproductive isolation.
(1) Mitochondrial DNA control region data show strong divergence between ocean basins, with only three haplotypes shared between the ocean basins in the 465 bp dataset. Although there are no diagnostic sites, nor reciprocal monophyly of haplotypes between ocean basins, coalescent-based measurements of inter-oceanic gene flow by females are less than one migrant per generation between some oceans, with a maximum of less than four migrants per generation.
(2) Nuclear DNA shows evidence of differentiation of allele frequencies and mutational divergence, although no diagnostic differences between ocean basins are present. The latter is unsurprising considering the slow rates of mutation estimated for these loci (0.05% Myr−1 [25]). However, nuclear loci can be used to assign individuals to ocean basins (more than 70%), and estimates of nuclear (bi-parental) gene flow are less than one whale per generation between some oceans, and a maximum of less than 2 migrant whales per generation in all comparisons, despite the relatively slower rate of genomic drift compared with mtDNA. These low rates suggest that populations in different ocean basins have been reproductively isolated, as well as isolated by maternal traditions within oceans.
Based on our results, and given the potential revision into oceanic subspecies, we propose the following names: M. n. kuzira (Gray, 1850) for the North Pacific, M. n. novaeangliae (Borowski, 1781) for the North Atlantic and M. n. australis (Lesson, 1828) for the Southern Hemisphere [61].
(d). Pleistocene divergence and expansion
While the radiation of current worldwide humpback lineages lies within the Pleistocene, more precise dates remain uncertain. Phylogenetic substitution rates [25] place these divergences within the time frame of the last million years, with colonization of the northern oceans by modern mtDNA lineages within the last 200 000 years. Relative clade ages suggest that modern lineage divergence into the North Pacific came earliest—the AE clade is estimated to radiate ca 175 000 years ago, but diverges over 500 000 years ago from other haplogroups. Multiple significant incursions are then made into both northern oceans in the last 100 000 years, once into the North Pacific and at least twice in the North Atlantic. Only in the North Atlantic IJ clade is there evidence for northern population expansion; the data indicate a spatial (slow advance) expansion, rather than a rapid radiation, suggesting that this clade may have been the first to expand into the North Atlantic, possibly after retreating ice in the past. This is consistent with reports that the North Atlantic IJ haplotype clade is more broadly distributed across the North Atlantic than the CD clade [3,7]. Differences in female reproductive success may however also influence clade patterns in this ocean basin (e.g. [28]).
Despite the low mtDNA diversity of North Pacific humpbacks [1,5], analyses of divergence times suggest an early split of the North Pacific AE clade from other humpback lineages worldwide, estimating the earliest divergences within the AE group more than 150 000 years ago. Population expansion metrics show no signs of a radiation, although none is rejected either. The low diversity of this clade may therefore be driven by non-age-related factors. Prolonged periods at small population size may increase apparent population divergence due to genetic drift, although humpback whale substitution rates are low [25], so this effect would have to persist over many thousands of years. It has been suggested that whaling led to the reductions in genetic diversity currently observed in other matrilineal species such as right whales [56,62] and bowheads [63]. A recent bottleneck due to whaling and/or deeper historical factors (such as reduction of feeding areas during glacial periods) are possible explanations.
There is a general debate over whether phylogenetic substitution rates are biased downwards when considering within-species radiations (e.g. [64]). No calibrations within the species are available to test whether this is the case for humpback whales. Applying a within-species rate derived from bowhead whales via ancient DNA [40] yields much more recent divergence times among humpback clades. If such rates turn out to be more accurate, the time frame of radiations would be much more recent, with the North Atlantic IJ clade estimated to ca 55 000 years before present, for example. A more representative collection of mitogenomic sequences (e.g. [25]) and additional estimates of population-level humpback mutation rates are required to resolve this uncertainty. Sea surface temperatures and ice sheet reconstructions of the Last Glacial Maximum (LGM) suggest that in the North Atlantic the Norwegian Sea was reliably free of ice during the summer months [65], whereas the Gulf of Maine and Scotia Shelf regions show evidence of grounded ice reaching to the continental shelf edge during that period (ca 21–22 000 years ago) [59]. Refugia in the eastern North Atlantic may therefore have been more extensive, although it is also possible that primary foraging areas were just shifted south during LGM periods. In the western North Pacific, cores from the Sea of Okhotsk suggest sea ice cover may have been perennial during the LGM [60], but data from areas to the east are patchy. Confidence intervals on our population expansion statistics are broad and do not exclude the possibility of post-LGM re-colonization, but considered in concert this evidence suggests de novo colonization of the northern oceans by humpbacks after the LGM (more than 12 000 years ago) is unlikely and that humpback persistence in these regions has a much longer history.
Acknowledgements
We thank the South Pacific Whale Research Consortium and many regional collaborators for access to DNA samples, the Taxonomy Working Group of the Marine Mammal Society for advice on subspecies criteria, Murdoch Vant for generating and analysing intronic data, and Claus-Dieter Hillenbrand for palaeo-oceanographic discussions. We thank George Amato and Rob DeSalle for support of the Sackler Institute's facilities, Steve Palumbi for stimulating discussion on the genetic diversity of exploited whales and two anonymous reviewers for helpful comments on this manuscript.
Funding statement
Primary funding was provided by a Marsden grant (no. 01-UOA-070) from the New Zealand Royal Society to C.S.B., and a sub-award from the Lenfest Ocean Foundation to C.S.B. and H.R. (award no. 2004-001492-023 to S. R. Palumbi). P.B. was partially funded by US National Science Foundation grant no. DEB-1145999.
References
- 1.Baker CS, et al. 1993. Abundant mitochondrial DNA variation and world-wide population structure in humpback whales. Proc. Natl Acad. Sci. USA 90, 8239–8243. ( 10.1073/pnas.90.17.8239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker CS, Palumbi SR, Lambertsen RH, Weinrich MT, Calambokidis J, O'Brien SJ. 1990. Influence of seasonal migration on geographic distribution of mitochondrial DNA haplotypes in humpback whales. Nature 344, 238–240. ( 10.1038/344238a0) [DOI] [PubMed] [Google Scholar]
- 3.Palsbøll P, Clapham PJ, Mattila DK, Larsen F, Sears R, Siegismund HR, Sigurjonsson J, Vasquez O, Arctander P. 1995. Distribution of mtDNA haplotypes in North Atlantic humpback whales: the influence of behaviour on population structure. Mar. Ecol. Prog. Ser. 116, 1–10. ( 10.3354/meps116001) [DOI] [Google Scholar]
- 4.Larsen AH, Sigurjonsson J, Oien N, Vikingsson G, Palsbøll P. 1996. Population genetic analysis of nuclear and mitochondrial loci in skin biopsies collected from central and northeastern North Atlantic humpback whales (Megaptera novaeangliae): population identity and migratory destinations. Proc. R. Soc. Lond. B 263, 1611–1618. ( 10.1098/rspb.1996.0236) [DOI] [PubMed] [Google Scholar]
- 5.Baker CS, et al. 1998. Population structure of nuclear and mitochondrial DNA variation among humpback whales in the North Pacific. Mol. Ecol. 7, 695–707. ( 10.1046/j.1365-294x.1998.00384.x) [DOI] [PubMed] [Google Scholar]
- 6.Valsecchi E, et al. 1997. Microsatellite genetic distances between oceanic populations of the humpback whale (Megaptera novaeangliae). Mol. Biol. Evol. 14, 355–362. ( 10.1093/oxfordjournals.molbev.a025771) [DOI] [PubMed] [Google Scholar]
- 7.Baker CS, Medrano-González L. 2002. Worldwide distribution and diversity of humpback whale mitochondrial DNA lineages. In Molecular and cell biology of marine mammals (ed. Pfeiffer CJ.), pp. 84–99. Malabar, FL: Krieger Publishing Company. [Google Scholar]
- 8.Baker CS, et al. 1994. Hierarchical structure of mitochondrial DNA gene flow among humpback whales Megaptera novaeangliae, worldwide. Mol. Ecol. 3, 313–327. ( 10.1111/j.1365-294X.1994.tb00071.x) [DOI] [PubMed] [Google Scholar]
- 9.Darling JD, McSweeney DJ. 1985. Observations on the migrations of North Pacific humpback whales (Megaptera novaeangliae). Can. J. Zool. 63, 308–314. ( 10.1139/z85-047) [DOI] [Google Scholar]
- 10.Clapham PJ, Mayo CA. 1987. Reproduction and recruitment in individually identified humpback whales (Megaptera novaeangliae) observed in Massachusetts Bay: 1979–1985. Can. J. Zool. 65, 2853–2863. ( 10.1139/z87-434) [DOI] [Google Scholar]
- 11.Clapham PJ, Baraff LS, Carlson CA, Christian MA, Mattila DK, Mayo CA, Murphy MA, Pittman S. 1993. Seasonal occurence and annual return of humpback whales, Megaptera novaeangliae, in the Southern Gulf of Maine. Can. J. Zool. 71, 440–443. ( 10.1139/z93-063) [DOI] [Google Scholar]
- 12.Garrigue C, Dodemont R, Steel D, Baker CS. 2004. Organismal and ‘gametic’ capture-recapture using microsatellite genotyping confirm low abundance and reproductive autonomy of humpback whales on the wintering grounds of New Caledonia. Mar. Ecol. Prog. Ser. 274, 251–262. ( 10.3354/meps274251) [DOI] [Google Scholar]
- 13.Olavarría C, et al. 2007. Population structure of South Pacific humpback whales and the origin of the eastern Polynesian breeding grounds. Mar. Ecol. Prog. Ser. 330, 257–268. ( 10.3354/meps330257) [DOI] [Google Scholar]
- 14.Baker CS, Rosenbaum HC, Anderson EC, Engel M, Rothschild A, Baker CS, Palumbi SR. 2013. Strong maternal fidelity and natal philopatry shape genetic structure in North Pacific humpback whales. Mar. Ecol. Prog. Ser. 494, 291–306. ( 10.3354/meps10508) [DOI] [Google Scholar]
- 15.Ruegg K, et al. 2013. Long-term population size of the North Atlantic humpback whale within the context of worldwide population structure. Conserv. Genet. 14, 103–114. ( 10.1007/s10592-012-0432-0) [DOI] [Google Scholar]
- 16.Rasmussen K, Palacios DM, Calambokidis J, Saborío MT, Dalla Rosa L, Secchi ER, Steiger GH, Allen JM, Stone GS. 2007. Southern Hemisphere humpback whales wintering off Central America: insights from water temperature into the longest mammalian migration. Biol. Lett. 3, 302–305. ( 10.1098/rsbl.2007.0067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bamy IL, Van Waerebeek K, Bah SS, Dia M, Kaba B, Keita N, Konate S. 2011. Species occurrence of cetaceans in Guinea, including humpback whales with southern hemisphere seasonality. Mar. Biodivers. Rec. 3, e48 ( 10.1017/S1755267210000436) [DOI] [Google Scholar]
- 18.Rosenbaum HC, Maxwell SM, Kershaw F, Mate B. 2014. Quantifying long-range movements and potential overlap with anthropogenic activity of humpback whales in the South Atlantic Ocean. Conserv. Biol. 28, 604–615. ( 10.1111/cobi.12225) [DOI] [PubMed] [Google Scholar]
- 19.Perrin WF, Mead JG, Brownell RL., Jr 2009. Review of the evidence used in the description of currently recognized cetacean subspecies. NOAA Technical Memorandum NMFS-SWFSC-450. Washington, DC: National Oceanic and Atmospheric Administration.
- 20.Tomilin AG. 1946. Thermoregulation and the geographical races of cetaceans. Dokl. Akad. Nauk SSSR 54, 465–472. [Google Scholar]
- 21.Rice DW. 1998. Marine mammals of the world: systematics and distribution. Mar. Mamm. Sci. 4, 67–78. [Google Scholar]
- 22.Reeves RR, Perrin WF, Taylor BL, Baker CS, Mesnick ML. 2004. Report of the workshop on shortcomings of cetacean taxonomy in relation to needs of conservation and management. NOAA Technical Memorandum NMFS-SWFSC-363. Washington, DC: National Oceanic and Atmospheric Administration.
- 23.Davis JI, Nixon KC. 1992. Populations, genetic variation and the delimitation of phylogenetic species. Syst. Biol. 41, 421–435. ( 10.1093/sysbio/41.4.421) [DOI] [Google Scholar]
- 24.Edwards SV. 2009. Is a new and general theory of molecular systematics emerging? Evolution 63, 1–19. ( 10.1111/j.1558-5646.2008.00549.x) [DOI] [PubMed] [Google Scholar]
- 25.Jackson JA, Baker CS, Vant M, Steel DS, Medrano-González L, Palumbi SR. 2009. Big and slow: phylogenetic estimates of molecular evolution in baleen whales (Suborder Mysticeti). Mol. Biol. Evol. 26, 2427–2440. ( 10.1093/molbev/msp169) [DOI] [PubMed] [Google Scholar]
- 26.Engel MH, Fagundes NJR, Rosenbaum HC, Leslie MS, Ott PH, Schmitt R, Secchi E, Dalla Rosa L, Bonatto SL. 2008. Mitochondrial DNA diversity of the Southwestern Atlantic humpback whale (Megaptera novaeangliae) breeding area off Brazil, and the potential connections to Antarctic feeding areas. Conserv. Genet. 9, 1253–1262. ( 10.1007/s10592-007-9453-5) [DOI] [Google Scholar]
- 27.Rosenbaum HC, et al. 2009. Population structure of humpback whales from their breeding grounds in the South Atlantic and Indian oceans. PLoS ONE 4, e7318 ( 10.1371/journal.pone.0007318) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenbaum HC, Weinrich MT, Stoleson SA, Gibbs JP, Baker CS, DeSalle R. 2002. The effect of differential reproductive success on population genetic structure: correlations of life history with matrilines in humpback whales of the Gulf of Maine. J. Hered. 93, 389–399. ( 10.1093/jhered/93.6.389) [DOI] [PubMed] [Google Scholar]
- 29.Sasaki T, et al. 2005. Mitochondrial phylogenetics and evolution of mysticete whales. Syst. Biol. 54, 77–90. ( 10.1080/10635150590905939) [DOI] [PubMed] [Google Scholar]
- 30.Maddison DR, Maddison WP. MacClade 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 31.Posada D, Crandall KA. 1998. Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818. ( 10.1093/bioinformatics/14.9.817) [DOI] [PubMed] [Google Scholar]
- 32.Hurvich CM, Tsai C-L. 1989. Regression and time series model selection in small samples. Biometrika 76, 297–307. ( 10.1093/biomet/76.2.297) [DOI] [Google Scholar]
- 33.Nei M. 1987. Molecular evolutionary genetics. New York, NY: Columbia University Press. [Google Scholar]
- 34.Excoffier L, Laval G, Schneider S. Arlequin ver 3.01: an integrated software package for population genetics data analysis. See http://cmpg.unibe.ch/software/arlequin3. [PMC free article] [PubMed] [Google Scholar]
- 35.Beerli P. 2006. Comparison of Bayesian and maximum likelihood inference of population genetic parameters. Bioinformatics 22, 341–345. ( 10.1093/bioinformatics/bti803) [DOI] [PubMed] [Google Scholar]
- 36.Beerli P, Felsenstein J. 1999. Maximum likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronquist F, Huelsenbeck JP. 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. ( 10.1093/bioinformatics/btg180) [DOI] [PubMed] [Google Scholar]
- 38.Rambaut A, Drummond AJ. Tracer: MCMC trace analysis tool v. 1.4. See http://tree.bio.ed.ac.uk/software/tracer. [Google Scholar]
- 39.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 1–8. ( 10.1186/1471-2148-7-214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho SYW, Saarma U, Barnett R, Haile J, Shapiro B. 2008. The effect of inappropriate calibration: three case studies in molecular ecology. PLoS ONE 3, e1615 ( 10.1371/journal.pone.0001615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tajima F. 1996. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 143, 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu Y-X. 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jombart T, Devillard S, Balloux F. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 1–15. ( 10.1186/1471-2156-11-94) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. ( 10.2307/2408641) [DOI] [PubMed] [Google Scholar]
- 46.Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene genalogies. Mol. Ecol. 9, 1657–1659. ( 10.1046/j.1365-294x.2000.01020.x) [DOI] [PubMed] [Google Scholar]
- 47.Excoffier L, Smouse PE, Quattro JM. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial-DNA restriction data. Genetics 131, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenbaum HC, et al. Submitted. Population genetic structure of Southern Hemisphere humpback whales throughout their range.
- 49.Palumbi SR, Baker CS. 1994. Contrasting population structure from nuclear intron sequences and mtDNA of humpback whales. Mol. Biol. Evol. 11, 426–435. [DOI] [PubMed] [Google Scholar]
- 50.Spilliaert R, Vikingsson G, Árnason Ú, Palsdottir A, Sigurjonsson J, Árnason A. 1991. Species hybridization between a female blue whale (Balaenoptera musculus) and a male fin whale (B. physalus): molecular and morphological documentation. J. Hered. 82, 269–274. [DOI] [PubMed] [Google Scholar]
- 51.Lyrholm T, Leimar O, Gyllensten U. 1996. Low diversity and biased substitution patterns in the mitochondrial control region of sperm whales: implications for estimates of time since common ancestry. Mol. Biol. Evol. 13, 1318–1326. ( 10.1093/oxfordjournals.molbev.a025578) [DOI] [PubMed] [Google Scholar]
- 52.Rosenbaum HC, et al. 2000. World-wide genetic differentiation of Eubalaena: questioning the number of right whale species. Mol. Ecol. 9, 1793–1802. ( 10.1046/j.1365-294x.2000.01066.x) [DOI] [PubMed] [Google Scholar]
- 53.Bakke I, Johansen S, Bakke Ø, El-Gewely MR. 1996. Lack of population subdivision among the minke whales (Balaenoptera acutorostrata) from Icelandic and Norwegian waters based on mitochondrial DNA sequences. Mar. Biol. 125, 1–9. ( 10.1007/BF00350755) [DOI] [Google Scholar]
- 54.Roman J, Palumbi SR. 2003. Whales before whaling in the North Atlantic. Science 301, 508–510. ( 10.1126/science.1084524) [DOI] [PubMed] [Google Scholar]
- 55.Alter SE, Rynes E, Palumbi SR. 2007. DNA evidence for historic population size and past ecosystem impacts of gray whales. Proc. Natl Acad. Sci. USA 104, 15 162–15 167. ( 10.1073/pnas.0706056104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patenaude NJ, et al. 2007. Mitochondrial DNA diversity and population structure among southern right whales (Eubalaena australis). J. Hered. 98, 147–157. ( 10.1093/jhered/esm005) [DOI] [PubMed] [Google Scholar]
- 57.Archer FI, Morin PA, Hancock-Hanser BL, Robertson KM, Leslie MS, Bérubé M, Panigada S, Taylor BL, Janke A. 2013. Mitogenomic phylogenetics of fin whales (Balaenoptera physalus spp.): genetic evidence for revision of subspecies. PLoS ONE 8, e63396 ( 10.1371/journal.pone.0063396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karl SA, Toonen RJ, Grant WS, Bowen BW. 2012. Common misconceptions in molecular ecology: echoes of the modern synthesis. Mol. Ecol. 21, 4171–4189. ( 10.1111/j.1365-294X.2012.05576.x) [DOI] [PubMed] [Google Scholar]
- 59.Shaw J, Piper DJW, Fader GBJ, King EL, Todd BJ, Bell T, Batterson MJ, Liverman DGE. 2006. A conceptual model of the deglaciation of Atlantic Canada. Q. Sci. Rev. 25, 2059–2081. ( 10.1016/j.quascirev.2006.03.002) [DOI] [Google Scholar]
- 60.Gorbarenko SA, Psheneva OY, Artemova AV, Matul AG, Tiedemann R, Nurnberg D. 2010. Paleoenvironment changes in the NW Okhotsk Sea for the last 18 kyr determined with micropaleontological, geochemical, and lithological data. Deep Sea Res. I 57, 797–811. ( 10.1016/j.dsr.2010.04.004) [DOI] [Google Scholar]
- 61.Clapham PJ, Mead JG. 1999. Megaptera novaeangliae Mamm. Species 604, 1–9. ( 10.2307/3504352) [DOI] [Google Scholar]
- 62.Carroll E, et al. 2011. Population structure and individual movement of southern right whales around New Zealand and Australia. Mar. Ecol. Prog. Ser. 432, 257–268. ( 10.3354/meps09145) [DOI] [Google Scholar]
- 63.Alter SE, et al. 2012. Gene flow on ice: the role of sea ice and whaling in shaping Holarctic genetic diversity and population differentiation in bowhead whales (Balaena mysticetus). Ecol. Evol. 2, 2895–2911. ( 10.1002/ece3.397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ho SY, Phillips MJ, Cooper A, Drummond AJ. 2005. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 22, 1561–1568. ( 10.1093/molbev/msi145) [DOI] [PubMed] [Google Scholar]
- 65.Kucera M, et al. 2005. Reconstruction of sea-surface temperatures from assemblages of planktonic foraminifera: multi-technique approach based on geographically constrained calibration data sets and its application to glacial Atlantic and Pacific Oceans. Q. Sci. Rev. 24, 951–998. ( 10.1016/j.quascirev.2004.07.014) [DOI] [Google Scholar]