

Summary
A hexanucleotide repeat expansion (HRE), (GGGGCC)n, in C9orf72 is the most common genetic cause of the neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Here we identify a molecular mechanism by which structural polymorphism of the HRE leads to ALS/FTD pathology and defects. The HRE forms DNA and RNA G-quadruplexes with distinct structures and promotes RNA•DNA hybrids (R-loops). The structural polymorphism causes a repeat length-dependent accumulation of transcripts aborted in the HRE region. These transcribed repeats bind to ribonucleoproteins in a conformationdependent manner. Specifically, nucleolin (NCL), an essential nucleolar protein, preferentially binds the HRE G-quadruplex, and patient cells show evidence of nucleolar stress. Our results demonstrate that distinct C9orf72 HRE structural polymorphism at both DNA and RNA levels initiates molecular cascades leading to ALS/FTD pathologies, and provide the basis for a mechanistic model for repeat-associated neurodegenerative diseases.
Keywords: ALS, FTD, C9orf72, G-quadruplex, R-loop, nucleolin, hnRNP, nucleolar stress, abortive transcription, tandem repeats, and repeat expansions
Introduction
Nucleotide repeat elements, including microsatellites and short tandem repeats, are common in eukaryotic genomes1. Recently, a hexanucleotide repeat expansion (HRE), (GGGGCC)n, in a noncoding region of C9orf72 was linked to the neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)2,3. ALS is characterized by a loss of motor neurons, with 90% of ALS cases being sporadic and the other ~10% having a family history4; the C9orf72 HRE represents the most common genetic cause of both familial and sporadic ALS5. FTD is characterized by degeneration of the frontal and temporal lobes of the brain and is the second most common type of dementia in people younger than 656. Again, the C9orf72 HRE is one of the most common genetic causes of FTD6. Increasing evidence suggests that ALS and FTD are two related diseases in a continuous clinical spectrum7, and there is a possibility that the C9orf72 HRE also contributes to Alzheimer’s and Huntington’s diseases8–11.
Normal human C9orf72 alleles have 2 to 25 intronic GGGGCC repeats, with the majority having fewer than eight repeats and more than half having two repeats12. The expanded repeats associated with ALS/FTD are thought to have variable lengths, ranging from tens to thousands of hexanucleotide repeats2,3, but correlations between the repeat lengths and clinical onset or progression have yet to be established. Although a molecular understanding of C9orf72 HRE pathological phenotypes has begun to emerge, the mechanisms by which the GGGGCC repeat expansion causes ALS/FTD pathology is unknown and our understanding of nucleotide repeats in the context of human disease is still in its infancy13–23.
Here we report that the C9orf72 HRE DNA/RNA sequence is structurally polymorphic; it can fold into stable G-quadruplex secondary structures and form transcriptionally induced RNA•DNA hybrids known as R-loops. The DNA of the C9orf72 HRE forms both antiparallel-and parallel-stranded G-quadruplexes, whereas the RNA adopts only parallel-stranded G-quadruplex conformations. These structural features of the C9orf72 HRE lead to truncated HRE-containing abortive transcripts. We identified ribonucleoproteins bound to the repeat RNA in an RNA conformation-dependent manner. Nucleolin (NCL), an essential nucleolar protein that binds specifically to C9orf72 HRE G-quadruplexes, is mislocalized in patient cells carrying the mutation. Accordingly, nucleolar function is impaired in patient cells. Furthermore, this nucleolar pathology can be recapitulated by introducing C9orf72 HRE-containing abortive transcripts into wild-type cells. These results point to the structural conformations of both the DNA and RNA hexanucleotide repeats as fundamental determinants of the pathogenic mechanisms of C9orf72 HRE-linked ALS/FTD
Results
G-quadruplexes Formed by HRE DNA/RNA
To understand how expansion of the GGGGCC repeat in the C9orf72 gene impedes cellular functions and leads to the associated diseases, we began by examining the repeat sequence for unique structural characteristics. The C9orf72 HRE GGGGCC DNA repeat sequence has properties that would allow it to form G-quadruplexes, which are stacks of planar tetramers consisting of four guanines connected by Hoogsteen hydrogen bonds24. To look for such quadruplexes we first examined the secondary structures formed by the GGGGCC hexanucleotide repeats using circular dichroism (CD) absorptivity. The (GGGGCC)4 DNA shows a characteristic spectrum for antiparallel G-quadruplexes in 100 mM KCl25, with a maximum absorbance at 295 nm and a minimum at 260 nm (Figure 1a). This conformational change at physiologically relevant levels of KCl (100 mM) is also apparent in the decreased mobility of the DNA in a native polyacrylamide gel electrophoresis (PAGE) gel shift assay (Figure 1b).
Figure 1. DNA and RNA of the C9orf72 HRE form G-quadruplexes.

a) The CD absorptivity shows characteristic K+-dependent spectra for antiparallel DNA G-quadruplexes with (GGGGCC)4. b) The presence of K+ during annealing induces a conformational change that decreases the mobility of (GGGGCC)4 DNA in a gel mobility shift assay. c) A DMS footprinting assay on (GGGGCC)4 DNA shows protection of the N7 positions on all the guanines when a G-quadruplex is formed. d) The proposed topology for the antiparallel DNA G-quadruplex formed by (GGGGCC)4. Each gray plane represents a G-quartet, as shown in the lower corner. Four separate G-quartets are stacked 5´ to 3´ with two cytosines forming each loop region. e) The CD spectra identified the formation of a parallel G-quadruplex for the RNA (GGGGCC)4 in the presence of 100 mM KCl. f) (GGGGCC)4 RNA demonstrates a slower mobility in the presence of KCl compared to (CCCCGG)4 RNA. g) The RNase T1 protection assay identifies single-stranded guanine residues (denoted in black) not involved in the formation of the G-quadruplex. h) The proposed parallel G-quadruplex topology formed by the RNA of the C9orf72 HRE. Three stacks form the parallel G-quadruplex, with the RNase T1-sensitive guanines shown as black dots.
To provide further conformational insight into the DNA G-quadruplex formed by (GGGGCC)4, we used dimethyl sulfate (DMS) footprinting following classical Maxam and Gilbert sequencing methods26. On the sequencing gel (Figure 1c), the band pattern resulting from guanine depurination is markedly reduced in the presence of KCl, indicating that the guanine N7 positions are protected from DMS methylation and subsequent base cleavage as a result of hydrogen bonding in a G-quartet conformation. These results provide evidence that the (GGGGCC)4 DNA forms a four-stack antiparallel G-quadruplex motif, with the guanines on the exterior of the stacks being more accessible to chemical modifications than the guanines buried in the interior (Figure 1d). Through a series of spectroscopic characterizations, we observed that the GGGGCC sequence can adopt intermolecular, intramolecular, and also parallel G-quadruplex structures when the number of repeats is varied (Extended Data Figure 1a–f and Supplementary Results). However, the antiparallel G-quadruplex appears to be the most dominant and the most stable conformation at physiologically relevant conditions.
Extended Data Figure 1. Spectroscopic characterization of DNA/RNA structural polymorphism.

a) The decanucleotide GGGGCCGGGG from the C9orf72 HRE adopts a parallel G-quadruplex conformation indicated by the CD spectra in the presence of KCl (left). The proposed intermolecular parallel G-quadruplex topology (right) of two decanucleotide sequences that form three stacks of G-quartets 5´ to 3´ with three or four nucleotides, GCCG, forming a loop region. b) The complementary sequence strand for the C9orf72 HRE, (CCCCGG)4, shows no apparent structural differences ± KCl. c) The variability in CD spectra increases with the number of repeats (dotted lines), but the CD spectra can be recapitulated (lines) by fitting a linear combination of three component spectra for hairpin, antiparallel Gquadruplex, and parallel G-quadruplexes from the spectra corresponding to 0 mM KCl (GGGGCC)4, 100 mM KCl (GGGGCC)4, and 100 mM KCl GGGGCCGGGG, respectively. The residuals of the fit are plotted below. d) CD spectral analysis of the thermal stability of the DNA antiparallel G-quadruplex. CD spectra were obtained as described in Methods with temperatures ranging from 25–95°C in 10 mM Tris-HCl pH 7.4 with 50 mM KCl. This two-state transition was evaluated by singular value decomposition (SVD) of the CD melting spectra (Matlab). The two basis component spectra, shown as left coefficients, represent an antiparallel G-quadruplex and an unfolded single-stranded DNA oligo. The residuals show the difference between the original CD spectrum and the basis component spectra fit. e) Increasing KCl concentrations increase the abundance and stability of antiparallel DNA G-quadruplexes, as shown by UV-VIS melting curves. CD absorbance was normalized to the equation (Abst – min)*(max – min)−1, where Abst is the absorbance at a given temperature, max is the observed maximum absorbance at 295 nm at that [KCl], and min is the minimum value obtained for all [KCl]s. Data was fit to a Boltzmann distribution (Prism) with a lower boundary constraint set to min. f) The decamer HRE sequence forms a parallel G-quadruplex that is less stable than the antiparallel conformation. The data obtained were normalized as described above, except the CD absorptivity was measured at 260 nm. g) CD melting spectrum shows a two-state transition from a folded parallel RNA G-quadruplex state to an unfolded linear state. The CD absorptivity data measured at 260 nm were normalized as described above and fit to a Boltzmann distribution without constraints. h) The (GGGGCC)4 forms a G-quadruplex that is unperturbed by a 3ʹ biotin label. CD spectra were obtained ± KCl as in Figure 1 to identify the stabilization of K+ in the formation of a G-quadruplex that had been chemically modified with the biotin tag. All calculated melting temperatures are provide in Extended Data Table 1 and results detailed further in Supplementary Results.
We next investigated the structural conformation and stability of the C9orf72 RNA HRE sequences. Through a series of experiments measuring CD absorptivity, the (GGGGCC)4 RNA shows a signature spectrum for parallel G-quadruplexes27, with a minimum absorbance at 240 nm and a maximum at 260 nm (Figure 1e), which is consistent with recent results for similar RNA HRE sequences15,16. When the (GGGGCC)4 RNA was examined by native PAGE, its mobility is decreased in the presence of 100 mM KCl, in agreement with a K+-dependent formation of G-quadruplexes (Figure 1f).
To provide further conformational insight, we performed an RNase protection assay. The RNA digestion pattern for endonuclease RNase T1, which cleaves single-stranded RNAs at the 3´ end of guanine residues, shows clear structural differences in the presence and absence of KCl. Without KCl, the (GGGGCC)4 RNA forms a secondary structure consistent with singlestranded bulges and hairpin regions as indicated by the RNase protection pattern (Figure 1g). In contrast, in the presence of 100 mM KCl the RNA adopts a G-quadruplex structure, in which every fourth guanine is highly sensitive to single-stranded cleavage. This provides evidence for a topology of a three-stacked parallel-stranded G-quadruplex with a guanine and two cytosines in the single-stranded loop region (Figure 1h). Together, these data indicate that the C9orf72 HRE RNA preferentially adopts a parallel G-quadruplex topology and is stable at physiological KCl concentrations, pH, and temperature (Extended Data Figure 1g).
Impeded transcription and abortive RNAs
To understand the functional consequence of structural polymorphism at the C9orf72 HRE, we examined transcription of the repeat region. First, we established an in vitro transcription assay by creating GGGGCC hexanucleotide repeats of varying lengths (3–70 repeats) de novo via repeat-primed PCR, and placing the repeats downstream of a T7 promoter as transcription templates. When in vitro transcriptional products are resolved by denaturing PAGE periodic abortive transcripts are observed, with longer repeats producing increasing amounts of truncated transcripts that correspond to the template hexanucleotide repeats (Figure 2a). Importantly, the increase in the abortive transcripts causes a concomitant decrease in full-length transcripts. To quantify this observation, we compared the levels of full-length transcripts, which contain regions downstream (3´) of the repeats, with the total levels of all transcripts, which contain regions upstream (5´) of the repeats (Figure 2a). The plot of this 3´/5´ ratio shows that there is a transition to more abortive transcripts relative to full-length products with increasing repeats (Figure 2b). Furthermore, we verified that the repeat templates were not extensively modified by depurination of the DNA sequence, excluding this potential modification as the cause of the observed abortive transcription (Extended Data Figure 2). Therefore, these in vitro results demonstrate that the C9orf72 HRE causes RNA polymerase processivity to be impaired in the repeat region, leading to an accumulation of repeat-containing abortive transcripts and loss of full-length transcripts.
Figure 2. Abortive transcription in the C9orf72 HRE.

a) Increasing lengths of GGGGCC repeats cause accumulation of abortive transcripts in a length-dependent manner in vitro. The transcriptional products were separated on a denaturing gel with a 500 nt ssRNA control (CTL). b) Transcripts levels shown in (a) were densitometrically quantified and then plotted as the ratio of full-length transcripts that contain regions 3´ of the repeat divided by all transcripts that contain 5´ regions. The curve was fit to a single exponential. Data are means ± s.d. n = 4. c) The C9orf72 HRE induces the formation of R-loops on C9orf72 HRE-containing plasmids with (GGGGCC)~70. Treatment of the in vitro transcription products with RNase A and H digests the RNA still hybridized with relaxed or supercoiled plasmid and reduces the smearing that was caused by the size heterogeneity of RNA•DNA hybrids. Genomic DNA (top band) serves as an internal loading control. d) Patients carrying the C9orf72 HRE have reduced pre-mRNA 3´/5´ratios relative to C9orf72 WT, consistent with the HRE-induced abortive transcription reducing full-length transcript levels. Data are means ± s.e.m. n = 5/6 (B lymphocytes), n = 12/10 (motor cortex), n = 8/5 (spinal cord) for C9orf72 WT/HRE samples, respectively. ***P < 0.001, **P < 0.01, *P < 0.05.
Extended Data Figure 2. Depurination of the HRE-containing plasmid template contributes minimally to abortive transcription.

a) Schematic digestion map for analyzing depurination levels present on the template plasmid. b) The same patterns are shown for the (CCCCGG)~70 repeat template and a GFP control template with piperidine treatment and prior acid treatment, confirming that the repeat tract in the repeat-containing template is depurinated basally at nonspecific/spontaneous levels similar to those of the GFP template. The ~135 nucleotide 5´-end-radiolabeled cleavage fragment in the pCR8-70 samples serves as a size marker for the lower boundary of the repeat region corresponding to the region where the repeats would begin to appear on the gel. More details concerning these findings and the experimental designs are described in Supplementary Results and Methods. c) Densitometric plot profiles of the gel bands from each lane in (b) are shown to illustrate the depurination results. The left panel shows the small depurination differences between the piperidine-treated template without prior treatment and those that went through the denaturing/annealing treatments ± KCl. The middle and right panels show that treatment with piperidine causes similar patterns of minimal cleavage for the pCR8-70 and control GFP templates, respectively, as compared to the acid-induced depurination controls, consistent with basal levels of nonspecifically/spontaneously cleaved products. The densitometry analysis was done using ImageJ and plotted in Prism.
Next, we investigated the molecular mechanisms underlying abortive RNA transcript production. We examined the structural features unique to the C9orf72 HRE region on the plasmid DNA and found that, consistent with data on the DNA oligonucleotides in Figure 1, the plasmid also forms stable G-quadruplexes in the presence of KCl, which was directly assessed by the preferential binding of the G-quadruplex-specific BG4 nanobody28 to the DNA G-quadruplex structure (Extended Data Figure 3a–b). The formation of these G-quadruplexes leads to transcripts being aborted earlier and to a further decrease in full-length transcripts in the in vitro transcription assay; these repeat-containing abortive transcripts accumulate over time (Extended Data Figure 3c–d). These results indicate that the formation of G-quadruplexes impairs polymerase processivity within the C9orf72 HRE region.
Extended Data Figure 3. G-quadruplexes increase abortive transcripts within the C9orf72 HRE region in vitro.

a) A newly developed BG4 nanobody directly binds the K+-dependent Gquadruplex formed by DNA (GGGGCC)4. BG4 was purified from E. coli, and an ELISA experiment using biotinylated DNA (GGGGCC)4, with or without 100 mM KCl during annealing, was used to determine the specificity of BG4, essentially as previously described28. Data show the mean ± S.D. n = 3. b) The ΔCt graph for 100 mM KCl vs 0 mM KCl shows that BG4 pulled down >50-fold more plasmid when the plasmid containing (GGGGCC)21 repeats was annealed in the presence of 100 mM KCl, directly demonstrating the presence of Gquadruplexes formed by the sequence. The DNA pulldown with BG4 is described in the Supplementary Methods. n = 1. c) Formation of G-quadruplexes in the coding region prior to transcription causes a shift to an earlier termination of abortive transcripts and further loss of the full-length products. The shift is represented below the densitometric plot profile by a significant change in the center of mass for the repeat region (n = 5). The further loss of full-length transcriptional product is shown on the graph to the right of the transcription profile (n = 3). P-values were calculated using a paired fit for each trial assuming a parametric distribution. Data are means ± s.e.m. *P < 0.05; **P < 0.01. d) Accumulation of the abortive transcripts indicates that the repeat-dependent impairment of transcription is not transient but persists over time ± KCl. Aliquots were removed from the in vitro transcription reactions at different time points and then visualized and analyzed as in (c).
Next, we examined the nascent RNA transcript to look for possible mechanistic contributions to abortive transcription. First, using a G-quadruplex•hemin complex-catalyzed peroxidase-like colorimetric assay29, we demonstrated that the repeat-containing transcripts also form RNA G-quadruplexes, but the formation of these structures on nascent RNA during transcription has negligible effects on the decrease in polymerase processivity (Extended Data Figure 4a–b and Supplementary Results). However, we noted that treatment with RNase H, which specifically digests RNA in an RNA•DNA hybrid, decreases the accumulation of abortive transcripts and increases longer transcripts during the assay (Extended Data Figure 4c). To test whether nascent RNA•DNA template hybrids (R-loops) form on the GGGGCC repeats, we performed the in vitro transcription assay, and then treated the samples with RNase H and RNase A to remove the RNA in this hybrid state and the excess single-stranded RNA, respectively. In the absence of the RNases, the mobility of the plasmid decreases, and consistent with the heterogeneity of their sizes as a result of R-loop formation, the plasmids migrate as a smear above the previously compact supercoiled/circular plasmid band (Figure 2c and Supplementary Results). However, upon simultaneous treatment with RNase H and RNase A, the plasmids then migrate as distinct compact bands. The control plasmid shows a minimally altered mobility when compared to the HRE-containing plasmid after treatment with RNase H and/or RNase A (Extended Data Figure 4d–e). Together, these results provide evidence that the formation of R-loops is an additional structurally polymorphic feature of the C9orf72 HRE that contributes to the abortive transcription mechanism.
Figure 4. Nucleolar stress is a result of repeat-containing RNA transcripts from the C9orf72 HRE.

a) In the control C9orf72 WT B lymphocytes, NCL (green) is localized to the condensed nucleolus. In contrast, the cells of patients with the C9orf72 HRE show an increased NCL diffusion and fractured nucleoli in the nucleus (Hoechst, blue). A heat map of NCL intensities marks the difference between cells. b) iPS motor neurons derived from patients carrying the C9orf72 HRE also demonstrate NCL mislocalization. β-III tubulin (Tuj1) (red) was used to identify neurons. c) NCL colocalizes with RNA foci (red) formed in motor cortex tissue from patients carrying the C9orf72 HRE. A (CCCCGG)2.5 probe was used to detect the (GGGGCC)n RNA foci, a previously identified pathological feature of the C9orf72 HRE tissues. d) Transfection of (GGGGCC)21 abortive transcripts (Figure 2a) recapitulates NCL pathological features observed in patients cells with the C9orf72 HRE.
Extended Data Figure 4. R-loops, and not G-quadruplex formation on nascent RNA transcripts, increase abortive transcription within the C9orf72 HRE region in vitro.
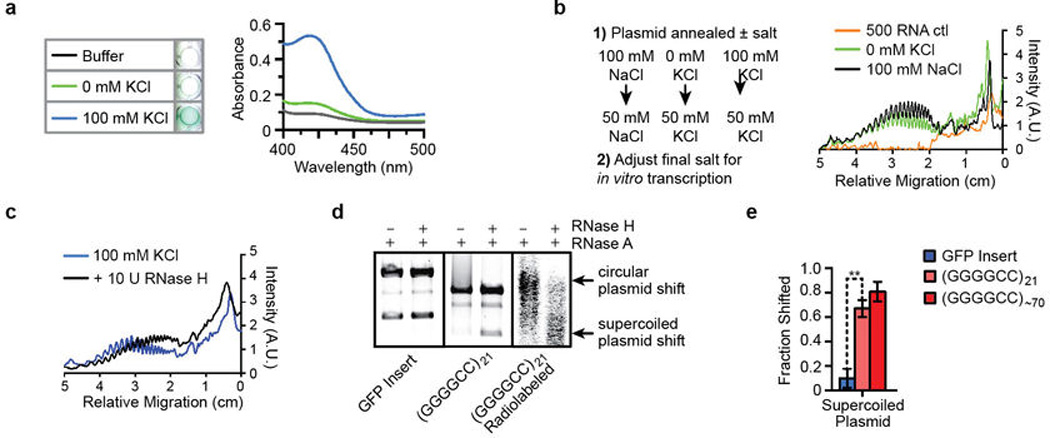
a) RNA transcripts containing many GGGGCC repeats form G-quadruplexes under physiologically relevant KCl concentrations. A colorimetric assay was performed to identify the formation of RNA G-quadruplexes utilizing the enzyme-like peroxidase activity of G-quadruplex•hemin complexes29. b) Workflow considerations for the transcriptional assay. The linear plasmid was first annealed ± 100 mM KCl or 100 mM NaCl in 10 mM Tris-HCl, pH 7.4. To prevent salt concentration-dependent effects on the in vitro transcriptional assay, a second adjustment was made to adjust the salts to a final 50 mM concentration in the assay. Reducing the effects on RNA polymerase allowed us to disambiguate the effects of salt on the conformation of DNA versus the nascent RNA. Comparison of DNA annealed in 0 mM KCl and 100 mM NaCl shows similar reduced polymerase processivity, suggesting that possible formation of G-quadruplexes on the nascent RNA transcripts makes a negligible contribution, but does not exclude intrinsic RNA hairpin-induced termination. c) RNase H treatment during transcription reduces the periodic accumulation of abortive transcripts and increases full-length transcripts. Treatment with RNase H, which specifically cleaves RNA•DNA hybrids, during transcription of the C9orf72 repeats causes a shift from truncated transcripts to full-length transcripts, suggestive of the formation of alternative secondary structures, known as R-loops, caused by increased nonduplex DNA during transcription. d) R-loop formation is observed for the plasmid containing the HRE insert, (GGGGCC)21, but not for a GFP insert in a control. Treatment with RNase A removes ssRNA but does not affect R-loops. As shown in Figure 2c, the HREs induce the formation of R-loops that cause a decrease in the plasmid mobility but can be relieved with RNase H treatment, which specifically cleaves RNA•DNA hybrids. The addition of RNase A and RNase H has little effect on the mobility of the plasmid containing the GFP insert. Radiolabeling the transcriptional products during in vitro transcription confirms the formation of R-loops, demonstrated by the shift consistent with the supercoiled and relaxed plasmids having altered mobility, which is relieved by treatment with both RNases. The transcripts were bodylabeled by including 20 μCi of α-[32P]UTP (Perkin Elmer) and then performing the in vitro transcription as previously described. e) There is a significant increase in the R-loop-induced plasmid mobility shift for the (GGGGCC)21 and (GGGGCC)~70 containing plasmids when compared to the GFP control insert. The shifted supercoiled plasmid bands were quantified by densitometrically measuring each band intensity after treatment with RNase A versus those after treatment with both RNase A and Rnase H (ImageJ, NIH). The overlapping densitometric signal of the supercoiled R-loop smear with the circular plasmid band (Figure 2b) prevented accurate quantification and was excluded. Data are means ± s.e.m. n = 3. *P < 0.05; **P < 0.01.
To further extend our observations concerning in vitro abortive transcription to possible truncation of the endogenous C9orf72 transcript, we examined the production of pre-mRNA transcripts at different positions along the C9orf72 gene in patient cells. Primers targeting intronic regions immediately upstream (5´) of the HRE versus downstream (3´) of the HRE region were used with quantitative reverse transcription polymerase chain reactions (qRT-PCR) to measure pre-mRNA levels. These results indicate that patient B lymphocytes carrying the C9orf72 HRE show a significant decrease in the ratio of 3´/5´ pre-mRNA levels relative to the controls (Figure 2d). This observation was extended to pathologically relevant tissue from patients such as motor cortex and cervical spinal cord. Quantitative measurements of pre-mRNA levels employing NanoString technology20 show a similar decrease in the ratio of 3´/5´ pre-mRNA levels in patients carrying the HRE. These results indicate that the endogenous C9orf72 HRE in patients induces abortive transcription and results in unequal transcriptional efficiency between regions upstream and downstream of the HRE, in agreement with our in vitro abortive transcript results (Figure 2a–b).
Structure-dependent HRE-binding proteins
Since the C9orf72 HRE can generate both full-length and abortive transcripts capable of forming RNA G-quadruplexes, we explored conformation-dependent protein interactions associated with the RNA repeat sequences. Biotinylated (GGGGCC)4 RNAs, in either G-quadruplex or hairpin conformations, and antisense (CCCCGG)4 hairpin RNAs were conjugated to streptavidin beads (Extended Data Figure 1h). In order to comprehensively and quantitatively identify the conformation-dependent ribonucleoprotein complexes formed by these HRE RNAs, we employed stable isotope labeling by amino acids in cell culture (SILAC)30 using HEK293T cells grown in medium containing normal, medium, or heavy amino acids prior to performing the RNA pulldown as shown in Extended Data Figure 5. In brief, labeled cell lysates were incubated with RNA-conjugated streptavidin beads and the isolated ribonucleoprotein complexes were washed with increasing KCl concentrations to remove proteins that were loosely associated with the RNA or with other proteins. Then the final eluted fractions were analyzed for complexes binding the (GGGGCC)4 G-quadruplex, (GGGGCC)4 hairpin, and (CCCCGG)4 hairpin with the respective labels by mass spectrometry. The SILAC analysis provided a list of 288 proteins with quantitation of their binding preferences (Supplementary Table S1) for the various RNA structures formed by the sense GGGGCC or the antisense CCCCGG sequences (Extended Data Table 1).
Figure 5. A model for the molecular cascade resulting from the C9orf72 HRE structural polymorphism.

The DNA and RNA•DNA structures formed in the GGGGCC repeat region impede RNA polymerase transcription, which results in transcriptional pausing and abortion. This leads to a loss of full-length products and an accumulation of abortive transcripts. Abortive transcripts that contain the hexanucleotide repeats form G-quadruplexes and hairpins and bind essential proteins in a conformation-dependent manner. Sequestration of these proteins leads to nucleolar stress and other downstream defects. The repeat-containing transcripts can also escape the nucleus and be bound by ribosomal complexes, thereby increasing repeat-associated non-ATG-dependent translation that results in aggregative polydipeptides.
Extended Data Table 1.
Sequences of primers, Nanostring probes, and characterized oligomers
| Primer/ Identity | Sequence (5–3) | ||||
|---|---|---|---|---|---|
| (GGCCCC)5 | GGCCCCGGCCCCGGCCCCGGCCCCGGCCCC | ||||
| (GGGGCC)8 | GGGGCCGGGGCCGGGGCCGGGGCCGGGGCCGGGGCCGGGGCCGGGGCC | ||||
| Intron 3 C9orf72 + | AGATCCGTGTTAAATTTCCTTCC | ||||
| Intron 3 C9orf72- | CAAAATTATTCTTCCCGAAAAGG | ||||
| 5’ C9orf72-HRE + | GTTTTTCCCACCCTCTCTCC | ||||
| 5’ C9orf72-HRE - | CGACTCCTGAGTTCCAGAGC | ||||
| 45S rRNA forward | GAACGGTGGTGTGTCGTT | ||||
| 45S rRNA reverse | GCGTCTCGTCTCGTCTCACT | ||||
| 28S rRNA forward | AGAGGTAAACGGGTGGGGTC | ||||
| 28S rRNA reverse | GGGGTCGGGAGGAACGG | ||||
| 18S rRNA forward | GATGGTAGTCGCCGTGCC | ||||
| 18S rRNA reverse | GCCTGCTGCCTTCCTTGG | ||||
| 5.8S rRNA forward | ACTCGGCTCGTGCGTC | ||||
| 5.8S rRNA reverse | GCGACGCTCAGACAGG | ||||
| Spect. forward | AGAACATAGCGTTGCCTTGG | ||||
| Spect. reverse | TACTGCGCTGTACCAAATGC | ||||
| GAPDH Nanostring Probe | GGGGAAGGTGAAGGTCGGAGTCAACGGATTTGGTCGTATTGGGCGCCTGGTCACCAGGGCTGCTTTTAACTCTGGTAAAGTGGATATTGTTGCCATCAAT | ||||
| GUSB Nanostring Probe | CGGTCGTGATGTGGTCTGTGGCCAACGAGCCTGCGTCCCACCTAGAATCTGCTGGCTACTACTTGAAGATGGTGATCGCTCACACCAAATCCTTGGACCC | ||||
| OAZ1 Nanostring Probe | GGTGGGCGAGGGAATAGTCAGAGGGATCACAATCTTTCAGCTAACTTATTCTACTCCGATGATCGGCTGAATGTAACAGAGGAACTAACGTCCAACGACA | ||||
| 5’ C9orf72-HRE Nanostring Probe | ATAAAGATTAACCAGAAGAAAACAAGGAGGGAAACAACCGCAGCCTGTAGCAAGCTCTGGAACTCAGGAGTCGCGCGCTAGGGGCCGGGGCCGGGGCCGG | ||||
| Intron 5 C9orf72 Nanostring Probe | AGCCTCTTTCTTCTGTTCTGTTCATAGCACAGTGCCTCATACGCAGTAGGTTATTATTACATGGTAACTGGCTACCCCAACTGATTAGGAAAGAAGTAAA | ||||
| Spectroscopic DNA/RNA analysis | % of each species when annealed in 100 mM KCI | Calculated melting temperatures | |||
| DNA oligonucleotide | Hairpin | Antiparallel G4 | Parallel G4 | 50 mM KCI | 100 mM KCI |
| GGGGCCGGGG | 0 | 0 | 100 | - | 79 |
| (GGGGCC)3 | 25 | 32 | 43 | - | - |
| (GGGGCC)4 | 0 | 100 | 0 | 81 | ≥91 |
| (GGGGCC)5 | 10 | 72 | 18 | - | - |
| (GGGGCC)6 | 26 | 54 | 20 | - | - |
| (GGGGCC)8 | 31 | 61 | 8 | 80 | - |
| (GGGGCC)10 | 26 | 61 | 13 | - | - |
| (CCCCGG)4 | - | - | - | 46 | - |
| RNA oligonucleotide | Hairpin | Antiparallel G4 | Parallel G4 | 50 mM KCI | 100 mM KCI |
| GGGGCCGGGG | 0 | 0 | 100 | - | - |
| (GGGGCC)4 | 0 | 0 | 100 | 82 | 83 |
| (GGGGCC)4-biotin | - | - | - | - | 83 |
| (CCCCGG)4 | - | - | - | 77 | - |
To confirm the specificity of the major proteins in binding to the various RNA structures identified by SILAC, we performed Western blot analysis on the increasingly stringent fractions from the RNA pulldown (Figure 3a). NCL and hnRNP U preferentially recognize the RNA G-quadruplex motif (Figure 3a–c and Extended Data Figure 6). The protein hnRNP F and RPL7, a, heterogeneous nuclear ribonucleoprotein and a ribosomal protein, respectively, prefer guanine-rich RNA and bind both the G-quadruplex and the alternative hairpin of the GGGGCC repeat. The protein hnRNP K, an RNA-binding protein that binds cytosine-rich RNA, shows preferential binding to the antisense CCCCGG repeat. Differential KCl salt elution indicates that while the ribonucleoprotein complexes with hnRNP U or hnRNP F are more easily destabilized with increasing KCl, the interactions with NCL or RPL7 are more robust and survive high KCl concentrations. These results are in accordance with the SILAC quantitative analysis. To further demonstrate that NCL binds directly to the G-quadruplex motif, we used an in vitro RNA pulldown with a recombinant GST-NCL fusion protein. NCL was shown to preferentially pull down the (GGGGCC)4 RNA G-quadruplexes (Figure 3c). These results establish NCL as a specific and enriched interactor that directly and preferentially binds the RNA G-quadruplexes formed by the C9orf72 HRE.
Figure 3. Identification of conformation-dependent C9orf72 HRE RNA-binding proteins.

a) Western blotting analyses of the sequential fractions eluted from a biotinylated-RNA pulldown of HEK293T cells with increasing KCl concentrations. The RNA-binding proteins identified using LC-MS were differentiated among those that recognize different structural motifs, including the RNA antisense (AS) hairpin, (CCCCGG)4; the RNA sense (S) hairpin (GGGGCC)4; and the sense G-quadruplex (G). NCL and hnRNP U have binding preferences for the G motif of the sense RNA. hnRNP F and RPL7 bind both guanine-rich sense sequences, regardless of the underlying RNA structure. hnRNP K prefers binding to the cytosine-rich AS. b) A representative spectrum from SILAC analysis is shown for the preferential binding of NCL to the G motif formed by (GGGGCC)4 compared to the hairpin motifs formed by the sense or antisense sequences. c) An RNA pulldown performed with GST-NCL demonstrates that NCL directly binds (GGGGCC)4 RNA in vitro with highest affinity for the G-quadruplex. Data are means ± s.d. n = 3.
Extended Data Figure 6. Example of mass spectrometry spectra using SILAC obtained from the eluted fractions from the RNA pulldown.

Four sets of spectra representing unique peptide sequences identified for hnRNP U, hnRNP F, RPL7, and hnRNP K show the quantitative RNA structural preferences that can be identified by using the SILAC methodology. In agreement with the work presented in Figure 3, each protein represented by the unique peptide spectrum here shows an individual preference for either RNA structure dependence, such as NCL (Figure 3) and hnRNP U, or sequence dependence, as in the cases of hnRNP K, hnRNP F, and RPL7, as shown by the abundance of the different isotope labels provided by the SILAC method. In Supplementary Table S1, the quantified differences are provided.
The C9orf72 HRE causes nucleolar stress
To investigate the consequences of the specific binding of NCL, a principal component of the nucleolus31 to G-quadruplex complexes formed by the C9orf72 HRE RNA, we examined phenotypic differences in the cellular localization of NCL in C9orf72 HRE patient cells and controls. In control B lymphocyte cells, endogenous NCL immunofluorescent (IF) staining is condensed in the nucleolus. In contrast, the nucleolus appears more fractured and NCL is more dispersed throughout the nucleus in patient cells carrying the C9orf72 HRE, as shown in heat maps indicating the NCL density (Figure 4a). Quantitation of NCL distribution in the nuclei of patient and control cells confirms a significant shift of NCL away from dense nucleoli to a more dispersed localization (Extended Data Figure 7a). This NCL pathology is also observed in C9orf72 patient fibroblasts, but not in fibroblasts from non-ALS or non-C9orf72 ALS controls (Extended Data Figure 7a). Next, we examined the NCL localization pattern in iPS motor neuron cells derived from patient fibroblasts20. As seen for the B lymphocytes, IF staining for NCL in disease-relevant motor neurons from patients with the C9orf72 HRE shows NCL dispersion that occupies a significantly increased area of the nucleus (Figure 4b, Extended Data Figure 7a). No obvious phenotypic differences were observed for hnRNP F, hnRNP U, or hnRNP K between C9orf72 HRE B lymphocytes and those of controls (Extended Data Figure 7d).
Extended Data Figure 7. Nucleolin defects observed in C9orf72 HRE-linked ALS patients are caused by HRE-containing transcripts.

a) Quantification of nucleolin (NCL) in the nucleus shows significant NCL nuclear dispersion in B lymphocytes, iPS motor neurons, and fibroblasts from C9orf72 HRE patients. A single focal plane was obtained through the center of the nucleus (Hoechst staining; blue). To quantify the area differences in NCL relative to the size of the nucleus a single lower threshold setting in ImageJ (NIH), ranging from 25–75 (which provided quantification of both dispersed NCL and dense nucleolar NCL), was used to measure the pixel area of NCL relative to the area of the nucleus outlined by the Hoechst staining (depicted in the cartoon). The measurements for the C9orf72 HRE cells were then normalized to those for controls (Prism). Data are means ± s.e.m. n = 110 and n = 112 for 3 cell lines examined for the C9orf72 WT and C9orf72 HRE B lymphocytes, respectively. n = 29 for iPS motor neurons from 2 C9orf72 HRE patients and for the iPS motor neuron controls. A representative fibroblast line for C9orf72 WT, SOD1 D90A ALS, and C9orf72 HRE was quantified. n = 21. *P < 0.05; **P < 0.01; ****P < 0.0001. b) There are no significant changes in NCL protein levels between patients with and without the C9orf72 HRE, despite its mislocalization. The Western blotting analysis was performed as described in the Methods, following a typical protocol with the B lymphocyte lysates from patients and controls. The bands were visualized using a quantitative LI-COR imager, and the intensities for NCL and tubulin protein were quantified using Odyssey software. NCL levels were normalized against those of tubulin. Data are means ± s.e.m. n = 6. c) RNA foci are a unique feature of the C9orf72 HRE ALS pathology. Motor cortex tissue from C9orf72 WT (non-ALS) and non-C9orf72 sALS show no RNA foci or NCL colocalization, which is observed in motor cortex tissue from patients carrying the C9orf72 HRE (Figure 4c). All images were obtained as described in Methods. d) The protein hnRNP F, K, and U, identified from the pulldown (Figure 3a and Extended Data Figure 6), show no phenotypic differences in localization between patients carrying the C9orf72 HRE and controls, as previously observed in iPS motor neurons from FTD patients46. n = 3. e) A representative HEK293T cell transfected with the control transcript shows the typical NCL localization (green), in contrast to the nucleolar dispersion observed in cells transfected with (GGGGCC)21 (Figure 4d). f) NCL is significantly mislocalized when HEK293T cells are transfected with (GGGGCC)21 transcripts, as compared to (GGGGCC)3 or control transcripts. RNA transfections were performed in duplicate, except for No RNA and (GGGGCC)3. Data are means ± s.e.m. n = 27, 50, 37, and 60 for the No RNA, CTL RNA, (GGGGCC)3, and (GGGGCC)21, respectively. ****P < 0.0001. g) There is a significantly decreased cell viability in HEK293T cells transfected with the (GGGGCC)21 abortive RNA transcripts when compared to a control RNA transcript. Cytotoxicity measurements were performed in a 96-well plate with HEK293T cells that had been transfected with 0–500 ng/mL of control RNA or (GGGGCC)21 repeat-containing RNAs generated from the in vitro transcription as described earlier and presented in Figure 2a. Cell viability was measured using the Cytotox Glo kit (Promega), with the fluorescence (viable cells) and luminescence (dead) measured on a Synergy H1 microplate reader. Data are means ± s.d. n = 3. *P < 0.05.
To confirm the association of NCL with C9orf72 repeat RNA in pathologically relevant patient tissues of ALS/FTD patients with the C9orf72 HRE, we performed RNA fluorescence in situ hybridization (FISH) in combination with NCL immunostaining in motor cortex tissues from these patients. The distribution of NCL in postmortem tissues appears variable; however, it is evident that NCL frequently colocalizes with the GGGGCC RNA foci in the neurons of the motor cortex only in C9orf72 HRE ALS patients (Figure 4c, Extended Data Figure 7c). These results support the interaction of NCL and the GGGGCC repeat transcripts in vivo and suggest the possible occurrence of functional defects associated with nucleolar stress in ALS/FTD patients.
Next, we examined whether the NCL mislocalization and nucleolar stress in the patient cells were caused by a gain in toxicity of the aberrant C9orf72 RNA. To directly address this question, we transfected HEK293T cells with the abortive transcripts generated in the aforementioned in vitro reactions. Treatment with the 21-repeat-containing abortive transcripts recapitulates the NCL pathology observed in patient cells carrying the C9orf72 HRE: NCL is significantly more dispersed in the nucleus (Figure 4d and Extended Data Figure 7f). Furthermore, the transfected HRE transcriptional products show a concentration-dependent decrease in cell viability compared to a control transcript (Extended Data Figure 7g). Thus, our results establish a direct link between abortive C9orf72 HRE transcripts, cytotoxicity, and patient pathology.
The mislocalization of another nucleolar component, nucleophosmin/B23, in B lymphocytes carrying the C9orf72 HRE further indicates general nucleolar stress (Extended Data Figure 8a–b). To determine whether the functions of the nucleolus are impaired in patient cells and tissues, we used qRT-PCR to measure the processing and maturation of the 45S rRNA (Extended Data Figure 8c). The results show a decrease in the processing of the precursor 45S rRNA into the mature cleaved 28S, 18S, and 5.8S rRNAs in C9orf72 HRE patient B lymphocytes (Extended Data Figure 8d). Furthermore, this rRNA maturation is significantly decreased in the motor cortex tissues from ALS patients carrying the C9orf72 HRE (Extended Data Figure 8d). Taken together, these results identify a functional defect involving the biochemical effects of the C9orf72 HRE in nucleolar stress and directly link this defect to disease pathology.
Extended Data Figure 8. Patients carrying the C9orf72 HRE show phenotypes indicative of nucleolar stress.

a) Another nucleolar component, nucleophosmin/B23 (green), shows a dispersed localization as seen for NCL in B lymphocytes from C9orf72 HRE patients. B23 (mouse monoclonal, B0556, Sigma) IF staining followed the manufacturer’s recommendations and the protocol described in Methods. B lymphocytes images were obtained as described in Methods. b) There is a significant increase in the total nuclear area occupied by B23 in B lymphocytes from a patient carrying the C9orf72 HRE when quantified similarly to NCL (Extended Data Figure 7a). The data are means ± s.e.m. n = 38 and n = 49 for representative C9orf72 WT and C9orf72 HRE B lymphocytes, respectively. *P < 0.05. c) The processing of the 45S pre-rRNA into the mature 28S, 18S, and 5.8S rRNAs that occurs in the nucleolus is depicted. d) Maturation of the 45S pre-rRNA to the 28S, 18S, and 5.8S rRNAs is impaired in patients carrying the C9orf72 HRE. B lymphocytes from patients (n = 6) show a decrease in 45S rRNA processing, but the processing is significantly decreased in the motor cortex tissues of patients carrying the C9orf72 HRE relative to controls (n = 4, 3, and 6 for C9orf72 WT, non-C9orf72 ALS, and C9orf72 HRE, respectively). Furthermore, ALS patients that do not carry the C9orf72 HRE show no significant impairment in 45s maturation (non-C9orf72 HRE ALS, n = 3). The primers for 45S, 28S, 18S, and 5.8S rRNAs (Extended Data Table 1) were previously described47. The linear-fold change in mature rRNA levels was normalized relative to the parent 45S rRNA levels. Data are means ± s.e.m. *P < 0.05.
Next, to examine other RNA-related functional defects that could arise in addition to the chronic nucleolar stress identified in C9orf72 HRE patients, we measured the abundance of processing bodies (P bodies) in patient-derived motor neurons. P bodies, which are composed of ribonucleoprotein complexes involved in the degradation of untranslated mRNAs32, are significantly increased in number, but not size, in iPS motor neurons from patients carrying the C9orf72 HRE relative to controls (Extended Data Figure 9a – b). This increase in P bodies is consistent with observations of decreased ribosomal maturation caused by nucleolar stress, which can then lead to increased untranslated mRNA in the cytoplasm and to a global perturbation in RNA processing.
Extended Data Figure 9. iPS motor neurons from patients carrying the C9orf72 HRE show an increased number of P bodies and increased sensitivity to tunicamycin.

a) The number of processing bodies (P bodies), but not the size, is significantly increased in iPS motor neurons from patients carrying the C9orf72 HRE. There is an almost two-fold increase in the total number of P bodies per iPS neuron from C9orf72 HRE patients. The number and size of P bodies from 30 fields of view were quantified in two independent patient iPS motor neuron lines and two control lines. Data are means ± s.e.m. n = 43. *P < 0.05. b) The representative images of P bodies in iPS neurons were visualized and quantified using a DCP1A antibody (Abnova H00055802-M06, green), and the imaging and analysis are described in Supplementary Methods. c) The percent change in cell death with increasing doses of tunicamycin indicates that it takes lower concentrations of tunicamycin to induce significant toxicity in C9orf72 HRE iPS motor neurons than in control neurons. Moreover, there is a significant difference in tunicamycin responses at 0.3 μM when compared between C9orf72 HRE and C9orf72 WT iPS motor neurons. Each iPS line was differentiated twice and the tunicamycin sensitivity at varying concentrations (0.0, 0.1, 0.3, 0.6, 1.0 and 3.0 μM) was analyzed twice. The mean number of PIpositive iPS neurons was normalized to that of untreated iPS neurons from the same line. Data are means ± s.e.m. n = 69/58 (0.0), 62/69 (0.1), 75/55 (0.3), 54/49 (0.6), 55/52 (1.0), and 60/53 (3.0) for C9orf72 WT/HRE samples, respectively. **P < 0.01. d) Representative iPS motor neuron images showing the increased PI staining (white) in response to increasing tunicamycin concentrations (Supplementary Method).
Finally, to test whether the cells of patients harboring the C9orf72 HRE are more sensitive to proteotoxic stress, we treated the iPS motor neurons with tunicamycin, which induces endoplasmic reticulum (ER) stress and unfolded protein response. C9orf72 HRE patient neurons show a dose-dependent increase in sensitivity to tunicamycin compared to controls (Extended Data Figure 9c–d). These results are consistent with the perturbation in protein homeostasis, which is possibly linked to the nucleolar stress identified in ALS patients carrying the C9orf72 HRE.
Discussion
The biological activity of nucleic acids is determined not only by their linear sequence of nucleotides but also by their structural diversity. Here we found that the generation of distinct polymorphic structures of the C9orf72 HRE DNA and RNA, such as G-quadruplexes and RNA•DNA hybrids (R-loops), underlie HRE-dependent molecular events, including abortive transcription and sequestration of unique RNA-binding proteins. The identification of conformation-dependent ribonucleoprotein complexes and a specific nucleolar pathology provides a cohesive mechanism for the disease (Figure 5 and Supplementary Discussion): An impairment in transcriptional processivity by the HRE leads to accumulation of abortive transcripts, and these repeat-containing RNAs sequester proteins that recognize their distinctive conformations, sensitizing cells for chronic neurodegenerative damage. Our findings concerning NCL represent a direct link between the characteristic G-quadruplexes of the C9orf72 HRE and the resulting cascade of pathological defects in ALS/FTD patients.
The nucleolus is a central hub in cellular stress responses33, and NCL has been shown to play a critical role in the long-term maintenance of mature neurons34. Our results demonstrate that motor neurons harboring the C9orf72 HRE show defects in rRNA processing and increased sensitivity to stresses related to protein homeostasis, suggesting that affected patient neurons are more vulnerable to age-dependent chronic stresses. Together, these results point to the targeting of these toxic nucleic acid conformations as a possible intervention at the root of the pathogenic cascade and also suggest a mechanistic model for similar repeat expansion neurodegenerative diseases that share nucleic acid features of the structurally polymorphic C9orf72 HRE
Methods Summary
CD analysis of nucleic acid structures was performed as described35, EMSAs were performed as recommended36,37, and DMS protection assays followed a previously described protocol26,36. The RNase protection assays were performed following Ambion’s recommendations. Plasmids were constructed in a pCR8 TOPO vector (Invitrogen), and GGGGCC HRE inserts were generated using a self-templating PCR protocol38. in vitro transcription reactions were performed with these plasmids and analyzed on sequencing gels. R-loop assays were adapted from previously described methods39. The cDNA from B lymphocytes or RNA from human tissues with or without the C9orf72 HRE were generated from total RNA following manufacturer’s protocols and relative levels were then measured. NanoString RNA analysis followed standard protocols as previously described20. The RNA pulldown with isotopically labeled HEK293T lysates and biotinylated RNA conjugated to streptavidin beads followed a previously described protocol40, with an additional KCl gradient wash. Quantitative mass spectrometry was performed by employing a three-state SILAC analysis using a filter-aided sample preparation (FASP) method followed by analysis on an LTQ-Orbitrap Elite mass spectrometer30,41. Peptides were identified using the Mascot search algorithm. Western blotting was performed on RNA pulldown fractions according to the manufacturer’s recommendations for each antibody. IF staining of lymphocytes, HEK293T cells, fibroblasts, and iPS motor neurons followed a standard protocol described in detail in Methods. RNA FISH with IF on human motor cortex tissue was performed essentially as previously described20.
Methods
Circular dichroism spectroscopy measurements of DNA and RNA
The circular dichroism (CD) absorptivity spectra for the oligonucleotides were obtained in 10 mM Tris-HCl pH 7.4 with 0, 5, 25, 50 or 100 mM KCl. All DNA and RNA oligonucleotides were used at 4 µM, except for the decanucleotides GGGGCCGGGG at 8 µM of concentration. The Jasco J-1810 parameters were set to the following: scan range 220–320 nm, scan speed 50 nm/min, 2 s response time, 1 nm bandwidth, and 5 acquisitions. All CD spectra were obtained at 25°C unless otherwise specified. CD melting curves were performed by measuring the absorptivity at a single wavelength, 295 nm for antiparallel G-quadruplexes and 260 nm for parallel G-quadruples, with a temperature range of 25°C to 95°C. The full CD spectrum melting curves are described in Extended Data Figure 1. When necessary, as in Extended Data Figure 1c, spectra were normalized to molar ellipticity for CD spectral comparisons of varying sizes of repeats using the equation, , where θobs is the observed ellipticity signal at a wavelength,λ,n is the molecular weight, l is the pathlength, and is the concentration. A list of DNA/RNA oligonucleotides is provided in Extended Table 1.
Spectral decomposition of the CD spectra was performed using the assumption that each experimentally observed spectrum (Exp_spectrum) was a linear combination of component spectra. We assumed three different components, DNA hairpin (H), antiparallel G-quadruplex (Qa), and parallel G-quadruplexes (Qp) corresponding to 0 mM KCl (GGGGCC)4, 100 mM KCl (GGGGCC)4, and 100 mM KCl GGGGCCGGGG, respectively, in the equation below.
Exp_spectrum = CH (H) +CQa (Qa) +CQp (Qp)
The coefficients for each component, (Ch), (CQa), and (CQp), for DNA hairpin, antiparallel G-quadruplex, and parallel G-quadruplexes respectively, were then calculated using nonlinear regression in Matlab. The relative distributions of each species were then determined from the coefficients. Residuals were calculated by subtracting the experimentally observed CD spectra from the calculated fit.
Electrophoresis mobility shift assay and DMS/RNase footprinting
Electrophoretic mobility shift assays (EMSA) were performed on 16% native polyacrylamide gel electrophoresis (PAGE) with the addition of 50 mM KCl in 1×TBE. The DNA and RNA EMSAs followed the same protocol.
For DMS footprinting, DNA end-labeling was performed using 100 pmol of DNA and 35 µCi of [32P]ATP (Perkin Elmer) with T4 polynucleotide kinase. Unincorporated [32P] ATP was removed using a microspin 30 Column (Biorad). The DMS protection assay was performed as previously described37. In brief, DNA samples were annealed by heating at 95°C for 5 min in 10 mM Tris-HCl, pH 7.4, with or without 100 mM KCl, and allowed to slowly cool to room temperature (RT). Following a DMS treatment, the DNA samples were subject to depurination and hydrolysis following Maxam and Gilbert sequencing methods26. The DNA samples were loaded onto sequencing gels at 5000 cpm, electrophoresed, exposed to a phosphor screen, and imaged on a Fuji Imager.
The ribonuclease (RNase) protection assay was performed with reagents described by Ambion. First, the RNAs were radiolabeled and annealed as described above for the DNAs. The evaluation of the hairpin/G-quadruplex structures was performed by annealing 2 pmol of radiolabeled RNA ± 100 mM KCl, which was incubated in 1×RNA structure buffer (10 mM Tris, pH 7.0, with 100 mM KCl and 10 mM MgCl2) at RT for 10 min. To stop the cleavage, five volumes of cold stop solution (30% β mercaptoethanol, 0.3 M sodium acetate, and 100 µg/mL of yeast tRNA) were added, followed by an ethanol precipitation. Samples were lyophilized to dryness, resuspended in formamide buffer to yield 5000 cpm per 3 µL, and heated at 95°C for 2 min. The RNA samples were electrophoresed on 8 M urea, 15% denaturing PAGE (40:1) at a constant 55 W for 2 h. The radiolabeled RNA bands were then imaged as described above.
Hexanucleotide repeat and plasmid construction
Hexanucleotide GGGGCC repeats were generated by self-templating PCR, using a pair of oligonucleotide primers (GGGGCC)8 and (GGCCCC)5 (Extended Data Table 1). The PCR reaction contained 1µM of each primer, 2.5U FailSafe PCR enzyme mix, and PCR buffer mix J (Epicentre). The PCR reaction was cycled 40x, with a denaturation step at 98˚C for 30 sec; annealing/extension at 72˚C, starting at 1 min, and extending 12 sec after each cycle. The PCR fragments were separated on agarose gel, excised at varying fragment lengths, and cloned into the Gateway pCR8/GW/Topo vector (Invitrogen) between the attL1 and attL2 sites. This vector contains a T7 promoter and was used in the in vitro transcription assays. The number of repeats inserted into the plasmid was determined by analyzing the EcoRI-digested fragment size relative to a DNA molecular weight standard ladder or by counting the number of abortive transcript bands.
in vitro transcription and R-loop assays
All in vitro transcription was performed using a HiScribe T7 transcription kit (NEB, E2030S). Prior to transcription, 0.5 µg of the plasmid containing between 3 and ~70 hexanucleotide repeats was linearized using ApaI or HpaI as recommended by the manufacturer (NEB). The transcription mix was incubated at 37°C and aliquots were removed at either 0.5 or 4 h after the addition of T7 polymerase. The transcription reactions were performed with or without denaturing the template plasmids. The reactions in Figure 2a were performed without the denaturation. Adding the denaturation step did not change the transcriptional profiles. In the additional experiments to determine the effects of G-quadruplex formation on transcription (Extended Data Figures 3c–d and 4b–c), the template plasmid was denatured and annealed with or without KCl. In this optional denaturation step, the plasmid was denatured in 10 mM Tris-HCl, pH 7.4, ± 100 mM KCl or 100 mM NaCl by heating to 95°C for 10 min and then annealed by slowly cooling to RT. After transcription, DNA template was removed by treatment with DNase for 20 min at RT and then cleaned using a Bio-spin column. The RNA samples were mixed with formamide buffer, heated at 95°C for 5 min, and then separated by 7.5% (acrylamide/bis-acrylamide 37.5:1) denaturing PAGE with 8 M urea. The samples were electrophoresed at 55 W for 1 h, stained with SYBR® gold (Invitrogen), and imaged on a Fuji Imager or Typhoon scanner. Gel lanes were analyzed in ImageJ (NIH) and plotted and analyzed using GraphPad Prism 6.
R-loop assays were performed as previously described39, with modifications. In brief, undigested template plasmid containing (GGGGCC)21 or (GGGGCC)~70 in 10 mM Tris-HCl, pH 7.4, with 100 mM KCl was denatured for 5 min at 100°C and slowly cooled to RT. Buffer was brought to a final concentration of 13 mM Tris-HCl, pH 7.4., with 67 mM KCl. in vitro transcription was performed as described above. Products were purified by phenol chloroform extraction followed by ethanol precipitation. They were resuspended in RNase H buffer (50 mM Tris-HCl, pH 8.3, with 75 mM KCl, 3 mM MgCl2, and 10 mM DTT) and digested with 1 U RNase H (NEB) and/or 1 µg RNase A (Invitrogen) for 1 h per transcription product of 300 ng template plasmid. The transcription mix was then phenol chloroform-extracted and ethanolprecipitated to remove proteins and salts then resuspended in water. The 300 ng template and transcription product were resolved by agarose gel electrophoresis (0.8% agarose, 1×TBE) at 60V for 2 h, and the bands were post-stained with ethidium bromide (0.5ug/mL) and imaged as described above.
Quantitative measurements of transcripts in patient cells
B lymphocytes were obtained from the NINDS Repository (ccr.coriell.org/NINDS) and the presence of the C9orf72 HRE in the patient cells was confirmed with fluorescence repeat-primer PCR2. For qRT-PCR assays, total RNA was extracted by Trizol extraction according to the manufacturer’s recommendations (QIAzol®, Qiagen). RNA was extracted from the human tissue using an RNeasy Kit (Qiagen). The cDNA was generated by using 0.2–1 µg of total RNA with a Quantitect Reverse Transcriptase kit (Qiagen). Two primer sets were designed using Primer 3 (v.0.4.0) to recognize an intronic region upstream (5´) of the repeats and an intronic region downstream (3´), respectively, one corresponding to a position between exon 1A and the repeats, and the other located in intron 3 (Extended Data Table 1). qRT-PCR was performed using an IQ™ Sybr®Green kit (Biorad) and following manufacturer’s recommended thermocycler settings. Fluorescence was measured on a mini-Opticon (Biorad) and CT values were analyzed using the accompanying software. The 3´ CT values were normalized to the 5´ CT values following qRT-PCR analysis. This 3´/5´ normalization was then used to compare the relative ratio differences between controls and C9orf72 HRE patients. The relative linear-fold change between human cells with and without the C9orf72 HRE was quantified by using the Livak method42 and normalized to the levels in the pre-repeat intron for analysis of abortive transcription. All calculations were performed in Numbers/Excel and Graphpad Prism.
NanoString quantification of the C9orf72 pre mRNA levels in the motor cortex and cervical spinal cord from patients was performed using the NanoString detection system and a customdesigned codeset as previously described20. In brief, approximately 0.1–0.2 µg of RNA was used to quantify gene expression via the nCounter Analysis System according to the manufacturer’s protocol. RNA counts were normalized using the nCounter program (NanoString), and the endogenous controls, GAPDH, GUSB, and OAZ1, were used to normalize for RNA quantity (Extended Data Table 1). The RNA counts for the 3´ levels were divided by the 5´ levels (Figure 2), and then the patients carrying the C9orf72 HRE were compared to controls. Data were plotted and analyzed in Prism.
Quantitative mass spectrometry by SILAC
The RNA pulldown assays were performed as previously described40. Annealing of 10 µg of 3´-biotinylated RNA oligomers, (GGGGCC)4 and (CCCCGG)4, was carried out in 10 mM Tris, pH 7.4, ± 100 mM KCl by heating at 95°C for 10 min and cooling slowly to RT. The RNA was incubated with ~ 20 µL of Pureproteome streptavidin magnetic beads (Millipore) in lysis buffer (10 mM HEPES, pH 7.0, with 200 mM KCl, 1% Triton X-100, and 10 mM MgCl2) for 20–30 min on a rotary shaker. The beads were pre-washed twice in lysis buffer at 4°C for 5 min each. HEK293T cells were labeled using the SILAC method30. Cells were cultured for at least six doublings with SILAC medium supplemented either with medium lysine (42D1) and medium arginine (613C6) or heavy lysine (613C6 215N7) and heavy arginine (613C6 415N7), along with 10% dialyzed fetal bovine serum (Life Technologies) and penicillin/streptomycin. Adequate SILAC labeling was confirmed prior to use for immunoprecipitation with desired RNA molecules. Cells were pelleted and washed twice with cold 1×PBS, resuspended in lysis buffer, and then gently sonicated. The cell lysate was centrifuged at 16,000 g for 10 min at 4°C, and the supernatant was removed and analyzed via BCA assay (Pierce). Egg white avidin (10 µg) and yeast tRNA (0.5 mg) per 1 mg of protein were added to the lysate and incubated with gentle shaking at 4° C for 20 min. The lysate was centrifuged, and the supernatant was treated with 200 U/mL of the RNasin (Promega). The cleared lysate supernatant was incubated with different RNA molecules: (GGGGCC)4 G-quadruplex, (GGGGCC)4 hairpin, and (CCCCGG)4 hairpin, which were mixed with cell lysates of heavy, medium, or light SILAC labels, respectively. The RNA-conjugated beads or beads alone were incubated with HEK293T cell cleared lysates on a rotary shaker at 4°C for 2 h. The beads were then washed three times with lysis buffer. Increasing concentrations of KCl in lysis buffer (0.4, 0.8, and 1.6 M) were sequentially used for washing. The beads were then boiled in elution buffer (100 mM Tris-HCl, pH 7.4, with 1% SDS and 100 mM DTT) and combined. The buffer was exchanged with 8 M urea in 20 mM HEPES, pH 8.0, using the FASP method41. Proteins were then reduced with 10 mM DTT at 60°C for 30 min and alkylated with 55 mM iodoacetamide at RT for 30 min. The samples were incubated with sequencing grade trypsin (Promega) at 37°C overnight. The trypsin-digested samples were then purified on an SDS-removal column (Thermo Fisher), and the peptides were desalted using a C18 StageTip43. The peptide samples were loaded onto a trap column (2 cm, 75 µm diameter) and separated by an analytical column (20 cm, 75 µm diameter) packed with C18 materials (Magic C18AQ, 5 µm, 100 Å pore) using a 100 min gradient on an EASY nLC (Thermo Scientific) online system connected to an LTQ-Orbitrap Elite mass spectrometer (Thermo Scientific). High-resolution data were acquired twice in a top-15 data-dependent acquisition mode using HCD fragmentation. MS data were processed using the Proteome discoverer platform and searched against the human protein database using the Mascot algorithm. Oxidation at methionine and acetylation at protein N-termini were set as variable modifications along with SILAC labels, while carbamidomethylation at cysteine was set as a fixed modification. Duplicate LC-MS/MS runs of SILAC samples were performed to identify peptide sequences at a 1% false-discovery rate.
Western blot analyses and NCL pulldown
All samples for Western blotting were boiled in 1×SDS loading buffer then electrophoresed on 4–20% SDS PAGE Criterion gels (Biorad) and transferred to nitrocellulose membranes (Biorad). After protein transfer, nitrocellulose membranes were blocked overnight at 4°C in 5% milk in 1×phosphate-buffered saline with Tween 20 (PBST). Membranes were washed three times with 1×PBST for 5 min and then incubated at RT with one of the following antibodies: 1:1000 dilution of hnRNP U (anti-rabbit polyclonal, Bethyl, A300–690A), NCL (anti-rabbit polyclonal, Proteintech Group,10556-1-AP), hnRNP K (anti-mouse polyclonal, Santa Cruz, sc-28380), a 1:5000 dilution of hnRNP F and hnRNP H (anti-mouse monoclonal, a generous gift from Michael Matunis, 10A544) or RPL7 (anti-rabbit polyclonal, AbCam, ab72550). The primary antibodies were diluted in 1×PBST, 5% bovine serum albumin, and 0.03% sodium azide. Membranes were washed three times at RT for 15 min in 1×PBST and then incubated with the following secondary antibodies: goat anti-rabbit IgG IRDye (680 LT, 926–68021 or 800 CW, 926–32211, LI-COR) or donkey anti-mouse IgG (680 LT, 926–68022; or 800 CW, 926–32212). The secondary antibodies were diluted 1:30,000 with 5% milk in 1×PBST and incubated at RT for 1.5 h. Membranes were again washed three times with 1×PBST at RT for 15 min and washed with 1×PBS for 15 min before imaging on an Odyssey instrument (LI-COR).
The preferential binding of NCL to the different RNA conformations formed by the (GGGGCC)4 repeats was validated in vitro using a recombinant GST-NCL fusion protein (Abnova, H00004691-P01) following previously described protocol45. In brief, Glutathioneconjugated magnetic beads (Pierce) were washed three times in lysis buffer supplemented with 50 µg/mL BSA and then incubated for 30 minutes at 4°C with 300 ng of GST-NCL. The GST-NCL-bound beads were washed three times and incubated overnight at 4°C with 10 pmol of radiolabeled (GGGGCC)4 RNA, either hairpin or G-quadruplex, or (CCCCGG)4 RNA hairpin, in lysis buffer supplemented with 80 U of RNaseOUT™ (Invitrogen) and a 1:200 dilution of Protease Inhibitor Cocktail (Sigma, P8340). The GST-NCL-bound beads were then washed three times with lysis buffer and the counts per minute measured using a scintillation counter.
Transfection of GGGGCC RNA and cell microscopy with HEK293T cells or B lymphocytes
For transfection of the HRE transcriptional products, 2 × 104 HEK293T cells were plated onto polyethyleneimine-coated glass cover slips and grown overnight at 37°C and 5% CO2 in DMEM medium supplemented with 10% FBS and 1× penicillin and streptomycin antibiotics. The growing medium was replaced with OMEM, and the cells were transfected using Lipofectamine 2000 (Invitrogen) with the HRE transcriptional products prepared as described above, following the manufacturer’s recommendations. After 4 h, the medium was replaced with the DMEM supplemented as described above and grown an additional 44 h before processing for immunostaining or cytotoxicity assays as described below.
B lymphocytes from patients with or without the C9orf72 HRE were grown at 37°C and 5% CO2 in RPMI medium supplemented with 15% FBS and 1× penicillin and streptomycin. For indirect immunofluorescent imaging of B lymphocytes, 1 × 106 cells were plated onto polyethyleneimine-coated glass cover slips and incubated for 3 h. The medium was aspirated, and the cells were washed three times with 1×PBS. The cells were fixed for 10 min in 4% paraformaldehyde in 1×PBS, washed three times with 1×PBS, and blocked in 1×PBS with 0.1 % Triton X-100 and 5% BSA at 37°C overnight. The antibodies (described in the methods for the Western blotting above) were diluted according to the manufacturer’s suggestions in blocking buffer and incubated with the slides at 37°C for 1 h. The slides were washed three times with 1×PBS-Triton and overlaid with a goat anti-rabbit IgG (Alexa 488; Invitrogen) diluted in 1:1000 in blocking buffer at 37°C for 1 h. The slides were washed three times in 1×PBS-Triton and post-fixed in 4% paraformaldehyde in 1×PBS, followed by staining with Hoechst 33342 (Sigma) at RT for 5 min. Immunofluorescent images were obtained on a Zeiss microscope with Apotome. Additional images were obtained on a Leica SP8 confocal microscope using a single representative cross section through the Hoechst signal. The images were obtained with matched exposure settings and in a blinded fashion.
iPS neuron and motor cortex tissue immunostaining
C9orf72 patient iPS cells were generated from ALS patients’ fibroblasts and differentiated into neurons as previously described20. The fibroblasts were collected at the Johns Hopkins Hospital with the patient consent (IRB protocol: NA_00021979) or by Dr. Pentti Tienari at the Helsinki University Central Hospital. The induced-pluripotent stem cells (iPSC) were validated by immunostaining for the stem cell marker TRA-1–60. iPS neuronal differentiation was validated by immunostaining for β-III tubulin (Tuj1) and HB920. Cultures used for subsequent immunostaining were fixed on day 48 of differentiation and exhibited ~90% Tuj1-positive cells and neuronal morphology.
iPS cells were grown on 12 mm coverslips with poly-L-lysine and laminin. At day 48 of neuronal differentiation, cells were fixed with 4% paraformaldehyde in 1×PBS and processed for immunostaining. Cells were washed three times with 1×PBS, then permeabilized in 0.3% Triton X-100 in 1×PBS for 15 min at RT. After three washes with 1×PBS, the cells were blocked with 10% goat serum (Jackson Labs) in 1×PBS for 1 h at RT. The NCL antibody (described above) was then applied with blocking buffer (1:6000) overnight at 4°C. iPS neurons were then washed four times with 1×PBS and an anti-rabbit Cy3 secondary antibody was applied (1:200, Jackson Labs) for 1 h at RT. Once the secondary antibody was removed, cells were washed five times with 1×PBS for 5 min each, and then rinsed once with distilled water prior to mounting on coverslips with ProLong Gold Antifade with DAPI (Invitrogen). Cells were imaged on a Zeiss Axio Imager. Z-stack sections were obtained to encompass the iPS neuron DAPI signal at 0.5 µm intervals. All images were obtained in a blinded fashion at matched exposure and post-processing settings.
The use of human tissue and demographic information was approved by the Johns Hopkins University Institutional Review Board and ethics committee (HIPAA Form 5 exemption, Application #11-02–10-01RD). For FISH-IF of human autopsied tissue, motor cortex tissue was fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, and embedded and frozen in optimum cutting temperature (OCT) compound; 10 µm sections were cut using a cryostat and placed onto glass slides. Tissue were postfixed in 4% paraformaldehyde, washed three times for 5 min each in 1×PBS, treated with ice-cold acetone for 10 min, and then 1% Triton X-100 for 15 min. A 5′-digoxigenin-labeled locked nucleic acid FISH probe generated by Exiqon and targeting the GGGGCC repeat, (CCCCGG)2.5 was hybridized to the tissue in 40% formamide with 40 nM of probe for 3 h at 40°C20. Following hybridization, the tissue was washed two times with 40% formamide at 45°C for 15 min each, and then stringently washed five times with 1×SSC then Tris-glycine for 5 min each. Tissue was then processed for IF staining in blocking buffer. IF buffers for incubation with antibodies consisted of 10× protease-free and 5% heatshocked BSA fraction V (Roche) in RNase-free 1×Tris-buffered saline. Tissue was blocked for 2 h at RT, and the NCL (Abcam), β-III tubulin (Chemicon), and digoxigenin (Jackson) antibodies were incubated overnight at 4°C. The respective secondary antibodies were applied for 1 h at RT, and the tissue was then washed four times with 1×PBS.
Statistical Analysis
All P-values were calculated by applying a t-test with an unpaired fit and assuming a parametric distribution unless otherwise noted.
All patient information and experimental analyses are provided in Supplementary Table S2. All restriction enzymes, DNA polymerases, kinases, ligases, and RNases were obtained from New England Biolabs unless otherwise noted. Integrated DNA Technologies synthesized all oligonucleotides. All figures were assembled in Adobe Illustrator. RNA hairpin cloverleaf structures were determined using the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi).
Supplementary Material
Extended Data Figure 5. Workflow for the SILAC methodology used for identification and quantification of proteins that bind the hexanucleotide repeats in a structurally dependent manner.

HEK293T cells that had complete incorporation of either heavy-, medium-, or lightlabeled isotopes were incubated with either the (GGGGCC)4 G-quadruplex, the (GGGGCC)4 hairpin, or the (CCCCGG)4 hairpin, respectively. The samples were then processed as described in Methods and presented in Figures 3. The final eluted samples were combined and prepared for trypsin digestion using filter-aided sample preparation (FASP). The buffer was exchanged to remove salts and SDS. The sample was then subject to LC-MS/MS analysis as described in the Methods.
Acknowledgments
We would like to thank Bryan Traynor for the information on the ALS patient cells, Mariely DeJesus-Hernandez and Rosa Rademakers for the genotyping PCR protocols on patient cells, Pentti J. Tienari for providing additional fibroblast/iPS cell lines, Michael Matunis for the hnRNP F/H antibody, Raja Sekhar Nirujogi for his assistance with the SILAC LC-MS analysis, Tushar Chakravarty for his assistance with transfection of HEK293T cells, the Shankar Balasubramanian lab for generously providing the BG4 plasmid, Peter Steinwald for purification and assistance in the validation of the BG4 protein, Regina M. Powers for assistance in the staining and quantification of P bodies in iPS neurons, and Olga Pletnikova for processing human tissues. This work was supported by NIH grants NS07432 (J.W.) and NS085207 (R.S. and J.D.R.), the Robert Packard Center for ALS Research at Johns Hopkins and the Muscular Dystrophy Association (J.W.), Target ALS (J.D.R.), the ALS Association (J.W.), the Johns Hopkins Alzheimer’s Disease Research Center NIH P50AG05146 (J.C.T.), NCI training grant 5T32CA009110-36 (A.R.H and E.A.J.S.), Maryland Stem Cell Research Fund (C.J.D.), Judith & Jean Pape Adams Charitable Foundation (R.S.), and the Samuel I. Newhouse Foundation (J.C.T.). We thank Pierre Coulombe, Valeria Culotta, Michael Matunis, Paul Miller, Brian Learn, and members of the Jiou Wang lab for manuscript advice and insightful discussions.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- CD
circular dichroism
- DMS
dimethyl sulfate
- FTD
frontotemporal dementia
- HRE
hexanucleotide repeat expansion
- hnRNP
heterogeneous nuclear ribonucleoprotein
- IP
immunoprecipitation
- iPS
induced pluripotent stem
- LC-MS
liquid chromatography-mass spectrometry
- NCL
nucleolin
- PAGE
polyacrylamide gel electrophoresis
- qRT-PCR
quantitative reverse transcriptase polymerase chain reaction
- P bodies
processing bodies
- and ER stress
endoplasmic reticulum stress
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information Reprints and permissions information are available at www.nature.com/reprints.
Author Contributions A.R.H. performed the nucleic acid structure analysis and the in vitro transcription studies. G.P. constructed plasmids for in vitro transcription studies. E.A.J.S. and A.R.H performed and analyzed the R-loop assay. A.R.H. performed and analyzed the RNA pulldown. M.K. and P.S. performed MS analysis on pulldown fractions. A.R.H. performed all B lymphocyte IF and analysis. C.J.D. R.S. and J.D.R. performed all patient-derived iPS motor neuron and human tissue analysis. N.J.M. maintained patient cell cultures. J.D.R and J.C.T. provided patient tissue samples. A.R.H and J.W. designed the studies, developed the model, and wrote the paper with insight from G.P and C.J.D. A.R.H developed and arranged the figures with insight from J.W. All authors discussed the results and commented on the manuscript.
References
- 1.Richard GF, Kerrest A, Dujon B. Comparative Genomics and Molecular Dynamics of DNA Repeats in Eukaryotes. Microbiology and Molecular Biology Reviews. 2008;72:686–727. doi: 10.1128/MMBR.00011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9orf72 Causes Chromosome 9p–Linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renton AE, et al. A Hexanucleotide Repeat Expansion in C9orf72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiernan MC, et al. Amyotrophic lateral sclerosis. The Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 5.Majounie E, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8:423–434. doi: 10.1038/nrneurol.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Langenhove T, van der Zee J, van Broeckhoven C. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med. 2012;44:817–828. doi: 10.3109/07853890.2012.665471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rollinson S, et al. Analysis of the hexanucleotide repeat in C9orf72 in Alzheimer’s disease Neurobiol. Aging. 2012;33:e5–e6. doi: 10.1016/j.neurobiolaging.2012.01.109. 1846. [DOI] [PubMed] [Google Scholar]
- 9.Kohli MA, et al. Repeat expansions in the C9orf72 gene contribute to Alzheimer’s disease in Caucasians Neurobiol. Aging. 2013;34:1519. doi: 10.1016/j.neurobiolaging.2012.10.003. e5-1519.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majounie E, et al. Repeat Expansion in C9orf72 in Alzheimer’s Disease. N Engl J Med. 2012;366:283–284. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moss DJH, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neuorology. 2014 doi: 10.1212/WNL.0000000000000061. Ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutherford NJ, et al. Length of normal alleles of C9orf72 GGGGCC repeat do not influence disease phenotype Neurobiol. Aging. 2012;33:2950. doi: 10.1016/j.neurobiolaging.2012.07.005. e5-2950.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 14.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nature. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fratta P, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy K, Zamiri B, Stanley SYR, Macgregor RB, Pearson CE. The Disease-associated r(GGGGCC)n Repeat from the C9orf72 Gene Forms Tract Length-dependent Uni- and Multimolecular RNA G-quadruplex Structures. Journal of Biological Chemistry. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, et al. Expanded GGGGCCrepeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad Sci U.S.A. 2013;110:7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mori K, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 19.Ash PEA, et al. Unconventional Translation of C9orf72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donnelly CJ, et al. RNA Toxicity from the ALS/FTD C9orf72 Expansion Is Mitigated by Antisense Intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagier-Tourenne C, et al. Targeted degradation of sense antisense C9orf72 RNA foci as therapy for ALS frontotemporal degeneration. Proc. Natl Acad Sci U.S.A. 2013;110:E4530–9. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sareen D, et al. Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9orf72 Repeat Expansion. Science Translational Medicine. 2013;5 doi: 10.1126/scitranslmed.3007529. 208ra149-208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zu T, et al. RAN proteins and RNA foci from antisense transcripts in C9orf72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U.S.A. 2013 doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gellert M, Lipsett MN, Davies DR. Helix Formation By Guanylic Acid. Proc. Natl. Acad. Sci. U.S.A. 1962;48:2013. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huppert JL. Four-stranded nucleic acids: structure, function and targeting of G-quadruplexes. Chem. Soc. Rev. 2008;37:1375. doi: 10.1039/b702491f. [DOI] [PubMed] [Google Scholar]
- 26.Maxam AM, Gilbert W. A new method for sequencing. DNA. Proc. Natl. Acad. Sci. U.S.A. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumari S, Bugaut A, Huppert JL, Balasubramanian S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nature Chemical Biology. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biffi G, Tannahill D, McCafferty J, Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nature Chemistry. 2013;5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, et al. Ultrasensitive label-free amplified colorimetric detection of p53 based on G-quadruplex MBzymes. Biosensors and Bioelectronic. 2013;50:180–185. doi: 10.1016/j.bios.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 30.Ong S-E, et al. Stable isotope labeling by amino acids in cell culture and SILAC as a simple and accurate approach to expression proteomics. Mol.Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 31.Abdelmohsen K, Gorospe M. RNA-binding protein nucleolin in disease. rnabiology. 2012;9:799–808. doi: 10.4161/rna.19718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nature reviews Molecular cell biology. 2007;8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- 33.Boulon S, Westman BJ, Hutten S, Boisvert F-M, Lamond AI. The nucleolus under stress. Molecular Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hetman M, Pietrzak M. Emerging roles of the neuronal nucleolus. Trends in Neurosciences. 2012;35:305–314. doi: 10.1016/j.tins.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hazel P, Huppert J, Balasubramanian S, Neidle S. Loop-Length-Dependent Folding of G-Quadruplexes. J.Am.Chem.Soc. 2004;126:16405–16415. doi: 10.1021/ja045154j. [DOI] [PubMed] [Google Scholar]
- 36.Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun D, Hurley LH. Methods in Molecular Biology. Vol. 608. Humana Press; 2009. pp. 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu R-J, et al. Long Tract of Untranslated CAG Repeats Is Deleterious in Transgenic Mice. PLoS ONE. 2011;6:e16417. doi: 10.1371/journal.pone.0016417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reddy K, et al. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Research. 2011;39:1749–1762. doi: 10.1093/nar/gkq935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iioka H, Loiselle D, Haystead TA, Macara IG. Efficient detection of RNA-protein interactions using tethered RNAs. Nucleic Acids Research. 2011;39:e53–e53. doi: 10.1093/nar/gkq1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Rappsilber J, Ishihama Y, Mann M. Stop go extraction tips for matrix-assisted laser desorption/ionization nanoelectrospray and LC/MS sample pretreatment in proteomics. Anal.Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 44.Matunis MJ, Xing J, Dreyfuss G. The hnRNP F protein: unique primary structure, nucleic acid-binding properties, and subcellular localization. Nucleic Acids Research. 1994;22:1059–1067. doi: 10.1093/nar/22.6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsoi H, Lau TC-K, Tsang S-Y, Lau K-F, Chan HYE. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc. Natl.Acad.Sci.U.S.A. 2012;109:13428–13433. doi: 10.1073/pnas.1204089109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almeida S, et al. Modeling key pathological features of frontotemporal dementia with C9orf72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uemura M, et al. Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation. Oncogene. 2011;31:1254–1263. doi: 10.1038/onc.2011.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.