

Summary
Objective
Elucidating molecular mechanisms underlying limbic epileptogenesis may reveal novel targets for preventive therapy. Studies of TrkB mutant mice led us to hypothesize that signaling through a specific phospholipase (PLC), PLCγ1, promoted development of kindling.
Methods
To test this hypothesis, we examined the development of kindling in PLCγ1 heterozygous mice. We also examined the cellular and subcellular location of PLCγ1 in adult wild type mice.
Results
The development of kindling was impaired in PLCγ1 heterozygous mice compared to wild type controls. PLCγ1 immunoreactivity was localized to the soma and dendrites of both excitatory and inhibitory neurons in hippocampus of adult mice.
Significance
This study implicates PLCγ1 signaling as the dominant pathway by which TrkB activation promotes limbic epileptogenesis. Its cellular localization places PLCγ1 in a position to modify the efficacy of both excitatory and inhibitory synaptic transmission. These findings advance PLCγ1 as a novel target for therapies aimed at preventing temporal lobe epilepsy induced by status epilepticus.
Keywords: Epilepsy, Seizure, TrkB, PLCγ1
Introduction
Temporal lobe epilepsy (TLE) is a common and often devastating form of human epilepsy that currently lacks preventive therapy.1 Elucidating the molecular mechanisms of epileptogenesis can provide targets for preventive therapies. A number of attractive candidates have recently emerged including mTOR2, REST3, EP2 receptor4, and mir-134.5
Work from multiple laboratories including our own has advanced the idea that enhanced activation of the brain-derived neurotrophic factor (BDNF) receptor, TrkB, promotes development of TLE in animal models and humans.6 Contrary to this idea, Paradiso et al7 reported that viral-mediated delivery of fibroblast growth factor-2 and BDNF following status epilepticus reduced spontaneous seizures and cell death in the pilocarpine model7; whether similar results ensue from viral-mediated delivery of BDNF alone is presently unclear. Using a chemical-genetic approach, Liu et al developed proof-of-concept evidence that inhibiting TrkB kinase and its downstream signaling transiently following status epilepticus prevented TLE.8 This provides a rationale for developing inhibitors of TrkB signaling for preventive therapies. Crystal structure of the ATP binding domain of TrkB reveals striking similarity to other tyrosine kinases9, underscoring the challenge of developing selective inhibitors.
An alternative strategy approach to limiting TrkB signaling would be to identify the signaling mechanisms by which TrkB activation promotes epileptogenesis. Activation of TrkB induces two major signaling pathways: phosphorylation of Y515 leads to binding of the adaptor protein, SHC; phosphorylation of Y816 leads to binding and activation of the enzyme PLCγ1. Studies of TrkB mutant mice in which phenylalanine was substituted for tyrosine at residue 515 or 816 revealed impairment of development of kindling in TrkBY816F but not TrkBY515F.10,11 This suggested that the pro-epileptogenic consequences of TrkB activation are mediated by its activation of PLCγ1. That said, multiple adaptor proteins and enzymes can bind a given motif of a receptor tyrosine kinase, underscoring the importance of determining whether PLCγ1 signaling per se promotes epileptogenesis. Null mutations of PLCγ1 result in death at embryonic day 9.5. Therefore, to test the hypothesis that PLCγ1 promotes epileptogenesis, we examined the development of kindling in Plcγ1 heterozygous mice.
Experimental Procedures
Mice
Plcγ1 mutant mice were generated by targeted deletion of genomic sequences encoding the X domain and both SH2 domains of PLCγ1 as described previously.12 Note that line generated with replacement vector TV-112 was used in these experiments. Homozygous disruption of PLCγ1 (−/−) results in embryonic lethality at approximately embryonic day 9.0–9.5. Immunoblot analysis of mouse embryonic fibroblasts (MEF) revealed no detectable full length Plcγ1 protein in Plcγ1−/− MEF and reduced Plcγ1 protein content in Plcγ1 heterozygous (+/−) MEF.12 Thirteen heterozygotes of PLCγ1 and 11 wild type (+/+) littermates were used for the kindling experiment. The genotype of each animal was assessed twice using PCR of genomic DNA isolated from tails (before and after experiments). No overt differences were detected in appearance of +/− and +/+ mice. The average body weights of +/− (22.15 ± 1.10 g) and +/+ (23.45 ± 0.73 g) were not significantly different. We crossed inbred strains of Plcγ1 mutant mice on a 129/SvJ background to C57BL/6 for 3 generations.
To verify the specificity of PLCγ1 antibody, we generated syn-cre+ PLCγ1flox/flox (syn-cre+ PLCγ1f/f) mice. Crossing PLCγ1 floxed mouse13 to syn-cre mouse14 generated progeny in which expression of the floxed gene was selectively eliminated in a subset of CNS neurons. The syn-cre+ PLCγ1f/f mice are viable and exhibit no overt differences from their syn-cre− PLCγ1f/f littermates.
Procedures involving animals followed National Institutes of Health guidelines for the care and use of experimental animals. All experiments conformed to local guidelines on the ethical use of animals.
Western blot
Both +/− and +/+ mice were decapitated under deep pentobarbital anesthesia (250 mg/kg, i.p.). Following decapitation, the mouse head was dipped into liquid nitrogen for 4 seconds to rapidly cool the brain. The hippocampi, amygdala and cortex were rapidly dissected on ice and homogenized in lysis buffer [20 mM Tris (pH 8.0), 137 mM NaCl, 1% NP40, 10% glycerol, 1 mM phenylmethylsulfonylfuoride (PMSF), and 1 Complete Mini protease inhibitor tablet (Mini, Roche, Mannheim, Germany)/10 ml]. The supernatant was saved following centrifugation at 16,000 × g for 10 min, aliquoted and stored at −80°C for further biochemical analysis.
Western blotting was performed to analyze PLCγ1 content using procedures as described previously.11,14 Antibodies to PLCγ1 (Cell Signaling, Danvers, MA) and β-actin (Sigma, St Louis, MO) were used in these experiments. Results from Western blotting were quantified as described previously.15 Briefly, the immunoreactivity of individual bands on Western blots was measured by Image J software and β-actin was used to control for loading and transfer. Western blotting of brain homogenates prepared from 4 +/+ and 4 +/− mice revealed reductions of 37%, 45%, and 44% in hippocampus, amygdala, and cortex respectively (Fig 1), thereby confirming reduced expression of PLCγ1 protein in the PLCγ1 +/− mice.
Figure 1. Efficacy of targeted deletion of PLCγ1 gene in PLCγ1+/−.

(A) Representative Western blots of PLCγ1 in cortex, hippocampus and amygdala from 2 pairs of +/− and +/+ mice. β-actin was used to control for loading and transfer. (B) Quantitative analysis of Western blot from 4 pairs of +/− and +/+ mice. The ratios of PLCγ1 vs actin in +/− mice were significantly decreased from all 3 regions comparing with those from +/+ mice. Data are presented as mean ± SEM, Student t test, ** p<0.01; *** p<0.001.
PLCγ1 immunohistochemistry
PLCγ1 immunohistochemistry was performed using a protocol described previously.11 Briefly, under pentobarbital anesthesia, mice were perfused with 4% paraformaldehyde in PBS and the brains were removed, post-fixed for 4h and cryoprotected until brains sank. Forty μm coronal sections were cut and used for immunofluorescent staining. The following antibodies were used for immunofluorescence staining: PLCγ1 (Cell Signaling, Danvers, MA), NeuN (Millipore, Temecula, CA), GFAP (Sigma, Louis, MO), GAD67 (Millipore, Temecula, CA), Parvalbumin (Sigma, St Louis, MO), MAP2 (Sigma, St Louis, MO), Tau1 (Millipore, Temecula, CA), Synapsin 1 (Synaptic Systems, Geottingen, Germany). After 1h incubation with blocking solution (5%NGS, 0.5%NP40 in PBS buffer), the primary antibodies were applied to floating sections overnight at 4°C. Alexa Fluor 594 goat anti-rabbit and Alexa Fluor488 goat anti-mouse secondary antibodies (Invitrogen, Eugene, Oregon) were used to visualize the immunofluorescent staining. Images were captured using a Leica (Nussloch, Germany) TCS SL confocal system. To quantify the colocalization of PLCγ1 with GAD67, 2 images under 63x objective were taken from CA1 oriens and stratum lacunosum-moleculare (SLM) respectively and the cells with PLCγ1-ir+ or GAD67-ir+ alone or both. The results are presented as the percentage of PLCγ1-ir+ or GAD67-ir+ cells averaged from 2 adjacent sections in 2 naive mice. The specificity of PLCγ1 antibody was verified by the reductions of immunoreactivity in CA3 and dentate gyrus in syn-cre+ PLCγ1 flox/flox mice compared with the WT controls (Fig. 3).
Figure 3. Specificity of PLCγ1 antibody for immunohistochemistry.
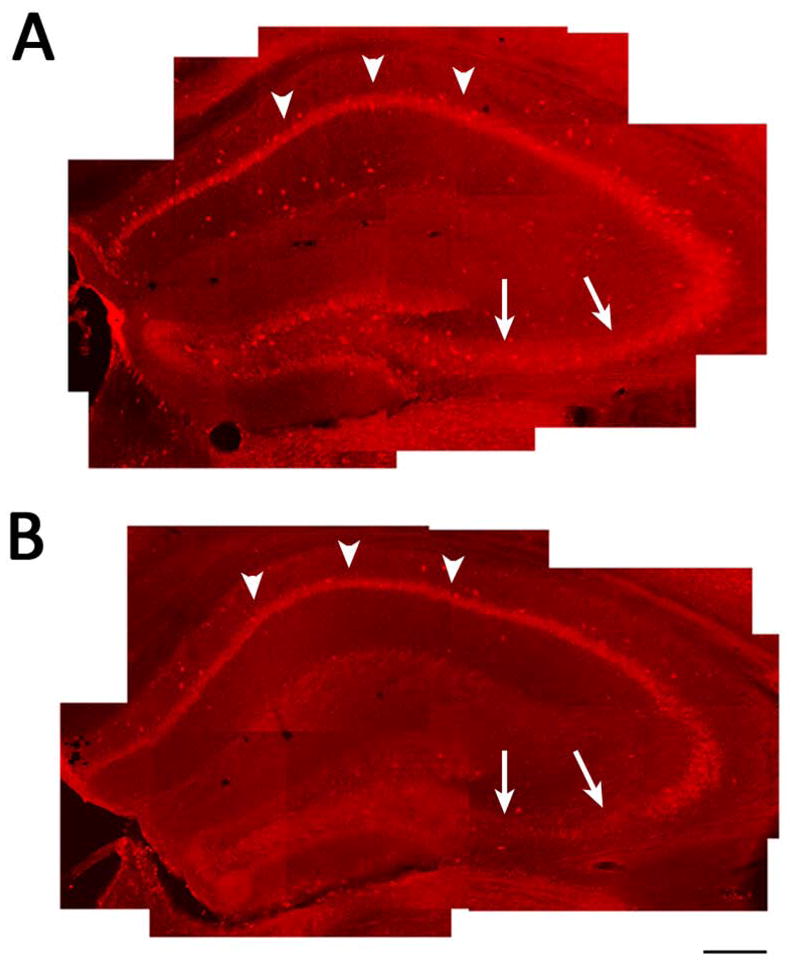
Representative images in low magnification of hippocampus of PLCγ1-ir in sections prepared from Syncre−PLCγ1f/f (A) and Syncre+PLCγ1f/f (B) mice. Note although PLCγ1-ir persists in CA1 (arrow heads), PLCγ1-ir is decreased in syncre+PLCγ1f/f compared to syncre−PLCγ1f/f mice, particularly in dentate granule cells and CA3 pyramidal cells (arrows), thereby establishing the specificity of antibody for PLCγ1. Scale bar: 300 μm.
Electrode implantation and kindling procedures
Eleven +/+ and 13 +/− mice were included in the kindling experiment. A bipolar electrode for stimulation and recording was stereotactically implanted in the right amygdala under pentobarbital anesthesia (60mg/kg) using the following coordinates with bregma as the reference: 1.2 mm posterior, 2.9 mm lateral, 4.6 mm below dura. After a post-operative recovery period of 1 week, the electrographic seizure threshold (EST) was determined by administering a 1 sec train of 1 msec biphasic rectangular pulses at 60 Hz beginning at 40 μA. Additional stimulations increasing by 10 μA were administered at one minute intervals until an electrographic seizure lasting at least 5 seconds was detected on the electroencephalogram (EEG) recorded from the amygdala. The behavioral manifestations of seizures were classified according to a modification of the description of Racine (1972).16 Stimulations were subsequently administered twice per day at an intensity of the EST until the animals exhibited 3 consecutive seizures of Class 4 or greater with limb clonus and/or tonus lasting at least 12 sec. The surgery and kindling procedures were performed by an individual blinded to genotype of the animals.
After a stimulation-free period of 2 weeks following the 3rd consecutive seizure of Class 4 or greater, persistence of the hyperexcitability was examined. The animals were perfused transcardially under deep pentobarbital anesthesia with ice-cold 4% paraformaldehyde in 1x PBS for 5 min at a flow rate of 7.5 ml/min. The brains were removed, post fixed in the same solution overnight at 4 °C and cryoprotected in 30% sucrose in 1x PBS until they sank. Coronal sections of 40 μm were cut in a cryostat at −20°C, mounted in PBS on slides, air-dried, frozen and stored at −80°C until use. The sections were stained with methyl green pyronine-Y (MGPY) and analyzed for electrode placement by a person without knowledge of genotype. Only animals with correct electrode placement in the amygdaloid complex were included in the statistical analysis for kindling experiment.
Statistical analysis
The results from Western Blot and kindling development were analyzed by two-tailed Student’s t test. Datasets are presented as mean ± S.E.M. for the designated number of experiments.
Results
Kindling development was inhibited in PLCγ1+/−
We hypothesized that the development of kindling would be inhibited in PLCγ1+/− mice for two reasons. First, the development of kindling was impaired in TrkB mutant mice in which phenylalanine was substituted for tyrosine residue 816 of TrkB; one protein bound by the motif of TrkB containing phosphorylated tyrosine 816 is PLCγ1. Second, use of a surrogate measure of PLCγ1 activation revealed its enhanced activation in both kindling and status epilepticus models.11 That said, because multiple adaptor proteins and enzymes can bind a given motif of a receptor tyrosine kinase, we sought to test whether signaling through PLCγ1 in particular affected development of kindling. To address this question, we examined the development of kindling in PLCγ1+/− mutant mice. PLCγ1+/− mice exhibited a significant impairment of limbic epileptogenesis, as evident in the increased number of stimulations required to elicit behavioral seizures compared with +/+ (Fig. 2B and D). The number of stimulations required to evoke a limbic seizure termed Class 1 or 2 was increased twofold in +/− (4.23 ± 0.62, n=13) compared with +/+ (2.09 ± 0.28, n=11, p<0.01). Likewise, the number of stimulations required to induce the 1st Class 4 or 5 seizure was significantly increased in +/− (7.38 ± 0.75) compared with +/+ controls (4.64 ± 0.41, p<0.01). Similarly, the number of stimulations required to induce the third consecutive class 4 or greater (fully kindled stage) was increased in +/− (14.69 ± 0.93) compared with +/+ controls (9.45 ± 0.65, p<0.001, Student’s t test). No significant difference was detected in the current required to evoke an initial electrographic seizure (179 ± 27 μA for +/− and 143 ± 20 μA for +/+) (Fig. 2A). Nor was a significant difference detected in the duration of electrographic seizure between +/− and +/+, during either the first evoked electrographic seizure or third consecutive Class 4 or above seizure (Fig. 2C). Interestingly, the cumulative duration of evoked electrographic seizure prior to the first seizure Class 4 or 5 for +/+ was 49.18 ± 4.90 s in comparison to 90.92 ± 12.33 s for +/− mice, an increase of almost twofold (p=0.006). That is, a greater duration of electrographic seizure activity is required prior to the first generalized seizure in +/− compared to +/+ mice. To determine whether the disruption of PLCγ1 influenced the persistence of the hyperexcitability after completion of kindling, +/− and +/+ mice were subjected to a single stimulation following a stimulation-free period of 2 weeks after kindling. All wild type and all PLCγ1 heterozygotes exhibited an electrographic seizure of similar duration (33 ± 2 s for +/−; 31 ± 2 s for +/+) in response to the initial stimulation and the proportion of each exhibiting seizures of Class 4 or 5 were similar (WT 82%; PLCγ Hets 92%), thereby excluding any substantial elevation of threshold. In summary, kindling development was impaired but the persistence of kindling was unaffected in the PLCγ1+/− compared to +/+ mice.
Figure 2. Partial inhibition of development of kindling in PLCγ1+/− mice.

(A) Electrographic seizure thresholds (EST) of +/− (filled circle, n=13) and +/+ (opened circle, n=11) mice are similar. Each symbol represents result of a single animal. Kindling development is presented as behavioral seizure class (B and D) and electrographic seizure duration (C) (y axis). Stimulation number (x axis in C,D) refers to the number of stimulation-evoked electrographic seizures with duration of at least 5 s. (B) Number of stimulations required to reach different seizure classes in +/− and +/+ mice. Fully kindled stage is defined by the occurrence of three consecutive seizures of class 4 or greater. All data are presented as mean ± SEM, Student t test, ** p<0.01; *** p<0.001.
Cellular localization of PLCγ1 in hippocampus
Understanding how PLCγ1 signaling could promote limbic epileptogenesis would be facilitated by knowing its cellular localization within the CNS of adult mice, leading us to perform immmunohistochemical analyses. Meaningful interpretation of these experiments requires establishing the specificity of the PLCγ1 antibody. Towards this end, we performed PLCγ1 immunohistochemistry in conditional PLCγ1 knockout mice (syn-cre+ PLCγ1f/f) and wild type controls which included syn-cre− PLCγ1f/f and syn-cre− PLCγ1+/+ mice. The PLCγ1f/f mice were crossed to a transgenic mouse in which Cre recombinase is driven by a synapsin1 promoter, a line in which Cre recombinase is expressed in CA3 pyramidal cells and dentate granule cells but to a lesser extent in CA1 pyramidal cells.14 A reduction of PLCγ1-immunoreactivity (PLCγ1-ir) in CA3 pyramidal and dentate granule cell body layers was evident in syn-cre+ PLCγ1f/f compared to control mice. Note the relative preservation of PLCγ1-ir+ in CA1 (arrow heads) compared to CA3 (arrows) or dentate gyrus in syn-cre+ PLCγ1f/f (Fig 3B) compared to control mice (Fig. 3A). The anatomic distribution of the reduced PLCγ1-ir+ in the syn-cre+ PLCγ1f/f compared to control mice parallels the expression of syn-cre14 and supports the specificity of the PLCγ1 antibody used in this experiment.
Consistent with the pattern of its mRNA distribution17, 18, PLCγ1 was widely expressed in most brain regions of wild type mice. Within the hippocampus, staining of the principal cell bodies, such as pyramidal neurons of CA1 and CA3 as well as dentate granule cells was prominent (Fig. 3A). In addition, some PLCγ1-ir+ cells were scattered throughout dentate hilus, stratum lucidum of CA3, and stratum radiatum, oriens and lacunosum-moleculare in both CA3 and CA1, a distribution consistent with GABA interneurons (Fig. 3A and Fig. 4C and 4D). Notably, the staining intensity of these cells was stronger than that of principal neurons.
Figure 4. PLCγ1-immunoreactivity is localized in hippocampal neurons.

(A, B) PLCγ1-ir is localized in hippocampal principal neurons. Representative images in high magnification of CA3a. PLCγ1-ir is localized to NeuN-ir+ (B) but not GFAP-ir+ (A) cells. (C, D) PLCγ1-ir is also localized in hippocampal interneurons. (C) Representative images from CA1 stratum radiatum with PLCγ1 (red) and GAD67 (green). PLCγ1-ir colocalized with GAD67-stained cells (arrows). (D) Representative images from CA3a, showing most PLCγ1-ir+ cells are Parvalbumin-ir+ (arrows). Scale bar: 50 μm.
To examine the nature of PLCγ1-ir+ cells, we conducted 3 immunohistochemical experiments. For the first step, double staining was performed using antibody to PLCγ1 together with antibodies to either a neuronal marker, NeuN, or an astrocyte marker, Glial Fibrillary Acidic Protein (GFAP). PLCγ1-ir+ cells (red), including principal cells and scattered cells, coincided with NeuN-ir+, but not with GFAP-ir+ (green, Fig 4B and A, respectively), establishing the neuronal localization of PLCγ1-ir+ cells. Next, to further analyze the type of PLCγ1-ir+ neurons, glutamate decarboxylase 67 (GAD67) was used as a marker of GABA containing neurons. The double staining revealed that more than half of scattered PLCγ1-ir+ neurons were GAD67-ir+: 62% in stratum lacunosum-moleculare (SLM) and 73% in stratum oriens of CA1. Likewise, the majority of GAD67-ir+ neurons exhibited PLCγ1 immunoreactivity: 95% in SLM and 90% in stratum oriens (Fig. 4C). To begin to examine which subsets of GABA interneurons exhibit PLC-ir+, colocalization of parvalbumin, a marker of fast-spiking basket cells, with PLCγ1-ir+ was examined. Parvalbumin-ir+ cells distribute broadly within hippocampus and most of them exhibited PLCγ1-ir+ (Fig 4D). Parvalbumin-ir was cytoplasmic and dendritic; however, PLCγ1-ir was mainly somatic (Fig. 4D). In sum, PLCγ1-ir is localized to both principal neurons and GABA interneurons in adult hippocampus, including parvalbumin subtype of interneurons.
To further explore the subcellular loci of the PLCγ1-ir within hippocampal neurons, sections from wild type mice were colabeled with an antibody to PLCγ1 and markers of axons (Tau) or dendrites (MAP-2). PLCγ1-ir prominently localized in the soma of neurons (Fig. 5A, arrow heads) and short processes in subsets of cells in dentate hilus (Fig. 5A, arrows). The colocalization of these processes with MAP2-ir+ supports their identity as proximal dendrites (Fig. 5A). Similarly, PLCγ1-ir prominently localized in CA1 pyramidal cells. Within CA1 pyramidal cells, PLCγ1-ir is mainly evident in soma and proximal portion of apical dendrites (not shown). Axon marker, tau, nicely labeled the axons of the dentate granule cells, named mossy fiber. Surprisingly, no overlap of tau- and PLCγ1-ir was evident in mossy fiber axons within stratum lucidum of CA3 (Fig. 5B, arrows). These findings demonstrate that PLCγ1-ir is largely confined to neuron soma, some within proximal dendrites but no detectable axonal localization within the axons of dentate granule cells.
Figure 5. PLCγ1-ir is localized to soma and proximal dendrites of hippocampal neurons.

(A) Representative images from dentate gyrus with PLCγ1 (red) and MAP2 (green). Note PLCγ1-ir is localized not only to soma (arrow heads) and but also proximal dendrites (arrows) in a sub set of neurons. (B)PLCγ1-ir does not colocalize with Tau, an axon specific marker. Note that a presumed mossy fiber immunoreactive for tau lacks PLCγ1-ir (arrows) but converse is true for soma. Scale bar: 25 μm.
Discussion
Our prior studies led us to hypothesize that signaling mediated by PLCγ1 promotes limbic epileptogenesis. To test this hypothesis, we examined the rate of kindling development in mice heterozygous for PLCγ1. We also ascertained the cellular distribution of PLCγ1 in hippocampus of adult mice using a PLCγ1-specific antibody. Two principal findings emerged. 1) The rate of development of kindling was impaired in PLCγ1 heterozygotes in comparison to wild type control mice. 2) PLCγ1-immunoreactivity was evident in neurons but not astrocytes and expressed in both principal excitatory neurons and GAD-67 interneurons. PLCγ1-immunoreactivity was detectable in soma and dendrites but not axons of these neurons. These findings implicate a causal role for PLCγ1 signaling in limbic epileptogenesis and provide anatomic locales at which to pursue the cellular mechanisms responsible for its pro-epileptogenic actions.
We hypothesized that the development of kindling would be inhibited in PLCγ1+/− mice for two reasons. First, the development of kindling was impaired in TrkB mutant mice in which phenylalanine was substituted for tyrosine residue 816 of TrkB, a motif that can bind PLCγ1 and promote its activation. Second, use of a surrogate measure of PLCγ1 activation revealed its enhanced activation in the kindling model.11 That said, because multiple adaptor proteins and enzymes can bind a given motif of a receptor tyrosine kinase, we sought to test whether signaling through PLCγ1 in particular affected development of kindling. The lack of a drug that selectively inhibits PLCγ1 necessitated a genetic approach to test this hypothesis. The embryonic lethality of PLCγ1 null mutants led us to test this hypothesis in PLCγ1 heterozygotes. Although reductions of PLCγ1 protein approximated only 40%, clear impairments of development of kindling were evident in the PLCγ1+/− mice. The magnitude of the impairments was less than that of trkBPLC/PLC mice11, the differences likely due to residual PLCγ1 available for activation in the PLCγ1+/− mice. That said, the present results support the conclusion that the dominant signaling pathway mediating the pro-epileptogenic consequences of TrkB activation in the kindling model involves PLCγ1.
As noted above, enhanced activation of PLCγ1 has been identified in the kindling model as revealed by enhanced immunoreactivity of the surrogate marker, p-PLCγ1 (pY783); importantly, no differences in expression of PLCγ1 protein were detected.11 What cellular and molecular consequences of PLCγ1 activation might promote the development of kindling? The cellular and subcellular locale of PLCγ1 provides a context within which to consider this question. The present findings confirm and extend analyses of PLCγ1 immunoreactivity in rat brain, analyses revealing predominant neuronal localization including dentate granule cells and pyramidal cells within both CA3 and CA1 of hippocampus.19 The present studies revealed PLCγ1 immunoreactivity in CNS neurons including both principal excitatory neurons and inhibitory interneurons, a pattern similar to that of TrkB itself.20 No PLCγ1 immunoreactivity colocalized with an astrocyte-specific marker, GFAP. Within neurons PLCγ1 immunoreactivity was most prominent within soma and to a lesser extent within proximal dendrites; whether an antibody with increased sensitivity would detect PLCγ1 immunoreactivity within axon terminals awaits further study.
What molecular and cellular consequences of PLCγ1 activation might promote epileptogenesis? Addition of BDNF to acutely isolated hippocampal slices produces a striking reduction of the K-Cl cotransporter, KCC2, the predicted consequence of which is accumulation of [Cl−]i and a shift of EGABA in a depolarizing direction21, an effect predicted to reduce synaptic inhibition. Importantly, the TrkB-mediated reduction of KCC2 expression presumably involves PLCγ1 because this effect required Y816 of TrkB.21 Findings from humans22, 23 and diverse in vivo and in vitro models 21, 24–28 advance reduced KCC2 expression as one mechanism underlying hyperexcitability of epileptic tissue. With respect to the kindling model in particular, striking reductions of KCC2 mRNA and protein have been identified in the dentate gyrus and CA3 and CA1 regions of hippocampus at multiple times following kindling.24 The localization of PLCγ1 to dendrites positions it to mediate such an effect. PLCγ1 activation might also promote the efficacy of excitatory synaptic transmission. That is, impairments of LTP of both mossy fiber-CA3 and CA3-CA1 synapses11, 29 have been identified in hippocampal slices isolated from trkBPLC/PLC mice. In sum, PLCγ1 activation may contribute to enhanced excitability in epileptic tissue by both enhancing excitatory and limiting inhibitory synaptic efficacy.
The present findings in the kindling model raise the question as whether PLCγ1 activation might promote limbic epileptogenesis in other models, specifically those induced by a brief episode of status epilepticus. Indeed, status epilepticus induced by pilocarpine results in a time dependent increase of PLCγ1 activation without change in its expression.11 Transient inhibition of TrkB signaling was recently shown to prevent epilepsy induced by status epilepticus in a different model, one induced by microinfusion of kainic acid into the amygdala.8 Whether the pro-epileptogenic consequences of TrkB activation in the kainic acid status epilepticus model are mediated by activation of PLCγ1 is presently unclear. Activation of PLCγ1 results in hydrolysis of phosphatidylinositol 4, 5-bisphosphate (PIP2), producing diacylglycerol (DAG) and inositol 1, 4, 5-trisphosphate (IP3). IP3 binds to IP3 receptors in the endoplasmic reticulum, resulting in a transient increase of intracellular free Ca2+.30, 31 Interestingly, Pal et al implicated calcium release from endoplasmic reticulum as a causal event in epileptogenesis in the cultured hippocampal neuron model32; it seems plausible that hydrolysis of PIP2 in this model may have been mediated by PLCγ1.
In sum, the present findings establish PLCγ1 as a molecular mechanism underlying development of kindling and implicate PLCγ1 signaling as the dominant signaling pathway mediating the pro-epileptogenic consequences of TrkB activation in this model. These findings advance PLCγ1 as a novel target for development of preventive agents for limbic epileptogenesis. While PLCγ1 inhibitors might induce some impairment of associative learning33, the brief period of exposure following status epilepticus likely required for prevention8 would minimize such unwanted effects.
Supplementary Material
Acknowledgments
This work was supported by NINDS grants NS56217 and NS060728 (J.O.M.).
Footnotes
Disclosure: We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. None of the authors has any conflict of interest to disclose.
References
- 1.Engel J, Williamson P, Wieser HG. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven; 1998. Mesial temporal lobe epilepsy; pp. 2417–2426. [Google Scholar]
- 2.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51(1):27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McClelland S, Flynn C, Dubé C, et al. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol. 2011;70(3):454–64. doi: 10.1002/ana.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang J, Quan Y, Ganesh T, et al. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc Natl Acad Sci U S A. 2013;110(9):3591–6. doi: 10.1073/pnas.1218498110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jimenez-Mateos EM, Engel T, Merino-Serrais P, et al. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat Med. 2012;18(7):1087–94. doi: 10.1038/nm.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boulle F, Kenis G, Cazorla M, et al. TrkB inhibition as a therapeutic target for CNS-related disorders. Prog Neurobiol. 2012;98(2):197–206. doi: 10.1016/j.pneurobio.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Paradiso B, Marconi P, Zucchini S, et al. Localized delivery of fibroblast growth factor-2 and brain-derived neurotrophic factor reduces spontaneous seizures in an epilepsy model. Proc Natl Acad Sci U S A. 2009;106(17):7191–6. doi: 10.1073/pnas.0810710106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu G, Gu B, He XP, et al. Transient Inhibition of TrkB Kinase after Status Epilepticus Prevents Development of Temporal Lobe Epilepsy. Neuron. 2013;79(1):31–8. doi: 10.1016/j.neuron.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertrand T, Kothe M, Liu J, et al. The crystal structures of TrkA and TrkB suggest key regions for achieving selective inhibition. J Mol Biol. 2012;423(3):439–53. doi: 10.1016/j.jmb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 10.He XP, Minichiello L, Klein R, et al. Immunohistochemical evidence of seizure-induced activation of trkB receptors in the mossy fiber pathway of adult mouse hippocampus. J Neurosci. 2002;22(17):7502–8. doi: 10.1523/JNEUROSCI.22-17-07502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He XP, Pan E, Sciarretta C, et al. Disruption of TrkB-mediated phospholipase Cgamma signaling inhibits limbic epileptogenesis. J Neurosci. 2010;30(18):6188–96. doi: 10.1523/JNEUROSCI.5821-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji QS, Winnier GE, Niswender KD, et al. Essential role of the tyrosine kinase substrate phospholipase C-gamma1 in mammalian growth and development. Proc Natl Acad Sci U S A. 1997;94(7):2999–3003. doi: 10.1073/pnas.94.7.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu G, Chen Y, Yu M, et al. Phospholipase Cγ1 is essential for T cell development, activation, and tolerance. J Exp Med. 2010;207(2):309–18. doi: 10.1084/jem.20090880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He XP, Kotloski R, Nef S, et al. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43(1):31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 15.Huang YZ, Pan E, Xiong ZQ, et al. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57(4):546–58. doi: 10.1016/j.neuron.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 16.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 17.Homma Y, Takenawa T, Emori Y, et al. Tissue- and cell type-specific expression of mRNAs for four types of inositol phospholipid-specific phospholipase C. Biochem Biophys Res Commun. 1989;164(1):406–12. doi: 10.1016/0006-291x(89)91734-8. [DOI] [PubMed] [Google Scholar]
- 18.Ross CA, MacCumber MW, Glatt CE, et al. Brain phospholipase C isozymes: differential mRNA localizations by in situ hybridization. Proc Natl Acad Sci U S A. 1989;86(8):2923–7. doi: 10.1073/pnas.86.8.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerfen CR, Choi WC, Suh PG, et al. Phospholipase C I and II brain isozymes: immunohistochemical localization in neuronal systems in rat brain. Proc Natl Acad Sci U S A. 1988;85(9):3208–12. doi: 10.1073/pnas.85.9.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan Q, Radeke MJ, Matheson CR, et al. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378(1):135–57. [PubMed] [Google Scholar]
- 21.Rivera C, Voipio J, Thomas-Crusells J, et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24(19):4683–91. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen I, Navarro V, Clemenceau S, et al. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298(5597):1418–21. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- 23.Huberfeld G, Wittner L, Clemenceau S, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27(37):9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rivera C, Li H, Thomas-Crusells J, et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159(5):747–52. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo NS, Lu J, England R, et al. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12(2):258–68. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- 26.Pathak HR, Weissinger F, Terunuma M, et al. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27(51):14012–22. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Zhou J, Chen Z, et al. Long-term expressional changes of Na+-K+-Cl− co-transporter 1 (NKCC1) and K+-Cl− co-transporter 2 (KCC2) in CA1 region of hippocampus following lithium-pilocarpine induced status epilepticus (PISE) Brain Res. 2008;1221:141–6. doi: 10.1016/j.brainres.2008.04.047. [DOI] [PubMed] [Google Scholar]
- 28.Blaesse P, Airaksinen MS, Rivera C, et al. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61(6):820–38. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Minichiello L, Calella AM, Medina DL, et al. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36(1):121–37. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- 30.Nishizuka Y. Studies and perspectives of protein kinase C. Science. 1986;233(4761):305–12. doi: 10.1126/science.3014651. [DOI] [PubMed] [Google Scholar]
- 31.Berridge MJ, Irvine RF. Inositol phosphates and cell signalling. Nature. 1989;341(6239):197–205. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- 32.Pal S, Sun D, Limbrick D, et al. Epileptogenesis induces long-term alterations in intracellular calcium release and sequestration mechanisms in the hippocampal neuronal culture model of epilepsy. Cell Calcium. 2001;30(4):285–96. doi: 10.1054/ceca.2001.0236. [DOI] [PubMed] [Google Scholar]
- 33.Gruart A, Sciarretta C, Valenzuela-Harrington M, et al. Mutation at the TrkB PLCγ-docking site affects hippocampal LTP and associative learning in conscious mice. Learning & Memory. 2007;14(1):54–62. doi: 10.1101/lm.428307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.