

Abstract
Alcohol abuse; the most common and costly form of drug abuse, is a major contributing factor to many disease categories. The alcohol-attributable disease burden is closely related to the average volume of alcohol consumption, with dose-dependent relationships between amount and duration of alcohol consumption and the incidence of diabetes mellitus, hypertension, cardiovascular disease, stroke, and pneumonia. The frequent occurrence of alcohol use disorders in the adult population and the significant and widespread detrimental organ system effects highlight the importance of recognizing and further investigating the pathophysiological mechanisms underlying alcohol-induced tissue and organ injury.
Moderate alcohol (ethanol) consumption, defined as up to 1 drink/day for women and up to 2 drinks/day for men is a socially accepted behavior, endorsed by >50% of adults in the United States (25a). Harmful alcohol use, encompassing both alcohol binge drinking [consuming >4–5 drinks on a single occasion, generally under a 2-h period, and elevating blood alcohol concentration (BAC) levels to 0.08% (legal limits) or higher] and chronic heavy alcohol consumption (>7 drinks/wk for women and >14 drinks/wk for men), remains the most common and costly form of drug abuse. Approximately 7% of the adult U.S. population meets diagnostic criteria for alcohol abuse and/or alcoholism (52). Alcohol abuse is a major factor contributing to many disease categories, including cancer, cardiovascular disease, liver cirrhosis, and traumatic injury.
Alcohol can permeate to virtually all tissues in the body, resulting in alterations in significant multi-systemic pathophysiological consequences. Approximately 3.4% of global noncommunicable disease-related burden of deaths, 5% of net years of life lost, and 2.4% of net disability-adjusted life years can be attributed to alcohol abuse, with higher burden for cancer and liver cirrhosis (86). Thus alcohol abuse is the third leading lifestyle-related cause of death in the United States. Dose-dependent relationships between alcohol consumption and incidence of diabetes mellitus, hypertension, ischemic heart disease, dysrhythmias, stroke, pneumonia, and fetal alcohol syndrome have been reported (95). However, recognition of alcohol as an underlying causal factor in comorbid conditions remains a challenge in the clinical setting (103). This review provides a brief summary of salient alcohol effects on nonneural tissues. Because often this is based on evidence derived from preclinical studies, it is important to take into consideration the context of alcohol administration (acute vs. chronic), the route of administration (oral, intraperitoneal, vapor), and the specific outcome studied under each condition. Thus the authors caution against generalizations on the effects of alcohol described in some preclinical studies to those resulting from years of alcohol abuse in the clinical setting. Moreover, the existing comorbid conditions, dietary habits, and additional drugs consumed by most individuals who abuse alcohol are not directly replicated in animal studies. This too should be taken into consideration when the existing preclinical literature is interpretted. When appropriate, this is highlighted in the review.
The fact that chronic alcohol abuse is conducive to tissue injury is a well accepted, evidence-based fact as described in this review. Several pathophysiological mechanisms have been identified as causative factors in tissue and organ injury resulting from alcohol abuse, including oxidative stress, inflammation, acetaldehyde generation and adduct formation, decreased barrier function, impaired anabolic signaling, upregulation of catabolic processes, fibroblast activation, mitochondrial injury, and cell membrane perturbations (FIGURE 1) (77). Some of these mechanisms are the result of direct alcohol-induced cell perturbations; others are the consequence of tissue alcohol metabolism. Thus a brief overview of salient aspects of alcohol metabolism and pharmacokinetics (reviewed in detail by Cederbaum and Khanna; Refs. 24, 61) is relevant to appreciate its significant role in organ injury. Neurological deficits and fetal alcohol syndrome resulting from alcohol abuse have been extensively reviewed by others. Here, we provide an overview of some of the critical, nonneuronal physiological systems impacted by alcohol abuse and their contribution to the pathophysiology underlying the most frequent comorbid conditions, and highlight critical areas in need of further research.
FIGURE 1.

Principal mechanisms of alcohol-induced pathophysiology
Alcohol abuse disrupts multiple cellular mechanisms, leading to altered organ function and disease. Among the most important pathophysiological mechanisms identified as causative factors in tissue and organ injury resulting from alcohol abuse include oxidative stress, inflammation, acetaldehyde generation and adduct formation, decreased barrier function, impaired anabolic signaling, upregulation of catabolic processes, fibroblast activation, mitochondrial injury, and cell membrane perturbations.
Alcohol Metabolism
The average rate at which alcohol is eliminated from the body is ∼7 g/h, which translates to ∼1 drink/hour (24, 61). Alcohol undergoes first-pass gastric metabolism by the enzyme alcohol dehydrogenase (ADH). Most tissues express ADH and are capable of alcohol metabolism, as reflected in Table 1. However, most alcohol oxidation occurs in the liver. Alcohol is metabolized to acetaldehyde primarily by ADH and the cytochrome P450 2E1 (CYP2E1). This latter pathway is particularly relevant following chronic alcohol abuse. Acetaldehyde is converted to acetate in the mitochondria by the enzyme acetaldehyde dehydrogenase type 2. Most of the acetate produced enters the systemic circulation and is activated to acetyl coenzyme A (CoA), a key intermediate metabolite in peripheral tissues. Acetaldehyde can form adducts that can produce injury through activation of immune responses (108). This is particularly relevant in alcoholic liver disease. During the oxidative process, both ADH and ALDH1 reactions reduce NAD+ to NADH, shifting the cellular redox ratio, thereby affecting several NAD+ requiring enzymes like lactate and pyruvate dehydrogenase and affecting pathways including glycolysis, citric acid cycle, fatty acid oxidation, and gluconeogenesis (39). In addition, the cytochrome P450 enzymes, particularly CYP2E1, contribute to the oxidation of alcohol to acetaldehyde, mainly at increasing alcohol concentrations as well as following their induction by chronic alcohol abuse. Because CYP2E1 is involved in oxidation of several drugs to their reactive intermediates (e.g., nitrosamines, acetaminophen, and halothane), their toxicity is enhanced in alcoholics. This pathway of alcohol oxidation results in the production of large amounts of reactive oxygen species (ROS) (25, 40) and is thought to be an important mechanism contributing to alcoholic liver injury (89). ROS are eliminated by antioxidants like glutathione (GSH) under normal conditions. Alcohol depletes cellular GSH stores, thereby further exacerbating ROS-mediated injury (50). ROS can interact with lipids, producing lipid peroxidation, leading to formation of reactive molecules such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE), which can in turn form protein adducts (22). A minor fraction of alcohol metabolism occurs in peroxisomes through catalase-dependent oxidation. Alcohol can also react with glucuronic acid to form ethyl-glucuronide, a soluble, non-volatile conjugate that is readily excreted and detected in body fluids, tissue, sweat, and hair for an extended time following alcohol consumption. Clearly, alcohol metabolism and the generation of ROS, depletion of reducing equivalents, particularly GSH, and the resulting alteration in cellular redox state contribute to tissue injury in several organ systems including the liver, lung, muscle, and brain (FIGURE 2).
Table 1.
Organ-specific rates of alcohol metabolism
| ADH Activity, mU/g of tissue | |
|---|---|
| Tissue | Rate |
| Liver | 260.00 |
| Stomach | 11.80 |
| Small intestine | 7.50–19.30 |
| Heart | 0.80 |
| Lungs | 3.60–8.10 |
| Skeletal muscle | 0.03 |
| Adipose | 6.70 |
| Brain | 0.05 |
| Testes | 26.20 |
| Ovaries | 7.00 |
Alcohol metabolism reflected by dehydrogenase (ADH) activity in rat tissues was compiled from Riveros-Rosas et al. and Raskin and Sokoloff. Rates of metabolism are expressed as milli-enzymatic units (mU) per gram (g) of tissue (94, 98). For comparison purposes, values from Raskin and Sokoloff were normalized to the ADH enzymatic rate of liver (factor of 1.49). Absolute tissue-specific rates may vary between species.
FIGURE 2.
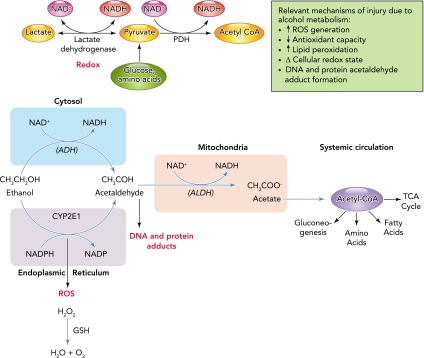
Alcohol metabolism and its contribution to tissue injury
The average rate at which alcohol is eliminated from the body is ∼7 g/h, which translates to ∼1 drink/h. Alcohol undergoes first pass gastric metabolism by the enzyme alcohol dehydrogenase (ADH). However, most alcohol oxidation occurs in the liver. Alcohol is metabolized to acetaldehyde primarily by alcohol dehydrogenase (ALD) and the cytochrome P450 2E1 (CYP2E1). This later pathway is particularly relevant following chronic alcohol abuse. Acetaldehyde is converted to acetate in the mitochondria by the enzyme acetaldehyde dehydrogenase (ALDH) type 2. Most of the acetate produced enters the systemic circulation and is activated to acetyl coenzyme A (CoA), a key intermediate metabolite in peripheral tissues. Acetaldehyde can form adducts that can produce injury through activation of immune responses. During the oxidative process, both ADH and ALDH reactions reduce NAD+ to NADH, shifting the cellular redox ratio, thereby affecting several NAD+ requiring enzymes like lactate and pyruvate dehydrogenase and affecting pathways including glycolysis, citric acid cycle, fatty acid oxidation, and gluconeogenesis. In addition, the cytochrome P450 enzymes, particularly CYP2E1, contribute to the oxidation of alcohol to acetaldehyde, particularly at increasing alcohol concentrations as well as following their induction by chronic alcohol abuse. Because CYP2E1 is involved in oxidation of several drugs to their reactive intermediates (e.g., nitrosamines, acetaminophen, and halothane), their toxicity is enhanced in alcoholics. This pathway of alcohol oxidation results in the production of large amounts of reactive oxygen species (ROS) and is thought to be an important mechanism contributing to alcoholic liver injury. ROS are eliminated by antioxidants like glutathione (GSH) under normal conditions. Alcohol depletes cellular GSH stores, thereby further exacerbating ROS-mediated injury. ROS can interact with lipids, producing lipid peroxidation, leading to formation of reactive molecules such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE), which can in turn form protein adducts. A minor fraction of alcohol metabolism occurs in peroxisomes through catalase-dependent oxidation. Alcohol can also react with glucuronic acid to form ethyl-glucuronide, a soluble, non-volatile conjugate that is readily excreted and detected in body fluids, tissue, sweat, and hair for an extended time following alcohol consumption.
Alcohol and the Neuroendocrine System
Alcohol intoxication, dependence, and withdrawal profoundly affect endocrine regulation and disrupt the body's ability to maintain and restore homeostasis following a challenge (28). One of the most sensitive pathways to the acute and chronic effects of alcohol abuse is the hypothalamo-pituitary-adrenal (HPA) axis (FIGURE 3). Responsiveness of the HPA axis to psychological and physical stressors can be heightened or blunted depending on duration of alcohol abuse (97). Alcohol-mediated disruption in HPA function has been implicated in the pathophysiology of pseudo-Cushing's syndrome (19), addiction (48), dependence (48), and relapse of recovering alcoholics (3). Alcohol produces dose-, frequency-, and duration-specific effects on arginine vasopressin (AVP), leading to alterations in water balance and mean arterial blood pressure homeostasis (115). Acute alcohol intoxication increases magnocellular and parvocellular neuronal activity; whereas chronic alcohol abuse significantly reduces the number of AVP-producing neurons in the supraoptic nuclei (SON) (29) and suppresses circulating AVP levels (29). Our studies have shown that acute alcohol intoxication augments paraventricular nucleus nitric oxide inhibitory tone and suppresses the hypovolemia- but not hyperosmolarity-induced AVP release (127). These acute effects of alcohol have the potential of affecting homeostatic mechanisms essential for restoring circulating blood volume following insults such as hemorrhagic shock. Although new set-points in osmolarity or hypovolemic responsiveness may be established following chronic alcohol abuse, persistent alterations in regulation of AVP release have been reported in abstinent alcoholics (35).
FIGURE 3.

Alcohol and the neuroendocrine system
Alcohol disrupts responsiveness of the hypothalamo-pituitary-adrenal (HPA) axis to psychological and physical stressors, and this has been implicated in the pathophysiology of pseudo-Cushing's syndrome, addiction, dependence, and relapse of recovering alcoholics. Alcohol produces dose-, frequency-, and duration-specific effects on arginine vasopressin (AVP), leading to alterations in water balance and mean arterial blood pressure homeostasis. Alcohol decreases the responsiveness of the hypothalamo-pituitary-thyroid (HPT) axis to central stimulation, decreases circulating levels of triiodothyronine (T3) and thyroxine (T4), and deiodination of T4 to T3. Chronic alcohol consumption disrupts the hypothalamo-pituitary-gonadal (HPG) axis and results in decreased testosterone levels, abnormal menstrual cycles, and infertility. GH, growth hormone; LH, luteinizing hormone; FSH, follicle stimulating hormone. The potential clinical consequences of alcohol abuse and its impact on the endocrine system are shown in the box.
Acute and chronic alcohol abuse blunts responsiveness of the hypothalamo-pituitary-thyroid (HPT) axis to central stimulation (132), decreases circulating levels of triiodothyronine (T3) and thyroxine (T4) (54), deiodination of T4 to T3 (83), and thyroid gland volume (53), and increases hepatic thyroid hormone uptake (56). Thyroid axis function can be further compromised in alcoholics with comorbid conditions and can contribute to behavioral manifestations of alcohol abuse-like depression (54). Chronic alcohol consumption disrupts the hypothalamo-pituitary-gonadal (HPG) axis, manifesting in decreased gonadotropin release, abnormal menstrual cycles, infertility, and impotence (33, 37). In addition, alcohol abuse markedly diminishes the growth hormone (GH) insulin-like growth factor (IGF-I) axis at multiple levels, including release, signaling, and cellular responses (109). This suppression in the GH/IGF-I axis is critical during adolescence, a period of widespread alcohol abuse, as well as during disease states (104). Thus the acute and chronic effects of alcohol abuse can lead to a multitude of endocrine-related disorders. Alcohol abuse contributes to an impaired ability of the host to respond to challenges and maintain homeostasis, affecting the ability to respond to stress. In addition, the cumulative impact of alcohol on disease burden results in detrimental effects on thyroid, gonadal, and somatotropic axis functions that can contribute to conditions including hypothyroidism, decreased reproductive function, and growth retardation.
Alcohol and the Gastrointestinal System
The gastrointestinal system participates in alcohol absorption and metabolism, and is an important target for alcohol-induced pathophysiology including esophageal and gastric dysmotility, altered acid secretion, impaired nutrient absorption, and disrupted intestinal barrier function. Esophageal dysmotility and delayed gastric emptying facilitating acid regurgitation increases the risk for postemetic lacerations of the distal esophagus induced by vomiting (Mallory-Weiss Syndrome) and for the development of esophageal varicosities resulting from increased intrahepatic pressure of liver fibrosis (73). Collectively, these factors promote acid injury and mucosal damage, increasing the risk of esophageal cancer. Gastric acid secretion and motility vary according to the alcohol content and to the fed state of the individual at the time of ingestion. Beer and wine (low alcohol %) stimulate gastric acid secretion and gastrin release and increase gastric emptying (118). These effects may have little to no consequence following isolated episodes of alcohol ingestion. However, the direct effects of repeated high level alcohol exposure on the gastric mucosa promote chronic gastritis, characterized by inflammatory cell infiltration and mucosal hypertrophy during the acute phase followed by decreased mucosal thickness and atrophy during the chronic phase (99). Moreover, chronic alcohol abuse impairs intestinal essential amino acid and vitamin absorption, leading to increased frequency of nutritional deficiencies, including vitamins A, B1, B2, B6, C, and folic acid (70) (FIGURE 4).
FIGURE 4.
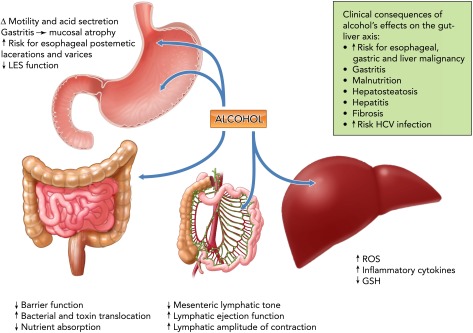
Alcohol and the gut-liver axis
Alcohol abuse produces marked alterations in the gastrointestinal tract. Esophageal and gastric dysmotility facilitate acid regurgitation and contribute to postemetic lacerations of the distal esophagus induced by vomiting (Mallory-Weiss Syndrome). Liver fibrosis and the resulting intrahepatic pressure increase leads to development of esophageal varicosities. Alcohol promotes chronic gastritis followed by decreased mucosal thickness and atrophy during the chronic phase. Chronic alcohol abuse impairs intestinal essential amino acid and vitamin absorption. In the liver, alcohol metabolism increases the production of ROS and lowers antioxidant levels, which contributes to liver injury. ROS generation leads to lipid peroxidation, alterations in plasma and intracellular membranes, and release of proinflammatory and profibrotic mediators. Alcohol and its metabolites disrupt intestinal barrier function by affecting the integrity of tight junctions, promoting the dissociation and redistributing proteins like ZO-1, claudin, and occludin. Increased paracellular permeability leads to increased bacterial toxin translocation from the gut lumen and disseminated to the systemic circulation via the portal vein and the lymphatic route. This later route of dissemination may be significant, since alcohol intoxication has been shown to promote lymphatic pumping. GSH, reduced glutathione; ROS, reactive oxygen species; HCV, hepatitis C virus; LES, lower esophageal sphincter. The potential clinical consequences of alcohol abuse and its impact on the endocrine system are shown in the box.
Alcohol and its metabolites increase intestinal epithelial permeability (60) through disruption of the integrity of tight junctions formed by transmembrane proteins (i.e., claudin, occludin, etc.) cross-linked to the actin cytoskeleton and intercellular signaling components by adaptor proteins (e.g., ZO-1/2/3, PATJ, PAR-3, and PAR-6) (11, 113). Alcohol-mediated disruption of the intestinal barrier integrity leads to gut bacterial toxin [i.e., lipopolysaccharide (LPS)] translocation and dissemination to the systemic circulation (44). The majority of LPS from the GI tract is delivered to the liver through the portal circulation, where only a minor portion escapes detoxification. In contrast, LPS delivered by the collecting lymphatic vessels directly enters the systemic circulation by the thoracic duct (12). Studies from our group have shown that acute alcohol intoxication impairs mesenteric collecting lymphatic pump function, decreasing mesenteric lymphatic tone and increasing lymphatic ejection fraction (110). Overall, these modifications appear to enhance the ability of lymphatics to transport lymph during the alcohol intoxicated state, which we predict would favor LPS lymphatic dissemination. Whether this altered lymphatic pumping persists following chronic alcohol exposure is not known. However, because transport of LPS through the lymphatic route escapes hepatic detoxification, a considerable amount of bioactive LPS could be delivered into the systemic circulation through this system (126) and may potentially contribute to the alcohol-associated LPS-induced tissue and organ inflammatory injury. Bacterial toxin translocation has been proposed as an important mechanism contributing to generalized inflammation and alcohol-induced liver disease characterized by fat accumulation, inflammation, and fibrosis (1). Although the contribution of alcohol-induced gut leakiness to systemic inflammation and liver injury has been well recognized, the contribution of lymphatic vs. portal delivery of LPS to the liver and extra-hepatic organs remains to be elucidated.
The liver, the principal organ involved in alcohol metabolism, is considered one of the main targets of alcohol-induced pathology. Liver disease is one of the most salient pathophysiological conditions resulting from alcohol abuse and a major cause of alcohol-related morbidity and mortality. Alcohol abuse is the principal cause of chronic liver disease in Western countries and afflicts ∼15% of alcoholics in the United States (71). Three mechanisms underlie the stages of alcoholic liver disease: hepatosteatosis, steatohepatitis, and cirrhosis. Although most heavy drinkers develop fatty liver, only 10–35% develop hepatitis and <20% progress to cirrhosis (49), suggesting the complexity of factors involved in alcoholic liver disease (ALD) progression.
Hepatosteatosis results from hepatocyte accumulation of cholesterol esters, phospholipids, and triglycerides (TG) due to alcohol-induced alterations in lipid metabolism. Among the mechanisms proposed are increased fatty acid mobilization from adipose tissues, increased hepatic lipid uptake, alteration of fat metabolism-associated transcription factors, and disruption of enzymes involved in fat metabolism. Chronic alcohol consumption results in progressive mitochondrial dysfunction characterized by decreased fatty acid oxidation leading to free fatty acid (FFA) accumulation (14). In the early stages of the disease, the liver adjusts to higher FFA levels to avoid TG accumulation but fails over time, leading to TG accumulation, steatosis, and lipotoxicity. The latter contribute to disruption of mitochondrial function and dysregulate numerous redox-sensitive signaling pathways, leading to apoptotic and necrotic cell death and additional TG accumulation (39). Steatosis is associated with an increased sensitivity to LPS, attributed to an imbalance of pro-inflammatory/oxidative and cytoprotective mechanisms (102). Steatosis is usually reversible with abstinence and sustained moderation of alcohol consumption, preventing the progression to chronic liver disease.
Steatohepatitis is characterized by hepatic infiltration of inflammatory cells, expression of pro-inflammatory cytokines, and oxidative injury resulting in hepatocellular impairment (46). Kupffer cells, liver-specific macrophages that play an important role in host defense mechanisms and have an essential role in maintaining liver homeostasis and preservation, have been implicated as being central players in liver damage induced by alcohol (31). Increased LPS delivery, resulting from a leaky intestinal barrier described above, can directly affect hepatocyte function but most importantly leads to Kupffer and hepatic stellate cell (HSC) activation, also resulting in hepatocyte injury (106, 113). LPS activation of Toll-like receptor 4 (TLR-4) leads to transcriptional activation and generation of potent innate immune responses (116), generation of pro-inflamatory cytokines and ROS, induction of apoptosis, and hepatocellular damage (113). However, the complexity of the etiology of alcoholic hepatitis is reflected in the failure of TNF-α antibody to improve outcomes in alcoholic hepatitis (120). In addition to activation of innate immunity by gut-derived LPS, alcohol abuse also has been shown to stimulate complement C3 and C5, which can in turn activate Kupffer cells (93). This pathway has been explored as a possible target for inducing anti-inflammatory and hepatoprotective cytokines to reduce alcohol-induced hepatocellular damage and to treat ALD (46). Activation of adaptive immunity also contributes to the pathogenesis of ALD. Elevated levels of IgG, T-lymphocytes, and antibodies against lipid peroxidation have been reported in patients with advanced ALD (5). Oxidative stress has also been implicated as a major factor in the development of ALD. Acetaldehyde impairs hepatocyte mitochondria functionality, and promotes lipid peroxidation and glutathione depletion (108), promoting oxidative stress and sensitizing the hepatocyte to oxidative injury (38). Nevertheless, clinical trials examining the benefit of antioxidant therapy for treatment of ALD have not shown improved outcomes targeting this system (43).
The progression of steatohepatitis to liver fibrosis involves excessive accumulation of extracellular matrix (ECM) proteins including collagen (84) and disruption of hepatic structure forming a fibrous scar with subsequent development of nodules of regenerating hepatocytes (15). Acetaldehyde forms adducts with proteins and DNA, impairing cellular function and gene expression (108). Proteins such as collagen are targeted by acetaldehyde, leading to adduct formation and induction of HSC collagen I synthesis, contributing to the onset and maintenance of fibrogenesis (23). Activation of HSCs, the main producers of ECM in the injured liver, is the result of gut-derived LPS (8), ROS, and cytokines released from neighboring Kupffer cells (39), and alcohol-mediated depletion of natural killer (NK) T-cells (58). NK cells induce HSCs apoptosis during liver fibrosis and thereby play an antifibrotic role. Once activated, HSCs not only increase ECM synthesis and deposition but also increase their own proliferation rates (45). In addition, ALD is associated with increased risk for hepatitis C virus infection, cirrhosis, and development of hepatocellular carcinoma (80).
Chronic heavy alcohol consumption is the most important risk factor for chronic pancreatitis (107, 130). The local metabolism of alcohol has been suggested to contribute to its toxic effects (9). The course of alcoholic pancreatitis is initiated as an acute process that with repeated episodes of acute injury promotes inflammation, acinar atrophy, and fibrosis, resulting in exocrine and endocrine dysfunction (10). Similar to the cellular mechanisms involved in alcoholic cirrhosis, pancreatic stellate cells become activated during alcohol oxidative metabolism and contribute to fibrogenic changes (124). The alcohol-associated risk for development of chronic pancreatitis is further exacerbated in smokers, contributing further to the overall risk of pancreatic cancer (130).
Alcohol and the Cardiopulmonary System
In contrast to the overall detrimental effects of alcohol on other organ systems, evidence indicates that low to moderate alcohol consumption is associated with a lower risk of coronary heart disease (30). In contrast, chronic heavy alcohol use increases risk for cardiovascular and pulmonary disease (125), including hypertension and non-ischemic dilated alcoholic cardiomyopathy (ADC) characterized by reduced ejection fraction, left ventricular dilation, and extensive interstitial cardiac fibrosis (87). Moreover, chronic alcohol abuse can exacerbate cardiac injury resulting from myocardial infarction, diabetes, hypertension, or pressure overload (4, 112). Several mechanisms have been proposed to contribute to alcohol-induced myocardial dysfunction, including oxidative stress, cardiomyocyte mitochondrial and sarcoplasmic reticulum damage, altered calcium dynamics, and cardiac fibrosis.
Cardiomyocyte damage resulting from chronic alcohol abuse is mediated by multiple mechanisms, including oxidative stress, alterations in calcium handling, and mitochondrial dysfunction. Alcohol abuse induces myocardial oxidative stress (89, 123) and depletes mitochondrial GSH, decreasing antioxidant capacity and enhancing myocyte susceptibility to oxidant injury and apoptosis (123). Alcohol-induced GSH depletion is not cardiac specific and is seen in liver and lung (40). Alcohol abuse disrupts cardiomyocyte contraction by damaging contractile proteins and interfering with calcium signaling and homeostasis through upregulation of L-type calcium channel expression and function, which can promote calcium overloading (51) and result in cardiomyocyte apoptosis and necrosis. Moreover, chronic alcohol abuse reduces myofiber calcium sensitivity and alters cellular calcium transients, resulting in reduced contractile function (88). Alcohol-mediated alterations in calcium handling have been implicated in sudden cardiac death and cardiac arrhythmias caused by binge drinking (65). Acetaldehyde has also been implicated as a causal factor in ethanol-induced cardiomyocyte damage by inhibiting calcium ATPases, leading to impaired excitation-contraction coupling and mitochondrial and sarcoplasmic reticulum toxicity (96).
The development of cardiac fibrosis appears to be a key mechanism of ADC dysfunction and is manifested in the initial stages as cardiac diastolic dysfunction (87). The extensive cardiac fibrosis impairs ventricular filling by decreasing ventricular compliance. Oxidative stress is considered one of the principal mechanisms underlying alcohol-induced fibrosis. Oxidative stress stimulates the production of collagen by fibrogenic cells, including cardiac fibroblasts, leading to both interstitial and perivascular fibrosis (89). Studies from our group have shown that alcohol-induced transformation of fibroblasts to myofibroblasts results in excess deposition of collagen (36). In addition to the activation of fibrogenic cells, alcohol may promote fibrosis indirectly through cardiomyocyte apoptosis or necrosis and their replacement by collagen.
Cessation or decreased alcohol consumption is associated with a reduction in blood pressure in hypertensive patients (129). The mechanisms of alcohol-mediated hypertension include potentiation of the renin-angiotensin-aldosterone system (RAAS) (27). This is reflected in significantly elevated circulating angiotensin II levels (27), elevated cardiac angiotensin converting enzyme expression (63), and increased cardiac expression of angiotensin type 1 (AT1) receptors (27). AT1 receptors have been implicated in ventricular dysfunction, elevations of end-diastolic pressure, and alcohol-induced vascular injury (13, 27). Signaling through the AT2 receptor antagonizes the effects of AT1 signaling. Its contribution to alcohol-induced modulation of blood pressure is debatable (27).
As in the heart and other organs, chronic alcohol abuse causes oxidative injury of the lungs. Alcohol decreases GSH, which leaves pulmonary cells susceptible to oxidative stress injury (50). As mentioned above, alcohol activates the RAAS, which promotes superoxide production by NADPH-oxidases in the lung (90). Chronic alcohol also interferes with the production of pulmonary surfactant by disrupting the composition of dipalmitoyl-lecithin (50). As a result, susceptible alveolar type II cells are lost to oxidative stress-induced apoptosis and necrosis, which reduces barrier function and increases alveolar-capillary permeability.
Alcohol abuse is a well established risk factor for acute respiratory distress syndrome (ARDS) and pneumonia (41, 79). Alcohol negatively affects the pulmonary response to injury, infection, and inflammation, and increases ARDS susceptibility and mortality almost twofold over that of non-alcoholic patients (79). This striking difference appears to be due to the adverse effects of ethanol on the lung's ability to respond to bacterial and inflammatory insult (41). The mechanisms responsible for alcohol-mediated impaired immune response to infection include altered balance between proinflammatory and anti-inflammatory cytokines by alveolar macrophages (131), impaired neutrophil function including phagocytosis and chemotaxis (21), attenuation of granulocyte-macrophage colony-stimulating factor (GM-CSF) release (81), and impaired airway ciliary function (128). The net result of these effects is suppression of the pulmonary immune response (76) (FIGURE 5).
FIGURE 5.
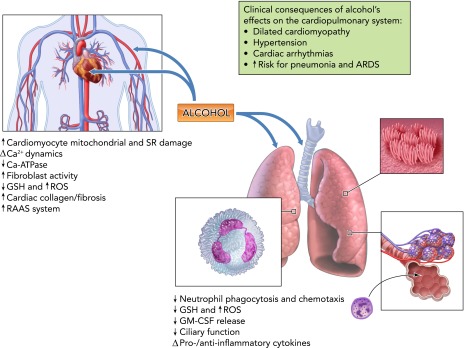
Alcohol and the cardiopulmonary system
Chronic alcohol abuse increases risk for cardiovascular and pulmonary disease. Alcohol-induced myocardial dysfunction results from oxidative stress, cardiomyocyte mitochondrial and sarcoplasmic reticulum damage, altered calcium dynamics, and cardiac fibrosis. Alcohol abuse induces myocardial oxidative stress and depletes mitochondrial GSH, decreasing antioxidant capacity and enhancing myocyte susceptibility to oxidant injury and apoptosis, and in addition disrupts cardiomyocyte contraction by damaging contractile proteins and interfering with calcium signaling and homeostasis through upregulation of L-type calcium channel expression and function. Alcohol-mediated hypertension results from potentiation of the RAAS reflected in elevated circulating angiotensin II levels, cardiac angiotensin converting enzyme, and angiotensin type 1 (AT1) receptor expression. Chronic alcohol abuse produces marked alterations in pulmonary function resulting from decreased GSH, increased ROS production, and marked alterations in lung host defense mechanisms. GM-CSF, granulocyte-macrophage colony-stimulating factor; RAAS, renin-angiotensin-aldosterone system; ARDS, acute respiratory distress syndrome. The most relevant clinical manifestations of alcohol-induced alterations in cardiopulmonary function are shown in the box.
Impact of Alcohol on Body Composition
In addition to the well recognized effects on nutritional state of the individual, chronic alcohol abuse disrupts multiple factors involved in the balance between anabolic and catabolic mechanisms. Alcohol abuse is associated with an ∼50% incidence of skeletal muscle myopathy (92), which is greater than the incidence of alcoholic cirrhosis (10–15%) in chronic alcoholics (46). Alcoholic myopathy has been shown to be the result of decreased muscle protein synthesis (85) and accelerated muscle proteolysis (117). Alcohol can alter the nutritional state of the individual either by decreasing food consumption and/or by producing malabsorption, resulting in decreased micronutrient availability and consequently modulation of circulating and tissue growth factors (75). Chronic alcohol abuse has been shown to increase whole body proteolysis and rates of amino acid oxidation (17), and to decrease the rate of skeletal muscle protein synthesis (91). Both capacity of muscle protein synthesis and translational efficiency are impaired by alcohol. In addition to adversely affecting multiple sites involved in regulating translational efficiency (66), chronic alcohol results in decreased circulating and tissue levels of androgens and IGF-I (69), upregulation of myostatin, a negative regulator of skeletal muscle growth (119), and increased proteolysis through the ubiquitin proteasome pathway (122). The detrimental impact of chronic alcohol abuse on skeletal muscle mass balance is further accentuated in diseased states. Studies from our group have demonstrated that chronic alcohol consumption markedly exacerbates the end-stage muscle wasting in a simian immunodeficiency virus-infected rhesus macaques (78), and this is the result of an inflammatory, oxidative milieu that promotes dysregulation of the ubiquitin proteasome pathway (67).
Alcohol and Bone
Alcohol abuse is associated with altered bone metabolism, decreased bone mineral density and mass (20, 105), and increased risk of fractures (16) despite lack of liver failure. Overall, the prevalence of osteoporosis in alcoholics has been estimated at >40% (111). Both direct and indirect mechanisms are likely factors in decreased bone health in chronic alcoholics (64). Among the most important factors regulating bone, adipose, and skeletal muscle mass are the anabolic hormones, particularly testosterone. Androgens play a critical role in the control of bone remodeling by suppressing osteoclastogenesis and promoting osteoblastogenesis (121), as well as by antagonizing the effect of pro-inflammatory cytokines (101) and promoting osteoprotegerin synthesis, reducing the activation and maturation of osteoclasts (55). The attenuation of the HPG axis in chronic alcoholics and the associated decreased circulating levels of testosterone (37) likely aggravates the pro-inflammatory cytokine's impact on bone loss (64). The detrimental impact of alcohol on bone health is not limited to an increased risk for osteoporotic fractures and delayed fracture repair. It is compounded by the greater risk for falls during acute alcohol intoxication (resulting from altered gait and balance), coupled with alcoholic myopathy and decreased bone mineral content and density, which can collectively significantly aggravate the burden of bone disease in this population (59).
Alcohol and Adipose Tissue
The impact of chronic alcohol abuse on adipose tissue mass and phenotype has not been investigated in a systematic way. Studies have reported that chronic alcohol consumption is associated with decreased fat mass (2), and this has been attributed to altered neuroendocrine function resulting in increased cortisol release (68). In contrast, others have reported a high incidence of dyslipidemia and increased fat mass in alcoholics, with >20% of patients meeting criteria for metabolic syndrome (57) (FIGURE 6). However, some recent studies suggest that alcohol may not only alter fat mass but, in addition, disrupt adipokine profiles such as that of leptin (82) and adiponectin (100). These alterations in adipose tissue phenotype are likely to result in marked metabolic dysregulation, favoring an insulin-resistant state. These findings raise the question of whether chronic alcohol abuse by obese individuals may further contribute to risk of metabolic syndrome. Based on the known effects of chronic alcohol abuse, we predict that a significantly greater metabolic disregulation would prevail, further disrupting glycemic control and possibly enhancing the risk of liver disease.
FIGURE 6.
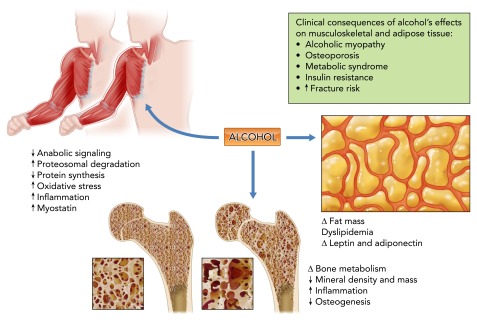
Alcohol and the musculoskeletal and adipose tissues
Chronic alcohol abuse disrupts multiple factors involved the balance between anabolic and catabolic mechanisms in bone and muscle. The underlying mechanisms include nutritional deficiencies, decreased growth factor availability and responsiveness, increased ubiquitin proteasome pathway activation, upregulation of negative regulators of skeletal muscle growth, and disruption of bone remodeling. Chronic alcohol abuse produces marked alterations in adipocyte function, resulting in fat mass redistribution, dyslipidemia, and altered pattern of adipokine release. The potential clinical implications of alcohol's effects on skeletal muscle, bone, and adipose tissue are summarized in the box.
Perspectives and Translational Implications
The systemic impact of alcohol abuse is reflected in the greater incidence of significant comorbid conditions, with a significant disease-related burden that spans all ages in alcohol-abusing individuals. The increasingly recognized relationships between chronic alcohol abuse and incidence of diabetes mellitus, hypertension, ischemic heart disease, dysrhythmias, stroke, and pneumonia, adds to the previously recognized risk for liver disease and fetal alcohol syndrome. The contribution of alcohol metabolism to organ injury is not trivial, as seen by the marked disruption in cellular processes resulting from ROS generation and altered cellular redox state. Among the critical pathophysiological mechanisms underlying the most frequent comorbid conditions are inflammation and oxidative stress. Oxidative stress resulting from either an excess production of ROS or a reduction in reducing antioxidant equivalents has been consistently demonstrated to be an overall mechanism of tissue injury resulting from chronic alcohol abuse (25, 40). Although acute alcohol intoxication has been consistently seen to reduce inflammation in response to infectious challenges, chronic alcohol consumption favors a pro-inflammatory milieu that plays an important role in tissue injury (114).
Overall, much remains to be learned regarding the mechanisms of alcohol-induced tissue injury, the possibility of reversibility, and ultimately the effectiveness of behavioral and pharmacological interventions to ameliorate alcohol abuse and its untoward consequences. Abstinence and nutritional therapy are still the first choice of intervention for ALD. Anti-inflammatory drugs, including corticosteroids, pentoxifylline (32), TLR-4 antagonists (72), and interleukin-22 (IL-22; an anti-inflammatory cytokine) (62), have failed to result in consistent improvement for ALD but may still confer protection from injury of other organs. Novel approaches in the treatment of ALD that remain to be explored include modulation of gut microbiota (42). In addition to measures aimed at reducing cardiac workload, ALDH type 2 has recently been proposed as a viable therapeutic target for alcoholic cardiomyopathy. Novel studies suggest that overexpression or activation of ALDH type 2 prevents alcohol-mediated cardiac dilation, dysfunction, and fibrosis (34, 47), and reduces ischemia-reperfusion injury (26). N-acetyl cysteine and GM-CSF administration and inhibition of the RAAS are identified as potential therapies for reducing alcohol-induced pulmonary injury and associated ARDS (18). However, these approaches remain to be proven effective in large clinical trials. The annual costs ($235 billion) to our nation related to crime, lost work productivity, and healthcare of alcohol abuse is greater than that of tobacco ($193 billion) or illicit drugs ($193 billion) (80a). The alcohol-attributable disease burden is closely related to the average volume of alcohol consumption and particularly affects disadvantaged subgroups of the population. Prevention of alcohol use disorders can effectively curtail this healthcare burden. Other than cessation or significant decrease in alcohol consumption, there is no specific treatment for alcohol-related comorbid conditions.
Footnotes
The authors are grateful for editorial support from Betsy Giaimo and research support provided by the National Institute on Alcohol Abuse and Alcoholism (AA-07577, AA-09803, UAA-021995A, and AA-11290, AA-021049).
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions: P.E.M., J.D.G., F.M.S.-S., and A.M.W. conception and design of research; P.E.M. analyzed data; P.E.M., J.D.G., F.M.S.-S., and A.M.W. prepared figures; P.E.M., J.D.G., F.M.S.-S., and A.M.W. drafted manuscript; P.E.M., J.D.G., F.M.S.-S., and A.M.W. edited and revised manuscript; P.E.M., J.D.G., F.M.S.-S., and A.M.W. approved final version of manuscript.
References
- 1.Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108: 218–224, 1995 [DOI] [PubMed] [Google Scholar]
- 2.Addolorato G, Capristo E, Marini M, Santini P, Scognamiglio U, Attilia ML, Messineo D, Sasso GF, Gasbarrini G, Ceccanti M. Body composition changes induced by chronic ethanol abuse: evaluation by dual energy X-ray absorptiometry. Am J Gastroenterol 95: 2323–2327, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Adinoff B, Junghanns K, Kiefer F, Krishnan-Sarin S. Suppression of the HPA axis stress-response: implications for relapse. Alcohol Clin Exp Res 29: 1351–1355, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguilar D, Skali H, Moyé LA, Lewis EF, Gaziano JM, Rutherford JD, Hartley LH, Randall OS, Geltman EM, Lamas GA, Rouleau JL, Pfeffer MA, Solomon SD. Alcohol consumption and prognosis in patients with left ventricular systolic dysfunction after a myocardial infarction. J Am Coll Cardiol 43: 2015–2021, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Albano E, Vidali M. Immune mechanisms in alcoholic liver disease. Genes Nutr 5: 141–147, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aoyama T, Paik YH, Seki E. Toll-like receptor signaling and liver fibrosis. Gastroenterol Res Pract 2010: 192543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Apte MV, Pirola RC, Wilson JS. Molecular mechanisms of alcoholic pancreatitis. Dig Dis 23: 232–240, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Apte MV, Wilson JS. Alcohol-induced pancreatic injury. Best Pract Res Clin Gastroenterol 17: 593–612, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Atkinson KJ, Rao RK. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am J Physiol Gastrointest Liver Physiol 280: G1280–G1288, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Azuma K, Akiyama M, Ebata T, Totsuka M, Hayasaka H. Endogenous endotoxin absorption and the role of intestinal lymphatics. Jpn J Surg 13: 535–539, 1983 [DOI] [PubMed] [Google Scholar]
- 13.Bai Y, Tan Y, Wang B, Miao X, Chen Q, Zheng Y, Cai L. Deletion of angiotensin II type 1 receptor gene or scavenge of superoxide prevents chronic alcohol-induced aortic damage and remodelling. J Cell Mol Med 16: 2530–2538, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baraona E, Lieber CS. Effects of ethanol on lipid metabolism. J Lipid Res 20: 289–315, 1979 [PubMed] [Google Scholar]
- 15.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 115: 209–218, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg KM, Kunins HV, Jackson JL, Nahvi S, Chaudhry A, Harris KA, Malik R, Arnsten JH. Association between alcohol consumption and both osteoporotic fracture and bone density. Am J Med 121: 406–418, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernal CA, Vazquez JA, Adibi SA. Leucine metabolism during chronic ethanol consumption. Metabolism 42: 1084–1086, 1993 [DOI] [PubMed] [Google Scholar]
- 18.Bernard GR, Wheeler AP, Arons MM, Morris PE, Paz HL, Russell JA, Wright PE. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest 112: 164–172, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Besemer F, Pereira AM, Smit JW. Alcohol-induced Cushing syndrome. Hypercortisolism caused by alcohol abuse. Neth J Med 69: 318–323, 2011 [PubMed] [Google Scholar]
- 20.Bikle DD. Alcohol-induced bone disease. World Rev Nutr Diet 73: 53–73, 1973 [DOI] [PubMed] [Google Scholar]
- 21.Boé DM, Nelson S, Zhang P, Bagby GJ. Acute ethanol intoxication suppresses lung chemokine production following infection with Streptococcus pneumoniae. J Infect Dis 184: 1134–1142, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Brieger K, Schiavone S, Miller FJ, Krause KH. Reactive oxygen species: from health to disease. Swiss Med Wkly 142: w13659, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Casini A, Cunningham M, Rojkind M, Lieber CS. Acetaldehyde increases procollagen type I and fibronectin gene transcription in cultured rat fat-storing cells through a protein synthesis-dependent mechanism. Hepatology 13: 758–765, 1991 [PubMed] [Google Scholar]
- 24.Cederbaum AI. Alcohol metabolism. Clin Liver Dis 16: 667–685, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cederbaum AI. Introduction-serial review: alcohol, oxidative stress and cell injury. Free Radic Biol Med 31: 1524–1526, 2001 [DOI] [PubMed] [Google Scholar]
- 25a.Center for Disease Control. Summary Health Statistics for U.S. Adults: National Health Interview Survey, table 27 (Online) Atlanta, GA: Center for Disease Control; http://www.cdc.gov/nchs/fastats/alcohol.htm, 2011 [Google Scholar]
- 26.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321: 1493–1495, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng CP, Cheng HJ, Cunningham C, Shihabi ZK, Sane DC, Wannenburg T, Little WC. Angiotensin II type 1 receptor blockade prevents alcoholic cardiomyopathy. Circulation 114: 226–236, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Cicero TJ. Neuroendocrinological effects of alcohol. Annu Rev Med 32: 123–142, 1981 [DOI] [PubMed] [Google Scholar]
- 29.Collins GB, Brosnihan KB, Zuti RA, Messina M, Gupta MK. Neuroendocrine, fluid balance, and thirst responses to alcohol in alcoholics. Alcohol Clin Exp Res 16: 228–233, 1992 [DOI] [PubMed] [Google Scholar]
- 30.Costanzo S, Di Castelnuovo A, Donati MB, Iacoviello L, de Gaetano G. Alcohol consumption and mortality in patients with cardiovascular disease: a meta-analysis. J Am Coll Cardiol 55: 1339–1347, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Cubero FJ, Nieto N. Kupffer cells and alcoholic liver disease. Rev Esp Enferm Dig 98: 460–472, 2006 [DOI] [PubMed] [Google Scholar]
- 32.De BK, Gangopadhyay S, Dutta D, Baksi SD, Pani A, Ghosh P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol 15: 1613–1619, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dees WL, Skelley CW, Hiney JK, Johnston CA. Actions of ethanol on hypothalamic and pituitary hormones in prepubertal female rats. Alcohol 7: 21–25, 1990 [DOI] [PubMed] [Google Scholar]
- 34.Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation 119: 1941–1949, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Döring WK, Herzenstiel MN, Krampe H, Jahn H, Pralle L, Sieg S, Wegerle E, Poser W, Ehrenreich H. Persistent alterations of vasopressin and N-terminal proatrial natriuretic peptide plasma levels in long-term abstinent alcoholics. Alcohol Clin Exp Res 27: 849–861, 2003 [DOI] [PubMed] [Google Scholar]
- 36.El Hajj EC, El Hajj MC, Voloshenyuk TG, Mouton AJ, Khoutorova E, Molina PE, Gilpin NW, Gardner JD. Alcohol modulation of cardiac matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs favors collagen accumulation. Alcohol Clin Exp Res 38: 448–456, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emanuele MA, Emanuele N. Alcohol and the male reproductive system. Alcohol Res Health 25: 282–287, 2001 [PMC free article] [PubMed] [Google Scholar]
- 38.Farfán Labonne BE, Gutiérrez M, Gómez-Quiroz LE, Konigsberg Fainstein M, Bucio L, Souza V, Flores O, Ortíz V, Hernández E, Kershenobich D, Gutiérrez-Ruíz MC. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol Toxicol 25: 599–609, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Feldstein AE, Bailey SM. Emerging role of redox dysregulation in alcoholic and nonalcoholic fatty liver disease. Antioxid Redox Signal 15: 421–424, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández-Checa JC, Kaplowitz N, Colell A, García-Ruiz C. Oxidative stress and alcoholic liver disease. Alcohol Health Res World 21: 321–324, 1997 [PMC free article] [PubMed] [Google Scholar]
- 41.Fernández-Solá J, Junqué A, Estruch R, Monforte R, Torres A, Urbano-Márquez A. High alcohol intake as a risk and prognostic factor for community-acquired pneumonia. Arch Intern Med 155: 1649–1654, 1995 [DOI] [PubMed] [Google Scholar]
- 42.Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol 43: 163–172, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frazier TH, Stocker AM, Kershner NA, Marsano LS, McClain CJ. Treatment of alcoholic liver disease. Therap Adv Gastroenterol 4: 63–81, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol 12: 162–169, 1991 [DOI] [PubMed] [Google Scholar]
- 45.Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, Casini A. Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells. Hepatology 31: 101–108, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141: 1572–1585, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ge W, Guo R, Ren J. AMP-dependent kinase and autophagic flux are involved in aldehyde dehydrogenase-2-induced protection against cardiac toxicity of ethanol. Free Radic Biol Med 51: 1736–1748, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res 32: 1535–1542, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gramenzi A, Caputo F, Biselli M, Kuria F, Loggi E, Andreone P, Bernardi M. Review article: alcoholic liver disease–pathophysiological aspects and risk factors. Aliment Pharmacol Ther 24: 1151–1161, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Guidot DM, Roman J. Chronic ethanol ingestion increases susceptibility to acute lung injury: role of oxidative stress and tissue remodeling. Chest 122: 309S–314S, 2002 [DOI] [PubMed] [Google Scholar]
- 51.Guppy LJ, Crabbe JC, Littleton JM. Time course and genetic variation in the regulation of calcium channel antagonist binding sites in rodent tissues during the induction of ethanol physical dependence and withdrawal. Alcohol Alcohol 30: 607–615, 1995 [PubMed] [Google Scholar]
- 52.Hasin DS, Stinson FS, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry 64: 830–842, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Hegedüs L. Thyroid size determined by ultrasound. Influence of physiological factors and non-thyroidal disease. Dan Med Bull 37: 249–263, 1990 [PubMed] [Google Scholar]
- 54.Hermann D, Heinz A, Mann K. Dysregulation of the hypothalamic-pituitary-thyroid axis in alcoholism. Addiction 97: 1369–1381, 2002 [DOI] [PubMed] [Google Scholar]
- 55.Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res 15: 2–12, 2000 [DOI] [PubMed] [Google Scholar]
- 56.Israel Y, Walfish PG, Orrego H, Blake J, Kalant H. Thyroid hormones in alcoholic liver disease. Effect of treatment with 6-n-propylthiouracil. Gastroenterology 76: 116–122, 1979 [PubMed] [Google Scholar]
- 57.Jarvis CM, Hayman LL, Braun LT, Schwertz DW, Ferrans CE, Piano MR. Cardiovascular risk factors and metabolic syndrome in alcohol- and nicotine-dependent men and women. J Cardiovasc Nurs 22: 429–435, 2007 [DOI] [PubMed] [Google Scholar]
- 58.Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology 134: 248–258, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kelly KN, Kelly C. Pattern and cause of fractures in patients who abuse alcohol: what should we do about it? Postgrad Med J 89: 578–583, 2013 [DOI] [PubMed] [Google Scholar]
- 60.Keshavarzian A, Fields JZ, Vaeth J, Holmes EW. The differing effects of acute and chronic alcohol on gastric and intestinal permeability. Am J Gastroenterol 89: 2205–2211, 1994 [PubMed] [Google Scholar]
- 61.Khanna JM, Israel Y. Ethanol metabolism. Int Rev Physiol 21: 275–315, 1980 [PubMed] [Google Scholar]
- 62.Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, Gao B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology 52: 1291–1300, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim SD, Beck J, Bieniarz T, Schumacher A, Piano MR. A rodent model of alcoholic heart muscle disease and its evaluation by echocardiography. Alcohol Clin Exp Res 25: 457–463, 2001 [PubMed] [Google Scholar]
- 64.Kimble RB. Alcohol, cytokines, and estrogen in the control of bone remodeling. Alcohol Clin Exp Res 21: 385–391, 1997 [DOI] [PubMed] [Google Scholar]
- 65.Kodama S, Saito K, Tanaka S, Horikawa C, Saito A, Heianza Y, Anasako Y, Nishigaki Y, Yachi Y, Iida KT, Ohashi Y, Yamada N, Sone H. Alcohol consumption and risk of atrial fibrillation: a meta-analysis. J Am Coll Cardiol 57: 427–436, 2011 [DOI] [PubMed] [Google Scholar]
- 66.Lang CH, Frost RA, Kumar V, Wu D, Vary TC. Impaired protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4E in muscle and eIF2B in liver. Alcohol Clin Exp Res 24: 322–331, 2000 [PubMed] [Google Scholar]
- 67.LeCapitaine NJ, Wang ZQ, Dufour JP, Potter BJ, Bagby GJ, Nelson S, Cefalu WT, Molina PE. Disrupted anabolic and catabolic processes may contribute to alcohol-accentuated SAIDS-associated wasting. J Infect Dis 204: 1246–1255, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leggio L, Malandrino N, Ferrulli A, Cardone S, Miceli A, Gasbarrini G, Capristo E, Addolorato G. Is cortisol involved in the alcohol-related fat mass impairment? A longitudinal clinical study. Alcohol Alcohol 44: 211–215, 2009 [DOI] [PubMed] [Google Scholar]
- 69.Lester R, Van Thiel DH. Gonadal function in chronic alcoholic men. Adv Exp Med Biol 85A: 399–413, 1977 [DOI] [PubMed] [Google Scholar]
- 70.Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health 27: 220–231, 2003 [PMC free article] [PubMed] [Google Scholar]
- 71.Mann RE, Smart RG, Govoni R. The epidemiology of alcoholic liver disease. Alcohol Res Health 27: 209–219, 2003 [PMC free article] [PubMed] [Google Scholar]
- 72.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut 58: 704–720, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Michel L, Serrano A, Malt RA. Mallory-Weiss syndrome. Evolution of diagnostic and therapeutic patterns over two decades. Ann Surg 192: 716–721, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA 291: 1238–1245, 2004 [DOI] [PubMed] [Google Scholar]
- 75.Molina P, Fan J, Gelato M, Lang C, Abumrad N. Modulation of insulin-like growth factor-I: A specific role for vitamin B1 (thiamine). J Nutr Biochem 7: 207–213, 1996 [Google Scholar]
- 76.Molina PE, Happel KI, Zhang P, Kolls JK, Nelson S. Focus on: alcohol and the immune system. Alcohol Res Health 33: 97–108, 2010 [PMC free article] [PubMed] [Google Scholar]
- 77.Molina PE, Hoek JB, Nelson S, Guidot DM, Lang CH, Wands JR, Crawford JM. Mechanisms of alcohol-induced tissue injury. Alcohol Clin Exp Res 27: 563–575, 2003 [DOI] [PubMed] [Google Scholar]
- 78.Molina PE, McNurlan M, Rathmacher J, Lang CH, Zambell KL, Purcell J, Bohm RP, Zhang P, Bagby GJ, Nelson S. Chronic alcohol accentuates nutritional, metabolic, and immune alterations during asymptomatic simian immunodeficiency virus infection. Alcohol Clin Exp Res 30: 2065–2078, 2006 [DOI] [PubMed] [Google Scholar]
- 79.Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA 275: 50–54, 1996 [PubMed] [Google Scholar]
- 80.Mueller S, Millonig G, Seitz HK. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol 15: 3462–3471, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80a.National Institute on Drug Abuse. Trends and Statistics; Cost of Substance Abuse (Online) Bethesda, MD: National Inst. on Drug Abuse: http://www.drugabuse.gov/related-topics/trends-statistics, 2012 [Google Scholar]
- 81.Nelson S, Summer W, Bagby G, Nakamura C, Stewart L, Lipscomb G, Andresen J. Granulocyte colony-stimulating factor enhances pulmonary host defenses in normal and ethanol-treated rats. J Infect Dis 164: 901–906, 1991 [DOI] [PubMed] [Google Scholar]
- 82.Nicolás JM, Fernández-Solà J, Fatjó F, Casamitjana R, Bataller R, Sacanella E, Tobías E, Badía E, Estruch R. Increased circulating leptin levels in chronic alcoholism. Alcohol Clin Exp Res 25: 83–88, 2001 [PubMed] [Google Scholar]
- 83.Nomura S, Pittman CS, Chambers JB, Buck MW, Shimizu T. Reduced peripheral conversion of thyroxine to triiodothyronine in patients with hepatic cirrhosis. J Clin Invest 56: 643–652, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orman ES, Odena G, Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol 28, Suppl 1: 77–84, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pacy PJ, Preedy VR, Peters TJ, Read M, Halliday D. The effect of chronic alcohol ingestion on whole body and muscle protein synthesis: a stable isotope study. Alcohol Alcohol 26: 505–513, 1991 [DOI] [PubMed] [Google Scholar]
- 86.Parry CD, Patra J, Rehm J. Alcohol consumption and non-communicable diseases: epidemiology and policy implications. Addiction 106: 1718–1724, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Piano MR. Alcoholic cardiomyopathy: incidence, clinical characteristics, pathophysiology. Chest 121: 1638–1650, 2002 [DOI] [PubMed] [Google Scholar]
- 88.Piano MR, Rosenblum C, Solaro RJ, Schwertz D. Calcium sensitivity and the effect of the calcium sensitizing drug pimobendan in the alcoholic isolated rat atrium. J Cardiovasc Pharmacol 33: 237–242, 1999 [DOI] [PubMed] [Google Scholar]
- 89.Poli G, Parola M. Oxidative damage and fibrogenesis. Free Radic Biol Med 22: 287–305, 1997 [DOI] [PubMed] [Google Scholar]
- 90.Polikandriotis JA, Rupnow HL, Elms SC, Clempus RE, Campbell DJ, Sutliff RL, Brown LA, Guidot DM, Hart CM. Chronic ethanol ingestion increases superoxide production and NADPH oxidase expression in the lung. Am J Respir Cell Mol Biol 34: 314–319, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Preedy VR, Peters TJ, Patel VB, Miell JP. Chronic alcoholic myopathy: transcription and translational alterations. FASEB J 8: 1146–1151, 1994 [DOI] [PubMed] [Google Scholar]
- 92.Preedy VR, Salisbury JR, Peters TJ. Alcoholic muscle disease: features and mechanisms. J Pathol 173: 309–315, 1994 [DOI] [PubMed] [Google Scholar]
- 93.Pritchard MT, McMullen MR, Stavitsky AB, Cohen JI, Lin F, Medof ME, Nagy LE. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology 132: 1117–1126, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raskin NH, Sokoloff L. Enzymes catalysing ethanol metabolism in neural and somatic tissues of the rat. J Neurochem 19: 273–282, 1972 [DOI] [PubMed] [Google Scholar]
- 95.Rehm J, Baliunas D, Borges GL, Graham K, Irving H, Kehoe T, Parry CD, Patra J, Popova S, Poznyak V, Roerecke M, Room R, Samokhvalov AV, Taylor B. The relation between different dimensions of alcohol consumption and burden of disease: an overview. Addiction 105: 817–843, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ren J, Davidoff AJ, Brown RA. Acetaldehyde depresses shortening and intracellular Ca2+ transients in adult rat ventricular myocytes. Cell Mol Biol (Noisy-le-grand) 43: 825–834, 1997 [PubMed] [Google Scholar]
- 97.Richardson HN, Lee SY, O'Dell LE, Koob GF, Rivier CL. Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci 28: 1641–1653, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Riveros-Rosas H, Julian-Sanchez A, Pinã E. Enzymology of ethanol and acetaldehyde metabolism in mammals. Arch Med Res 28: 453–471, 1997 [PubMed] [Google Scholar]
- 99.Roberts DM. Chronic gastritis, alcohol, and non-ulcer dyspepsia. Gut 13: 768–774, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rogers CQ, Ajmo JM, You M. Adiponectin and alcoholic fatty liver disease. IUBMB Life 60: 790–797, 2008 [DOI] [PubMed] [Google Scholar]
- 101.Romas E, Gillespie MT, Martin TJ. Involvement of receptor activator of NFkappaB ligand and tumor necrosis factor-alpha in bone destruction in rheumatoid arthritis. Bone 30: 340–346, 2002 [DOI] [PubMed] [Google Scholar]
- 102.Romics L, Mandrekar P, Kodys K, Velayudham A, Drechsler Y, Dolganiuc A, Szabo G. Increased lipopolysaccharide sensitivity in alcoholic fatty livers is independent of leptin deficiency and toll-like receptor 4 (TLR4) or TLR2 mRNA expression. Alcohol Clin Exp Res 29: 1018–1026, 2005 [DOI] [PubMed] [Google Scholar]
- 103.Saitz R. Clinical practice. Unhealthy alcohol use. N Engl J Med 352: 596–607, 2005 [DOI] [PubMed] [Google Scholar]
- 104.Sampson HW, Perks N, Champney TH, DeFee B. Alcohol consumption inhibits bone growth and development in young actively growing rats. Alcohol Clin Exp Res 20: 1375–1384, 1996 [DOI] [PubMed] [Google Scholar]
- 105.Saville PD. Alcohol-related skeletal disorders. Ann NY Acad Sci 252: 287–291, 1975 [DOI] [PubMed] [Google Scholar]
- 106.Schaffert CS, Duryee MJ, Hunter CD, Hamilton BC, DeVeney AL, Huerter MM, Klassen LW, Thiele GM. Alcohol metabolites and lipopolysaccharide: roles in the development and/or progression of alcoholic liver disease. World J Gastroenterol 15: 1209–1218, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schneider A, Singer MV. Alcoholic pancreatitis. Dig Dis 23: 222–231, 2005 [DOI] [PubMed] [Google Scholar]
- 108.Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev 3: 178–185, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Soszynski PA, Frohman LA. Inhibitory effects of ethanol on the growth hormone (GH)-releasing hormone-GH-insulin-like growth factor-I axis in the rat. Endocrinology 131: 2603–2608, 1992 [DOI] [PubMed] [Google Scholar]
- 110.Souza-Smith FM, Kurtz KM, Molina PE, Breslin JW. Adaptation of mesenteric collecting lymphatic pump function following acute alcohol intoxication. Microcirculation 17: 514–524, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Spencer H, Rubio N, Rubio E, Indreika M, Seitam A. Chronic alcoholism. Frequently overlooked cause of osteoporosis in men. Am J Med 80: 393–397, 1986 [DOI] [PubMed] [Google Scholar]
- 112.Stranges S, Wu T, Dorn JM, Freudenheim JL, Muti P, Farinaro E, Russell M, Nochajski TH, Trevisan M. Relationship of alcohol drinking pattern to risk of hypertension: a population-based study. Hypertension 44: 813–819, 2004 [DOI] [PubMed] [Google Scholar]
- 113.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol 16: 1321–1329, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Szabo G, Mandrekar P, Oak S, Mayerle J. Effect of ethanol on inflammatory responses. Implications for pancreatitis. Pancreatology 7: 115–123, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taivainen H, Laitinen K, Tähtelä R, Kilanmaa K, Välimäki MJ. Role of plasma vasopressin in changes of water balance accompanying acute alcohol intoxication. Alcohol Clin Exp Res 19: 759–762, 1995 [DOI] [PubMed] [Google Scholar]
- 116.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 17: 1–14, 2005 [DOI] [PubMed] [Google Scholar]
- 117.Teschner M, Schaefer RM, Weissinger F, Kulzer P, Duelk MJ, Peter G, Heidland A. Chronic ethanol ingestion enhances catabolism and muscle protease activity in acutely uremic rats. Nephron 50: 338–344, 1988 [DOI] [PubMed] [Google Scholar]
- 118.Teyssen S, González-Calero G, Korn A, Singer MV. Action of ethanol and some alcoholic beverages on gastric acid secretion in anaesthetized rats. Alcohol Alcohol 32: 23–31, 1997 [DOI] [PubMed] [Google Scholar]
- 119.Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, Kambadur R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem 275: 40235–40243, 2000 [DOI] [PubMed] [Google Scholar]
- 120.Tilg H, Jalan R, Kaser A, Davies NA, Offner FA, Hodges SJ, Ludwiczek O, Shawcross D, Zoller H, Alisa A, Mookerjee RP, Graziadei I, Datz C, Trauner M, Schuppan D, Obrist P, Vogel W, Williams R. Anti-tumor necrosis factor-alpha monoclonal antibody therapy in severe alcoholic hepatitis. J Hepatol 38: 419–425, 2003 [DOI] [PubMed] [Google Scholar]
- 121.Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C. Androgens and bone. Endocr Rev 25: 389–425, 2004 [DOI] [PubMed] [Google Scholar]
- 122.Vary TC, Frost RA, Lang CH. Acute alcohol intoxication increases atrogin-1 and MuRF1 mRNA without increasing proteolysis in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 294: R1777–R1789, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vendemiale G, Grattagliano I, Altomare E, Serviddio G, Portincasa P, Prigigallo F, Palasciano G. Mitochondrial oxidative damage and myocardial fibrosis in rats chronically intoxicated with moderate doses of ethanol. Toxicol Lett 123: 209–216, 2001 [DOI] [PubMed] [Google Scholar]
- 124.Vonlaufen A, Wilson JS, Pirola RC, Apte MV. Role of alcohol metabolism in chronic pancreatitis. Alcohol Res Health 30: 48–54, 2007 [PMC free article] [PubMed] [Google Scholar]
- 125.Walsh CR, Larson MG, Evans JC, Djousse L, Ellison RC, Vasan RS, Levy D. Alcohol consumption and risk for congestive heart failure in the Framingham Heart Study. Ann Intern Med 136: 181–191, 2002 [DOI] [PubMed] [Google Scholar]
- 126.Wang HJ, Zakhari S, Jung MK. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J Gastroenterol 16: 1304–1313, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Whitaker AM, Sulzer JK, Molina PE. Augmented central nitric oxide production inhibits vasopressin release during hemorrhage in acute alcohol-intoxicated rodents. Am J Physiol Regul Integr Comp Physiol 301: R1529–R1539, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wyatt TA, Sisson JH. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 281: L575–L581, 2001 [DOI] [PubMed] [Google Scholar]
- 129.Xin X, He J, Frontini MG, Ogden LG, Motsamai OI, Whelton PK. Effects of alcohol reduction on blood pressure: a meta-analysis of randomized controlled trials. Hypertension 38: 1112–1117, 2001 [DOI] [PubMed] [Google Scholar]
- 130.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144: 1252–1261, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang P, Nelson S, Summer WR, Spitzer JA. Acute ethanol intoxication suppresses the pulmonary inflammatory response in rats challenged with intrapulmonary endotoxin. Alcohol Clin Exp Res 21: 773–778, 1997 [PubMed] [Google Scholar]
- 132.Zoeller RT, Fletcher DL, Simonyl A, Rudeen PK. Chronic ethanol treatment reduces the responsiveness of the hypothalamic-pituitary-thyroid axis to central stimulation. Alcohol Clin Exp Res 20: 954–960, 1996 [DOI] [PubMed] [Google Scholar]