

Abstract
The idiopathic inflammatory myopathies or myositis syndromes, the most common forms of which are polymyositis, dermatomyositis and inclusion body myositis, are systemic autoimmune diseases defined by chronic muscle weakness and inflammation of unknown etiology resulting in significant morbidity and mortality. We describe a dermatomyositis patient who represents a newly recognized phenotype defined by anti-p155 autoantibodies. She presented with photosensitive skin rashes and had a chronic illness course with widespread skin disease and generalized lipodystrophy. Research suggests that categorizing the heterogeneous myositis syndromes into mutually exclusive and stable phenotypes by using clinical and immune response features is useful for predicting clinical signs and symptoms, associated genetic and environmental risk factors, responses to therapy and prognosis. Knowledge of myositis phenotypes should enhance clinicians’ ability to recognize and manage these rare disorders.
CASE PRESENTATION
An 11-year-old white girl developed scalp flaking and a red, pruritic facial rash, followed by scaling, palpable erythema over the knuckles and erythema over her shoulders, arms and legs. A reaction to poison ivy was initially diagnosed, but her rashes progressed despite topical therapies and antibiotics. At illness onset the patient was experiencing stress due to the birth of a baby sister, and her rashes were noted to be exacerbated by sun exposure. By the time she was referred to a pediatric rheumatologist, she had developed progressive fatigue and experienced difficulty climbing stairs and carrying her school book bag. She was found to have mild proximal muscle weakness, a mildly elevated creatine kinase level of 270 U/L and a positive antinuclear antibody at a 1:1280 dilution. Magnetic resonance imaging of the thigh muscle revealed diffuse increased muscle edema bilaterally on short tau inversion recovery images. Juvenile dermatomyositis was diagnosed. Despite therapy with multiple immunosuppressive medications, including tacrolimus, cyclophosphamide, anti-tumor necrosis factor therapies and rituximab, disease activity increased with attempted tapering of therapy. She continues to have persistent skin and muscle disease for more than a decade, which includes photosensitive facial rashes (Figure 1A) as well as rashes in the V of the neck and around the shoulders in the distribution of a shawl, widespread erythema, subcutaneous edema, and severe nailfold and gingival capillary changes. She was recently found to have the anti-p155 autoantibody that is directed against transcription inhibitory factor 1 gamma (TIF-1γ).
Figure 1. Characteristic findings associated with clinical and autoantibody phenotypes in myositis.
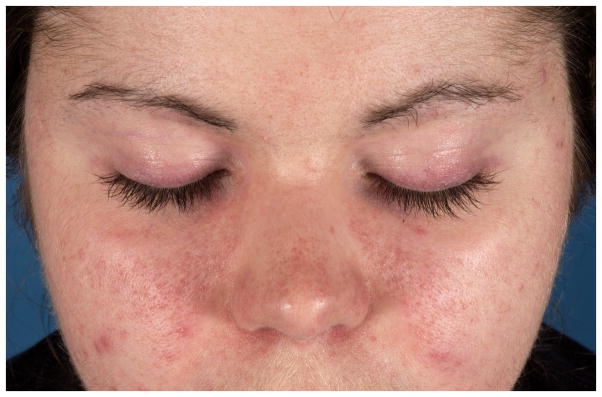
A. Heliotrope, malar rash and facial erythema in pattern of photosensitivity, as seen in this 11-year-old juvenile dermatomyositis patient with anti-p155 autoantibodies.
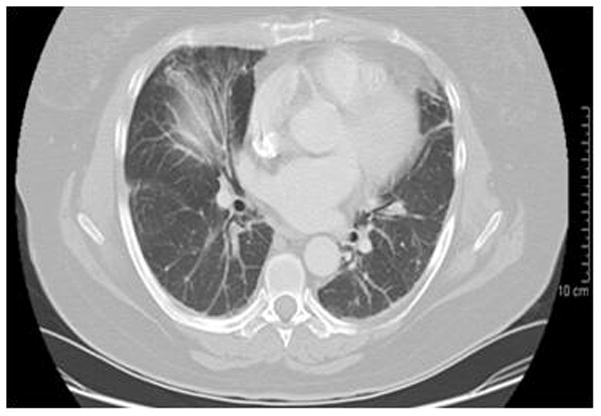
B. Interstitial lung disease, defined by high-resolution computed tomographic (CT) scanning, is seen frequently in patients with polymyositis, as well as with the anti-synthetase autoantibody phenotype. More recently, rapidly progressive interstitial lung disease has been associated with the CADM-140 autoantibody. The chest CT scan depicted here is from a 55 year old female patient with anti-Jo-1 autoantibodies and interstitial lung disease.
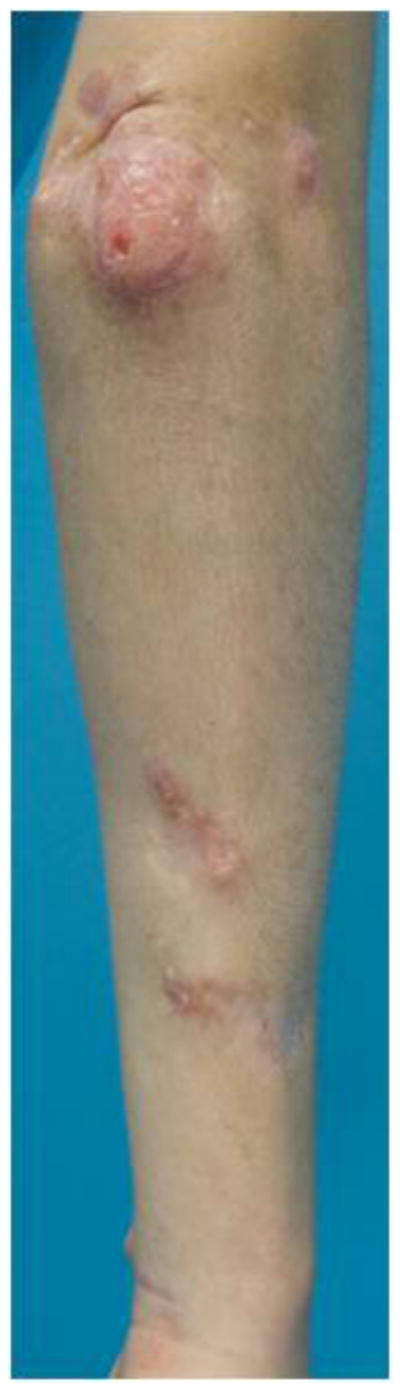
C. Dystrophic calcification, as present around the elbow and forearm here in this 10.5-year-old girl with juvenile dermatomyositis and anti-MJ autoantibodies, is seen in up to 25% of juvenile-onset dermatomyositis and less frequently in adult-onset disease. Calcinosis is present in > 60% of patients with the anti-MJ autoantibody phenotype.
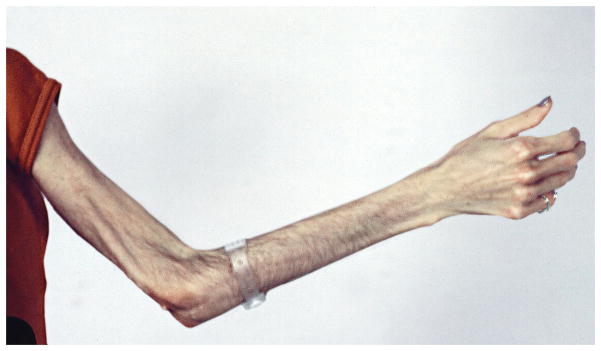
D. Lipodystrophy, loss of subcutaneous fat with frequently associated insulin resistance, diabetes and hypertriglyceridemia, are associated with juvenile-onset dermatomyositis. Generalized lipodystrophy, a widespread loss of fat from the trunk and extremities, has been associated with anti-p155 autoantibodies. A loss of fat from the arm is seen in this 20-year-old woman with juvenile onset dermatomyositis since age 8 years, who has anti-p155 autoantibodies and lipodystrophy.
THE IDIOPATHIC INFLAMMATORY MYOPATHIES
Diagnosis and Epidemiology
These systemic autoimmune diseases are currently diagnosed by a combination of clinical and laboratory features that include the presence of symmetric, progressive muscle weakness of the shoulder and hip muscles; elevated serum levels of muscle enzymes (including creatine kinase, aldolase, lactate dehydrogenase and transaminases); altered electrical activity on electromyography consisting of a triad of polyphasic, short, small motor-unit potentials; fibrillations, positive sharp waves, increased irritability and high-frequency repetitive discharges; myofiber degeneration and regeneration, chronic mononuclear cell infiltration, or perifascicular atrophy on muscle biopsy; and, for dermatomyositis, the presence of characteristic skin rashes, including the heliotrope rash (a red or purplish discoloration of the eyelids) or Gottron’s papules (raised erythematous plaques over the joint extensor surfaces).1 The presence of at least three of these features suggests that the patient has probable dermatomyositis or polymyositis (PM), and the presence of at least four features suggests that the patient has definite dermatomyositis or PM.1 The characteristic rashes are required for a diagnosis of dermatomyositis. Recently a collaborative myositis group has clarified the requirement for a muscle biopsy in the absence of the characteristic skin rashes.2
A diagnosis of myositis requires exclusion of a number of mimicking conditions. For patients presenting with weakness, the differential diagnosis includes muscular dystrophies, particularly the limb-girdle and facioscapulohumeral dystrophies, metabolic and mitochondrial myopathies, endocrine myopathies (including thyroid disease) and drug-induced myopathies (http://www.neuro.wustl.edu/neuromuscular/index.html). For patients presenting with rash and weakness, a number of infectious myopathies and systemic autoimmune diseases must be considered. Many skin conditions, including psoriasis, eczema and verrucae vulgaris can mimic the characteristic Gottron’s papules of dermatomyositis, and allergies can mimic the heliotrope rash.3
Myositis syndromes are the most common causes of acquired muscle disease in adults but are still rare disorders. The annual incidence of myositis is estimated at 5–10 cases per million in adults and 1–5 cases per million in children, with an estimated overall prevalence of 50–100 cases per million.4–6 Polymyositis (PM) and dermatomyositis peak in prevalence throughout childhood, with a mean age of 7 years,5 and in midlife, with a peak age between 30 and 50 years,7 whereas inclusion body myositis (IBM) peaks after age 50 years.8 Epidemiologic studies of US populations suggest that these disorders are increasing in frequency, perhaps due to environmental influences.4;6
Phenotypes and their importance
Dermatomyositis is the predominant form of myositis seen in children, constituting 80–85% of cases,3;9 whereas in adults about 35–50% of myositis cases are dermatomyositis.7 In children, PM is seen in 2–8% of myositis cases, and myositis overlapping with another connective tissue disease constitutes 3–10% of childhood idiopathic inflammatory myopathy cases.3;9 In adults, these subgroups are more frequent, with PM representing about 30–45% of adult idiopathic inflammatory myopathy cases and overlap myositis constituting approximately 20% of adult cases.10–12 IBM and cancer-associated myositis are very unusual in children but are more common among adult inflammatory myopathy patients and are seen in about 10–20% and 5–10% of patients, respectively.3;8 Other phenotypes based on pathologic or clinical features, including macrophagic, eosinophilic, granulomatous, focal and orbital myositis, are rare in both children and adults.3
The clinical phenotypes differ in their key clinical features and prognoses (Table 1). Polymyositis, for example, is characterized by moderate to severe weakness, with a high frequency of interstitial lung disease (Figure 1B) and cardiac disease, including heart failure, arrhythmias and ventricular dysfunction in up to 50% of patients, which negatively impact survival.10;13 Patients with adult and juvenile dermatomyositis have mild to moderate weakness, with dystrophic calcification seen in approximately 25% of children and less frequently in adults (Figure 1C).3 Other characteristic features of dermatomyositis include cutaneous and gastrointestinal ulcerations, which develop from underlying vasculopathy and tissue ischemia, and lipodystrophy (Figure 1D), both of which are more common in children.3 Survival ranges from 75–90% in patients with adult dermatomyositis and is greater than 95% in those with juvenile dermatomyositis.13;14 The risk of cancer in association with myositis is increased two to four fold, particularly in patients with adult dermatomyositis, as well as in males over 50 years of age.15 Myositis patients with cancer tend to have more severe weakness and rashes and lower serum levels of creatine kinase but higher sedimentation rates. The associated malignancies are most commonly adenocarcinomas of the lungs, gastrointestinal tract, breast and ovaries, as well as lymphoma, with a 5-year survival rate of 60%.16 Patients with IBM tend to be older men with slowly progressive proximal and distal weakness, which can be asymmetric and especially involves the quadriceps and finger flexors, rapid development of muscle atrophy and frequent dysphagia. Although the 5-year survival rate is close to 100%, many of these patients develop severe functional impairment and become wheelchair dependent.8
Table 1.
Clinical and Autoantibody Phenotypes in Myositis
| Clinical or Autoantibody Phenotype | Demographics | Clinical Phenotype | Associated Clinical Features | Response to Therapy | Prognosis | References |
|---|---|---|---|---|---|---|
| Polymyositis | Seen mainly in adults, peak onset age 30–50 years | Absence of Gottron’s papules and Heliotrope rashes | Moderate to severe weakness, interstitial lung disease, cardiac dysfunction, arrhythmias | Moderate, frequent use of cytotoxic therapy, frequent flare with reduction of therapy | 75–94% 5 year survival in adults | Miller7 Love10 |
| Adult and juvenile dermatomyositis | Peak age at onset 7 years in children, 30–50 years in adults | Dermatomyositis is relatively more frequent in children than adults | Mild to moderate weakness, calcinosis, ulcerations, lipodystrophy (JDM) | Good | 75–90% 5-year survival in adult dermatomyositis; >95% in JDM | Love10 Feldman3 Wedderburn 9 |
| Cancer-associated myositis | Male > female, age > 60 years, adenocarcinomas and lymphomas most common | Dermatomyositis > PM | Severe weakness and rashes, high sedimentation rate, low creatine kinase | Variable, frequent administration of other immununosuppressive therapies | 60–66% 5-year survival in adults | Hill15 Madan16 |
| Inclusion body myositis | Older white men | Asymmetric proximal and distal weakness, early thigh atrophy, finger flexor weakness, dysphagia | Poor, lower-dose prednisone often with other immunosuppressive therapies, slow rate of progression | 100% 5-year survival, but high-level functional disability | Needham 8 Needham8 |
|
| Anti-synthetase autoantibodies | Seen mainly in adults; in 25% of all myositis patients | PM, adult and juvenile dermatomyositis, overlap myositis | Interstitial lung disease, arthritis, fevers, Raynaud’s, mechanics hands | Moderate, flare with taper of therapy | 75% 5-year survival in adults | Love10 |
| Anti-signal recognition particle autoantibodies | Seen in 3–5% of all myositis patients | PM | Acute, severe proximal and distal weakness, myalgias, cardiac involvement | Poor, slow rate of progression with multiple immunosuppressive therapies | 20% 5-year survival in adults | Love10 Kao20 Wedderburn9 |
| Anti-Mi-2 autoantibodies | Seen in 6% of all myositis patients | Adult and juvenile dermatomyositis | Mild to moderate weakness, dermatomyositis rashes, V and shawl rashes, cuticular overgrowth | Good | 90% 5-year survival in adults | Love10 |
| Anti-p155 (TIF-1γ) autoantibodies | Seen in 23–29% of dermatomyositis, patients | Adult and juvenile dermatomyositis, overlap myositis with dermatomyositis, cancer-associated dermatomyositis | Moderate to severe weakness, V and shawl rashes, erythroderma, ulcers, edema, generalized lipodystrophy | Unknown | Unknown | Gunawardena 17 Wedderburn9 |
| Anti-MJ (NXP-2) autoantibodies | Seen in 13–23% of dermatomyositis patients | Adult and juvenile dermatomyositis | Calcinosis, joint contractures, no trunk rash | Unknown | Unknown | Gunawardena 17 Wedderburn9 |
| Anti-CADM140 (MDA-5) autoantibodies | Reported in Japanese patients | Amyopathic dermatomyositis and dermatomyositis, | Rapidly progressive interstitial lung disease, | Unknown | Unknown, but > 50% mortality in reported series | Fischer19 Sato21 |
Abbreviations: PM, polymyositis; JDM, juvenile dermatomyositis; TIF-1γ, transcription inhibitory factor 1 gamma; NXP-2, nuclear matrix protein 2; MDA-5, melanoma differentiation–associated gene 5
Each clinical and autoantibody group is mutually exclusive.
The heterogeneity of these disorders can be decreased not only by clustering patients using clinical and pathologic features but also by the presence of certain autoantibodies. The myositis autoantibodies have been traditionally defined by their capacity to immunoprecipitate characteristic proteins or RNAs, and new solid phase-based methods are not considered as validated. Immunoprecipitation–western blotting or immunoprecipitation-immunodepletion can detect autoantigens of lower abundance, such the proteins targeted by the recently recognized p155 and MJ autoantibodies.17;18 Currently only a few commercial laboratories perform validated immunoprecipitation and immunoprecipitation-blotting assays for these autoantibodies.18
Patients with anti-aminoacyl-tRNA synthetase autoantibodies, seen in up to 25% of adult patients and 5% of juvenile-onset patients, have a high frequency of moderate to severe myositis associated with characteristic extra-muscular manifestations that include interstitial lung disease, low-grade fevers, arthritis, Raynaud’s phenomenon and mechanic’s hands.3;10 The most common of these autoantibodies is called the anti-Jo-1 autoantibody, and it is directed against histidyl-tRNA synthetase. However, five other anti-aminoacyl tRNA synthetases are targeted in myositis, and other autoantibodies, such as those to alanyl- and threonyl-tRNA synthetase, have been seen in association with idiopathic interstitial lung disease in the absence of myositis.19 Patients with anti-signal recognition particle autoantibodies tend to have an acute onset of severe PM involving both proximal and distal muscle groups, with a high frequency of myocarditis and arrhythmias in some series that negatively impact morbidity and mortality; many of these patients become wheelchair dependent and require extensive rehabilitation.9;10;20 Anti-Mi-2 autoantibodies, seen in approximately 6% of patients, occur exclusively in dermatomyositis and are associated with more mild muscle involvement, rashes in the V region of the neck and over the shoulders in a shawl distribution (V- and shawl-sign rashes, respectively) and cuticular overgrowth.10
More recently identified myositis autoantibodies are also associated with distinct clinical features. Children with dermatomyositis have been found frequently to have anti-p155 or anti-MJ autoantibodies, thus accounting for a large proportion of the autoantibody phenotypes associated with juvenile dermatomyositis. Anti-p155 autoantibodies, which target TIF-1γ, have been identified in up to 25% of dermatomyositis and juvenile dermatomyositis patients, and in up to 75% of dermatomyositis patients with malignancy.9;17 Data remain limited on the clinical phenotype associated with this autoantibody, but moderate to severe muscle weakness with classic dermatomyositis skin rashes, as well as more severe cutaneous involvement with a high frequency of ulcerations, erythroderma, subcutaneous edema and generalized lipodystrophy (Figure 1D) have been seen frequently.9 Anti-MJ autoantibodies, which have been found to target nuclear matrix protein 2 (NXP2), have been seen in up to 25% of adult and juvenile dermatomyositis patients and are associated with a high frequency of calcinosis (Figure 1C), joint contractures, arthritis and an absence of truncal rashes.9;17 A third emerging phenotype is that associated with anti-CADM-140 autoantibodies, which are directed against an RNA helicase known as melanoma differentiation–associated gene 5 (MDA-5); this phenotype has been identified mainly in Japanese patients with amyopathic dermatomyositis and are associated with rapidly progressive interstitial lung disease (Figure 1B).19;21
Pathogenesis
By definition the causes of the idiopathic inflammatory myopathies remain unknown, yet data from similar autoimmune diseases support the hypothesis that these conditions result from chronic immune activation after exposure to environmental risk factors in individuals with a predisposing genetic background.7
Genetic risk factors include polymorphisms of many genes that regulate responses to environmental agents, particularly human leukocyte antigens (HLA), cytokine and immunoglobulin genes (Table 2).22 Certain polymorphic loci, such as HLA DRB1*0301, TNFα-308A, and Gm 3 23 5, 13 are risk factors for all of the major clinical groups. Other alleles are specific to particular autoantibody phenotypes, and often the genetic associations are stronger with particular autoantibodies (Table 2). It appears that a possible explanation for the mutual exclusivity and stability of myositis phenotypes is that genes that are risk factors for one phenotype are often protective for another.22 Interactions of environmental and genetic risk factors, in the relative absence of protective factors, are considered an initial step in the process leading to immune activation and autoantibody formation in autoimmune diseases. One example is the interaction of cigarette smoking with HLA DRB1 shared epitope alleles, PTPN22, and anti-citrullinated peptide autoantibodies in patients with rheumatoid arthritis.23
Table 2.
Possible factors involved in the pathogenesis of myositis phenotypes*
| Factor | Phenotype | Odds Ratio (range) | Comments† | References |
|---|---|---|---|---|
| Polymorphic genes | ||||
| HLA DRB1*0301 | PM, dermatomyositis, JDM, IBM, anti-synthetase autoantibodies | 1.9–18.5 | Stronger association with autoantibodies than clinical phenotypes. Other HLA alleles are associated with other autoantibodies and other ethnic groups. | O’Hanlon11;22; Chinoy46 |
| HLA DRB1*0701 | Anti-Mi-2 autoantibodies | 4.9–11.1 | Stronger association with autoantibody than clinical phenotypes | O’Hanlon11;22 |
| HLA DQA1*0301 | Anti-p155 autoantibodies | 5.4 | O’Hanlon22 | |
| TNFα-308A | PM, dermatomyositis, JDM; JDM with calcinosis, ulcerations and a chronic illness course | 2.5–5.0 | O’Hanlon47 | |
| Gm 3 23 5, 13 | PM, dermatomyositis, JDM, IBM, anti-synthetase autoantibodies | 2.2–3.4 | O’Hanlon47 | |
| Gm13 | JDM | 3.9 | O’Hanlon47 | |
| Environmental Agents: | ||||
| Infectious agents | ||||
| Group A Streptococcus | JPM/JDM | 2.8 | Evidence based on a case-control study | Gourley24 |
| Echovirus | JDM | NA | Many case series, particularly in patients with agammaglobulinemia | Reed48 |
| Drugs | ||||
| D-penicillamine | PM | NA | Multiple cases with dechallenge, several cases with dechallenge-rechallenge | Miller7 |
| Human growth hormone | JDM | NA | Multiple cases with dechallenge, several cases with dechallenge-rechallenge | Reed48 |
| Therapeutic cytokines | ||||
| Interferon alpha | PM/dermatomyositis | NA | Multiple cases with dechallenge | Miller7 |
| Interferon gamma | PM/dermatomyositis | NA | Multiple cases with dechallenge | Miller7 |
| Medical devices | ||||
| Bovine collagen implants | PM/dermatomyositis | 5.1 – 18.8 | Associated with a delayed-type hypersensitivity response to collagen | Miller7 |
| Other exposures | ||||
| Physical exertion | PM/dermatomyositis | 3.9 | Evidence based on a case-sibling study | Lyon49 |
| Ultraviolet radiation | Dermatomyositis, anti-Mi-2 autoantibodies in women | 3,8–17.3 | Global and U.S. epidemiologic studies support this association | Love50 |
Abbreviations: JPM, juvenile polymyositis; JDM, juvenile dermatomyositis; NA, not available; PM, polymyositis; ; IBM, inclusion body myositis; HLA, human leukocyte antigen; SRP, signal recognition particle.
Data from Caucasians. Some genes are both risk factors for one phenotype and protective for another phenotype. Genes other than those listed are likely important.
Dechallenge refers to clinical improvement of the myositis after removing the agent, and rechallenge refers to reoccurrence of the myositis after reinstituting the agent.
Although the evidence is limited, different environmental exposures, in particular, infections, therapeutic agents, physical exertion and collagen implants, have been reported to be involved in the development of certain myositis phenotypes (Table 2).24 Additional, albeit indirect, evidence supporting a role for the environment in myositis includes clinical improvement after removing the agent (dechallenge) and reoccurrence of the myositis after reinstituting the agent (rechallenge).7 A spring season of onset of myositis has been described for adult patients with anti-synthetase autoantibodies, and a summer season for onset in adults without defined myositis autoantibodies,25 suggesting that different environmental factors may be associated with each phenotype. Further evidence for this comes from the findings that ultraviolet radiation intensity at the location of disease onset is associated with dermatomyositis and anti-Mi-2 autoantibodies, particularly in women.12;26 Of interest, children without a defined myositis autoantibody had a higher frequency of documented infections within 6 months of illness onset compared to those with myositis autoantibodies.27
The evidence supporting a role for the immune system in the pathogenesis of idiopathic inflammatory myopathy includes the findings that: some myositis patients have multiple autoimmune disorders;7 autoantibodies are characteristic of certain phenotypes;17 T cell-mediated myocytotoxicity or complement-mediated microangiopathy are seen in particular clinical phenotypes;28 polymorphic immune response genes are primary genetic risk factors;22 and immunotherapies often result in clinical responses.9;29
Current work suggests that different immune processes are likely at play in different myositis phenotypes. In dermatomyositis, initial processes include activation of the complement cascade through C3 and deposition of the complement C5b-9 membrane attack complex on the endomysial vasculature, with resultant capillary destruction, muscle ischemia and dilatation of the remaining capillaries.7;28 These processes lead to infiltration of B lymphocytes, CD4+ helper T cells and plasmacytoid dendritic cells in perimysial areas of muscle fascicles and small blood vessels. MHC Class I antigen and intracellular adhesion molecule are upregulated on the cell surfaces of damaged fibers and in perivascular areas, respectively.30 In contrast, PM and IBM are characterized by a predominant cytotoxic T lymphocyte-mediated process involving perforin, with CD8+ T cells accompanied by smaller numbers of macrophages surrounding and invading otherwise normal-appearing myocytes in endomysial areas.31 MHC Class I antigen is upregulated on the surface of the majority of muscle fibers, even those not affected by inflammation.32 Other pathogenic processes include the likely role of type I interferons in dermatomyositis3 and the possible role of autoantigens whose fragments have chemokine activity.33 Additional factors include the endoplasmic reticulum stress response.34 These mechanisms appear to converge in the upregulation of NF-κB and activation of positive feedback loops to maintain the proinflammatory cascades that maintain chronic tissue inflammation.
Therapy
Although the pathophysiology of PM and dermatomyositis are distinct, current therapies for these disorders, derived primarily from anecdote and uncontrolled studies, are similar and based on broad immunosuppression. The core therapeutic approach remains high-dose daily oral corticosteroid therapy, along with adjunctive steroid-sparing immunosuppressive therapies, which are used to treat disease activity, prevent mortality, and attempt to reduce long-term disability (Box). The treatment of IBM with evidence of active disease is controversial, but some experts believe the primary goal of therapy is to slow the rate of disease progression by immunosuppression and physical therapy.8 The use of second-line therapies, such as high-dose intravenous pulse corticosteroids, methotrexate, intravenous gammaglobulin, azathioprine or cyclosporine, as part of initial therapy in juvenile patients with moderate to severe disease or adult patients with poor prognostic factors is supported by consensus among specialists and open-label studies that suggest a shorter course of illness, reduced frequency of calcinosis, and fewer corticosteroid side effects with early introduction of additional therapies.9;29;35 For patients with severe, refractory or corticosteroid-dependent disease, combinations of second-line therapies or the addition of newer third-line therapies are frequently used (Box). Among these third-line agents, uncontrolled trials support the use of mycophenolate mofetil for adult and juvenile myositis patients with severe disease36 and the use of intravenous monthly pulse cyclophosphamide for patients with severe or refractory juvenile dermatomyositis.37 Use of oral tacrolimus is supported by studies in patients with treatment-refractory disease, including those with difficult-to-treat disease associated with anti-synthetase and anti-signal recognition particle autoantibodies.38
Box. Therapeutic Agents Used to Treat Adult and Juvenile Dermatomyositis and Polymyositis.
| First Line Therapies* |
| Prednisone (daily oral) |
| Intravenous pulse methylprednisolone+ Methotrexate+ |
| Adjunctive Therapies: Physical therapy, hydroxychloroquine, topical therapies for skin rashes, photoprotective measures, calcium and vitamin D for bone protection |
| Second Line Therapies |
| Azathioprine |
| Cyclosporine |
| Intravenous gammaglobulin+ |
| Combinations of the above |
| Third Line Therapies |
| Mycophenolate mofetil |
| Tacrolimus |
| Cyclophosphamide Rituximab |
| Anti-tumor necrosis alpha agents |
| Other biologics: anakinra, alemtuzumab |
| Stem cell therapy |
Case series and small open-label trials suggest that rituximab, a monoclonal antibody directed against B lymphocytes, is often effective in adult and juvenile dermatomyositis patients with severe, refractory disease, although improvement in skin disease activity is not clear.39 Adults with anti-synthetase and anti-signal recognition particle autoantibodies also experienced a dramatic improvement in muscle strength and associated interstitial lung disease after rituximab therapy, with a decline in anti-synthetase autoantibody titers.40;41 Responses to the anti-tumor necrosis factor alpha therapies have been mixed, with a more consistent response in juvenile dermatomyositis patients. All five patients with severe juvenile dermatomyositis improved 8–30 months after open-label treatment with infliximab, and in some cases calcinosis also improved.42 Responses to infliximab and etanercept have varied in adult patients with dermatomyositis or PM, with disease progression in some patients.43 Case reports of the development of myositis after anti-tumor necrosis alpha therapy also suggest caution in the use of these agents for therapy.44 Alemtuzumab, a monoclonal antibody directed against T lymphocytes and monocytes, resulted in improved strength and function paralleling the decline in peripheral blood and muscle T cells in a majority of patients with IBM.45 Stem cell transplantation is suggested as a final option for severe, unremitting disease, with case reports of success.9
The response to treatment differs among the clinical and autoantibody phenotypes. For example, patents with dermatomyositis tend to be maintained on a lower dose of prednisone, with < 50% requiring other therapies, and although their disease activity often increases during prednisone taper, 13% do not require continued therapy.10 In contrast, patients with PM more frequently require cytotoxic agents, and in 40% their disease activity increases during reduction of therapy. Patients with IBM are often treated with lower doses of prednisone and fewer cytotoxic agents but generally require ongoing therapy.10 Among the autoantibody subgroups, patients with anti-synthetase autoantibodies require a moderate dose of prednisone, with 60% increasing disease activity during tapering of therapy, and most patients require treatment most of the time. Patients with anti-signal recognition particle autoantibody require a higher dose of prednisone, generally require concomitant therapy with other agents, and usually cannot be taken off therapy for years.10 Patients with anti-Mi-2 autoantibodies use the lowest dose of prednisone and spend approximately 20% of their time off therapy; fewer require treatment with other agents, and only 25% experience increased disease activity during reduction of therapy.10
CONCLUSION
Understanding the many mutually exclusive and stable myositis phenotypes aids in deciphering the mechanisms by which these conditions arise, interpreting and anticipating their diverse clinical presentations and managing their care. Given the differences in genetic and environmental risk factors, pathology and prognosis among myositis phenotypes, future studies should incorporate phenotype status into investigations of their pathogenesis and therapy.
Acknowledgments
Funding/Support: This research was supported by the Intramural Research Program of the NIEHS, National Institutes of Health.
Footnotes
Author Contributions: Dr. Rider had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Rider, Miller.
Acquisition of data: Rider, Miller.
Analysis and interpretation of data: Rider, Miller.
Drafting of the manuscript: Rider, Miller.
Critical revision of the manuscript for important intellectual content: Rider, Miller.
Obtained funding: Miller.
Administrative, technical, or material support: Rider, Miller.
Financial Disclosures: No authors reported financial disclosures.
Additional Contributions: We thank Mark Gourley M.D. and Fred Ognibene M.D. at the National Institutes of Health, Bethesda, Maryland, and Kathleen Coyle M.D. at the Food and Drug Administration for their critical review of the manuscript. None of these persons was compensated for his or her contribution.
Role of the Sponsor: The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; or in the preparation, review, or approval of the manuscript.
Contributor Information
Lisa G. Rider, Email: riderl@mail.nih.gov.
Frederick W. Miller, Email: millerf@mail.nih.gov.
References
- 1.Bohan A, Peter JB. Polymyositis and dermatomyositis. Parts 1 and 2. N Engl J Med. 1975;292:344–347. 3403–407. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 2.Oddis CV, Rider LG, Reed AM, et al. International consensus guidelines for trials of therapies in the idiopathic inflammatory myopathies. Arthritis Rheum. 2005;52:2607–2615. doi: 10.1002/art.21291. [DOI] [PubMed] [Google Scholar]
- 3.Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008:2201–2212. doi: 10.1016/S0140-6736(08)60955-1. [DOI] [PubMed] [Google Scholar]
- 4.Oddis CV, Conte CG, Steen VD, Medsger TA., Jr Incidence of polymyositis-dermatomyositis: A 20-year study of hospital diagnosed cases in Allegheny County, PA 1963–1982. J Rheumatol. 1990;17:1329–1334. [PubMed] [Google Scholar]
- 5.Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis, 1995–1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300–305. doi: 10.1002/art.11122. [DOI] [PubMed] [Google Scholar]
- 6.Wilson FC, Ytterberg SR, St Sauver JL, Reed AM. Epidemiology of sporadic inclusion body myositis and polymyositis in Olmsted County, Minnesota. J Rheumatol. 2008;35:445–447. [PubMed] [Google Scholar]
- 7.Miller FW. Inflammatory myopathies: Polymyositis, dermatomyositis, and related conditions. In: Koopman W, Moreland L, editors. Arthritis and Allied Conditions: A Textbook of Rheumatology. 15. Philadelphia: Lippincott Williams & Wilkins; 2005. pp. 1593–1620. [Google Scholar]
- 8.Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol. 2007;6:620–631. doi: 10.1016/S1474-4422(07)70171-0. [DOI] [PubMed] [Google Scholar]
- 9.Wedderburn LR, Rider LG. Juvenile dermatomyositis: new developments in pathogenesis, assessment and treatment. Best Pract Res Clin Rheumatol. 2009;23:665–678. doi: 10.1016/j.berh.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70:360–374. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 11.O’Hanlon TP, Carrick DM, Targoff IN, et al. HLA-A, -B, -DRB1 and -DQA1 allelic profiles for the idiopathic inflammatory myopathies: Distinct immunogenetic risk and protective factors distinguish European American patients with different myositis autoantibodies. Medicine. 2006;85:111–127. doi: 10.1097/01.md.0000217525.82287.eb. [DOI] [PubMed] [Google Scholar]
- 12.Okada S, Weatherhead E, Targoff IN, Wesley R, Miller FW. Global surface ultraviolet radiation intensity may modulate the clinical and immunologic expression of autoimmune muscle disease. Arthritis Rheum. 2003;48:2285–2293. doi: 10.1002/art.11090. [DOI] [PubMed] [Google Scholar]
- 13.Lundberg IE. Mortality in idiopathic inflammatory myopathies. Clin Exp Rheumatol. 2008;26 (suppl 51):S109–S114. [PubMed] [Google Scholar]
- 14.Rider LG, Lachenbruch PA, Monroe JB, et al. Damage extent and predictors in adult and juvenile dermatomyositis and polymyositis as determined with the myositis damage index. Arthritis Rheum. 2009;60:3425–3435. doi: 10.1002/art.24904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96–100. doi: 10.1016/S0140-6736(00)03540-6. [DOI] [PubMed] [Google Scholar]
- 16.Madan V, Chinoy H, Griffiths CE, Cooper RG. Defining cancer risk in dermatomyositis. Part I. Clin Exp Dermatol. 2009;34:451–455. doi: 10.1111/j.1365-2230.2009.03216.x. [DOI] [PubMed] [Google Scholar]
- 17.Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009;48:607–612. doi: 10.1093/rheumatology/kep078. [DOI] [PubMed] [Google Scholar]
- 18.Trieu EP, Gross JK, Targoff IN. Immunoprecipitation-western blot for proteins of low abundance. Methods Mol Biol. 2009;536:259–275. doi: 10.1007/978-1-59745-542-8_28. [DOI] [PubMed] [Google Scholar]
- 19.Fischer A, Swigris JJ, du Bois RM, et al. Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med. 2009;103:1719–1724. doi: 10.1016/j.rmed.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50:209–215. doi: 10.1002/art.11484. [DOI] [PubMed] [Google Scholar]
- 21.Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193–2200. doi: 10.1002/art.24621. [DOI] [PubMed] [Google Scholar]
- 22.O’Hanlon TP, Miller FW. Genetic risk and protective factors for the idiopathic inflammatory myopathies. Curr Rheumatol Rep. 2009;11:287–294. doi: 10.1007/s11926-009-0040-2. [DOI] [PubMed] [Google Scholar]
- 23.Mahdi H, Fisher BA, Kallberg H, et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet. 2009;41:1319–1324. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- 24.Gourley M, Miller FW. Mechanisms of disease: Environmental factors in the pathogenesis of rheumatic disease. Nat Clin Pract Rheumatol. 2007;3:172–180. doi: 10.1038/ncprheum0435. [DOI] [PubMed] [Google Scholar]
- 25.Sarkar K, Weinberg CR, Oddis CV, et al. Seasonal influence on the onset of idiopathic inflammatory myopathies in serologically defined groups. Arthritis Rheum. 2005;52:2433–2438. doi: 10.1002/art.21198. [DOI] [PubMed] [Google Scholar]
- 26.Love LA, Weinberg CR, McConnaughey DR, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. 2009;60:2499–2504. doi: 10.1002/art.24702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rider LG, Wu L, Mamyrova G, Targoff IN, Miller FW. Environmental factors preceding illness onset differ in phenotypes of the juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 2010;49:2381–2390. doi: 10.1093/rheumatology/keq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalakas MC. Mechanisms of disease. Signaling pathways and immunobiology of inflammatory myopathies. Nature Clinical Practice Rheumatology. 2006;2:219–227. doi: 10.1038/ncprheum0140. [DOI] [PubMed] [Google Scholar]
- 29.Hengstman GJ, van den Hoogen FH, van Engelen BG. Treatment of the inflammatory myopathies: update and practical recommendations. Expert Opin Pharmacother. 2009;10:1183–1190. doi: 10.1517/14656560902913815. [DOI] [PubMed] [Google Scholar]
- 30.Figarella-Branger D, Civatte M, Bartoli C, Pellissier JF. Cytokines, chemokines, and cell adhesion molecules in inflammatory myopathies. Muscle Nerve. 2003;28:659–682. doi: 10.1002/mus.10462. [DOI] [PubMed] [Google Scholar]
- 31.Goebels N, Michaelis D, Engelhardt M, et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest. 1996;97:2905–2910. doi: 10.1172/JCI118749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalakas MC. Muscle biopsy findings in inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:779–98. vi. doi: 10.1016/s0889-857x(02)00030-3. [DOI] [PubMed] [Google Scholar]
- 33.Howard OM, Dong HF, Yang D, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;196:781–791. doi: 10.1084/jem.20020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagaraju K, Casciola-Rosen L, Lundberg I, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- 35.Huber AM, Giannini EH, Bowyer SL, et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Children’s Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res (Hoboken ) 2010;62:219–225. doi: 10.1002/acr.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken ) 2010;62:1496–1501. doi: 10.1002/acr.20212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riley P, Maillard SM, Wedderburn LR, Woo P, Murray KJ, Pilkington CA. Intravenous cyclophosphamide pulse therapy in juvenile dermatomyositis. A review of efficacy and safety. Rheumatology (Oxford) 2004;43:491–496. doi: 10.1093/rheumatology/keh082. [DOI] [PubMed] [Google Scholar]
- 38.Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52:2439–2446. doi: 10.1002/art.21240. [DOI] [PubMed] [Google Scholar]
- 39.Rios FR, Callejas Rubio JL, Sanchez CD, Saez Moreno JA, Ortego CN. Rituximab in the treatment of dermatomyositis and other inflammatory myopathies. A report of 4 cases and review of the literature. Clin Exp Rheumatol. 2009;27:1009–1016. [PubMed] [Google Scholar]
- 40.Majmudar S, Hall HA, Zimmermann B. Treatment of adult inflammatory myositis with rituximab: an emerging therapy for refractory patients. J Clin Rheumatol. 2009;15:338–340. doi: 10.1097/RHU.0b013e3181bb8e70. [DOI] [PubMed] [Google Scholar]
- 41.Valiyil R, Casciola-Rosen L, Hong G, Mammen A, Christopher-Stine L. Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: a case series. Arthritis Care Res (Hoboken ) 2010;62:1328–1334. doi: 10.1002/acr.20219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riley P, McCann LJ, Maillard SM, Woo P, Murray KJ, Pilkington CA. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology (Oxford) 2008;47:877–880. doi: 10.1093/rheumatology/ken074. [DOI] [PubMed] [Google Scholar]
- 43.Dastmalchi M, Grundtman C, Alexanderson H, et al. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann Rheum Dis. 2008;67:1670–1677. doi: 10.1136/ard.2007.077974. [DOI] [PubMed] [Google Scholar]
- 44.Klein R, Rosenbach M, Kim EJ, Kim B, Werth VP, Dunham J. Tumor necrosis factor inhibitor-associated dermatomyositis. Arch Dermatol. 2010;146:780–784. doi: 10.1001/archdermatol.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dalakas MC, Rakocevic G, Schmidt J, et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain. 2009;132:1536–1544. doi: 10.1093/brain/awp104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chinoy H, Lamb JA, Ollier WE, Cooper RG. An update on the immunogenetics of idiopathic inflammatory myopathies: major histocompatibility complex and beyond. Curr Opin Rheumatol. 2009;21:588–593. doi: 10.1097/BOR.0b013e3283315a22. [DOI] [PubMed] [Google Scholar]
- 47.O’Hanlon TP, Rider LG, Schiffenbauer A, et al. Immunoglobulin gene polymorphisms are susceptibility factors in clinical and autoantibody subgroups of the idiopathic inflammatory myopathies. Arthritis Rheum. 2008;58:3239–3246. doi: 10.1002/art.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reed AM, Ytterberg SR. Genetic and environmental risk factors for idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:891–916. doi: 10.1016/s0889-857x(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 49.Lyon MG, Bloch DA, Hollak B, Fries JF. Predisposing factors in polymyositis-dermatomyositis: results of a nationwide survey. J Rheumatol. 1989;16:1218–1224. [PubMed] [Google Scholar]
- 50.Love LA, Weinberg CR, McConnaughey DR, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. 2009;60:2499–2504. doi: 10.1002/art.24702. [DOI] [PMC free article] [PubMed] [Google Scholar]