

Abstract
Novel drugs are designed against specific molecular targets, but almost unavoidably they bind non-targets, which can cause additional biological effects that may result in increased activity or, more frequently, undesired toxicity. Chemical proteomics is an ideal approach for the systematic identification of drug targets and off-targets, allowing unbiased screening of candidate interactors in their natural context (tissue or cell extracts).
E-3810 is a novel multi-kinase inhibitor currently in clinical trials for its anti-angiogenic and anti-tumor activity. In biochemical assays, E-3810 targets primarily vascular endothelial growth factor and fibroblast growth factor receptors. Interestingly, E-3810 appears to inhibit the growth of tumor cells with low to undetectable levels of these proteins in vitro, suggesting that additional relevant targets exist. We applied chemical proteomics to screen for E-3810 targets by immobilizing the drug on a resin and exploiting stable isotope labeling by amino acids in cell culture to design experiments that allowed the detection of novel interactors and the quantification of their dissociation constant (Kd imm) for the immobilized drug. In addition to the known target FGFR2 and PDGFRα, which has been described as a secondary E-3810 target based on in vitro assays, we identified six novel candidate kinase targets (DDR2, YES, LYN, CARDIAK, EPHA2, and CSBP). These kinases were validated in a biochemical assay and—in the case of the cell-surface receptor DDR2, for which activating mutations have been recently discovered in lung cancer—cellular assays.
Taken together, the success of our strategy—which integrates large-scale target identification and quality-controlled target affinity measurements using quantitative mass spectrometry—in identifying novel E-3810 targets further supports the use of chemical proteomics to dissect the mechanism of action of novel drugs.
The “target deconvolution” process, namely, the identification and characterization of proteins bound by a drug of interest (1), is a crucial step in drug development that allows definition of the compound selectivity and the early detection of potential side effects. Target deconvolution can be achieved by means of systematic in vitro biochemical assays measuring the ability of the drug to interact with candidate binders and, if they are enzymes, interfere with their activity. An alternative approach is chemical proteomics (chemoproteomics), which combines affinity chromatography and proteomic techniques (2, 3). Up-to-date chemical proteomics essentially consists of three main steps: (i) drug immobilization on a solid phase; (ii) drug affinity chromatography to capture drug targets in complex protein mixtures, such as cell or tissue lysates; and (iii) mass spectrometry (MS)-based1 identification of the proteins retained by the immobilized drug (4–6).
In chemical proteomics, the affinity chromatography step is typically performed under mild conditions, to allow the identification of all possible natural binders. The drawback of using mild, non-denaturing conditions is the significant number of proteins nonspecifically binding to the solid phase, which, once identified via MS, can be difficult to discern from genuine drug targets. The relatively high number of such nonspecific binders has limited the widespread use of this strategy.
More recently, the development and implementation of quantitative strategies in proteomics based on the use of differentially stable isotopes to label proteomes from distinct functional states, together with significant technological and instrumental developments in the MS field concerning sensitivity and throughput, have largely allowed this limitation to be overcome. One of the most popular labeling techniques is stable isotope labeling by amino acids in cell culture (SILAC) (7). In SILAC, dividing cells are cultured in media supplemented with amino acids containing stable isotopic variants of carbon (12C/13C), nitrogen (14N/15N), or hydrogen (1H/2H), which are incorporated into newly synthesized proteins during cell division. When extensive labeling (>98%) of cells is achieved upon the appropriate number of replications, light and heavy cells are differentially treated (e.g. exposed to drug versus vehicle), mixed in equal proportion, and subjected to proteomics analysis by means of liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Peptides from the two functional states can be distinguished by their specific delta mass values, and their intensity ratio in MS spectra is directly proportional to the relative abundance of the corresponding proteins in the initial protein extract. Robust analysis of SILAC data is possible with dedicated software, such as MaxQuant (8). The application of SILAC strategies to interactomic studies is an efficient means of discerning specific from background binders (9). When applied to chemical proteomics, quantitative proteomics is crucial, as it offers quality filters to discern genuine drug interactors from proteins binding to the solid phase, with the use of different experimental setups (4, 5).
In this study, we successfully coupled SILAC with chemical proteomics to carry out an unbiased screening of protein interactors of the anti-cancer drug E-3810, currently in Phase II clinical trials. E-3810 is a novel multi-kinase inhibitor, a class of targeted drug that comprises different molecules currently used in clinical practice (e.g. imatinib, dasatinib, sunitinib, sorafenib) (10). E-3810 exhibits both anti-tumor and anti-angiogenic properties (11). In preclinical studies, E-3810 showed broad anti-tumor activity in vivo, when used as monotherapy in a variety of human xenografts, or in conjunction with conventional chemotherapy (11, 12).
Cellular vascular endothelial growth factor receptors (VEGFRs) and fibroblast growth factor receptors (FGFRs) are the principal targets of E-3810, as previously demonstrated by in vitro kinase assays, which showed that E-3810 inhibited VEGFR-1, -2, and -3 and FGFR-1 and -2 in the nanomolar range (11). Studies performed on several kinase inhibitors demonstrated that these molecules can elicit pleiotropic effects not easily explained by the sole inhibition of their known targets (13, 14). These effects are in most cases due to an inhibitory activity of the drug on additional kinase targets not tested in vitro that may lead to synergistic anti-cancer effects or undesirable toxicity. This could also be the case for E-3810, which was shown to inhibit in vitro additional kinase targets with high affinity, and which is able to inhibit the growth of tumor cells expressing low to undetectable levels of VEGFRs/FGFRs, suggesting that its spectrum of target inhibition has not been fully explored (11).
We thus established a SILAC-based chemical proteomic platform composed of a set of affinity chromatography experiments using E-3810 immobilized on agarose resin and incubated with SILAC-labeled extract from the ovarian cancer cell line A2780. We identified proteins interacting with the resin via MS and took advantage of SILAC-based protein quantitation to discern genuine from background binders and derive quantitative information about the specific interactions. Our findings demonstrate that additional targets of E-3810 exist and that these targets may contribute to the anticancer effect of E-3810.
EXPERIMENTAL PROCEDURES
E-3810 Derivatization and Immobilization
E-3810 (kindly provided by EOS S.p.A., Milano, Italy) was derivatized at the amino moiety of the substituent in position 7 of the quinoline nucleus with a propyl N-[2-[(2-methoxyacetyl)amino]ethyl]carbamate linker and subsequently loaded onto agarose resin following the reaction scheme reported in supplemental Fig. S1. The primary amino group of E-3810 (compound 1) was initially alkylated by reaction with 3-iodopropyl benzoate. After removal of the benzoyl group through alkaline hydrolysis with LiOH and protection of the amino nitrogen as Fmoc carbamate, the resulting hydroxy derivative (compound 5) was reacted with N,N′-disuccinimidyl carbonate to give a mixed carbonate (compound 6), which in turn was coupled with Affi-Gel 102 resin (Bio-Rad). The resulting derivatized resin (compound 7) was finally incubated with piperidine in CH3CN to remove the Fmoc protecting group, providing the solid support immobilized E-3810 probe (compound 8) at an estimated concentration of 3 μmol/ml (for the 50% slurry in PBS).
Cell Culture and SILAC Labeling
Human ovarian carcinoma cells (A2780) and human non-small-cell lung carcinoma cells (HCC-366) (purchased from DSMZ, Braunschweig, Germany) were cultured in standard RPMI 1640 medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (both from Invitrogen). A2780 cells were SILAC-labeled using RPMI 1640 medium deficient in lysine and arginine (Invitrogen) supplemented with 10% dialyzed fetal bovine serum (Invitrogen) and the appropriate amino acids as follows: unlabeled l-lysine (Lys0) and l-arginine (Arg0) (both from Sigma) were used to obtain light-labeled cells, and 13C615N4 l-arginine (Arg10, Sigma, 608033) and 13C615N2 l-lysine (Lys8, Sigma, 608041) were used to obtain heavy-labeled cells. For the triple-SILAC experiment, 13C6 l-arginine (Arg6, Sigma, 643440) and 2H4 l-lysine (Lys4, Cambridge Isotope Laboratories, DLM-2640) were used to obtain medium-labeled cells. The final amino acid concentration corresponded to the standard RPMI 1640 composition (i.e. 200 mg/l arginine and 40 mg/l lysine). Cells were cultured for at least nine replications to achieve complete labeling. Full protein labeling was verified by geLC-MS analysis (15).
Preparation of Cell Extracts
Exponentially growing cells (either unlabeled or SILAC-labeled) were harvested at about 90% confluence. Cells were detached by trypsinization and washed twice in PBS. Cell pellets were lysed in ice-cold modified RIPA buffer containing 50 mm Tris-HCl, pH 7.5, 1% Nonidet P-40, 0.1% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, and protease inhibitor mixture (Complete tablet, Roche). Lysates were sonicated for 10 s on ice and centrifuged at 13,000 rpm for 10 min at 4 °C to pellet cellular debris. The supernatant was recovered and the protein content was quantified using the Bradford protein assay (Bio-Rad).
Human breast cancer cells (MDA-MD-231 and MDA-MD-134) were cultured in DMEM (Invitrogen), human ovarian cancer cells (OVCA-432) were cultured in minimum Eagle's medium (Invitrogen), and OVCAR-8 cells were cultured in RPMI 1640 medium, all supplemented with 10% FBS. To assess FGFR2 expression, cells were lysed in a buffer containing 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 20 mm Tris, 150 mm NaCl, 1 mm EDTA, and protease inhibitors. Protein concentration was quantified using the Bradford protein assay, and 25 mg of protein extracts were probed by immunoblotting with anti-FGFR2 (Santa Cruz, Biotechnology, Dallas, TX) and anti-vinculin (Sigma-Aldrich) antibodies followed by secondary peroxidase-conjugated antibodies (Thermo Scientific).
E-3810 Affinity Chromatography on Unlabeled Lysates Followed by Immunoblot against FGFR2
1 mg of protein extract obtained from unlabeled A2780 cells was incubated overnight at 4 °C with 15 μl of E-3810 resin or with 15 μl of nonderivatized agarose resin as a negative control. In the test performed to assess binding capacity, 1 mg of sample was incubated with 3, 15, or 40 μl of E-3810 resin slurry. For the competition assay performed with free E-3810, 1 mg of protein extract obtained from unlabeled A2780 cells was incubated overnight at 4 °C with 40 μl of E-3810 resin slurry in the presence of increasing amounts of free E-3810 (15, 60, and 240 nmol, corresponding to concentrations of 30, 120, and 480 μm in 0.5 ml of lysate volume); the molar ratios of free, competing E-3810 to the immobilized form of the drug were equivalent to 0.125-, 0.5-, and 2-fold. One sample was not subjected to competition, for use as a control.
After the incubation, the flow-through fractions were collected. The resins were washed three times using lysis buffer and two times using lysis buffer supplemented with 0.2% SDS (high-stringency buffer), as reported in Ref. 5. To elute proteins, we incubated the resin at 99 °C for 10 min in 60 μl of LDS Sample Buffer 4X (Invitrogen), DTT 50 mm. 20 μl of eluted proteins were loaded on 7% acrylamide gel. The same volumes of A2780 protein extract and flow-through were loaded on the same gel. After the electrophoresis, the gel was blotted on polyvinylidene fluoride membrane (100 V, 75 min). After blocking with 3% BSA in TBS-T solution (25 mm Tris, 0.15 m NaCl, 0.05% Tween-20, pH 7.4), the primary antibody directed against human FGFR2 sc-122 (Santa Cruz) was incubated overnight with the membrane. Secondary horseradish-peroxidase-conjugated antibody was from Pierce; signals were detected by using pico-ECL (Thermo Scientific). Images were scanned and exported as JPG or TIFF files. The brightness and contrast of the images were adjusted for clarity on the image as a whole.
Competition Assay Based on Quantitative Chemical Proteomics
1 mg of protein extract obtained from light-labeled or heavy-labeled (Arg10 and Lys8) A2780 cells was incubated overnight at 4 °C with 40 μl of E-3810 resin slurry. Free E-3810 was dissolved in dimethyl sulfoxide (DMSO) and spiked in the incubated lysates as a 100-fold concentrated solution to compete for resin binding (the amount of spiked E-3810 was equal to 15, 60, or 240 nmol, equivalent to a concentration of 30, 120, or 480 μm in 0.5 ml of lysate). As a control, an equivalent volume of DMSO was added in one incubation experiment in which no competition occurred. Competition with soluble E-3810 was performed in the heavy lysate in a forward setup or in the light lysate in a reverse setup. After incubation, the resin was washed three times using lysis buffer and twice using lysis buffer supplemented with 0.2% SDS (high-stringency buffer). The resins incubated with light and heavy lysates were mixed during the last washing step to form heavy/light (H/L) SILAC samples. We eluted proteins by incubating the mixed resin at 99 °C for 10 min in 60 μl of LDS Sample Buffer 4X (Invitrogen) supplemented with DTT 50 mm.
Estimation of Target Affinity for E-3810 via Chemoproteomic Kd Assay
1 mg of heavy and 1 mg of light protein extract was incubated with 40 μl of E-3810 resin slurry (corresponding to 120 nmol of immobilized E-3810). 1 mg of extract obtained from cells labeled with Arg6 and Lys4 (medium-labeled (M)) was incubated with 40 μl of nonderivatized resin slurry. In the forward setup, after an overnight incubation at 4 °C, the unbound fraction of the heavy sample was collected and re-incubated with 40 μl of E-3810-derivatized slurry; the same procedure was performed for the light sample in the reverse setup, with the heavy and medium proteins incubated with the E-3810 resin and nonderivatized agarose, respectively. After a second overnight incubation, the resin was washed three times using lysis buffer and twice with the high-stringency buffer. The resins incubated with light, medium, and heavy samples were mixed at the last washing step. To elute proteins, we incubated the resin at 99 °C for 10 min in 60 μl of LDS Sample Buffer 4X (Invitrogen), DTT 50 mm.
Saturation Assay
1 mg of protein extracts from cells labeled with Arg10 and Lys8 and 3 mg of protein extract from light-labeled A2780 cells were incubated overnight at 4 °C with 40 μl of resin slurry. The subsequent washing steps, light:heavy sample mixing, and elution of proteins bound to the resin were performed as described in the preceding paragraphs.
SDS-PAGE and Protein Digestion
30 μl of proteins eluted from the resin were separated in the 200–20-kDa range on a 4–12% gradient Bis-Tris mini gel (Invitrogen). For the shotgun analysis of A2780 cell lysates, 50 μg of light-labeled A2780 cell lysate were loaded on gel. After Coomassie staining (Colloidal Blue Staining Kit, Invitrogen), each lane was cut in slices corresponding to regions of different molecular weights (9 slices for chemical proteomic assays, 20 slices for the shotgun analysis of the A2780 cell lysates). Gel slices were subsequently digested with trypsin according to a previously described protocol (16). Briefly, gel pieces were destained via alternating cycles of incubation with 50% acetonitrile/25 mm ammonium bicarbonate (NH4HCO3) solution followed by 100% acetonitrile for dehydration. Gel pieces were incubated for 60 min at 56 °C with 10 mm dithiothreitol in 50 mm NH4HCO3 for cysteine reduction, and this was immediately followed by incubation with 55 mm iodoacetamide in 50 mm NH4HCO3 for 45 min at room temperature and in the dark for cysteine alkylation. After the iodoacetamide had been discarded, gel pieces were rinsed several times in 50 mm NH4HCO3 and finally incubated in 100% acetonitrile for dehydration. About 1 μg of trypsin (Promega, Madison, WI) dissolved in 50 mm NH4HCO3 was incubated with each gel slice. After overnight digestion at 37 °C, the tryptic mixture was acidified with 2 μl of 50% trifluoroacetic acid solution and collected. Gel pieces were incubated for 10 min with 30% acetonitrile, 3% trifluoroacetic acid, and the solution was collected and pooled with the initial peptide mixture. Gel pieces were incubated for an additional 10 min with 100% acetonitrile, and the solution was pooled with the initial one. Eluted peptides were evaporated on a vacuum concentrator to about 5 μl. The concentrated peptides were solubilized in 100 μl of 0.1% formic acid, desalted, and concentrated using reverse-phase C18 handmade nano-columns (StageTips) (17). Samples loaded on C18 StageTips were eluted with 80% acetonitrile, lyophilized, and resuspended in 10 μl of 0.1% formic acid for LC-MS/MS analysis.
LC-MS/MS Analysis
Nanocolumns (capillary columns; 15 cm long, 75-μm inner diameter, 350-μm outer diameter) were prepared by being packed with C18 resin (ReproSil C18-AQ, 3 μm, Dr. Maisch, Germany). 5 μl of desalted peptides were injected on column at 500 nl/min and separated via nanoflow liquid chromatography on an Agilent 1100 Series LC system (Agilent Technologies, Santa Clara, CA) online with an LTQ-FT Ultra mass spectrometer (Thermo Scientific). The gradient used for peptide separation was applied at a flow rate of 250 nl/min and consisted of 0%–40% solvent B (0.1% formic acid, 80% acetonitrile) over 90 min, followed by 40%–60% for 10 min and then 60%–80% over 5 min. The mass spectrometer was controlled by Xcalibur software (Thermo Scientific) and operated in data-dependent acquisition mode so as to automatically switch between MS and MS/MS scans. Survey full-scan MS spectra were acquired by the Fourier transform detector from 400 to 1500 m/z, and then the five most intense ions with charges of 2+ to 4+ were selected for fragmentation in the linear ion trap by means of collision-induced dissociation. The collision energy was set at 35 eV. In the LTQ-FT Ultra, full-scan MS spectra were acquired at a target value (automatic gain control) of 106 ions, with a resolution equal to 105 at 400 m/z.
Protein Identification and Quantification
Acquired spectra were matched in the human IPI database, version 3.68 (87,083 entries), by MaxQuant (8), version 1.2.0.18, which performed peak list generation, protein identification via the Andromeda search engine (18), and protein quantitation based on SILAC. The selected protease was trypsin (cleaving at the C terminus of Lys and Arg, unless followed by Pro), with a maximum of two missed cleavages allowed. Cysteine carbamidomethylation and methionine oxidation were set as fixed and variable modifications, respectively. The mass tolerance was set at 10 ppm for MS spectra (Fourier transform MS) and 0.5 Da for MS/MS spectra (ion trap MS). Arg10 and Lys8 were selected as “heavy” labels for standard two-channel SILAC (multiplicity = 2), with the addition of Arg6 and Lys4 as “medium” labels in the case of triple SILAC (multiplicity = 3). Protein and peptide false discovery rates, calculated as described previously (8), were set at 1%. Protein isoforms listed in the IPI human database v3.68 were considered as distinct, individual proteins.
Unless explicitly stated otherwise, each assay was performed in forward and reverse setups, and these were considered replicate analyses and indicated as separate experiments in the experimental design template of MaxQuant. For each assay, forward and reverse setups were analyzed together to increase the number of identified peptides, using the “match between runs” option. In the experimental design template, the different slices from one gel lane were indicated as “fractions” for automatic pooling by MaxQuant. The values set for the remaining parameters of MaxQuant were the default ones.
After MaxQuant analysis, proteins identified by at least two peptides, of which at least one was unique, were considered as high-quality identifications and were further analyzed. For the iBAQ calculation and the saturation assay, proteins identified based on at least two unique peptides were considered. In the case of redundancy in protein identification (peptides matching multiple protein groups), we reported the leading protein in the “protein groups” output table from MaxQuant. Normalized ratios were used for all assays, with the lone exception of the saturation assay, because a protein ratio distribution centered on 1 was not expected in such an experiment. The abundance of the proteins identified by the shotgun analysis of the A2780 cell lysates was estimated by enabling the iBAQ LogFit function present in MaxQuant (19). Perseus package version 1.2.0.16 (18) was used for the annotation of Gene Ontology terms (e.g. kinase activity, ATP binding) for the identified proteins.
Detection of E-3810 Targets via Competition Assay
Significant differences in ratios from samples A to D were evaluated using H/L ratio values in the forward setup and L/H ratio values in the reverse setup. A script for R software was developed to test trends of ratio variation based on linear regression methods. Linear trends of ratio variation (A > B > C > D) were detected via linear regression. Plateau trends of ratio variation (A > B ≈ C ≈ D) were detected by using a modified version of the linear regression method that computed a linear regression between the ratio value obtained in sample A and the average ratio values obtained in samples B, C, and D. Such a trend would highlight proteins already competed at low excess fold concentrations of free, nonderivatized E-3810, which were expected to show similar ratio values in samples B, C, and D. The linear and plateau tests evaluated the strength of the association and also the goodness of fit of the data to highlight either linear or plateau-shaped ratio variation from sample A to sample D. Proteins showing a decreasing ratio with p values of <0.05 in both forward and reverse setups that were quantified in at least three out of four samples were considered selectively competed by free E-3810 in the competition assay. The R code implemented for linear regression analysis is available upon request.
The ratio distribution in sample A was used to set the thresholds defining outlier ratio values. The distribution of the normalized ratio values for sample A, in which no competition occurred, was centered on 1, with 5th through 95th percentile values of 0.732–1.368 (H/L ratio) in the forward assay and 0.809–1.352 (L/H ratio) in the reverse assay (supplemental Fig. S5). Proteins characterized by ratio values between the 5th and 95th averaged percentiles (0.770 and 1.360, respectively) in sample A were considered as “non-outlier” proteins and were further analyzed (919 proteins; supplemental Data S1). Out of the 919 proteins present in the dataset, only proteins satisfying the following criteria were considered genuine E-3810 targets: (i) negative slope (either linear or plateau slope coefficient < 0) with an associated p value < 0.05; and (ii) outlier ratio value in the highest competition condition (sample D ratio value < 0.770 in both reverse and forward setups).
Clustering Analysis
The proteins detected as putative E-3810 targets in the competition assay were grouped via unsupervised clustering based on the fuzzy c-means algorithm using GProX, a graphic interface of the software R (20). The fuzzy c-means algorithm associates each protein with a cluster with a given likelihood (i.e. the membership value). The membership value indicates how well a given protein fits the consensus profile and allows color-coding of cluster graph items according to their goodness of fit to the cluster consensus profile. Protein names, together with the ratio values derived from forward and reverse experiments (respectively, H/L and L/H ratios), were loaded into GProX as two independent experiments. Forward and reverse experiments were analyzed together, with the number of clusters set at three, to detect proteins competed at the three different concentrations of free inhibitor used in the competition assay, as described above. The following parameters were selected: no value standardization required, limits for threshold regulation = 1.0. Other parameters were left as the defaults, such as fuzzification = 2 and number of algorithm iterations = 100.
Biochemical Assays
Kinase selectivity was measured using the “Kinase Profiler” service offered by Millipore, based on a radiometric filter-binding assay. Briefly, five different concentrations of E-3810 (0.1 μm, 0.3 μm, 1 μm, 3 μm, and 10 μm) were tested using an ATP concentration corresponding to the Michaelis–Menten constant (Km) of each selected kinase, as under the condition [ATP] = Km, the general formula to compute the dissociation constant for the inhibitor (Ki)—that is, Ki = IC50 × Km/([ATP] + Km)—can be simplified to Ki = IC50/2. Therefore, in our assay the measured IC50 was directly proportional to Ki. Kinase inhibition was expressed as the percentage of activity determined in the absence of inhibitor versus in the presence of inhibitor. Sigmoid concentration–response curves plotting percentages of effect versus the log concentration of E-3810 were analyzed using nonlinear regression analysis using GraphPad Prism (version 4.0) to obtain IC50 values, and Ki values were computed using the formula mentioned above.
Western Blot Analysis of DDR2 and Phosphotyrosine upon E-3810 Treatment of 293T Cells
DDR2 was overexpressed in 293T cells through the use of a pCDNA5 plasmid containing the human DDR2 gene (Origene). After transfection, cells were starved for 24 h and then treated for 120 min with collagen type I (20 μg/ml) to induce DDR2 phosphorylation and thus activation (21). After collagen treatment, proteins were extracted in lysis buffer containing 50 mm HEPES, pH 7.0, 250 mm NaCl, 1% Nonidet P-40. Protease inhibitors (Complete-EDTA Free, Roche) and phosphatase inhibitors (10 mm NaF, 10 mm sodium pyrophosphate, 4 mm sodium orthovanadate; Sigma) were freshly added to the buffer before use. Proteins were separated via SDS-PAGE on 8% acrylamide gel, immunoblotted on polyvinylidene fluoride membrane (100 V, 75 min), and incubated overnight with antibodies against phosphorylated tyrosine (Millipore, clone 4G10) or DDR2 (Santa Cruz, sc7455). Signals were detected and images were acquired as described above.
Cell Proliferation, Cell Cycle Progression, and Apoptosis Rate Evaluation
HCC-366 cells were cultured in 24-well plates (16,500 cells/ml/well) for the cell proliferation assay or in 100-mm cell culture dishes (5 × 105 cells/ml/well) for the cell cycle and apoptosis assays. Forty-eight hours after seeding, cells were treated with different concentrations of E-3810 dissolved in 0.25% v/v DMSO to final concentrations of 2, 4, 8, 16, and 32 μm. For each condition, the final concentration of DMSO was 0.25% v/v. Control cells were treated with 0.25% v/v DMSO in PBS. To assess cell proliferation, six days after treatment, 5× concentrated 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (2.5 mg/ml in PBS) was added to cultured cells at a final concentration of 0.5 mg/ml. After 3 h of incubation, the medium was discarded and cells were lysed in 1 ml of 100% DMSO. The absorbance of 250 μl of cell lysate was read using a Glomax luminometer (Promega) at a 565-nm wavelength in a standard 96-well plate. Data are reported as relative luminescence values (treated/control cells).
For the evaluation of cell cycle progression and apoptosis rate, the cells attached to the dishes and floating in the culture medium six days after treatment with 2, 4, 8, 16, or 32 μm E-3810 were harvested and washed in PBS. After centrifugation at 2000 rpm for 5 min at 4 °C, cell pellets were resuspended in 250 μl of PBS and fixed by the dropwise addition of 750 μl of ice-cold ethanol during vortexing. Cells were left in fixative at 4 °C overnight. After being washed with 1% BSA in PBS, cells were stained using 1 ml of propidium iodide (50 μg/ml) supplemented with RNase at a final concentration of 250 μg/ml. Stained cells were analyzed by FACScan (BD Biosciences) to evaluate the percentage of apoptotic cells and to profile cell phases.
RESULTS
Preparation of Immobilized E-3810 on Agarose Beads
The core of the E-3810 structure (Fig. 1A and supplemental Table S1, compound 1) is a quinoline nucleus presenting substitutes at positions 4, 6, and 7. Although no crystallographic data are available on E-3810–kinase complexes, structurally related kinase inhibitors are known to interact with a “hinge region” backbone NH (Cys 919 for VEGFR-2, Met1160 for c-MET) through a critical hydrogen bond with the quinoline nitrogen, pointing the substituent at position 7 toward the solvent front (22–25). Assuming a similar binding mode for E-3810, we attached the linker for solid support at this position, in order to minimize the impact on binding affinity to the ATP kinase pocket. Therefore, compound 4 (supplemental Fig. S1) was synthesized as the precursor for attachment to the solid support and tested in in vitro kinase assays to assess its inhibitory activity against the known targets VEGFR-1, VEGFR-2, VEGFR-3, FGFR-1, and FGFR-2. A negligible decrease of inhibitory potency was observed for all targets (e.g. IC50 equal to 130 nm, compared with 77 nm of E-3810 for FGFR2; supplemental Table S1), which enabled us to use the E-3810 derivative for immobilization on agarose resin (compound 8 in supplemental Fig. S1).
Fig. 1.

Characterization of immobilized E-3810. A, E-3810 structure, in free form and after immobilization on agarose resin. B, E-3810 maintains the ability to bind FGFR2 after immobilization on resin. Immunoblot analysis against FGFR2 in A2780 protein extract (input, IN), flow-through (FT), and eluates (EL) obtained by affinity chromatography with immobilized E-3810 (15 μl of resin slurry for 1 mg of extract). Non-derivatized agarose beads were used as a negative control (empty resin). FT contained proteins not retained by the E-3810 resin; EL contained proteins captured by the resin and subsequently eluted. C, increasing volumes of E-3810 resin captured increasing amounts of FGFR2. Immunoblot analysis against FGFR2 for IN, FT, and EL obtained from the affinity chromatography experiments performed by incubating 1 mg of protein extract with 3, 15, or 40 μl of E-3810 resin slurry.
We tested the target affinity of the immobilized E-3810 by assessing its binding to the known target FGFR2. Whole cell extracts from the A2780 cells, which express the receptor, were incubated with the conjugated resin. After incubation and washing, proteins retained on the resin were eluted and the presence of FGFR2 was assessed via immunoblotting (Fig. 1B and supplemental Fig. S3A). FGFR2 was efficiently and specifically retained on the E-3810 resin, confirming that the immobilization of the drug did not impair its target affinity. Six replicate incubation experiments captured very similar amounts of FGFR2, demonstrating the reproducibility of our affinity purification procedure (supplemental Figs. S3B and S3C). Increasing amounts of resin were then tested in order to assess the binding capacity of the resin, and increasing volumes of resin captured greater amounts of FGFR2 (Fig. 1C). On the basis of these results, we chose 40 μl of E-3810 resin slurry, corresponding to 120 nmol of E-3810, for the chemical proteomics assays.
In order to define the optimal conditions for a competition assay in which the standard affinity chromatography using the E-3810 resin was integrated with competition experiments using increasing doses of the free, soluble drug, we co-incubated protein extracts with E-3810 resin in the presence of increasing amounts of the competitor (supplemental Fig. S4A). We found that free E-3810 progressively reduced FGFR2 binding at increasing doses; this result allowed us to set the range of concentrations of soluble drug capable of competing with specific binders from the E-3810 resin (supplemental Fig. S4B).
Identification of E-3810 Targets via Serial Competition Assay
When chemical proteomics is used to perform competition assays in combination with SILAC (7), one is able to discern genuine drug targets from background binders that do not compete with the soluble drug (4, 5). We selected human ovarian carcinoma A2780 cells as an ideal model system for carrying out a comprehensive target-deconvolution analysis for E-3810, based on several observations: (i) they are highly responsive to E-3810 treatment in anti-proliferative in vitro assays (supplemental Fig. S2A), (ii) they express FGFR-2 at levels comparable to those of other transformed cell lines bearing FGFR-2 gene amplification (supplemental Fig. S2B), and (iii) they are amenable to efficient metabolic labeling with isotope-encoded amino acids, having a fast replication rate and growing in SILAC medium.
We performed a composite in-batch competition experiment in which light and heavy A2780 extracts where incubated with E-3810 resin in the presence of increasing concentrations of soluble E-3810 (Fig. 2A). The experimental design is illustrated in Fig. 2A: four parallel SILAC experiments were carried out, each of them containing a heavy and a light channel combination (samples A, B, C, and D). Light-labeled (L) cell extracts were incubated with 40 μl of E-3810 resin slurry, and heavy-labeled (H) cell extracts were incubated with the same volume of E-3810 resin in the presence of increasing amounts of free, soluble E-3810. In sample A, no competitor was added. Following sample incubation and the washing steps, the resins derived from the light and heavy channel pairs within each experiment were mixed, and the proteins were eluted, analyzed via geLC-MS (SDS-PAGE fractionation followed by LC-MS/MS), and quantified using MaxQuant software (8). Each multiplex competition assay was performed in duplicate, in forward and reverse setups, with label swapping. The acquired MS spectra matched 1566 proteins in the human IPI database (supplemental Dataset S1), reduced to 1305 proteins in common with the two experiments upon the application of filtering criteria to ensure identification quality (supplemental Dataset S1).
Fig. 2.

Identification of E-3810 targets via SILAC-based chemical proteomics competition assay. A, schematic view of the experimental design, in the forward SILAC setup. Light- and heavy-labeled samples are color-coded, respectively, in black and red. Cell extracts from SILAC-labeled A2780 cells were incubated with 40 μl of E-3810 resin slurry. Heavy lysates were co-incubated with increasing concentrations of E-3810, which competes for target binding; no competition was performed in sample A. After the incubation and washing steps, the resins of the light and heavy lysates were mixed to form four distinct SILAC samples. For each sample, eluted proteins were separated via SDS-PAGE, digested, and analyzed via LC-MS/MS. B, expected SILAC ratio readout in samples A–D for specific E-3810 interactors, as compared with unspecific background binders. In the forward experiment, specific interactors are expected to show a progressive H/L ratio decrease due to the competition with E-3810. Background binders are not competed by the drug and thus maintain a constant ratio value of about 1 throughout the serial competition. Heavy and light channels are swapped in the reverse experiment.
The trends of the SILAC ratio modulation upon competition were then used to discern genuine interactors from unspecific binders. Genuine interactors were expected to be specifically competed by increasing doses of E-3810, thus showing a progressive H/L ratio (L/H in the reverse assay) decrease from sample A to sample D (Fig. 2B). Conversely, background interactors bound the resin with similar efficiency, independently of the presence of increasing amounts of competing drug, and thus had protein H/L ratio values close to 1 across samples A–D (Fig. 2B). Statistically significant trends of protein ratio decrease were estimated using two different regression analysis methods, as described in “Experimental Procedures.”
The protein ratio distribution of sample A, in which no competition occurred in either the L or the H channel, was used to select as non-outliers those proteins with SILAC protein ratio values between the 5th and the 95th average percentiles of the normal distribution. A total of 919 candidates were thus selected whose ratio trends were monitored in competition experiments B–D (supplemental Dataset S1). Within this set, 22 proteins displayed a decreasing trend of SILAC ratio from sample A to sample D and a ratio value in sample D below the statistically significant threshold of two sigma levels (5%), so they were considered genuine E-3810 binders (Table I and supplemental Fig. S6). The remaining proteins were considered as background (supplemental Fig. S7).
Table I. Candidate E-3810 targets identified by the chemoproteomic competition assay. Proteins are grouped in three categories: protein kinases, proteins involved in nicotinamide/nicotinate catalysis, and other proteins. Each protein has an associated p value, used to evaluate the ratio trend from sample A to sample D as either linear (slope test) or logarithmic (plateau). Proteins also found to be putative E-3810 targets in the Kd assay (Table II) are marked with “+”.
| Gene name | UniProt I.D. | Protein name | Slope test | Simple p value | Plateau test | Plateau p value | Present in Table II |
|---|---|---|---|---|---|---|---|
| Protein kinases | |||||||
| PDGFRA | P16234–1 | Platelet-derived growth factor receptor α | −0.26 | 2.5E-02 | −0.86 | 6.8E-05 | + |
| CARDIAK | O43353–1 | Receptor-interacting serine/threonine-protein kinase 2 | −0.24 | 3.3E-02 | −0.82 | 1.9E-05 | + |
| DDR2 | Q16832 | Discoidin domain-containing receptor 2 | −0.27 | 1.6E-02 | −0.86 | 1.3E-07 | + |
| LYNa | Q6NUK7 | LYN protein | −0.31 | 2.9E-03 | −0.86 | 1.3E-04 | + |
| CSBP (1)a | Q16539–2 | Cytokine suppressive anti-inflammatory drug-binding protein | −0.39 | 8.9E-03 | −0.51 | 1.2E-01 | |
| YES | P07947 | Tyrosine protein kinase Yes | −0.28 | 5.9E-06 | −0.53 | 4.5E-02 | + |
| SRC | P12931–2 | Proto-oncogene tyrosine-protein kinase Src | −0.30 | 2.1E-06 | −0.55 | 4.8E-02 | |
| EPHA2 | P29317 | Ephrin type-A receptor 2 | −0.26 | 2.6E-04 | −0.43 | 1.1E-01 | |
| CSNK1E | B3KRV2 | Casein kinase I isoform ϵ | −0.28 | 1.1E-03 | −0.65 | 7.9E-03 | |
| MAP4K4 | O95819–3 | HPK/GCK-like kinase HGK | −0.20 | 9.5E-03 | −0.35 | 1.4E-01 | |
| CSBP (2) | Q16539–1 | Cytokine suppressive anti-inflammatory drug-binding protein | −0.33 | 3.8E-06 | −0.71 | 1.1E-02 | + |
| Nicotinamide/nicotinate catalysis | |||||||
| NMOR2 | P16083 | NAD(P)H dehydrogenase [quinone] 2 | −0.28 | 6.5E-05 | −0.46 | 1.1E-01 | + |
| NMOR1 | P15559 | NAD(P)H dehydrogenase [quinone] 1 | −0.38 | 2.1E-04 | −0.65 | 9.2E-02 | + |
| QPRT | Q15274 | Nicotinate-nucleotide pyrophosphorylase [carboxylating] | −0.21 | 4.0E-02 | −0.20 | 5.1E-01 | + |
| Other proteins | |||||||
| GBAS | O75323 | Protein NipSnap homolog 2 | −0.28 | 8.5E-04 | −0.47 | 1.2E-01 | |
| NIPSNAP1 | Q9BPW8 | Protein NipSnap homolog 1 | −0.30 | 2.3E-04 | −0.46 | 1.5E-01 | + |
| RIE2 | B3KWY9 | Highly similar to RING finger protein 10 | −0.14 | 2.1E-02 | −0.32 | 7.5E-02 | |
| MTIF2 | P46199 | Translation initiation factor IF-2, mitochondrial | −0.17 | 2.2E-02 | −0.15 | 5.0E-01 | |
| RABGGTA | Q92696 | Geranylgeranyl transferase type II subunit α | −0.15 | 8.1E-03 | −0.21 | 2.6E-01 | |
| UBF | P17480–1 | Nucleolar transcription factor 1 | −0.10 | 4.1E-02 | −0.08 | 6.2E-01 | |
| KIAA0197 | Q12769–1 | 160 kDa nucleoporin | −0.15 | 2.1E-03 | −0.20 | 2.3E-01 | |
| TRDX | P10599 | Thioredoxin | −0.10 | 2.9E-05 | −0.19 | 3.5E-02 | + |
a Fragmentation spectra for proteins identified based on two peptides, of which one unique peptide is reported in supplemental Fig. S10.
The ratio modulation across experiments A–D of the 22 candidates was compared using GProx (20), generating three clusters (Fig. 3 and supplemental Table S2). Cluster 1 contained proteins whose SILAC ratio decreased in the presence of the lowest dose of competitor (sample B), suggesting that they had the highest affinity for the non-immobilized form of E-3810. This group included platelet-derived growth factor receptor α (PDGFRA), DDR2, the tyrosine-protein kinase Lyn (LYN), and receptor-interacting serine/threonine-protein kinase 2 (CARDIAK). Cluster 2 contained eight proteins that were competed by an intermediate dose of the drug (sample C), including the proto-oncogene tyrosine-protein kinase Src (SRC), the tyrosine-protein kinase Yes (YES), ephrin type-A receptor 2 (EPHA2), and cytokine suppressive anti-inflammatory drug-binding protein (CSBP) (isoform 2). Non-kinase proteins also belonged to this group, such as NipSnap homolog-1 and -2 (NIPSNAP1 and NIPSNAP2). Cluster 3 contained seven proteins that either were competed at the highest dose of E-3810 (sample D) or showed a mild but constant ratio modulation; we considered these as low-affinity E-3810 interactors and found only one kinase within this class, the HPK/GCK-like kinase HGK (MAP4K4).
Fig. 3.
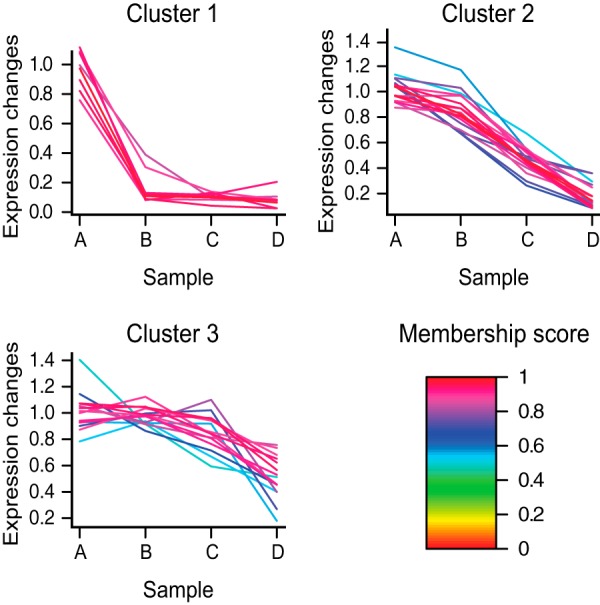
Unsupervised clustering analysis for the putative E-3810 targets selected through the competition assay. Proteins were grouped according to the trends of ratio modulation from sample A to sample D upon E-3810 competition. Cluster 1 contained proteins competed with the lowest concentration of E-3810, suggesting the highest affinity for the free form of the drug. Cluster 2 contained proteins competed at a medium concentration of E-3810, representing medium-affinity targets. Cluster 3 contained proteins competed only by the highest doses of free E-3810. Color-coded membership values, indicating the goodness of fit to the three clusters, are indicated in the legend. The members of each cluster are listed in supplemental Table S2.
Nineteen out of the 22 candidate targets showed highly reproducible trends of SILAC ratio modulation between the forward and reverse assays that resulted in identical clustering (supplemental Table S2).
Estimation of Target Affinities for E-3810 via Chemoproteomic Kd Assay
To validate the candidate targets detected in the competition assay and to further characterize their binding to the drug, we carried out a variant of the SILAC-based chemical proteomics assay referred to as the Kd assay, because it enabled calculation of the dissociation constant of the immobilized drug for each putative binder (4). Fig. 4A shows a schematic of the design of the triple SILAC experiment, which was carried out in two replicates, with isotope-labeled amino acid swapping: the light (L) protein extract from A2780 cells was incubated with the E-3810 resin, the medium (M) lysate was incubated with non-derivatized resin as a control, and the heavy (H) protein extract was subjected to two subsequent rounds of incubation with the E-3810-conjugated resin, so that the flow-through from the first incubation was re-incubated with fresh resin. The three samples were mixed, and then captured proteins were eluted and analyzed via geLC-MS. In the MS spectra, the peptides were present as L, M, and H SILAC triplets. With such an experimental design, the M/L ratio in the forward assay and the corresponding M/H ratio in the reverse assay enabled measurement of the binding specificity for E-3810 resin relative to the control resin; thus they were named “specificity ratios.” Similarly, the H/L ratio in the forward assay and the corresponding L/H ratio in the reverse assay enabled estimation of the affinity of each protein for the immobilized E-3810 and were termed “affinity ratios.”
Fig. 4.

Kd chemoproteomic assay with immobilized E-3810. A, schematic view of the experimental design used to calculate the dissociation constant of candidate E-3810 targets for the immobilized drug (Kd imm). Light-, medium-, and heavy-labeled proteins are color-coded in black, green, and red, respectively. In the forward experiment, the light protein extract was incubated with E-3810 resin, the medium-labeled lysate was incubated with non-derivatized agarose resin, and the heavy extract was subjected to two subsequent rounds of incubation with the E-3810 resin. After the incubation and washing steps, the resins from the three experiments were mixed to generate light:medium:heavy SILAC triplets. Genuine E-3810 targets are expected to show specificity ratios (M/L in the forward experiment) < 1, whereas the affinity ratios (H/L in the forward experiment) are used to compute the value of Kd imm E-3810. B, affinity ranking of the putative kinases targeted by E-3810, based on Kd imm E-3810.
Upon MaxQuant analysis of the two replicates, 2314 proteins were identified; this number was reduced to 1383 after the application of filtering criteria to ensure identification quality (supplemental Dataset S2). The distributions of the specificity and affinity ratio values for the proteins identified in the forward and reverse assays showed that the vast majority of the proteins had both specificity and affinity ratio values close to 1, including a subset of kinase proteins, highlighted as red squares in Figs. 5A and 5B. We distinguished genuine binders of the immobilized E-3810 from background contaminants by selecting only the proteins below the 10th percentile of the specificity ratio distribution and further analyzing them. Then, we applied an additional filter based on the affinity ratio distribution: assuming that genuine interactors bind the derivatized resin during the first round of incubation and thus, under conditions where the resin is not saturating, should bind the resin to a lesser extent in the second round of incubation (4), we selected as candidates those proteins with an affinity ratio less than 1.0. Twenty-four potential E-3810 interactors were selected using these two cutoffs with a broad range of calculated Kd values (Table II). Although the known target FGFR2 was quantified by only one ratio count (MaxQuant RC = 1), which provided borderline confidence, it was included in Table II as a reference both because it was identified with high confidence and because its specificity and affinity ratio values classified it as an E-3810 interactor. Overall, the putative E-3810 targets showed high consistency between the forward and reverse assays in terms of the specificity and affinity ratios, thus highlighting the robustness and reproducibility of our strategy (Figs. 5C and 5D). Remarkably, 11 out of 25 proteins were kinases, of which 60% were also identified as putative E-3810 targets in the competition assay (highlighted in Table I). Among the remaining 14 non-kinase proteins, 36% confirmed the result from the previous assay (NMOR2, QPRT, NIPSNAP1, NMOR1, and TRDX).
Fig. 5.

Scatterplot of protein ratios obtained from the Kd chemoproteomic assay. A, H/L and M/L ratios in the forward assay. B, corresponding L/H and M/H ratios in the reverse assay. C, Specificity ratio values for the 25 putative targets (M/L and corresponding M/H ratios in the forward and reverse assays, respectively). Lower values (M/L and M/H ≪ 1) indicate selective binding to immobilized E-3810 as compared with empty resin. D, affinity ratio values for the 25 putative targets (H/L and corresponding L/H from the forward and reverse assays, respectively). Lower protein ratio values (≪1) indicate strong affinity for the immobilized E-3810. Black dots represent the 1305 proteins identified and quantified in the chemical proteomics Kd assay; red diamonds indicate kinase proteins. Dotted lines specify the thresholds used for E-3810 target selection.
Table II. Putative E-3810 interactors identified through the Kd assay. Proteins are grouped in distinct categories: protein kinases, other ATP binders, proteins involved in nicotinamide/nicotinate catalysis or 5′-AMP catalysis, and other proteins. H/L and L/H ratios values respectively measured in the forward and reverse triple SILAC experiments are reported. The average protein ratio values with the associated standard deviations are also reported, together with the calculated Kd imm E-3810. Proteins are sorted by increasing Kd value within each group. FGFR2 has been included in the table as a reference because it is a known E-3810 target, although quantified only with a SILAC peptide pair.
| Gene name | UniProt I.D. | Protein name | H/L DIR ratio | L/H REV ratio | Average ratio | S.D. | Kd imm E-3810 (μm) | |
|---|---|---|---|---|---|---|---|---|
| Protein kinases | PDXK | O00764–1 | Pyridoxal kinase | 0.064 | 0.071 | 0.068 | 0.005 | 16.1 |
| DCK | P27707 | Deoxycytidine kinase | 0.090 | 0.107 | 0.099 | 0.012 | 24.4 | |
| CARDIAK | O43353–1 | CARD-containing IL-1 β-converting enzyme-associated kinase | 0.124 | 0.163 | 0.143 | 0.028 | 37.1 | |
| PDGFRA | P16234–1 | Platelet-derived growth factor receptor α | 0.157 | 0.218 | 0.188 | 0.043 | 51.3 | |
| FGFR2a,b | Q59F30 | Fibroblast growth factor receptor 2 | 0.314 | 0.416 | 0.365 | 0.072 | 127 | |
| DDR2 | Q16832 | Discoidin domain-containing receptor 2 | 0.283 | 0.520 | 0.401 | 0.168 | 148 | |
| DDR1 | Q08345–5 | Discoidin domain-containing receptor 1 | 0.226 | 0.629 | 0.428 | 0.285 | 166 | |
| LYN | Q6NUK7 | LYN protein | 0.410 | 0.488 | 0.449 | 0.055 | 181 | |
| YES | P07947 | Tyrosine protein kinase Yes | 0.449 | 0.601 | 0.525 | 0.107 | 245 | |
| CSBP (2) | Q16539–1 | Cytokine suppressive anti-inflammatory drug-binding protein | 0.689 | 0.902 | 0.795 | 0.151 | 864 | |
| Other ATP binders | GLNS | P15104 | Glutamine synthetase | 0.081 | 0.137 | 0.109 | 0.040 | 27.2 |
| ACTBL2a | Q562R1 | β-Actin-like protein 2 | 0.221 | 0.193 | 0.207 | 0.020 | 58.0 | |
| YARS | P54577 | Tyrosyl–tRNA ligase | 0.328 | 0.439 | 0.383 | 0.079 | 138 | |
| Nicotinamide/nicotinate catalysis | NMOR2 | P16083 | NAD(P)H dehydrogenase [quinone] 2 | 0.354 | 0.486 | 0.420 | 0.094 | 161 |
| QPRT | Q15274 | Nicotinate-nucleotide pyrophosphorylase [carboxylating] | 0.397 | 0.522 | 0.459 | 0.089 | 189 | |
| NMOR1 | P15559 | NAD(P)H dehydrogenase [quinone] 1 | 0.375 | 0.725 | 0.550 | 0.247 | 271 | |
| 5′-AMP catalysis | PDE12 | Q6L8Q7–1 | 2,5-phosphodiesterase 12 | 0.197 | 0.641 | 0.419 | 0.314 | 160 |
| DPDE3a | Q08499–10 | cAMP-specific 3,5-cyclic phosphodiesterase 4D | 0.346 | 0.562 | 0.454 | 0.153 | 185 | |
| Other proteins | EPLIN | Q53GG0 | Epithelial protein lost in neoplasm β variant | 0.113 | 0.101 | 0.107 | 0.009 | 26.6 |
| EFHD2 | Q96C19 | EF-hand domain-containing protein D2 | 0.154 | 0.092 | 0.123 | 0.044 | 31.2 | |
| BIT1 | Q96ME4 | Peptidyl-tRNA hydrolase 2, mitochondrial (EC 3.1.1.29) | 0.139 | 0.276 | 0.207 | 0.097 | 58.0 | |
| NIPSNAP1 | Q9BPW8 | Protein NipSnap homolog 1 | 0.458 | 0.612 | 0.535 | 0.108 | 255 | |
| GGTB | P53611 | Geranylgeranyl transferase type II subunit β | 0.839 | 0.904 | 0.871 | 0.046 | 1500 | |
| TRDX | P10599 | Thioredoxin | 0.926 | 0.969 | 0.948 | 0.031 | 4000 | |
| CGI-118 | Q96GC5 | 39S ribosomal protein L48, mitochondrial | 0.981 | 0.990 | 0.986 | 0.006 | 15,500 |
a Fragmentation spectra for proteins identified with two peptides, a unique one of which is presented in supplemental Fig. S11.
b Quantified by one ratio count in both forward (DIR) and reverse (REV) assays, for a total of two quantifications.
To estimate the affinity of the putative targets for the immobilized E-3810, we measured the dissociation constant for the immobilized E-3810 (Kd imm E-3810) using the ratio r, which corresponded to the average of the affinity H/L and L/H ratio values, indicating the amount of target protein retained in the second incubation relative to first. In this kind of chemoproteomic assay, when non-saturating and equilibrium conditions are reached and the exact concentration of the immobilized compound is known, the r ratio of each protein can be used to calculate the dissociation constant for the immobilized drug (Kd imm drug) of each binder (15). Calculated Kd imm E-3810 values are listed in Table II. Within the group of protein kinases, the Kd imm E-3810 values spanned the micromolar range (from 16.1 μm to 863.5 μm), with pyridoxal kinase and deoxycytidine kinase having the lowest Kd values, followed by CARDIAK, PDGFRA, FGFR2, DDR2, DDR1, LYN, YES, and CSBP (isoform 2). Non-kinase proteins showed a broader range of Kd imm E-3810 values varying from 26.6 μm to 15.5 mm.
To ensure that the immobilized E-3810 was present in molar excess relative to its targets in the Kd assay—a crucial condition for the correct estimation of the Kd imm E-3810 values—we carried out a double SILAC affinity chromatography experiment in which 40 μl of E-3810 resin were incubated with the same amount of protein extract used in the assay (1 mg, heavy labeled) and with a 3-fold excess (3 mg, light labeled) (supplemental Fig. S8A). Upon geLC-MS analysis, 673 proteins identified with at least two unique peptides and quantified with at least two SILAC ratios were considered (supplemental Dataset S3). The H/L ratio distribution was centered on an H/L value of 0.66 ± 0.20 (supplemental Fig. S8B), and all the putative E-3810 targets showed an average value of 0.34 ± 0.11 (supplemental Fig. S8C). Whereas H/L ratio values close to 1.0 would indicate that 1 mg of protein extract was already saturating the E-3810 resin, our result indicates that the immobilized E-3810 was in molar excess relative to its targets in the Kd assay.
To rule out potential biases toward the most abundant binders in the Kd calculation, we estimated the abundance of proteins for the A2780 proteome through an in-depth shotgun analysis on light-labeled cells and then conducted iBAQ score calculation, which was developed for protein quantification in large-scale experiments (19). The iBAQ scores (computed for 5967 of 7140 identified proteins in supplemental Dataset S4) spanned 6 orders of magnitude (supplemental Fig. S9A), with the subgroup of the protein kinases covering the same dynamic range of protein abundance (supplemental Fig. S9B). Because our candidate targets were scattered throughout the iBAQ range, we could rule out the possibility that the Kd assay was biased toward either the most abundant proteins or the most abundant kinases.
These control experiments overall support the reliability of the triple SILAC Kd assay and the soundness of the affinity ranking based on calculated Kd imm values, which indicated pyridoxal kinase and deoxycytidine kinase as the highest affinity kinase proteins, followed by PDGFRΑ, CARDIAK, DDR2, DDR1, and LYN.
Validation of Novel E-3810 Targets in Biochemical Assays
To validate E-3810 activity toward the kinase proteins identified as putative binders by chemical proteomics, we tested the novel putative targets DDR2, LYN, CARDIAK, CSBP (isoform 2), EPHA2, YES, and SRC, together with the known targets FGFR2 and PDGFRA, in a biochemical assay measuring E-3810's ability to inhibit the enzymatic activity of the selected kinases, which provided the compound half-maximal inhibitory concentration (IC50) and the inhibitor constant (Ki) values—two parameters referring to the inhibition potency. Inhibition was observed for nearly all the tested kinases, with the only exception being SRC, confirming the results from the chemical proteomics experiments (Figs. 6A and 6B). As expected, the compound potently inhibited FGFR2 activity (Ki < 0.05 μm), followed by PDGFRA activity (Ki = 0.11 μm) (Fig. 6A). The Ki values obtained for DDR2, LYN, CARDIAK, CSBP (isoform 2), EPHA2, and YES ranged between 0.26 and 8 μm (Fig. 6A). These results are consistent with those from the cluster analysis (Fig. 3), in which the kinases more potently inhibited by E-3810 (PDGFRA, FGFR2, LYN, and DDR2) were grouped in cluster 1, whereas the kinases moderately inhibited by E-3810 (CSBP, SRC, and EPHA2) were gathered in cluster 2. Together the evidence suggests that the stronger the interaction between the enzyme and E-3810, the greater the inhibitory effect.
Fig. 6.
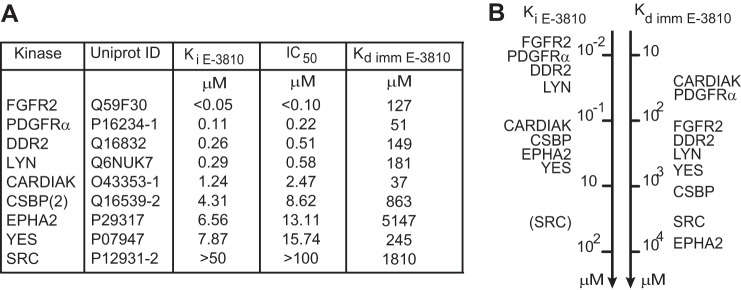
In vitro inhibition assay for selected E-3810 interactors. Eight kinase proteins were selected from among the candidate interactors identified in chemical proteomics experiments to assess whether E-3810 could inhibit their enzymatic activity; FGFR2 was included as a positive control. A, table showing inhibition constant (Ki E-3810) and half-maximal inhibitory concentration (IC50 E-3810) values calculated from the in vitro assay, with the corresponding dissociation constant calculated from the chemical proteomics assay (Kd imm E-3810). The value of Ki > 50 μm for SRC suggests that this kinase was not specifically inhibited by E-3810 at the concentrations tested in the assay. B, comparative ranking of the E-3810 targets based on Ki E-3810 and Kd imm E-3810 values, referring to the drug's efficacy and affinity, respectively.
DDR2 as a Novel E-3810 Target
DDR2, a tyrosine kinase receptor for collagen, was one of the highest affinity novel targets for E-3810 in our chemical proteomics study (Tables I and II) and was also validated in biochemical assays, which showed that E-3810 inhibited the enzymatic activity of the recombinant DDR2 with a lower Ki value relative to the other tested candidate kinase targets (Fig. 6). We therefore focused on DDR2 for follow-up experiments.
We overexpressed DDR2 in 293T cells via transient transfection and tested whether E-3810 in intact cells could directly inhibit receptor autophosphorylation, which is an indicator of receptor activation (26). The receptor was not phosphorylated in basal conditions, as previously shown (21), and stimulation with 20 μg/ml collagen for 2 h was sufficient to activate it (Fig. 7A). Co-incubation of 20 μg/ml collagen in the presence of increasing concentrations of E-3810 resulted in the reduction of DDR2 autophosphorylation, starting at the lowest tested concentration (2 μm), demonstrating that E-3810 is able to inhibit DDR2 activity in intact cells (Fig. 7A).
Fig. 7.

DDR2 is a novel target of E-3810. A, phosphorylation of DDR2 is inhibited by E-3810. In 293T cells overexpressing DDR2, collagen stimulation (20 μg/ml) induces activation of the receptor measured by the increase of its phosphorylated form, as shown by double immunoblot analysis using anti-DDR2 and anti-phosphotyrosine antibodies. Co-treatment with both collagen and E-3810 (2, 8, or 32 μm) for 120 min reduced DDR2 phosphorylation, confirming the inhibitory effect of the drug. Vinculin was used as a loading control. B, cell proliferation assay in HCC-366 cells. Treatment of HCC-366 cells with increasing concentrations of E-3810 (2, 4, 8, 16, and 32 μm) inhibited cell proliferation. C, cell cycle analysis of HCC-366 cells upon treatment with increasing doses of E-3810. D, percentage of apoptotic HCC-366 cells upon E-3810 treatment.
DDR2 has been involved in the control of cell proliferation and survival and is considered as a potential therapeutic target in cancer treatment (26–29). Activating mutations of DDR2 kinase activity have been found in different tumor types, and the knock-down of mutated DDR2 impairs proliferation and survival of the non-small-cell lung cancer HCC-366 cell line, suggesting that the inhibition of DDR2 can have a therapeutic effect (28). We evaluated the cell proliferation, apoptosis rate, and cell cycle progression of HCC-366 cells treated with different concentrations of E-3810. Consistent with its inhibitory effect on DDR2, E-3810 dramatically inhibited cell proliferation and induced apoptosis in HCC-366 cells in a dose-dependent manner, with a concomitant increase of the cell fraction in S and G2/M phases of the cell cycle (Figs. 7B–7D). These data support the hypotheses that DDR2 plays a key role in driving the proliferation and survival of tumor cells carrying its activating mutations and that pharmacological inhibition of DDR2 with E-3810 might have therapeutic potential in cancer treatment.
DISCUSSION
Methodologies based on quantitative chemical proteomics for the unbiased identification of proteins interacting with small molecules have been recently set up, but their application in the context of new drugs has been limited so far. Here, we applied a quantitative chemical proteomics approach to characterize the multi-kinase inhibitor E-3810, a novel anti-cancer drug currently undergoing Phase II clinical trials. As compared with more conventional in vitro kinase inhibition assays, chemical proteomics allows screening for E-3810 targets in more physiological conditions, where each potential target (wild type or mutated form) is present at physiological levels, bearing native post-translational modifications (e.g. phosphorylation) and with the possibility of interacting with its natural binders and cofactors at the appropriate stoichiometry. All these features modulate the final target activity, which in turn influences the inhibitory potential of the drug.
We used a SILAC approach to design composite chemoproteomic assays in which differentially labeled proteins interacting with the solid supported E-3810 were mixed and eluted simultaneously before undergoing identification via LC-MS. Compared with other stable-isotope labeling strategies, such as chemical labeling with iTRAQ or tandem mass tags, the advantage of SILAC relies on the fact that by omitting the protein/peptide chemical derivatization steps, it enables simpler workflows, with reduced risk of manipulation biases, increased accuracy in protein quantification, and, consequently, greater confidence in the discernment of specific binders.
Of the 22 proteins identified as genuine E-3810 interactors in the competition assay, 11 were kinase proteins, which we considered putative E-3810 targets. This group included the tyrosine kinases PDGFRA, DDR2, EPHA2, YES, LYN, and SRC and the serine/threonine kinases CARDIAK, CSNK1E, and CSBP (isoforms 1 and 2). Cluster analysis revealed that PDGFRA, DDR2, LYN, and CARDIAK were competed by the lowest concentrations of E-3810; hence they were classified as the highest affinity interactors. The same proteins, together with YES and CSBP (isoform 2), were confirmed as E-3810 interactors by the Kd assay.
FGFR2, the best-characterized target of E-3810 in in vitro biochemical assays and thus profiled as a positive control, was classified as an interactor in the Kd assay based on only one peptide pair, although it failed MS detection in the competition assay. Possible explanations for the low MS signal might be its sequence composition, membrane localization, and abundance; membrane proteins in general represent a challenge in chemical proteomics because proteins are extracted in non-denaturing conditions in order to preserve their native conformation and, consequently, their interaction with the drug, which is generally based on non-covalent interactions. Conversely, the extraction of membrane proteins requires dedicated solubilization conditions (such as the use of relatively harsh, denaturing detergents). To extract proteins from A2780 cells, we chose mild lysis conditions suitable for chemoproteomic assays (5) that were not devoted to the extraction of the membrane receptor FGFR2 and might have caused its partial loss during the lysis procedure. In addition, detection of FGFR2 might have been hampered by its low abundance in the A2870 lysate, also suggested by the lack of the receptor in the list of 6581 proteins identified via in-depth shotgun proteomics. Nevertheless, specific binding of FGFR2 to the E-3810 resin was confirmed by Western blotting on the same samples used for proteomics analysis.
In addition to kinase proteins, the chemoproteomic Kd assay confirmed as specific interactors the non-kinase proteins TRDX, QPRT, NMOR1, NMOR2, and NIPSNAP1. An interesting case is represented by the ribosyldihydronicotinamide dehydrogenase NMOR2, an enzyme catalyzing quinone oxidoreduction for biosynthetic processes and cellular detoxification. This enzyme has been reported to interact with the kinase inhibitors imatinib, dasatinib, and bosutinib (30). Inhibition of enzyme activity has been demonstrated in vitro for both imatinib and dasatinib; moreover, crystallography studies revealed that imatinib competes with the substrate for the active site of the enzyme (31). Considering the quinoline nucleus of E-3810, it is possible that NMOR2 could be a direct, non-kinase interactor of the inhibitor.
We hypothesize that most of the other non-kinase proteins represent indirect binders, as they lack the kinase domain for ATP binding that is supposed to sterically attract E-3810. The detection of potential indirect interactors is likely a consequence of the non-denaturing conditions used to maintain the proper protein folding, which also preserve protein–protein interactions. This feature can be turned into a useful tool for screening intact protein complexes targeted by inhibitors, as recently described for histone deacetylase inhibitors (6).
In our study, however, the affinity capture was performed in the presence of 0.1% Na-deoxycholate, and the resin was washed with a high-stringency buffer containing 0.2% SDS to reduce background interactions (5). The use of ionic detergents probably reduced protein–protein interactions, resulting in the identification of a relatively low number of indirect interactors.
The composite design of the Kd assay allowed us not only to recognize specific interactors, but also to estimate their interaction affinity with the immobilized form of the inhibitor (Figs. 4 and 5). The obtained Kd imm E-3810 values spanned a broad range in the micromolar range, with the kinase proteins pyridoxal kinase, deoxycytidine kinase, CARDIAK, and PDGFRA showing the lowest Kd values (less than 100 μm). The Kd imm E-3810 value for FGFR2 was 127 μm. The kinase proteins DDR2, DDR1, LYN, YES, and CSPB (isoform 2) showed less affinity (in the 148–863 μm range). Some non-kinase interactors, such as GLSN and EPLIN, showed Kd values less than or close to 100 mm, independently of the presence of the ATP binding pocket in their structure.
In vitro biochemical inhibition assays confirmed the chemical proteomics results and provided biochemical IC50 and Ki values for the newly identified targets that spanned the low micromolar range. The Kd imm E-3810 values obtained via chemical proteomics cannot be directly compared with the IC50 and Ki values obtained via conventional in vitro kinase inhibition assays, as Kd values are related to the target's affinity for E-3810, whereas the Ki and IC50 values refer to the efficacy of E-3810 in inhibiting the activity of kinase targets. Nevertheless, there was remarkable similarity among the Kd and Ki rankings (Fig. 6B), indicating that, overall, the kinases inhibited more potently by E-3810 corresponded to the proteins binding the immobilized drug with the greatest affinity in the chemoproteomic assays.
The Kd assay confirmed LYN, CARDIAK, YES, and CSBP isoform 2 as E-3810 interactors, together with the receptor EPHA2. It is possible that these lower-affinity interactors could have a relevant biological role if present in sufficient amounts in defined cellular compartments.
SRC was not confirmed as an E-3810 target by the in vitro kinase assay. As SRC has already been described as an interactor of DDR2 (27, 32), it is possible that it was indirectly captured by the E-3810 resin in complex with DDR2, thus representing an indirect binder.
Among the confirmed targets of E-3810, we focused on the tyrosine kinase DDR2, a receptor that has been recently suggested as an important therapeutic target in lung cancer. Three inhibitors of the tyrosine kinase ABL—dasatinib, imatinib, and, to a lesser extent, bostutinib—have been reported to inhibit DDR2 activity in biochemical assays (30). The observation that treatment with low micromolar doses of E-3810 inhibited DDR2 phosphorylation, and thus its activity, in cells overexpressing the receptor provided an independent validation of the results obtained via chemical proteomics. E-3810 treatment also impaired the proliferation of HCC-366 non-small-cell lung carcinoma cells, which endogenously express a mutated form of DDR2 and depend on DDR2 for their growth (26). Our data support the model of an oncogenic potential of DDR2 mutation and confirm its role as a driving force in promoting cell proliferation. As a perspective, preclinical models carrying gain-of-function mutations in DDR2 could be used to assess the effectiveness of E-3810 as an anticancer drug.
In this work, we implemented a strategy based on chemical proteomics and quantitative mass spectrometry for drug target deconvolution. Its application to E-3810, a drug currently in clinical phase trials, allowed us to evaluate the strengths and limits of our platform, as well as its robustness and ability to reveal novel targets in cellular lysates, with immediate practical implications for the development of the drug.
Supplementary Material
Acknowledgments
We thank Bruno Amati (IEO, IIT), Silvano Spinelli, and Gabriella Camboni, Ethical Oncology Science (EOS) S.p.A., Milan, for generous support, discussions, and critical reading of the manuscript. We also thank David Cairns for having developed the R-based script necessary to detect the trend of SILAC ratio variation and Alessandro Cuomo for technical support on the MS instruments.
Footnotes
Author contributions: S.M. and T.B. designed research; M.C., R.N., M.R., G.C., M.P., and D.F. performed research; M.C., R.N., and T.B. analyzed data; M.C. and T.B. wrote the paper; R.N. contributed to paper revision; M.V. is head of the medicinal chemistry unit; S.M. contributed to paper editing; T.B. coordinated work among different units.
* M.C. and R.N. developed this project in the TB group, which is supported by grants from the Giovanni Armenise-Harvard Foundation Career Development Program, the Association of International Cancer Research, the Italian Association for Cancer Research (AIRC), and the Italian Ministry of Health. M.R.'s work is supported by the Italian Association for Cancer Research (AIRC).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- geLC-MS
- SDS-PAGE followed by in-gel digestion and peptide analysis via mass spectrometry
- SILAC
- stable isotope labeling by amino acids in cell culture
- VEGFR
- vascular endothelial growth factor receptor
- FGFR
- fibroblast growth factor receptor
- Fmoc
- fluorenylmethoxycarbonyl
- DMSO
- dimethyl sulfoxide
- DDR
- discoidin domain-containing receptor
- PDGFRA
- platelet-derived growth factor receptor α
- LYN
- tyrosine-protein kinase Lyn
- CARDIAK
- receptor-interacting serine/threonine-protein kinase 2
- SRC
- tyrosine-protein kinase Src
- YES
- tyrosine-protein kinase Yes
- EPHA2
- ephrin type-A receptor 2
- CSBP
- cytokine suppressive anti-inflammatory drug-binding protein
- Kd
- dissociation constant
- Ki
- inhibitor constant
- IC50
- half-maximal inhibitory concentration
- H
- heavy
- L
- light
- M
- medium
- imm E-3810
- immobilized E-3810
- iBAQ
- intensity-based absolute quantification.
REFERENCES
- 1. Schirle M., Bantscheff M., Kuster B. (2012) Mass spectrometry-based proteomics in preclinical drug discovery. Chem. Biol. 19, 72–84 [DOI] [PubMed] [Google Scholar]
- 2. Rix U., Superti-Furga G. (2009) Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 5, 616–624 [DOI] [PubMed] [Google Scholar]
- 3. Godl K., Wissing J., Kurtenbach A., Habenberger P., Blencke S., Gutbrod H., Salassidis K., Stein-Gerlach M., Missio A., Cotten M., Daub H. (2003) An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 100, 15434–15439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharma K., Weber C., Bairlein M., Greff Z., Kéri G., Cox J., Olsen J. V., Daub H. (2009) Proteomics strategy for quantitative protein interaction profiling in cell extracts. Nat. Methods 6, 741–744 [DOI] [PubMed] [Google Scholar]
- 5. Ong S. E., Schenone M., Margolin A. A., Li X., Do K., Doud M. K., Mani D. R., Kuai L., Wang X., Wood J. L., Tolliday N. J., Koehler A. N., Marcaurelle L. A., Golub T. R., Gould R. J., Schreiber S. L., Carr S. A. (2009) Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad. Sci. U.S.A. 106, 4617–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bantscheff M., Hopf C., Savitski M. M., Dittmann A., Grandi P., Michon A.-M., Schlegl J., Abraham Y., Becher I., Bergamini G., Boesche M., Delling M., Dümpelfeld B., Eberhard D., Huthmacher C., Mathieson T., Poeckel D., Reader V., Strunk K., Sweetman G., Kruse U., Neubauer G., Ramsden N. G., Drewes G. (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265 [DOI] [PubMed] [Google Scholar]
- 7. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 8. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 9. Hubner N. C., Mann M. (2011) Extracting gene function from protein-protein interactions using Quantitative BAC InteraCtomics (QUBIC). Methods 53, 453–459 [DOI] [PubMed] [Google Scholar]
- 10. Grant S. K. (2009) Therapeutic protein kinase inhibitors. Cell. Mol. Life Sci. 66, 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bello E., Colella G., Scarlato V., Oliva P., Berndt A., Valbusa G., Serra S. C., D'Incalci M., Cavalletti E., Giavazzi R., Damia G., Camboni G. (2011) E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res. 71, 1396–1405 [DOI] [PubMed] [Google Scholar]
- 12. Bello E., Taraboletti G., Colella G., Zucchetti M., Forestieri D., Licandro S. A., Berndt A., Richter P., D'Incalci M., Cavalletti E., Giavazzi R., Camboni G., Damia G. (2013) The tyrosine kinase inhibitor e-3810 combined with Paclitaxel inhibits the growth of advanced-stage triple-negative breast cancer xenografts. Mol. Cancer Ther. 12, 131–140 [DOI] [PubMed] [Google Scholar]
- 13. Wesierska-Gadek J., Hajek S. B., Sarg B., Wandl S., Walzi E., Lindner H. (2008) Pleiotropic effects of selective CDK inhibitors on human normal and cancer cells. Biochem. Pharmacol. 76, 1503–1514 [DOI] [PubMed] [Google Scholar]
- 14. Ponticelli C. The pleiotropic effects of mTor inhibitors. (2014) J. Nephrol. 17, 762–768 [PubMed] [Google Scholar]
- 15. Schmidt F., Strozynski M., Salus S. S., Nilsen H., Thiede B. (2007) Rapid determination of amino acid incorporation by stable isotope labeling with amino acids in cell culture (SILAC). Rapid Commun. Mass Spectrom. 21, 3919–3926 [DOI] [PubMed] [Google Scholar]
- 16. Wilm M., Shevchenko A., Houthaeve T., Breit S., Schweigerer L., Fotsis T., Mann M. (1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379, 466–469 [DOI] [PubMed] [Google Scholar]
- 17. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 18. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 19. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 20. Rigbolt K. T. G., Vanselow J. T., Blagoev B. (2011) GProX, a user-friendly platform for bioinformatics analysis and visualization of quantitative proteomics data. Mol. Cell. Proteomics 10, O110.007450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leitinger B., Kwan A. P. L. (2006) The discoidin domain receptor DDR2 is a receptor for type X collagen. Matrix Biol. 25, 355–364 [DOI] [PubMed] [Google Scholar]
- 22. Albrecht B. K., Harmange J.-C., Bauer D., Berry L., Bode C., Boezio A. A., Chen A., Choquette D., Dussault I., Fridrich C., Hirai S., Hoffman D., Larrow J. F., Kaplan-Lefko P., Lin J., Lohman J., Long A. M., Moriguchi J., O'Connor A., Potashman M. H., Reese M., Rex K., Siegmund A., Shah K., Shimanovich R., Springer S. K., Teffera Y., Yang Y., Zhang Y., Bellon S. F. (2008) Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J. Med. Chem. 51, 2879–2882 [DOI] [PubMed] [Google Scholar]
- 23. Harmange J.-C., Weiss M. M., Germain J., Polverino A. J., Borg G., Bready J., Chen D., Choquette D., Coxon A., DeMelfi T., DiPietro L., Doerr N., Estrada J., Flynn J., Graceffa R. F., Harriman S. P., Kaufman S., La D. S., Long A., Martin M. W., Neervannan S., Patel V. F., Potashman M., Regal K., Roveto P. M., Schrag M. L., Starnes C., Tasker A., Teffera Y., Wang L., White R. D., Whittington D. A., Zanon R. (2008) Naphthamides as novel and potent vascular endothelial growth factor receptor tyrosine kinase inhibitors: design, synthesis, and evaluation. J. Med. Chem. 51, 1649–1667 [DOI] [PubMed] [Google Scholar]
- 24. La D. S., Belzile J., Bready J. V., Coxon A., DeMelfi T., Doerr N., Estrada J., Flynn J. C., Flynn S. R., Graceffa R. F., Harriman S. P., Larrow J. F., Long A. M., Martin M. W., Morrison M. J., Patel V. F., Roveto P. M., Wang L., Weiss M. M., Whittington D. A., Teffera Y., Zhao Z., Polverino A. J., Harmange J.-C. (2008) Novel 2,3-dihydro-1,4-benzoxazines as potent and orally bioavailable inhibitors of tumor-driven angiogenesis. J. Med. Chem. 51, 1695–1705 [DOI] [PubMed] [Google Scholar]
- 25. Qian F., Engst S., Yamaguchi K., Yu P., Won K.-A., Mock L., Lou T., Tan J., Li C., Tam D., Lougheed J., Yakes F. M., Bentzien F., Xu W., Zaks T., Wooster R., Greshock J., Joly A. H. (2009) Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 69, 8009–8016 [DOI] [PubMed] [Google Scholar]
- 26. Vogel W., Gish G. D., Alves F., Pawson T. (1997) The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell. 1, 13–23 [DOI] [PubMed] [Google Scholar]
- 27. Yang K., Kim J. H., Kim H. J., Park I.-S., Kim I. Y., Yang B.-S. (2005) Tyrosine 740 phosphorylation of discoidin domain receptor 2 by Src stimulates intramolecular autophosphorylation and Shc signaling complex formation. J. Biol. Chem. 280, 39058–39066 [DOI] [PubMed] [Google Scholar]
- 28. Hammerman P. S., Sos M. L., Ramos A. H., Xu C., Dutt A., Zhou W., Brace L. E., Woods B. A., Lin W., Zhang J., Deng X., Lim S. M., Heynck S., Peifer M., Simard J. R., Lawrence M. S., Onofrio R. C., Salvesen H. B., Seidel D., Zander T., Heuckmann J. M., Soltermann A., Moch H., Koker M., Leenders F., Gabler F., Querings S., Ansén S., Brambilla E., Brambilla C., Lorimier P., Brustugun O. T., Helland A., Petersen I., Clement J. H., Groen H., Timens W., Sietsma H., Stoelben E., Wolf J., Beer D. G., Tsao M. S., Hanna M., Hatton C., Eck M. J., Janne P. A., Johnson B. E., Winckler W., Greulich H., Bass A. J., Cho J., Rauh D., Gray N. S., Wong K.-K., Haura E. B., Thomas R. K., Meyerson M. (2011) Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 1, 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang K., Corsa C. A., Ponik S. M., Prior J. L., Piwnica-Worms D., Eliceiri K. W., Keely P. J., Longmore G. D. (2013) The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 15, 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bantscheff M., Eberhard D., Abraham Y., Bastuck S., Boesche M., Hobson S., Mathieson T., Perrin J., Raida M., Rau C., Reader V., Sweetman G., Bauer A., Bouwmeester T., Hopf C., Kruse U., Neubauer G., Ramsden N., Rick J., Kuster B., Drewes G. (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044 [DOI] [PubMed] [Google Scholar]
- 31. Winger J. A., Hantschel O., Superti-Furga G., Kuriyan J. (2009) The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct. Biol. 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ikeda K., Wang L.-H., Torres R., Zhao H., Olaso E., Eng F. J., Labrador P., Klein R., Lovett D., Yancopoulos G. D., Friedman S. L., Lin H. C. (2002) Discoidin domain receptor 2 interacts with Src and Shc following its activation by type I collagen. J. Biol. Chem. 277, 19206–19212 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.