

Abstract
The combination of immuno-based methods and mass spectrometry detection has great potential in the field of quantitative proteomics. Here, we describe a new method (immuno-SILAC) for the absolute quantification of proteins in complex samples based on polyclonal antibodies and stable isotope–labeled recombinant protein fragments to allow affinity enrichment prior to mass spectrometry analysis and accurate quantification. We took advantage of the antibody resources publicly available from the Human Protein Atlas project covering more than 80% of all human protein-coding genes. Epitope mapping revealed that a majority of the polyclonal antibodies recognized multiple linear epitopes, and based on these results, a semi-automated method was developed for peptide enrichment using polyclonal antibodies immobilized on protein A–coated magnetic beads. A protocol based on the simultaneous multiplex capture of more than 40 protein targets showed that approximately half of the antibodies enriched at least one functional peptide detected in the subsequent mass spectrometry analysis. The approach was further developed to also generate quantitative data via the addition of heavy isotope–labeled recombinant protein fragment standards prior to trypsin digestion. Here, we show that we were able to use small amounts of antibodies (50 ng per target) in this manner for efficient multiplex analysis of quantitative levels of proteins in a human HeLa cell lysate. The results suggest that polyclonal antibodies generated via immunization of recombinant protein fragments could be used for the enrichment of target peptides to allow for rapid mass spectrometry analysis taking advantage of a substantial reduction in sample complexity. The possibility of building up a proteome-wide resource for immuno-SILAC assays based on publicly available antibody resources is discussed.
Mass spectrometry–based proteomics is fast developing in the direction of clinical applications. Therefore, reliable quantification methods for absolute protein concentration determination are indispensible tools for future applications. So far, enzyme-linked immunosorbent assays and similar antibody-based methods excel in the sensitive detection of low levels of proteins in complex matrices, whereas mass spectrometry enables unbiased approaches and can provide unsurpassed specificity. The fact that most proteomes have a very high dynamic range between high and low abundant proteins, in particular for clinical samples, such as plasma and serum, often makes it necessary to use protein depletion of the most abundant proteins (1, 2) and/or elaborate fractionations (3–5) before running the mass spectrometry analysis. This has prompted several investigators to introduce a protein or peptide capture step using specific antibodies to allow for immunoaffinity enrichment prior to the MS analysis. In this way, a “sandwich” assay is obtained, but instead of having a readout in the analysis step based on a second antibody, the analysis step is performed using MS. In such an approach, either the intact protein is captured using an anti-protein antibody (6) or a peptide derived from the protein is captured using an anti-peptide antibody that has been raised to the target peptide of interest (7–11). This is the principle behind stable isotope standards and capture by anti-peptide antibodies (SISCAPA),1 developed by Anderson and co-workers (12–15). In immunoaffinity proteomics, it is preferable for the affinity of the anti-peptide capture antibody to be high, but the requirement for high selectivity is lower, because the mass spectrometer can readily distinguish and quantify the analyte peptide of interest despite the binding of other peptides in the digested sample.
A disadvantage with the immunoaffinity proteomics strategy is the limited availability of suitable antibodies that recognize peptides from the corresponding protein targets. The affinity enrichment of peptides usually requires the generation of custom antibodies for each target peptide, and this very time-consuming process makes high-throughput efforts very difficult to pursue. Most efforts so far have been aimed toward generating monoclonal antibodies against specific peptides selected as appropriate for mass spectrometric detection, which is a laborious and costly exercise. It would therefore be of great interest to explore whether antibodies generated toward native proteins or protein fragments could be used for the capture of peptides and in this way take advantage of the huge resource of already existing reagents for immunoproteomics.
Here, we investigated whether the publicly available resources on polyclonal antibodies could be used for immuno-enrichment followed by quantitative proteomics. According to the Antibodypedia portal, there exist more than a million publicly available antibodies toward human protein targets, and more than 70% of these antibodies are polyclonal antibodies. These antibodies are of course interesting starting points as a resource for immunoproteomics, although this application was not intended at the time when the antibodies were generated. More specifically, we have investigated the use of polyclonal antibodies from the Human Protein Atlas project, covering more than 80% of all human protein-coding genes. These antibodies have been raised against human recombinant proteins called protein epitope signature tags (PrESTs), and we have therefore investigated the direct use of this resource for quantitative proteomics.
An attractive strategy for quantitative proteomics using immuno-enrichment is to use stable isotope approaches, including methods based on adding stable isotope–labeled peptides (16, 17), proteins (18, 19), or protein fragments (20). These methods are built on the detection of peptides generated by protease cleavage of the proteins in the sample, and the quantification is achieved by reading out the ratio between the endogenous peptide and the heavy-labeled spiked-in peptide. Because the endogenous protein and the labeled internal standard behave identically throughout the sample preparation including the immuno-enrichment, the relative ratio provides quantitative information, as the peptides can be distinguished by the mass spectrometer because of the shift in mass. We recently described (20) a method for protein quantification making use of the large library of PrESTs that has been developed in the course of the Human Protein Atlas (21) project. Heavy isotope–labeled PrESTs were quantified against an ultrapurified and accurately quantified protein standard using the albumin binding protein (ABP) tag. Thereafter, known amounts of heavy PrESTs were spiked into cell lysates, and the SILAC ratios were used to determine the cellular quantities of the endogenous proteins. That approach sidesteps the quantification-, storage-, and digestion-related causes of quantification error that are inherent to peptide-based methods. The PrEST-SILAC principle was used to simultaneously determine the copy numbers of 40 proteins in HeLa cells demonstrating quantitative measurements over a wide range of protein abundances, from the highly abundant cytoskeletal protein Vimentin, with 20 million copies, down to the low abundant transcription factor FOS, with only 6000 copies per cell.
Here, we combined the use of polyclonal antibodies for immunocapture with quantitative proteomics using heavy isotope–labeled proteins. A semi-automated immuno-SILAC method was developed for multiplex analysis of protein targets, taking advantage of the linear epitopes of the antibodies. A special effort was made to decrease the amounts of antibodies used in the assay. Based on the results, a new strategy for rapid mass spectrometry readout for target-specific proteomics is outlined in which antibodies are used for the multiplex immunocapture of peptides generated via trypsin digestion of cell extracts spiked with isotope-labeled recombinant protein fragments corresponding to the protein targets.
EXPERIMENTAL PROCEDURES
Generation of Antibodies
Antigens were designed using the software PRESTIGE (22). Gene fragments were amplified from a pool of RNA isolated from human tissues, cloned into a vector, and expressed in Escherichia coli. To generate polyclonal antibodies, purified and validated recombinant protein fragments were used for the immunization of New Zealand White rabbits, and the polyclonal rabbit sera were purified using their corresponding antigens as affinity ligands (23).
Epitope Mapping Using High-density Peptide Array
High-density peptide arrays were designed to contain 12-mer peptides with an overlap of 11 amino acid residues, in total covering all the antigen sequences. Parallel in situ peptide synthesis on microscope slides and removal of side chain protecting groups were performed by Roche NimbleGen Inc. (Madison, WI). Each slide containing 12 identical subarrays was covered with a PX12-mixer mask (Roche NimbleGen Inc.) according to the manufacturer's instructions. The polyclonal antibodies were combined into pools of 20 antibodies and diluted in binding buffer (10 mm Tris, 0.45% NaCl, pH 7.4, alkali soluble casein 0.5%) to a final concentration of 0.5 μg/ml for each antibody. The samples were added to the peptide arrays and incubated overnight at room temperature in a NimbleGen Hybridization Station (Roche NimbleGen Inc.). After primary incubation, the mixer masks were removed and the slides were washed in coplin jars twice with TBSTT (20 mm Tris, 0.9% NaCl, pH 7.4, 0.1% Tween 20, 0.4% Triton X-100) and twice with TBS, with each wash lasting 10 min. Secondary DyLight649 conjugated anti-rabbit antibodies (Jackson ImmunoResearch, West Grove, PA) were diluted to 0.15 μg/ml in binding buffer in LockMailer jars, and the slides were incubated for three hours on a shaking table. The slides were washed again twice with TBSTT and twice with TBS as described above, quickly rinsed three times in filtered de-ionized water, and dried using a microarray slide centrifuge. The slides were scanned at 2 μm resolution using a NimbleGen MS200 scanner (Roche NimbleGen Inc.), and the median fluorescence intensities of the peptide features in the scanned images were analyzed using the NimbleScan software (Roche NimbleGen Inc.).
Preparation of PrEST Digest
PrESTs were mixed into three pools of 41, 42, and 44 targets, respectively. Samples containing 10 μg of each PrEST were first reduced with DTT and thereafter digested using the filter-aided sample preparation method (24). Briefly, the sample was added to a 30-kDa cutoff spin filter (Millipore, Bedford, MA), and the buffer was exchanged to denaturation buffer. The sample was alkylated with iodoacetamide and the buffer was changed to 50 mm NH4HCO3 before trypsin (Sigma-Aldrich, St. Louis, MO) was added, and the sample was incubated at 37 °C overnight.
Affinity Enrichment of Peptides Using Polyclonal Antibodies
Immunoaffinity enrichment of peptides from the trypsin-digested PrEST mixture was carried out with a subset of 127 polyclonal rabbit antibodies. A total of 250 ng of each antibody was pooled into 41-, 42-, and 44-plex pools, and the final volume was adjusted to 300 μl with PBS and Chaps detergent to yield a final concentration of 0.03% (w/v). In parallel, 5.3 mg of Protein A Dynabeads (Invitrogen, #10001D) was placed on a handheld magnet (Dynal, Oslo, Norway), and the storage buffer was removed before the beads were washed twice with wash buffer (1× PBS, 0.03% (w/v) CHAPS). Each subset of pooled antibodies was immobilized together with 150 μg of Protein A–coated beads per microgram of antibody and incubated for 30 min on a rotor mixer for 1 h at room temperature. A total of 200 ng of each trypsin-digested PrEST was diluted to 50 μl with PBS-supplemented CHAPS to a yield a final concentration of 1× PBS, 0.03% (w/v) CHAPS. All samples were prepared in duplicate and transferred to one 96-well standard microplate (ABgene, Hamburg, Germany) that was inserted into a Magnatrix 1200 (Magnetic Biosolutions AB, Stockholm, Sweden) automated bead processing system. Afterward, immobilized antibody–bead mixtures corresponding to 50 ng of antibody per target were transferred in triplicate to separate wells in a 96-well PCR plate (Thermo Scientific), and the plate was inserted into the Magnatrix 1200 system. The beads were washed twice with wash buffer and mixed with the peptide mixture from the trypsin-digested PrEST mixture using robotics. The 96-well standard microplate was manually covered with an opaque adhesive foil, and peptides were enriched for 16 h overnight at room temperature on a microtiter plate shaker at 1350 rpm. The following day, the plate was inserted into the Magnatrix 1200, and the beads were washed twice with wash buffer and then twice with 50 mm NH4HCO3. The enriched peptides were eluted with 10 μl of 0.1% formic acid (pH 2.5) for 2 min. All samples were heat treated at 96 °C for 5 min in order to denature antibodies that were eluted along with peptides from the solid bead support. Each sample was manually supplemented with 1 μl of 33% acetonitrile prior to storage at −20 °C until LC-MS analysis.
Production of Antigen Standards for Absolute Quantification
The expression vector pAff8c containing a fragment coding for the quantification standard HisABPOneStrep was transformed into E. coli Rosetta DE3 cells (Novagen, Merck, Darmstadt, Germany), and HisABPOneStrep was expressed according to the standard protocol used within the Human Protein Atlas project (25). After purification using immobilized metal ion affinity chromatography and buffer exchange to 1× PBS (10 mm NaP, 150 mm NaCl, pH 7.3) on a PD-10 desalting column (GE Healthcare, Uppsala, Sweden), a second purification step was performed using a StrepTrap™ HP column (GE Healthcare) on an ÄKTAexplorer system (GE Healthcare) according to the suggested protocol. The concentration of the purified protein was determined using amino acid analysis. Expression vectors containing PrEST fragments were transformed into an E. coli strain auxotrophic for lysine and arginine (26) for the production of heavy isotope–labeled PrESTs. A total of 41 targets were chosen for this purpose. Included PrESTs contained at least five theoretical tryptic peptides that had previously been detected in discovery proteomics experiments (data not presented here). These PrESTs were non-overlapping with the set used for the affinity enrichment screening. Cultivations of 10 ml were performed in 100-ml shake flasks using minimal autoinduction media as previously described (20, 27). Heavy isotope–labeled (13C and 15N) arginine and lysine (Cambridge Isotope Laboratories, Tewksbury, MA) and light versions of the remaining 18 amino acids (Sigma-Aldrich) were added to the medium to a final concentration of 200 μg/ml. After cultivation, the cells were lysed and the PrESTs were purified according to the standard Human Protein Atlas protocol (25).
Quantification of PrESTs
HisABPOneStrep and heavy isotope–labeled PrESTs were mixed in 50 mm NH4HCO3. The sample was reduced with DTT, alkylated with iodoacetamide, and digested with trypsin (Sigma-Aldrich) overnight. The samples were diluted in 5% acetonitrile, 0.1% formic acid, injected onto a 150 mm × 0.5 mm Zorbax 80SB-C18 column (Agilent, Santa Clara, CA), and separated using a 20-min gradient of 10%–40% acetonitrile with a flow rate of 20 μl/min on an Agilent 1200 capillary-LC system. The peptides were analyzed using an Agilent 6520 electrospray ionization quadrupole TOF mass spectrometer in a data-dependent manner; three precursor ions per cycle were chosen and fragmented via collision-induced dissociation. The isolation width was set at 4 m/z. Full-scan MS spectra were acquired between 300 and 2000 m/z, and product ion scans between 100 and 2000 m/z. Data analysis was performed using the software APP (unpublished), combining several MS analysis modules into one data analysis tool. The search engine X!Tandem (28) (version 2011.12.01.1) was used with the human UniProt database (70,555 entries, downloaded May 28, 2013) with the addition of the HisABPOneStrep sequence. Carbamidomethylation of cysteines was added as a fixed modification, and methionine oxidation was allowed as a variable modification. The minimum peptide length was five amino acids, and a maximum of two missed cleavages was allowed. Data from X!Tandem were further processed with PeptideProphet (29) and ProteinProphet (30), and SILAC ratios were determined with XPRESS software (31) (all from TPP v4.6 occupy rev 3). Three technical replicates were performed for each PrEST, and the median value was used when determining the protein concentration.
Preparation of HeLa Cell Lysates and Trypsin Digestion
HeLa cells (32) were cultivated in Dulbecco's modified Eagle's medium (Sigma-Aldrich) containing 10% fetal bovine serum (Sigma-Aldrich) and antibiotic and antimycotic solution (Sigma-Aldrich). Cells were released from the culture dish with a trypsin-EDTA solution (Sigma-Aldrich) and frozen at −80 °C in aliquots of 10 million cells per tube. Tubes were thawed on ice, and the cells were lysed with 1 ml of lysis buffer (0.1 m Tris-HCl, 4% SDS, 10 mm DTT, pH 7.6). Samples were incubated at 95 °C for 3 min and sonicated for 1 min. Aliquots of 100 μl corresponding to 1 million HeLa cells were used for tryptic digestion. For the first experiment, 1 pmol of each heavy isotope–labeled PrEST was spiked into the HeLa sample. Triplicate samples were then prepared in which the PrEST amounts had been adjusted to a ratio close to 1:1 relative to the corresponding endogenous protein. The correct amount of each PrEST was mixed and reduced with DTT before the PrEST mix was spiked into the HeLa lysate. The sample was diluted with denaturing buffer (8 m urea, 0.1 m Tris-HCl, pH 8.5) and centrifuged through a 0.65-μm spin filter (Millipore) to get rid of cell debris. Digestion was performed using the filter-aided sample preparation method (24) as described above.
Peptide Fractionation for PrEST-SILAC
Before MS analysis, 30 μg of the peptide mixture was divided into six fractions by means of strong anion exchange chromatography. This was done in a pipette-tip format as previously described (33). In brief, pipette tips were packed with strong anion exchange material (3M Bioanalytical Technologies, St. Paul, MN), and the peptide sample was loaded. Peptides were eluted according to isoelectric point with buffers of decreasing pH. After fractionation, eluted peptides were desalted using C18 StageTips.
Absolute Quantification of HeLa Cell Lysate
Immunoaffinity enrichment of peptides from trypsin-digested HeLa lysate, into which heavy PrESTs had been spiked prior to digestion, was carried out in the same way as described above for 41 polyclonal rabbit antibodies for which heavy-labeled PrESTs were available. Here, a total of 15 μg of trypsin-digested HeLa lysate with spiked-in heavy PrESTs was diluted to 50 μl with PBS-supplemented CHAPS to a yield a final concentration of 1× PBS, 0.03% (w/v) CHAPS. A total of 500 ng of each corresponding antibody was pooled (41-plex), and the final volume was adjusted to 500 μl with PBS and CHAPS detergent to yield a final concentration of 0.03% (w/v). All antibodies were immobilized onto 3.15 mg Protein A–coated magnetic beads. All samples were prepared in triplicate and processed in the same way as described above.
Liquid Chromatography and Mass Spectrometry
For PrEST-SILAC samples, 2 μg of peptides per fraction were analyzed, and for immuno-SILAC, only 50% of the sample was used. Peptides were first trapped on a Zorbax 300SB-C18 column (Agilent) and separated on an NTCC-360/100–5-153 (Nikkyo Technos Co., Ltd., Tokyo, Japan) column using a gradient of 6%–40% acetonitrile over 180 min (PrEST-SILAC) or 15 min (immuno-SILAC) with a flow rate of 0.4 μl/min on an Agilent 1200 nano-LC system. MS analysis was performed on a Q Exactive instrument (Thermo Fisher Scientific, San Jose, CA) operated in a data-dependent manner, with five precursors selected for fragmentation by higher energy collisional dissociation in each full MS scan. MS spectra were recorded between 300 and 1700 m/z at 70,000 resolution, and MS fragment ion spectra were recorded at 17,500 resolution.
Analysis of PrEST-SILAC and Immuno-SILAC MS Data
Data from Q Exactive MS runs were analyzed using MaxQuant software (34) (version 1.3.0.5) with the built-in search engine Andromeda (35). A human UniProt database (70,555 entries, downloaded May 28, 2013) was used in the search. The minimum peptide length was six amino acids, and two missed cleavages were allowed. Carbamidomethylation of cysteines was set as a fixed modification, and methionine oxidation and N-terminal acetylation were chosen as variable modifications. The initial MS mass tolerance for recalibration was 20 ppm, the initial mass deviation for the precursor ions was 4.5 ppm, and a maximum error of 20 ppm was allowed for MS/MS spectra. The false discovery rate was set at 0.01, and the match between runs option was used with a 2-min retention-time window. Identified peptides were grouped with their corresponding PrEST, and copy numbers were calculated for each peptide. The median peptide value was used as the copy number for the corresponding protein as well as the median value among the technical replicates.
RESULTS
Principle of the Targeted Immunoproteomics Method
The principle of immuno-SILAC is shown in Fig. 1. The method relies on the use of stable isotope–labeled recombinant protein fragment standards (PrESTs) produced in bacteria (Fig. 1A). In immuno-SILAC, a cell lysate is mixed with known amounts of accurately quantified heavy-labeled protein fragment standards, generated with heavy isotope–labeled versions (15N and 13C) of the amino acids arginine and lysine. The protein mixture is enzymatically digested, and the generated peptides are subsequently captured by antibodies immobilized onto Protein A–coated magnetic beads as illustrated in Fig. 1C. Following enrichment from the complex peptide mixture, target peptides are eluted with formic acid from the solid phase bead support. If the peptides are eluted in an MS-compatible buffer, only acetonitrile has to be added before LC-MS/MS analysis. The low complexity of the resulting sample enables very short analysis times. Here, a single 15-min HPLC gradient was sufficient for separation of a multiplexed sample in which up to 44 different antibodies were used for peptide enrichment. The ratios of light peptides originating from the endogenous digested proteins and peptides from the spiked-in heavy isotopic standards are compared, giving an absolute quantitative measurement of the studied proteins.
Fig. 1.

The principle of the immuno-SILAC method. A, absolute protein quantification using PrESTs as the internal standard. Peptides originating from the albumin binding protein (ABP) tag (yellow) are used to quantify each PrEST against an ultrapurified ABP protein standard. The PrEST can thereafter be used as an internal standard in unknown samples to quantify the corresponding endogenous protein (red) in LC-MS. B, schematic representation of the immuno-SILAC workflow. Highly purified and accurately quantified isotopic heavy-labeled PrESTs are spiked into cell lysates prior to trypsin digestion, thereby minimizing the risk of differences arising between samples and standards during sample preparation. Antibodies coupled to magnetic solid phase support enrich target peptides from the digested sample, and the endogenous protein concentration is calculated from the ratio of heavy to light peptides detected via MS. C, comparison between the SISCAPA technology (7) using heavy-labeled peptides (blue) and immuno-SILAC using heavy-labeled protein fragments (green) for absolute protein quantification.
A schematic overview comparing the workflows for the related SISCAPA and immuno-SILAC methods is shown in Fig. 1B. In SISCAPA, known amounts of heavy-labeled synthetic peptides are spiked in after trypsin digestion, whereas in immuno-SILAC the protein fragments are added prior to trypsin digestion. The addition of protein fragments as standards before enzyme digestion has the advantage of compensating for miscleavages or otherwise incomplete digestion, as the heavy protein standards undergo the same processing as the endogenous proteins (36). In this way, possible losses during sample preparation do not introduce quantification errors.
Analysis of Linear Epitopes of Polyclonal Antibodies Using High-density Peptide Arrays
In order to analyze the number of linear epitopes for polyclonal antibodies generated in a standardized manner using protein fragments (PrESTs), a high-density array with 175,000 overlapping synthetic 12-mer peptides with a single-amino-acid lateral shift was designed covering 941 protein fragment sequences. The target proteins, a majority of all human kinases and a number of interesting biomarkers for cancer, were selected by the 7th European Union framework project Affinomics (37) for the ultimate goal of generating corresponding affinity reagents. Parallel in situ peptide synthesis of the arrays was achieved via repeated cycles of selective activation using a UV-light source and a micromirror device followed by the incorporation of amino acids with a photolabile protective group. Each of the synthesized peptide arrays was incubated with a pool containing 20 of the selected polyclonal antibodies, and a fluorophore-conjugated secondary anti-rabbit antibody was used to detect antibody–peptide interactions. The small shift of only a single amino acid between the overlapping peptides allowed very detailed mapping of the linear epitopes recognized by the antibodies. Previous results for polyclonal antibodies epitope-mapped together with two separate pools of 29 unrelated antibodies showed very similar binding profiles, indicating limited cross-reactivity of the unrelated antibodies to the antigen sequence peptides (data not shown). Two examples of epitope mapping are shown in Figs. 2A and 2B, where the bars represent the median fluorescence intensity of each of the overlapping peptides. The anti-AGAP2 antibody HPA023474 showed two distinct linear epitopes (Fig. 2A), whereas the antibody HPA027341, targeting the protein fumarate hydratase, recognized four linear epitopes (Fig. 2B). The epitope mapping of the 941 antibodies showed that the number of linear epitopes varied for the different antibodies (Fig. 2C), but on average 2.9 linear epitopes were detected. The fact that most of the analyzed polyclonal antibodies recognized multiple linear epitopes suggests the possibility of using these polyclonal antibodies as capturing agents for the enrichment of peptides from trypsin-digested complex samples. It is noteworthy that ∼40% of the identified epitopes contained a trypsin cleavage site (data not shown), suggesting that they might not be functional for immuno-enrichment of the corresponding peptides.
Fig. 2.

Epitope mapping of polyclonal antibodies on planar peptide arrays. Linear epitopes of 941 polyclonal antibodies were analyzed using synthetic 12-mer peptides with an overlap of 11 amino acids, covering the antigen sequences. Two examples of the epitope mappings are shown in A (HPRA023474) and B (HPA027341), where each bar on the x-axis corresponds to one of the overlapping peptides and the height shows the median fluorescence intensity. C, distribution of the number of polyclonal antibodies recognizing 0 to 10 linear epitopes.
Immunocapture of Peptides for Mass Spectrometry Analysis
In order to investigate the performance of the epitope-mapped antibodies for immuno-enrichment of target protein peptides from complex digested samples, we chose a random subset of 150 antibodies. For 127 of these, the corresponding recombinant PrESTs were available. The protein fragments had previously been expressed in E. coli and purified as described before (25). The 127 PrESTs were pooled together in equimolar amounts and used for trypsin cleavage using a standardized protocol (24). The peptide mixtures were transferred to a robotic work station for magnetic bead handling (38), and immuno-enrichment of target peptides was performed using a multiplex mixture of 40 to 45 of the corresponding polyclonal antibodies captured on Protein A–coated magnetic beads, using only 50 ng of each antibody. After overnight affinity capture, the beads were thoroughly washed and the enriched peptides were eluted with formic acid suitable for the subsequent mass spectrometry analysis. The MS analysis revealed that 57 out of the 127 target proteins were successfully identified by at least one tryptic peptide (supplemental Table S1). A representative subset of identified peptides and the corresponding mapped linear epitopes for the antibody used can be seen in Fig. 3. As shown by the examples, predicted epitopes as determined by epitope mapping using overlapping 12-mer peptides often correlated with the sequences of enriched peptides in immuno-SILAC. However, in some cases (e.g. FLT1 and CAMK4 (Fig. 3)), some peptides were not predicted from the high-density array mapping. It is tempting to speculate that in many of these cases, the epitopes seen by the antibody were not covered within the 12-mer peptides displayed on the high-density array, implying that these epitopes needed longer peptides to form than available on the peptide arrays. It is also apparent from the examples that many of the linear epitopes predicted by the epitope mapping were not observed after the MS analysis. Many of these epitopes contain lysines or arginines, and the corresponding peptides are thus not expected to be captured, as the epitope is cleaved by trypsin. It is also likely that many other peptides were not detected in the MS analysis because of technical issues (39). In summary, the results presented here, based on 127 antibodies, suggest that approximately half of the polyclonal antibodies enriched at least one peptide that could subsequently be detected via mass spectrometry.
Fig. 3.

Comparison of mapped epitopes and peptides identified in immuno-SILAC screening of polyclonal antibodies against trypsin-digested PrESTs. The upper part of each panel shows the binding intensities in median fluorescence intensity to overlapping 12-mer peptides in the epitope mapping, where epitopes are shown as clusters of bound peptides. The blue horizontal bars in the lower part show the locations of peptides identified in immuno-SILAC screening on their corresponding antigen, and gray vertical lines indicate trypsin cleavage sites.
Comparative Analysis of PrEST-SILAC and Immuno-SILAC
In order to investigate the success rate of finding functional immunoproteomics pairs using corresponding recombinant PrESTs as internal standards, we analyzed a new set of 41 protein targets known to be present in HeLa cells based on RNA sequencing data (40). The targets were chosen based on sequences of the corresponding PrESTs, with a minimum of five tryptic peptides required. A HeLa cell lysate was analyzed using both the PrEST-SILAC method described earlier (20) and the immuno-SILAC protocol in parallel. Isotope-labeled protein fragments corresponding to 41 human protein targets were spiked into a cell lysate sample as schematically shown in Fig. 1C, and the mixture of heavy standards and light peptides was captured using the immobilized multiplex antibodies (n = 41). Of the 41 antibodies used in the multiplex analysis, 22 managed to capture peptide(s) corresponding to the correct target protein. Two targets were successfully quantified using two different antibodies, resulting in 20 quantified proteins. The targets are shown in Table I, and the cell lysate was an extract from the HeLa cell cultivation. The concentrations of the endogenous proteins were calculated based on the ratio between the light peptides from the sample and the corresponding heavy peptide from the protein standard. In parallel, the same protein targets were analyzed using the PrEST-SILAC protocol. For immuno-SILAC, 15 μg of a digested HeLa sample was used in the peptide enrichment and prior to MS analysis; a 15-min HPLC gradient was needed for sufficient peptide separation. For PrEST-SILAC, 30 μg of peptides was divided into six fractions, and then each fraction was further separated on an HPLC column using a 3-h gradient. The difference in sample complexity between the two methods is illustrated in Fig. 4, where example chromatograms from one immuno-SILAC run and one of the six fractions from a corresponding PrEST-SILAC run are shown. An example MS spectrum (at the retention time indicated by the arrow) showing a peptide from SERPINB6 is shown for both methods. The peptide intensities are similar in the two spectra, but the absence of interfering peaks in the immuno-SILAC spectra indicates a better separation of peptides along the HPLC gradient. To further demonstrate the difference in sample complexity between the two methods, intensities for peptides corresponding to the target proteins were compared with the total intensity of all identified endogenous peptides (common contaminants excluded). The proportion of target peptides in immuno-SILAC was 83%, as compared with 0.5% for PrEST-SILAC (data not shown).
Table I. Quantified protein copy number per HeLa cell for immuno-SILAC and PrEST-SILAC. Copy numbers are shown for each replicate, along with the median copy number, relative standard deviation (RSD), and number of peptides used for quantification.
| Gene name | Copy number replicate 1 | Copy number replicate 2 | Copy number replicate 3 | Median copy number | RSD (%) | Number of peptides |
|---|---|---|---|---|---|---|
| Immuno-SILAC | ||||||
| ACOT7 | 1283838 | 998876 | 498230 | 998876 | 42.9 | 2 |
| ANXA1 | 6664952 | 9833375 | 6362332 | 6664952 | 25.2 | 4 |
| ANXA3 | 2219240 | 2298319 | 4650459 | 2298319 | 45.2 | 1 |
| BLVRB | 2148052 | - | 3002937 | 2575495 | 23.5 | 1 |
| CANT1 | - | 70703 | 42667 | 56685 | 35.0 | 1 |
| CAPG | 3078134 | 2579694 | - | 2828914 | 12.5 | 1 |
| CLPP | 1208794 | 1087915 | 777082 | 1087915 | 21.7 | 1 |
| DAP3 | 1498865 | 1016070 | 900236 | 1016070 | 27.9 | 6 |
| DECR1 | - | - | 588225 | 588225 | - | 1 |
| DIMT1 | - | - | 235127 | 235127 | - | 1 |
| ERLIN1 | 529872 | 923508 | 1013719 | 923508 | 31.3 | 2 |
| P4HA1 | 277258 | 307834 | 431686 | 307834 | 24.1 | 1 |
| PDIA5 | 94844 | 166129 | 186790 | 166129 | 32.3 | 3 |
| PRPF4 | 306890 | 316067 | 342904 | 316067 | 5.8 | 1 |
| PTPN1 | 763589 | 1035203 | 686727 | 763589 | 22.1 | 2 |
| SERPINB6 | 885677 | 2539203 | 1477373 | 1477373 | 51.3 | 2 |
| SIL1 | - | 117997 | - | 117997 | - | 1 |
| SLC25A24 | 2317150 | 2099723 | 1239592 | 2099723 | 30.2 | 1 |
| STUB1 | 573646 | 474906 | 490305 | 490305 | 10.4 | 1 |
| UGDH | 193277 | 501192 | 605305 | 501192 | 49.5 | 1 |
| PrEST-SILAC | ||||||
| ACOT7 | 771360 | 1170086 | 955768 | 955768 | 20.7 | 4 |
| ANXA1 | 12516639 | 10077946 | 11725516 | 11725516 | 10.9 | 14 |
| ANXA3 | 1899959 | 2428880 | 4058745 | 2428880 | 40.2 | 12 |
| BLVRB | 779755 | 836484 | 624563 | 779755 | 14.7 | 4 |
| CANT1 | - | 67847 | - | 67847 | - | 2 |
| CAPG | 2238047 | 2535800 | 2762024 | 2535800 | 10.5 | 4 |
| CLPP | 1017569 | 705756 | 639815 | 705756 | 25.6 | 3 |
| DAP3 | 329798 | - | 638890 | 484344 | 45.1 | 2 |
| DECR1 | - | - | - | - | - | - |
| DIMT1 | 337769 | 408358 | 342916 | 342916 | 10.8 | 6 |
| ERLIN1 | 559997 | 963844 | 803794 | 803794 | 26.2 | 5 |
| P4HA1 | 1114521 | 627467 | 733443 | 733443 | 31.0 | 11 |
| PDIA5 | 134089 | 253862 | 152319 | 152319 | 25.5 | 4 |
| PRPF4 | 411509 | 274967 | 566398 | 411509 | 34.9 | 6 |
| PTPN1 | 520007 | 542602 | 597039 | 542602 | 7.2 | 5 |
| SERPINB6 | 576320 | 974111 | 1269089 | 974111 | 37.0 | 9 |
| SIL1 | 96363 | 155829 | 111533 | 111533 | 25.5 | 1 |
| SLC25A24 | 1077749 | 1015007 | 925753 | 1015007 | 7.6 | 4 |
| STUB1 | 719533 | 604201 | 692742 | 692742 | 9.0 | 4 |
| UGDH | 1000743 | 888750 | 746425 | 888750 | 14.5 | 5 |
Number of peptides: the total number of different peptides used for the quantification.
Fig. 4.
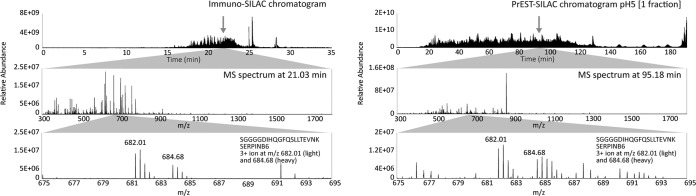
Comparison of HPLC chromatograms and MS spectra from one immuno-SILAC and one PrEST-SILAC sample. The upper panels show HPLC chromatograms for one immuno-SILAC sample (left) and one PrEST-SILAC fraction (right). The peptide SGGGGDIHQGFQSLLTEVNK from SERPINB6 was identified in both experiments as a triple-charged ion of m/z 682.01 (light version). The middle panels show extracted full MS spectra at the respective retention times, and the lower panels show the same MS spectra at m/z between 675 and 695.
In Fig. 5, a comparison of identified peptides from targets quantified via both immuno-SILAC (blue) and PrEST-SILAC (yellow) is shown for 6 of the 20 proteins together with the epitopes mapped on peptide arrays (green). In general, PrEST-SILAC identified, as expected, more unique peptides than immuno-SILAC, as no specific peptides were enriched. However, the peptide enrichment step reduces the complexity of the input material, consequently decreasing the analysis time drastically. For SLC25A24, four peptides were found using PrEST-SILAC, and one peptide was found when using the corresponding antibody for peptide enrichment, as this peptide contained the consensus epitope determined on the peptide array. The same peptide was also identified in PrEST-SILAC, indicating that it was present at sufficient levels for MS analysis even without peptide enrichment. This was the case for most of the analyzed proteins, as the target proteins were all moderate to highly abundant proteins in HeLa cells. Interestingly, some peptides were identified only in immuno-SILAC (see PRPF4 and STUB1). In the case of PRPF4, two epitope regions were detected during mapping, but the most C-terminal epitope contains a trypsin cleavage site (indicated by dotted vertical lines) and, as expected, the antibody failed to bind any of the two resulting tryptic peptides. The N-terminal epitope region probably consists of two overlapping epitopes, of which at least one is still intact after digestion and can be enriched. For SERPINB6, two different peptides can be identified.
Fig. 5.
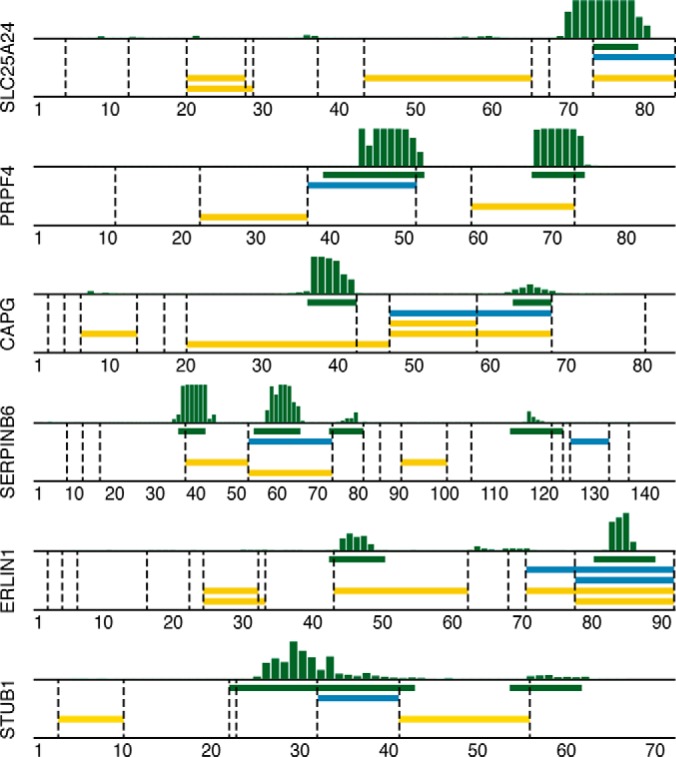
Comparison of epitope mapping, immuno-SILAC, and PrEST-SILAC peptides. Horizontal bars show the locations of linear epitopes (green) and peptides identified in immuno-SILAC (blue) and PrEST-SILAC (yellow) on their corresponding antigen; dashed vertical lines indicate trypsin cleavage sites. Above each plot, the raw epitope mapping binding intensities to overlapping 12-mer peptides are shown as vertical bars.
Quantitative Analysis of 20 Targets in HeLa Cell Lysates Using Immuno-SILAC and PrEST-SILAC
A quantitative analysis of 20 protein targets in a human HeLa cell lysate was performed. The same targets were analyzed using three technical replicates for both immuno-SILAC and PrEST-SILAC methods. Given the amount of HeLa cells used in the assay, absolute quantification of endogenous proteins as copy numbers per HeLa cell was determined from the ratio of heavy to light peptides. Heavy standard amounts were spiked into the lysate close to the level of endogenous protein in order to generate SILAC ratios close to 1:1 and hence more reliable quantitative data. Relative standard deviations ranged between 10% and 40% for most targets, with somewhat lower numbers for PrEST-SILAC (Table I).
Ratios obtained from the MS analysis for all peptides used for quantification in immuno-SILAC in comparison with PrEST-SILAC are shown in Fig. 6. A good correlation between the PrEST-SILAC and the immuno-SILAC can be observed for all 20 targets. Note that miscleaved peptides also can be used for quantitative analysis. Here, miscleaved peptides refer to peptides that still contained one or more potential cleavage sites after trypsin digestion. These miscleaved peptides become an issue in methods relying on spiked-in peptide standards for protein quantification (41), such as AQUA (17), in which peptides are spiked in after the enzymatic digestion. However, it is noteworthy that information from these miscleaved peptides can be used in the quantitative analysis in immuno-SILAC; here we took advantage of heavy-labeled protein fragments enzymatically digested along with the protein target. The observed ratios suggest that the enzymatic digestion was equal for both protein fragment standards and endogenous proteins, and these peptides add extra and valuable information rather than introducing ambiguity to the subsequent quantitative analysis.
Fig. 6.

Comparison of peptide ratios along the PrEST sequences for PrEST-SILAC (yellow) and immuno-SILAC (blue). Identified peptides from the two methods are plotted along the PrEST-sequence (x-axis) showing ratios (y-axis) between heavy and light peptides used for quantification of endogenous protein in HeLa cell lysate. The dotted line represents the median ratio for PrEST-SILAC. Overlapping peptide sequences come from missed cleavages. Two different heavy PrESTs were used for annexin 1 (ANXA1) and PDIA5.
The absolute quantifications for all 20 targets as determined via immuno-SILAC are shown in Fig. 7A. Among the 20 identified proteins, copy numbers ranged from 6.6 million copies per cell for annexin 1 down to 57,000 copies per cell determined for calcium activated nucleotidase, which represents moderately to highly abundant proteins. The absolute quantitative data given by immuno-SILAC was compared with that obtained via the previously described PrEST-SILAC method (20). Cell copy numbers determined via each respective absolute quantitative method (Table I) were plotted against each other as illustrated in Fig. 7B. In total, 17 and 18 proteins were quantified in at least two out of three replicates using the immuno-SILAC and PrEST-SILAC methods, respectively. Out of these, 59% of the targets (10 out of 17 proteins) were detected with only a single peptide using immuno-SILAC, whereas only 6% of the targets (1 out of 18) were detected with only one peptide using PrEST-SILAC. It should be noted that the data used for quantification with the two methods differed, as in general PrEST-SILAC identified more peptides per protein than immuno-SILAC. However, even though many proteins were quantified with only one proteotypic peptide in immuno-SILAC, good correlation could be observed between the two methods. To investigate whether the determined protein cell copy numbers showed any correlation to mRNA levels of the corresponding genes, we determined the transcript levels of all genes in the HeLa cell culture using deep sequencing of mRNA molecules using RNA sequencing (42). Fig. 7C shows a comparison between protein cell copy numbers as determined from the immuno-SILAC experiments and the absolute transcript levels for the 20 targets analyzed here. Even though a completely linear relationship between protein and RNA levels cannot be assumed and it is especially difficult to compare protein and RNA using data from only a single cell line, the analysis still showed a trend between RNA and protein levels. It is clear from Fig. 7C that the RNA levels to some extent indicated protein levels.
Fig. 7.
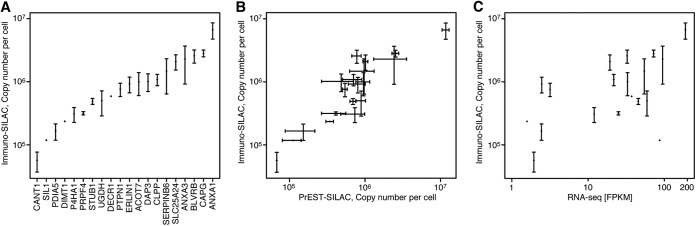
Immuno-MS results from antibodies toward 20 different target proteins in HeLa cell lysates. A, immuno-SILAC quantification of 20 target proteins as copy number per cell. The analysis was performed in triplicate, and median values were plotted with error bars showing the standard deviation. B, comparison of copy numbers per cell obtained using immuno-SILAC (y-axis) and PrEST-SILAC (x-axis). C, comparison of protein copy number (y-axis) and transcript abundance from RNA sequencing (x-axis).
DISCUSSION
Here, we show that polyclonal antibodies raised against recombinant protein fragments can be used for immunoproteomics analysis. The immuno-SILAC method takes advantage of the fact that the majority of the epitopes of the generated antibodies are directed to relatively short linear epitopes and are therefore useful for immunocapture of peptides generated after trypsin cleavage. In addition, the method takes advantage of the fact that protein fragments used for generating the antibodies can be efficiently expressed in bacteria (E. coli) using cultivation in minimal media with isotope-labeled arginines and lysines followed by a standardized protocol for affinity purification using an affinity tag. Thus, it is easy to generate protein standards, and these can be quantified in an exact manner using the PrEST-SILAC method previously described by us (20).
The method resembles other immunoproteomics methods, such as SISCAPA (7) and the use of AQUA peptides (17), but this new method has several important differences. First, the use of protein fragments often generates multiple peptides, and it is in many cases possible to obtain quantitative data from several independent peptides using the same antibody and protein standard. In addition, the fact that the protein fragments are added prior to the trypsin cleavage ensures that uncleaved endogenous peptides will not affect the quantification as long as the digestion efficiency of the protein standard is the same as that of the endogenous protein target. Furthermore, miscleaved peptides can be used as additional standards, and the problem of miscleaved peptides is therefore transformed into an opportunity to generate more data that can be used in the analysis. Finally, the generation of the protein standards does not involve peptide synthesis, and the quantification of the protein standards can be easily performed using the quantification tag included as a fusion on every protein fragment (20).
The publicly available antibodies from the Human Protein Atlas project constitute a huge antibody resource for the immuno-SILAC method described here. These polyclonal antibodies are generated in a standardized manner involving immunizations of animals using recombinant protein fragments selected for their low sequence identity to other human proteins. In addition, more than a million antibodies toward human targets are listed in the antibody portal Antibodypedia (43), and ∼70% of these antibodies are polyclonal antibodies. However, the fact that the method described here relies on polyclonal antibodies makes it important to decrease the consumption of antibody reagent in each assay. Although it is possible to renew polyclonal antibodies through re-immunization of the same antigen in more animals, batch-to-batch variations exist, and one can never be sure of an unlimited resource of renewable antibodies, as is the case with monoclonal antibodies. We have investigated various protocols to lower the consumption of the antibody reagent used in each assay. The protocol described here requires only 50 ng of antibody for each target, and these antibodies do not need to be chemically immobilized to a solid support and can be simply mixed with Protein A–coated magnetic beads in a multiplex manner. Although the amount of antibodies varies greatly for publicly available polyclonal antibodies, the mean concentration of the ∼18,000 polyclonal antibodies generated within the framework of the Human Protein Atlas program is ∼140 μg/ml (unpublished); thus an aliquot of 100 μl can be used for more than 250 individual assays, and the whole batch with ∼10 ml of affinity-purified antibodies would last for 25,000 assays. Furthermore, it is likely that the need for antibodies can be reduced even further through the use of smaller magnetic beads and altered capture procedures, such as the use of microfluidics. Thus, the publicly available polyclonal antibodies covering more than 90% of the human protein-coding genes listed in Antibodypedia are an attractive resource for efforts to develop new immunoproteomics assays.
Here, we have screened polyclonal antibodies raised against human protein fragments to investigate their functionality for immunoproteomics. In total, the immuno-enrichment experiments generated 79 new immunoproteomics pairs, each with an antibody suitable for immuno-SILAC. Here, approximately half of the analyzed antibodies (57/127) yielded functional antibodies for peptide immuno-enrichment in the initial screening, and the subsequent quantitative HeLa analysis had a similar success rate (22/41). It is noteworthy that the high-density microarray assay identified approximately three linear epitopes per polyclonal antibody on average, but the results presented here suggest that only a fraction of these epitopes are suitable for immuno-SILAC.
The rapid turn-around times coupled with the ease of automating all the unit operations of sample preparation makes the immuno-SILAC method ideal for both research applications and future clinical diagnostic assays. In total, the PrEST-SILAC method described here required an MS analysis time of around 24 h for the six fractions of the sample. To compare, the MS analysis time for the 40-plex immuno-SILAC experiments took ∼40 min. It is noteworthy that more than 80% of the total intensities from the mass spectrometry analysis corresponded to the target peptides for the immuno-SILAC experiment, suggesting that the analysis time can be reduced even further. The method allows for multiplex analysis, and we have used it for the simultaneous analysis of 20 protein targets using both PrEST-SILAC and immuno-SILAC. As a large number of antibodies are publicly available, it might not be unrealistic to increase the multiplexing to several hundreds of protein targets. Particularly for samples with a huge dynamic range between abundant proteins and proteins to be diagnosed, such as plasma and serum analysis, the immunocapture step prior to the mass spectrometry analysis is an attractive way forward. In summary, we have shown that small amounts of polyclonal antibodies can be used for efficient multiplex analysis of quantitative levels of proteins, opening up the possibility of building up a proteome-wide resource for immuno-SILAC reagents based on already available public antibody resources.
Supplementary Material
Acknowledgments
We acknowledge the entire staff of the Human Protein Atlas program and the Science for Life Laboratory for valuable contributions.
Footnotes
Author contributions: M.M. and M.U. designed research; F.E., T.B., and B.F. performed research; M.Z., H.J., and J.L. contributed new reagents or analytic tools; F.E., T.B., and B.F. analyzed data; F.E., T.B., B.F., M.M., and M.U. wrote the paper; E.L. and S.H. provided intellectual input.
* Funding was provided by the Knut and Alice Wallenberg Foundation, the ProNova center through a grant from VINNOVA, and PROSPECTS and Affinomics 7th Framework grants from the European Directorate.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SISCAPA
- stable isotope standards and capture by anti-peptide antibodies
- PrEST
- protein epitope signature tag
- ABP
- albumin binding protein
- SILAC
- stable isotope labeling by amino acids in cell culture.
REFERENCES
- 1. Bjorhall K., Miliotis T., Davidsson P. (2005) Comparison of different depletion strategies for improved resolution in proteomic analysis of human serum samples. Proteomics 5, 307–317 [DOI] [PubMed] [Google Scholar]
- 2. Steel L. F., Trotter M. G., Nakajima P. B., Mattu T. S., Gonye G., Block T. (2003) Efficient and specific removal of albumin from human serum samples. Mol. Cell. Proteomics 2, 262–270 [DOI] [PubMed] [Google Scholar]
- 3. Jin W. H., Dai J., Li S. J., Xia Q. C., Zou H. F., Zeng R. (2005) Human plasma proteome analysis by multidimensional chromatography prefractionation and linear ion trap mass spectrometry identification. J. Proteome Res. 4, 613–619 [DOI] [PubMed] [Google Scholar]
- 4. Pieper R., Gatlin C. L., Makusky A. J., Russo P. S., Schatz C. R., Miller S. S., Su Q., McGrath A. M., Estock M. A., Parmar P. P., Zhao M., Huang S. T., Zhou J., Wang F., Esquer-Blasco R., Anderson N. L., Taylor J., Steiner S. (2003) The human serum proteome: display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics 3, 1345–1364 [DOI] [PubMed] [Google Scholar]
- 5. Pernemalm M., Lewensohn R., Lehtio J. (2009) Affinity prefractionation for MS-based plasma proteomics. Proteomics 9, 1420–1427 [DOI] [PubMed] [Google Scholar]
- 6. Berna M., Ott L., Engle S., Watson D., Solter P., Ackermann B. (2008) Quantification of NTproBNP in rat serum using immunoprecipitation and LC/MS/MS: a biomarker of drug-induced cardiac hypertrophy. Anal. Chem. 80, 561–566 [DOI] [PubMed] [Google Scholar]
- 7. Anderson N. L., Anderson N. G., Haines L. R., Hardie D. B., Olafson R. W., Pearson T. W. (2004) Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 3, 235–244 [DOI] [PubMed] [Google Scholar]
- 8. Whiteaker J. R., Zhao L., Abbatiello S. E., Burgess M., Kuhn E., Lin C., Pope M. E., Razavi M., Anderson N. L., Pearson T. W., Carr S. A., Paulovich A. G. (2011) Evaluation of large scale quantitative proteomic assay development using peptide affinity-based mass spectrometry. Mol. Cell. Proteomics 10, M110.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neubert H., Gale J., Muirhead D. (2010) Online high-flow peptide immunoaffinity enrichment and nanoflow LC-MS/MS: assay development for total salivary pepsin/pepsinogen. Clin. Chem. 56, 1413–1423 [DOI] [PubMed] [Google Scholar]
- 10. Hoofnagle A. N., Becker J. O., Wener M. H., Heinecke J. W. (2008) Quantification of thyroglobulin, a low-abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin. Chem. 54, 1796–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ahn Y. H., Lee J. Y., Lee J. Y., Kim Y. S., Ko J. H., Yoo J. S. (2009) Quantitative analysis of an aberrant glycoform of TIMP1 from colon cancer serum by L-PHA-enrichment and SISCAPA with MRM mass spectrometry. J. Proteome Res. 8, 4216–4224 [DOI] [PubMed] [Google Scholar]
- 12. Razavi M., Frick L. E., LaMarr W. A., Pope M. E., Miller C. A., Anderson N. L., Pearson T. W. (2012) High-throughput SISCAPA quantitation of peptides from human plasma digests by ultrafast, liquid chromatography-free mass spectrometry. J. Proteome Res. 11, 5642–5649 [DOI] [PubMed] [Google Scholar]
- 13. Anderson N. L., Jackson A., Smith D., Hardie D., Borchers C., Pearson T. W. (2009) SISCAPA peptide enrichment on magnetic beads using an in-line bead trap device. Mol. Cell. Proteomics 8, 995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Whiteaker J. R., Zhao L., Zhang H. Y., Feng L. C., Piening B. D., Anderson L., Paulovich A. G. (2007) Antibody-based enrichment of peptides on magnetic beads for mass-spectrometry-based quantification of serum biomarkers. Anal. Biochem. 362, 44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whiteaker J. R., Zhao L., Anderson L., Paulovich A. G. (2010) An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics 9, 184–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barr J. R., Maggio V. L., Patterson D. G., Jr., Cooper G. R., Henderson L. O., Turner W. E., Smith S. J., Hannon W. H., Needham L. L., Sampson E. J. (1996) Isotope dilution–mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin. Chem. 42, 1676–1682 [PubMed] [Google Scholar]
- 17. Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brun V., Dupuis A., Adrait A., Marcellin M., Thomas D., Court M., Vandenesch F., Garin J. (2007) Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol. Cell. Proteomics 6, 2139–2149 [DOI] [PubMed] [Google Scholar]
- 19. Singh S., Springer M., Steen J., Kirschner M. W., Steen H. (2009) FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. J. Proteome Res. 8, 2201–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeiler M., Straube W. L., Lundberg E., Uhlen M., Mann M. (2012) A Protein Epitope Signature Tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol. Cell. Proteomics 11, O111.009613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S., Wernerus H., Bjorling L., Ponten F. (2010) Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 28, 1248–1250 [DOI] [PubMed] [Google Scholar]
- 22. Berglund L., Bjorling E., Jonasson K., Rockberg J., Fagerberg L., Al-Khalili Szigyarto C., Sivertsson A., Uhlen M. (2008) A whole-genome bioinformatics approach to selection of antigens for systematic antibody generation. Proteomics 8, 2832–2839 [DOI] [PubMed] [Google Scholar]
- 23. Agaton C., Falk R., Hoiden Guthenberg I., Gostring L., Uhlen M., Hober S. (2004) Selective enrichment of monospecific polyclonal antibodies for antibody-based proteomics efforts. J. Chromatogr. A 1043, 33–40 [DOI] [PubMed] [Google Scholar]
- 24. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 25. Tegel H., Steen J., Konrad A., Nikdin H., Pettersson K., Stenvall M., Tourle S., Wrethagen U., Xu L., Yderland L., Uhlen M., Hober S., Ottosson J. (2009) High-throughput protein production—lessons from scaling up from 10 to 288 recombinant proteins per week. Biotechnol. J. 4, 51–57 [DOI] [PubMed] [Google Scholar]
- 26. Matic I., Jaffray E. G., Oxenham S. K., Groves M. J., Barratt C. L., Tauro S., Stanley-Wall N. R., Hay R. T. (2011) Absolute SILAC-compatible expression strain allows Sumo-2 copy number determination in clinical samples. J. Proteome Res. 10, 4869–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 28. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 29. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 30. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 31. Han D. K., Eng J., Zhou H., Aebersold R. (2001) Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat. Biotechnol. 19, 946–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scherer W. F., Syverton J. T., Gey G. O. (1953) Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 97, 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wisniewski J. R., Zougman A., Mann M. (2009) Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 [DOI] [PubMed] [Google Scholar]
- 34. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 36. Kuhn E., Whiteaker J. R., Mani D. R., Jackson A. M., Zhao L., Pope M. E., Smith D., Rivera K. D., Anderson N. L., Skates S. J., Pearson T. W., Paulovich A. G., Carr S. A. (2012) Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol. Cell. Proteomics 11, M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoevesandt O., Taussig M. J. (2012) European and international collaboration in affinity proteomics. Nat. Biotechnol. 29, 511–514 [DOI] [PubMed] [Google Scholar]
- 38. Uhlen M., Hultman T., Wahlberg J., Lundeberg J., Bergh S., Pettersson B., Holmberg A., Stahl S., Moks T. (1992) Semi-automated solid-phase DNA sequencing. Trends Biotechnol. 10, 52–55 [DOI] [PubMed] [Google Scholar]
- 39. Mirzaei H., Regnier F. (2006) Enhancing electrospray ionization efficiency of peptides by derivatization. Anal. Chem. 78, 4175–4183 [DOI] [PubMed] [Google Scholar]
- 40. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Paabo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Glatter T., Ludwig C., Ahrne E., Aebersold R., Heck A. J., Schmidt A. (2012) Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 11, 5145–5156 [DOI] [PubMed] [Google Scholar]
- 42. Danielsson F., Wiking M., Mahdessian D., Skogs M., Ait Blal H., Hjelmare M., Stadler C., Uhlen M., Lundberg E. (2013) RNA deep sequencing as a tool for selection of cell lines for systematic subcellular localization of all human proteins. J. Proteome Res. 12, 299–307 [DOI] [PubMed] [Google Scholar]
- 43. Bjorling E., Uhlen M. (2008) Antibodypedia, a portal for sharing antibody and antigen validation data. Mol. Cell. Proteomics 7, 2028–2037 [DOI] [PubMed] [Google Scholar]
- 44. Malm, et al. (2014), unpublished [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.