

Abstract
Streptavidin is one of the most important hubs for molecular biology, either multimerizing biomolecules, bridging one molecule to another, or anchoring to a biotinylated surface/nanoparticle. Streptavidin has the advantage of rapid ultra-stable binding to biotin. However, the ability of streptavidin to bind four biotinylated molecules in a heterogeneous manner is often limiting. Here, we present an efficient approach to isolate streptavidin tetramers with two biotin-binding sites in a precise arrangement, cis or trans. We genetically modified specific subunits with negatively charged tags, refolded a mixture of monomers, and used ion-exchange chromatography to resolve tetramers according to the number and orientation of tags. We solved the crystal structures of cis-divalent streptavidin to 1.4 Å resolution and trans-divalent streptavidin to 1.6 Å resolution, validating the isolation strategy and explaining the behavior of the Dead streptavidin variant. cis- and trans-divalent streptavidins retained tetravalent streptavidin's high thermostability and low off-rate. These defined divalent streptavidins enabled us to uncover how streptavidin binding depends on the nature of the biotin ligand. Biotinylated DNA showed strong negative cooperativity of binding to cis-divalent but not trans-divalent streptavidin. A small biotinylated protein bound readily to cis and trans binding sites. We also solved the structure of trans-divalent streptavidin bound to biotin-4-fluorescein, showing how one ligand obstructs binding to an adjacent biotin-binding site. Using a hexaglutamate tag proved a more powerful way to isolate monovalent streptavidin, for ultra-stable labeling without undesired clustering. These forms of streptavidin allow this key hub to be used with a new level of precision, for homogeneous molecular assembly.
Abbreviations: LDLR, low-density lipoprotein receptor; MPD, 2-methyl-2,4-pentanediol; PDB, Protein Data Bank; PBS, phosphate-buffered saline
Keywords: avidin, protein design, bivalent, supramolecular, nanotechnology
Graphical Abstract
Highlights
-
•
Streptavidin is a widely used nanohub: often, tetravalency makes assembly imprecise.
-
•
We isolated divalent streptavidins with biotin-binding sites in cis or in trans.
-
•
cis- and trans-divalent streptavidins retain exceptional biotin binding and thermostability.
-
•
Binding large biotinylated ligands to adjacent binding sites is greatly disfavored.
-
•
cis- and trans-divalent streptavidins represent a simple way to bridge biomolecules precisely.
Introduction
Streptavidin is a tetrameric protein from Streptomyces avidinii [1] that, along with the structurally similar protein from chicken egg white, avidin, exhibits extraordinarily high affinity for the vitamin biotin [2,3]. The femtomolar affinity of streptavidin/avidin for biotinylated targets, combined with resilience to harsh conditions and an on-rate of ~ 107 M− 1 s− 1, is the basis for biotin's central role as a specific biological “glue” in diverse research methods and biotechnologies [1–3].
Although streptavidin has a long history of use for capture of biotinylated ligands from complex mixtures, it has gained many recent applications for precise and stable assembly in the emerging field of bionanotechnology [4–6]. Such assemblies can be centered on a single streptavidin, for example, streptavidin:MHC (major histocompatibility complex) tetramers, to analyze immune function [7]. Assemblies can also depend on multiple streptavidin molecules holding together networks, such as for amplification in histochemistry [8] or to bridge biomolecules to facilitate structure determination by electron microscopy, atomic force microscopy, and X-ray crystallography [9–11].
A central issue that needs addressing is to control the nature of the four binding sites in the streptavidin tetramer. While the binding of four ligands may be desirable in some applications, in other applications, it may be necessary to limit binding to only one, two, or three subunits to achieve the desired assemblies and geometries [11–13]. Splitting up the streptavidin tetramer into monomers or dimers is possible, but this has always been accompanied by orders of magnitude lower affinity for biotin [14–17], because of the contribution of the neighboring subunit to the biotin-binding site [1,18]. Such monomers or dimers give desirable reversibility for purification but forfeit much of the advantage of streptavidin's stability for assembly applications.
We previously generated streptavidin heterotetramers with exactly one, two, or three functional biotin-binding sites, through purifying tetramers containing defined numbers of non-biotin-binding “Dead” and His6-tagged wild-type subunits [19]. However, tetramers with two functional and two Dead subunits can assemble in three different arrangements. These arrangements are the 1,2, 1,3, and 1,4 divalent forms of streptavidin (Fig. 1). In the 1,2 arrangement the biotin-binding sites of streptavidin are in a cis configuration as the binding sites both point in the same direction (Fig. 1). However, in the 1,3 arrangement, both sites are in a trans configuration, pointing away from each other (Fig. 1). Taking a vector from N1 to the terminal carbon atom of biotin, in 1,2, both biotins point 23° from the two-fold axis in Fig. 1, while in 1,3, one biotin is 23° from this axis and the other is 160°. In 1,2, the terminal carbon atoms of the two biotins are 20 Å apart, and in 1,3, they are 35 Å apart (Fig. 1). Therefore, any biotinylated ligands to divalent streptavidin will adopt different distances and angles with respect to each other. Such mixed divalent species, containing all three arrangements, would not be suitable for applications where defined spatial control is necessary. For example, a cis arrangement may be suitable for capping biotinylated protein oligomers [20], resulting in molecules with a uniform length, while the trans arrangement may serve as a linker in the middle of such an assembly [20,21].
Fig. 1.

Principle of defined divalent streptavidins. Numbering of subunits in the streptavidin tetramer, with each monomer colored separately and biotin in space-fill (from PDB ID: 3RY2). Below are arrangements of 1,2 or 1,3 biotin-binding sites (biotin-bound monomers, green; non-binding monomers, blue), showing relative orientations and distances between the biotin carboxyl group carbons.
Control over subunit orientation could be achieved by generating single-chain dimers or tetramers [22], but the symmetry and folding characteristics of streptavidin mean that connection of subunits (using circular permutation) has been accompanied by impaired folding and affinity [16,23].
To generate a controlled and stable bridge for bionanotechnology, we demonstrate here a method for the simple isolation of streptavidin variants with precise cis-divalent or 1,3 trans-divalent arrangements. We confirmed the variant structures by crystallography and investigated the binding characteristics of these defined divalents, which helps to resolve decades of data about interaction between binding sites on streptavidin/avidin [24–28].
Results
Streptavidins with defined valencies and subunit orientation by engineering of charge
To prepare streptavidin tetramers with controlled pairs of biotin-binding sites (Fig. 1), we added charged tags at specific sites in the subunits to enable resolution of the tetramers by ion-exchange chromatography (Figs. 2 and 3). We refolded from inclusion bodies the mixture of charged and non-charged streptavidin variants and then separated the tetramers on a MonoQ column (anion exchange), since the tetramers with increasing negative charge required higher ionic strength for elution. Geometrically distinct species bearing the same number of charged tags can be separated from each other. On that basis, two different engineering strategies made it possible to isolate the cis-divalent and 1,3 trans-divalent forms (Fig. 1).
Fig. 2.

Generation of the 1,3 trans-divalent streptavidin. (a) Amino acid sequence of streptavidin-E6 (SAe) with E6 tag red and underlined, along with Dead (D) streptavidin showing residues impairing biotin binding in blue. (b) Generation of the different valency forms of SAe/D by mixed refold and ion exchange, with predicted pI of monomer and tetramer indicated. (c) Ion-exchange chromatogram of a mixture of tetramers containing different proportions of SAe and D. (d) Analysis of peaks from ion exchange by SDS-PAGE with Coomassie staining, unboiled (left) to show tetramer mobility, or boiled (right) to show subunit composition.
Fig. 3.

Generation of cis-divalent streptavidin. (a) Amino acid sequence of core streptavidin (SA), along with Dead (Dd) containing a polyaspartate insertion in the 3/4 loop (red and underlined) and residues impairing biotin binding in blue. (b) Generation of the different valency forms of SA/Dd by mixed refold and ion exchange, with predicted pI of monomer and tetramer indicated. (c) Ion-exchange chromatogram of a mixture of tetramers containing different proportions of SA and Dd. (d) Analysis of peaks from ion exchange by SDS-PAGE with Coomassie staining, unboiled (left) to show tetramer mobility, or boiled (right) to show subunit composition.
C-terminal polyglutamate tag allowed 1,3 trans-divalent isolation
In the first instance, we placed a hexaglutamate tag at the C-terminus of core streptavidin monomer (termed SAe) (Fig. 2a). First, these charges ought to allow easy isolation of forms containing 0–4 biotin-binding sites by ion-exchange chromatography, giving clearer separation than the previous method of separation using a C-terminal His6 tag and Ni-NTA, where peaks from different tetramer forms largely overlapped [19,29]. Second the C-termini are close together between subunits 1 and 3 (27 Å) but far apart between subunits 1 and 2 (57 Å) or subunits 1 and 4 (50 Å), so that elution from the ion-exchange resin would be predicted to occur later for the 1,3 divalent (Fig. 1).
SAe was refolded together with Dead (D) streptavidin (N23A, S27D, S45A) [19] (Fig. 2a). Seven different tetramer forms are possible: one nullivalent, one monovalent, one trivalent, one tetravalent, and three divalent (Fig. 2b). The ion-exchange chromatogram showed six peaks (Fig. 2c), consistent with our hypothesis that 1,2 and 1,4 divalents would co-elute, while the 1,3 divalent would elute later.
We analyzed the eluted peaks by SDS-PAGE, where streptavidin will remain a tetramer without boiling (Fig. 2d) [30]. The tetramers do not run according to their molecular weight: the increasing negative charge increases migration of tetramers containing a higher proportion of SAe. Upon boiling in SDS, the tetramer breaks up into monomers and the relative proportion of SAe and D subunits is shown (Fig. 2d). This gel analysis confirmed that peak 1 was D4, peak 2 was the monovalent SAe1D3, peak 5 was the trivalent SAe3D1, peak 6 was the tetravalent SAe4, and peaks 3 and 4 were both divalent. A titration with biotin-4-fluorescein (whose fluorescence is quenched dramatically upon streptavidin binding) [31,32] confirmed the expected number of biotin-binding sites for D4, SAe4, SAe1D3, and the putative 1,3 trans-divalent (Supplementary Data, Fig. S1a).
We analyzed the eluted peaks by SDS-PAGE, where streptavidin will remain a tetramer without boiling (Fig. 2d) [30]. The tetramers do not run according to their molecular weight: the increasing negative charge increases migration of tetramers containing a higher proportion of SAe. Upon boiling in SDS, the tetramer breaks up into monomers and the relative proportion of SAe and D subunits is shown (Fig. 2d). This gel analysis confirmed that peak 1 was D4, peak 2 was the monovalent SAe1D3, peak 5 was the trivalent SAe3D1, peak 6 was the tetravalent SAe4, and peaks 3 and 4 were both divalent. A titration with biotin-4-fluorescein (whose fluorescence is quenched dramatically upon streptavidin binding) [31,32] confirmed the expected number of biotin-binding sites for D4, SAe4, SAe1D3, and the putative 1,3 trans-divalent (Supplementary Data Fig. S1a).
Multiple aspartates in a surface loop allowed cis-divalent isolation
To isolate cis-divalent streptavidin (1,2 arrangement of biotin-binding subunits) (Fig. 1), we introduced a series of aspartates into the loop between β-strands 3 and 4 (L3,4) of the Dead subunit (Fig. 3a). After exploring a number of different insertions for optimal expression and separation, we chose the sequence termed Dd (Fig. 3a). Introduction of the charges in this position means that in the 1,3 and 1,4 arrangements, these charged regions should be far apart, while in the 1,2 arrangement, the charged loops should be apposed (Fig. 3b). Since L3,4 is important for biotin binding affinity [1,18,33], it was necessary to insert these residues on the Dead subunit, so that the cis-divalent streptavidin could maintain high biotin binding affinity. Ion-exchange chromatography of a mixed refold of SA/Dd again allowed efficient separation of tetramers, giving six peaks, two of which corresponded to divalent species (Fig. 3b and c).
We analyzed peaks by SDS-PAGE with and without boiling. For unboiled samples, the negative charge on Dd accelerated the mobility of the tetramers (Fig. 3d). Upon boiling, the varying intensities of SA and Dd bands clearly showed that the tetramers contained the expected ratios of each subunit (Fig. 3d). This gel analysis confirmed that peak 1 was tetravalent SA4, peak 2 was the trivalent SA3Dd1, peak 5 was the monovalent SA1Dd3, peak 6 was the non-binding Dd4, and peaks 3 and 4 were both divalent. A titration of the different tetramer forms with biotin-4-fluorescein also confirmed the expected number of biotin-binding sites (Supplementary Data Fig. S1b). Overall, the introduction of a series of negative charges into the surface-exposed 3,4-loop of streptavidin allowed the isolation of a second divalent species, cis-divalent.
We analyzed peaks by SDS-PAGE with and without boiling. For unboiled samples, the negative charge on Dd accelerated the mobility of the tetramers (Fig. 3d). Upon boiling, the varying intensities of SA and Dd bands clearly showed that the tetramers contained the expected ratios of each subunit (Fig. 3d). This gel analysis confirmed that peak 1 was tetravalent SA4, peak 2 was the trivalent SA3Dd1, peak 5 was the monovalent SA1Dd3, peak 6 was the non-binding Dd4, and peaks 3 and 4 were both divalent. A titration of the different tetramer forms with biotin-4-fluorescein also confirmed the expected number of biotin-binding sites (Supplementary Data Fig. S1b). Overall, the introduction of a series of negative charges into the surface-exposed 3,4-loop of streptavidin allowed the isolation of a second divalent species, cis-divalent.
Crystal structures of cis-divalent and 1,3 trans-divalent streptavidins
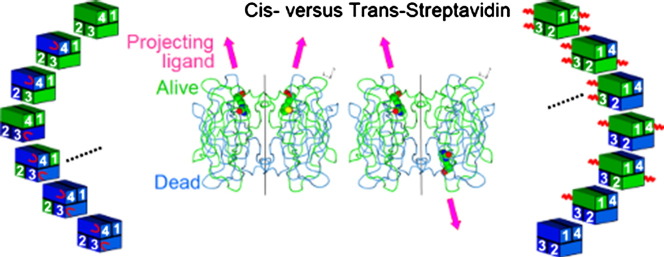
To confirm these divalent arrangements suggested by the ion-exchange elution profiles, we solved at high resolution the crystal structures of the 1,3 trans- and cis-divalent streptavidin (Fig. 4). Initially, all our crystal forms only contained two subunits in the asymmetric unit, which did not allow us to distinguish the possible divalent forms. However, one condition obtained using the Morpheus crystal screen [34] yielded the desired asymmetric unit for the cis-divalent streptavidin containing all four subunits. For the 1,3 trans-divalent form, crystallization with 2-methyl-2,4-pentanediol (MPD) gave us the desired asymmetric unit [35]. The obtained 1,3 trans-divalent crystal structure was solved at 1.6 Å resolution (Fig. 4b and Table 1), and cis-divalent streptavidin was solved at 1.4 Å resolution (Fig. 4d and Table 1), with the phases in each case solved using molecular replacement.
Fig. 4.

Crystal structures of divalent streptavidins. (a) Electron density (2Fo − Fc contoured at 1 rmsd) at residues 23, 27, and 45 in the 1,3 trans-divalent structure shown for chains b/c (SAe, green) and chains a/d (D, blue), with the side chain found indicated below. (b) Crystal structure of the 1,3 trans-divalent streptavidin, with green showing the biotin-binding SAe subunits and blue showing the non-biotin-binding D subunits. (c) Electron density in the cis-divalent structure, with chains b/d (SA, green) and chains a/c (Dd, blue). (d) Crystal structure of cis-divalent streptavidin, with green showing the biotin-binding SA subunits and blue showing the non-biotin-binding Dd subunits.
Table 1.
Data collection, refinement statistics, and structure validation for the 1,3 trans-divalent and cis-divalent streptavidin structures.
| 1,3 trans-divalent (4BX6) | cis-divalent (4BX5) | 1,3 trans-divalent with biotin-4-fluorescein (4BX7) | |
|---|---|---|---|
| Data collection | |||
| Space group | P1211 | P1211 | P3121 |
| Asymmetric unit | Tetramer | Tetramer | Dimer |
| Unit cell size (Å) | 47.04 × 81.59 × 65.12 | 46.22 × 84.24 × 58.25 | 64.41 × 64.41 × 103.13 |
| Unit cell angles (°) | 90 × 95.94 × 90 | 90 × 98.81 × 90 | 90 × 90 × 120 |
| Completeness (%) | 98.00 | 92.59 | 99.77 |
| Refinement | |||
| Resolution (Å) | 1.59–24.98 | 1.43–42.12 | 2.26–49.06 |
| No. of reflections | 64,074 | 75,108 | 12,059 |
| Rwork/Rfree | 0.1640/0.1866 | 0.1601/0.1873 | 0.1839/0.2369 |
| No. of atoms | |||
| Protein | 3756 | 3739 | 1768 |
| Heteroatoms | 24 | 42 | 54 |
| Water | 466 | 517 | 78 |
| B-factors (mean of all atoms, Å2) | |||
| Wilson plot | 20.98 | 14.65 | 42.43 |
| Mean B value | 28.40 | 22.00 | 45.10 |
| rmsd | |||
| Bond length (Å) | 0.011 | 0.01 | 0.01 |
| Bond angle (°) | 1.28 | 1.14 | 0.94 |
| Structure validation | |||
| MolProbity clash score | 3.65 (97th percentile) | 2.71 (98th percentile) | 2.29 (100th percentile) |
| Poor rotamers (%) | 2.01 | 0.57 | 2.33 |
| Ramachandran outliers (%) | 0.43 | 0.00 | 0.00 |
| Ramachandran favored (%) | 97.02 | 97.65 | 97.85 |
| MolProbity score | 1.55 (85th percentile) | 1.13 (98th percentile) | 1.32 (100th percentile) |
| Residues with bad bonds (%) | 0.00 | 0.00 | 0.00 |
| Residues with bad angles (%) | 0.00 | 0.00 | 0.00 |
We identified each subunit (whether SAe/D or SA/Dd) unambiguously by examining whether the electron density around residues 23, 27, and 45 corresponded to the residues of the wild-type or Dead subunit (N23A, S27D, S45A). For the putative 1,3 trans-divalent, the electron density closely fitted residues 23, 27, and 45 only if chains b/c were SAe and chains a/d were D (Fig. 4a), confirming that the biotin-binding subunits indeed had a 1,3 trans orientation (Fig. 4b). For the putative cis-divalent sample, the electron density closely fits if chains a/c are Dd and chains b/d are SA (Fig. 4c), also confirming that the biotin-binding subunits indeed had a cis orientation (Fig. 4d).
Furthermore, while the electron density around L3,4 could be clearly observed in the SA chains b and d of the cis-divalent form, the electron density of L3/4 was poorly defined in Dd chains a and c (Supplementary Data Fig. S2). The engineered L3,4 of Dd (chains a/c) is expected to be highly flexible, which would lead to weak diffraction in this region.
Furthermore, while the electron density around L3,4 could be clearly observed in the SA chains b and d of the cis-divalent form, the electron density of L3/4 was poorly defined in Dd chains a and c (Supplementary Data Fig. S2). The engineered L3,4 of Dd (chains a/c) is expected to be highly flexible, which would lead to weak diffraction in this region.
The rmsd of any of the chains in 1,3 trans-divalent and cis-divalent streptavidin is less than 0.5 Å to chain a of the 0.95-Å structure of streptavidin [Protein Data Bank (PDB) ID: 3RY2] [35]. The Dead streptavidin subunit has not been previously analyzed by crystallography, and the changes to the structure are localized to the mutated residues, consistent with the comparable thermostability of tetramers containing wild-type or Dead subunits [19,33]. The overlay of D and a wild-type subunit bound to biotin illustrates how the three mutations in D greatly reduced the number of polar contacts to biotin (Fig. 5a). In the overlay, Asp27 has van der Waals clash with biotin and there would be repulsion between the Asp27 carboxylate and biotin's carbonyl oxygen lone pair (Fig. 5a).
Fig. 5.

Structures of streptavidin interaction with ligands. (a) Structure of the biotin-binding pocket in the Dead subunit (N23A, S27D, and S45A), showing chain a of the 1,3 trans-divalent streptavidin (carbon atoms in blue) overlaid with biotin-bound wild-type streptavidin (PDB ID: 3RY2, carbon atoms in yellow). Hydrogen bonds from residues 23, 27, and 45 to biotin in wild-type streptavidin are shown as broken lines. (b) Crystal structure of the 1,3 trans-divalent streptavidin bound to biotin-4-fluorescein (space-fill, carbons in cyan), with green for SAe and blue for D subunits. (c) Residues surrounding the fluorescein tail (carbon atoms in cyan) in the 1,3 trans-divalent:biotin-4-fluorescein structure, labeled according to chain (b, carbon atoms in green; d, carbon atoms in blue), and with putative interactions as broken lines. Electron density for biotin-4-fluorescein is shown as blue mesh and contoured at 1 rmsd. (d) Clash for cis-bound biotin-4-fluorescein, generated from the 1,3 trans-divalent biotin-4-fluorescein structure, if two biotin-4-fluorescein molecules (carbon atoms of one in yellow and the other in cyan) were to bind in the same conformation in cis as they do in 1,3 trans-divalent.
Crystal structure of biotin-4-fluorescein bound to streptavidin
To understand interaction between ligand binding sites, we solved 1,3 trans-divalent streptavidin's crystal structure bound to biotin-4-fluorescein. Biotin-4-fluorescein represents the largest ligand where there is a crystal structure with streptavidin or avidin, as well as a widely used probe of the binding kinetics and capacity of biotin-binding proteins [19,31,32,36–38]. The fluorescence of biotin-4-fluorescein is quenched ~ 90% on binding to streptavidin/avidin, allowing a clear and sensitive test of ligand binding without any separation step [31,32]. Our 2.3-Å-resolution structure shows the 1,4 dimer in the asymmetric unit (one copy of SAe and one copy of D). Building the native streptavidin tetramer from this dimer shows that the two biotin-4-fluorescein molecules are in the expected trans (1,3) arrangement (Fig. 5b). All the contacts of the 1,3 trans-divalent streptavidin with the biotin moiety are as expected [18]. Fluorescein and the ethylene diamine spacer form several non-polar contacts to streptavidin not observed with biotin (Fig. 5c). Features that have been proposed to quench fluorescein are photoinduced electron transfer to nearby tryptophans or tyrosines [39], co-planarity of the xanthenone and benzoate rings [40], and hydrogen bonding to O3 of fluorescein [41]. Trp120 is 5 Å from fluorescein (Fig. 5c) and may explain part of the quenching of fluorescence.
Biotin-4-fluorescein bound in chain b reaches over and interacts with residues of chain d. However, we do not believe this conformation of biotin-4-fluorescein is the same as when four ligands are bound to the same tetramer: if the SAe:biotin-4-fluorescein complex were in a cis arrangement, there would be an obvious clash between the fluorescein moieties of neighboring ligands (Fig. 5d).
Low off-rate and high thermostability of defined divalents
Having confirmed the identity of the defined divalents, we initially assessed these proteins by measuring the off-rate for a biotin conjugate (Fig. 6a). The strong biotin-binding stability of wild-type streptavidin was retained by the defined divalents. Both divalents had dissociation rates for biotin-4-fluorescein comparable to the original SA4 or SAe4 tetramers. In fact, the binding stability for the 1,3 trans-divalent streptavidin was slightly superior to that of the cis-divalent one (P ≤ 0.001, n = 3) and that of the wild-type SA4 tetramer (P ≤ 0.001, n = 3, one-way ANOVA with Tukey's multiple comparison) (Fig. 6a).
Fig. 6.

Off-rate and thermostability of divalent streptavidins. (a) Biotin-4-fluorescein dissociation over time at 37 °C in the presence of competing biotin, for a tetramer of wild-type streptavidin subunits (SA4), a tetramer of SAe subunits (SAe4), and cis-divalent or 1,3 trans-divalent streptavidin (mean of triplicate + 1 SD). (b) Tetramer thermostability for streptavidin variants determined by SDS-PAGE with Coomassie staining, following incubation in PBS for 3 min at the indicated temperatures. In B, the protein was boiled in the presence of SDS to achieve complete conversion to monomers. The mobilities of the starting tetramer and the resulting monomers are marked.
We next compared the thermostability of the two divalent forms with tetramers containing only the constituent monomers. We incubated at varying temperatures and then analyzed the resulting protein forms by SDS-PAGE (Fig. 6b). Both cis-divalent and 1,3 trans-divalent forms started to denature to their constituent monomers above 70 °C, in a similar fashion to SA4, Dd4, SAe4, and D4. Some mixing of the divalent tetramers into new species could be observed at 70 °C before full denaturation into the monomer species (Fig. 6b). To check whether this mixing would be an issue at 37 °C or room temperature, we incubated cis-divalent and 1,3 trans-divalent streptavidins for 1–8 days, but we observed no detectable dissociation to rearranged tetramers or to constituent monomers (Supplementary Data, Fig. S3).
We next compared the thermostability of the two divalent forms with tetramers containing only the constituent monomers. We incubated at varying temperatures and then analyzed the resulting protein forms by SDS-PAGE (Fig. 6b). Both cis-divalent and 1,3 trans-divalent forms started to denature to their constituent monomers above 70 °C, in a similar fashion to SA4, Dd4, SAe4, and D4. Some mixing of the divalent tetramers into new species could be observed at 70 °C before full denaturation into the monomer species (Fig. 6b). To check whether this mixing would be an issue at 37 °C or room temperature, we incubated cis-divalent and 1,3 trans-divalent streptavidins for 1–8 days, but we observed no detectable dissociation to rearranged tetramers or to constituent monomers (Supplementary Data Fig. S3).
Binding of large biotinylated ligands by defined divalent streptavidins
To characterize further the interaction between the two biotin-binding sites of cis-divalent and 1,3 trans-divalent streptavidin, we examined the capture of larger biotinylated ligands. Binding of a monobiotinylated 439-bp PCR product was analyzed by a mobility shift in agarose gel electrophoresis (Fig. 7a). The 1,3 trans-divalent streptavidin bound two DNA molecules almost immediately. The cis-divalent form bound one DNA molecule rapidly, but binding of a second DNA molecule was very slow (Fig. 7a). A similar pattern was observed when titrating the biotinylated DNA with varying concentrations of each divalent streptavidin. While the 1,3 trans-divalent streptavidin preferentially bound two molecules of DNA, the cis-divalent form bound one or two molecules (Fig. 7b).
Fig. 7.

Differential binding of biotinylated molecules by divalent streptavidins. (a) A monobiotinylated PCR product (50 nM) was incubated with 1,3 trans- or cis-divalent streptavidin (100 nM) for the indicated times at 37 °C and then analyzed by agarose gel electrophoresis, with ethidium bromide visualization of DNA. C is a control of DNA without streptavidin. (b) Titration of the biotinylated PCR product (50 nM) with varying amounts of 1,3 trans-divalent (upper panel) or cis-divalent (lower panel) streptavidin for 16 h at 25 °C, analyzed by electrophoresis as in (a). (c) Native PAGE of monobiotinylated affibody titrated with 1,3 trans-divalent (upper panel) or cis-divalent (lower panel) streptavidin, analyzed by Coomassie staining. (d) Comparison of the relative sizes of an affibody (PDB ID: 2KZJ, space-fill), a short segment of B-form DNA (PDB ID: 1BNA, space-fill), and the separation between cis-binding sites on streptavidin (PDB ID: 3RY2, protein in gray, biotin in red, Connolly surface).
We also analyzed binding of a biotinylated protein, avoiding the high charge of polynucleotides. Titration of the divalents with a site-specifically biotinylated affibody [42], analyzed by the mobility shift in native PAGE, yielded a similar pattern (Fig. 7c). With streptavidin binding sites in excess, one or two molecules of the monobiotinylated affibody could bind to the 1,3 trans-divalent streptavidin, while for the cis-divalent one, only a single affibody bound. However, at higher concentrations of affibody, two molecules of affibody bound to the cis-divalent form (Fig. 7c). The sizes of the affibody and DNA double helix relative to streptavidin are illustrated in Fig. 7d.
Monovalent streptavidin isolated by ion-exchange chromatography allowed specific cellular imaging
Monovalent streptavidin has found diverse uses, such as a highlighter in electron microscopy [43], calibrating energies in protein folding [44], enhancing antibody detection sensitivity [45], and DNA origami-mediated nano-assembly [46]. The clean resolution of monovalent streptavidin SAe1D3 from other valency forms by ion-exchange chromatography (Fig. 2c) is superior to our previous Ni-NTA method, where elution of His6-tagged monovalent and divalent forms substantially overlapped [19,29]. Therefore, we validated that the new SAe1D3 form was still effective for cellular imaging and the altered charge did not lead to high non-specific cellular binding. HeLa cells were transfected with ER-resident biotin ligase and acceptor peptide-tagged low-density lipoprotein receptor (LDLR) [29] and then imaged with AlexaFluor 555-labeled SAe1D3. SAe1D3 clearly labeled cells expressing AP-GFP-LDLR but did not label nearby non-expressing cells or cells that were not co-transfected with biotin ligase (Supplementary Data Fig. S4), consistent with specific biotin detection.
Monovalent streptavidin has found diverse uses, such as a highlighter in electron microscopy [43], calibrating energies in protein folding [44], enhancing antibody detection sensitivity [45], and DNA origami-mediated nano-assembly [46]. The clean resolution of monovalent streptavidin SAe1D3 from other valency forms by ion-exchange chromatography (Fig. 2c) is superior to our previous Ni-NTA method, where elution of His6-tagged monovalent and divalent forms substantially overlapped [19,29]. Therefore, we validated that the new SAe1D3 form was still effective for cellular imaging and the altered charge did not lead to high non-specific cellular binding. HeLa cells were transfected with ER-resident biotin ligase and acceptor peptide-tagged low-density lipoprotein receptor (LDLR) [29] and then imaged with AlexaFluor 555-labeled SAe1D3. SAe1D3 clearly labeled cells expressing AP-GFP-LDLR but did not label nearby non-expressing cells or cells that were not co-transfected with biotin ligase (Supplementary Data Fig. S4), consistent with specific biotin detection.
Discussion
Herein, we describe the generation and analysis of tetramers of streptavidin with a defined orientation of twin biotin-binding sites. We were able to engineer cis-divalent streptavidin with comparably low biotin off-rate to wild-type streptavidin but 1,3 trans-divalent streptavidin showed even better binding stability. Defined divalent streptavidins should act as an efficient and simple bridge for diverse applications in bionanotechnology.
Although new in its application to streptavidin, the separation of multimers by ion-exchange chromatography according to the position of charged tags has been applied previously [47–49]. A similar orientation-dependent separation was observed for wild-type streptavidin tetramers containing two bound biotins because of biotin's charge. However, the biotin occupancy of such streptavidin tetramers changed over hours [27], whereas our defined divalents were stable for at least a week at 37 °C and should be stable indefinitely at − 80 °C. We found high thermostability of both divalent tetramers, with negligible dissociation into monomers until above 60 °C. The introduced mutations have also not reduced biotin-conjugate binding affinity, a problem frequently encountered when engineering streptavidin [14–17].
Despite testing several strategies to isolate 1,4 divalent streptavidins, we could never attain a clean separation. However, we anticipate that the most important thing is having one defined trans-divalent streptavidin, since the biotin–biotin separation distance for 1,3 and 1,4 is very close.
The work here also establishes a substantially improved route to monovalent streptavidin. Monovalent streptavidin with an E6 tag can be purified on a larger scale compared to the His6-tagged monovalent streptavidin, because its elution is well separated from divalent streptavidin. SAe1D3 is also easier to analyze for purity, because it is much better resolved on SDS-PAGE. The E6 tag did not impair the use of monovalent streptavidin for specific cellular imaging.
Divalent streptavidins without defined orientation have been used for generating monovalent quantum dots [50] and for studying mechanical interactions between MHC class I and T-cell receptors [51], illustrating areas where our defined divalents would improve control in nano-assembly.
Streptavidin and avidin are the most widely used biotin-binding proteins, but recently, several others have been found, including the dimers rhizavidin [52] and shwanavidin [53]. These relatives may find a range of applications, but the natural dimers contain disulfides and provide only one divalent orientation (equivalent to 1,4), and streptavidin appears to have stronger binding to biotin conjugates [53,54]. A further advantage of retaining the whole streptavidin tetramer is the possibility in future work to functionalize Dead subunits, such as by fusion to an antibody fragment [55] or via an exposed cysteine [56], to create a multifunctional hub.
The cooperativity that can exist for biotinylated ligand binding is not well appreciated by the wider community using streptavidin/avidin. For biotin itself, even though streptavidin has a conformational change and increased tetramer stability when biotin binds [18], there is no cooperativity of biotin binding [27]. However, for large biotinylated ligands, there can be substantial negative cooperativity in binding to streptavidin/avidin, as shown for biotin metal–ligand complexes [24]. Similarly, the binding of more than two biotinylated DNA molecules to wild-type streptavidin was not readily observed [5,57]. Our defined divalents allowed direct analysis of these ligand–ligand repulsions at cis or 1,3 trans binding sites. Binding of two large biotinylated ligands was efficient for the 1,3 trans-divalent streptavidin. Even with a ligand as small as biotin-4-fluorescein, our structure showed the interactions that would inhibit cis binding. For moderate-sized ligands, such as a biotinylated affibody, we found a degree of negative cooperativity for cis binding. For a larger and densely charged ligand such as biotinylated DNA, binding in cis was both slow and required substantial ligand excess. The two binding sites in the cis-divalent streptavidin are 20 Å apart, and the DNA double helix is 20 Å in diameter; hence, binding of the first DNA molecule provides substantial steric and electrostatic hindrance to binding of a second molecule. In particular, the 1,3 trans-divalent form should be preferable in generating precise DNA–streptavidin assemblies, as only fully loaded complexes (two DNA molecules per tetramer) were observed, rather than heterogeneous mixtures seen with wild-type streptavidin [5,57].
Numerous chemical (e.g., click chemistry) [58] and biological (e.g., SNAP-tag [59], HaloTag [60,61], SpyTag [62], and coiled coils [63]) approaches have been developed in recent years for stable protein assembly. However, streptavidin is still ubiquitous because of the wide-ranging biotinylated resources available and streptavidin's simple, nearly diffusion-limited, and high-yield binding, enabling “plug and play” construction. Our defined divalent streptavidins should be valuable modules for researchers to contrive novel nano-assemblies using nucleic acids, proteins, sugars, and non-biological building blocks [64].
Materials and methods
Constructs
pET21-Core streptavidin (SA) [65] and pET21-Dead streptavidin (D) [19] were used as templates for PCR with KOD Hot Start polymerase (EMD Millipore). For the addition of a C-terminal hexaglutamate tag to core streptavidin to give SAe (GenBank accession number KF378616), 5′-ATACATATGGCTGAAGCTGGTATCACCGGCACCTGG and 5′-CGCAAGCTTTTATTACTCTTCCTCTTCCTCTTCGGAAGCAGCGGACGGTTTAACTTTGG were used. The resulting PCR product was digested with NdeI and HindIII and subcloned into pET21. We inserted a polyaspartate loop into the Dead variant to give Dd (GenBank accession number KF378617) using the Site-directed Ligase-Independent Mutagenesis (SLIM) protocol [66] with 5′-GATGACGATGGTGACGATGACGGTGATGACGATGGAGCTGAATCTAGATACGTTCTGACC, 5′-GCTGAATCTAGATACGTTCTGACC, 5′-TCCATCGTCATCACCGTCATCGTCACCATCGTCATCACCAACAGCGGCTTCGTAGGT, and 5′-ACCAACAGCGGCTTCGTAGGT. pET21b-AP-Affibody was made by inverse PCR [67] from pET21b-ZSPA [68] with an N-terminal acceptor peptide (AP tag, GLNDIFEAQKIEWHE) for BirA-mediated biotinylation [69]. To introduce mutations to bind IGF1R (type 1 insulin-like growth factor receptor) [42], we generated AP-AffibodyIGF1R (GenBank accession number KF378618) using the forward primer 5′-AATCGAAAACAGTCTACCGCATTTATTTCTAGCCTTGAAGATGACCCAAGCCAAAGCGCT and the reverse primer 5′-TAGATTCGGTAATGCCAGGATTTCGATTGCAGCATAGAAACCTTCTTTGTTGAATTTGTTGTC.
Biotin ligase with a signal sequence and ER retention sequence (pDisplay-BirA-ER) and pEGFP-AP-GFP-LDLR (human LDLR containing an N-terminal acceptor peptide and GFP) were previously described [70].
All constructs were verified by sequencing. Plasmids for streptavidin constructs are available from Addgene:2 D Plasmid 20859, SAe 46367, and Dd 46368.
Protein expression and purification
SA, SAe, D, and Dd streptavidin variants were expressed in Escherichia coli and refolded from inclusion bodies by dilution into phosphate-buffered saline (PBS) as previously described [29]. After refolding, the protein mixtures of SA/Dd or SAe/D were first purified on a 5-mL iminobiotin-Sepharose affinity column (Affiland, S.A.) using 50 mM sodium borate and 300 mM NaCl, pH 11.0, as the binding buffer and 20 mM KH2PO4, pH 2.2, as the elution buffer, with a 5-mL/min flow rate. The eluate was then exchanged into 20 mM Tris–HCl, pH 8.0, by dialysis and loaded onto a 1-mL Mono-Q column (GE Healthcare). The different tetramers were then isolated using a 100-column-volume (i.e., 100 mL) linear gradient of 0–1 M NaCl, collecting 1-mL fractions with a 1-mL/min flow rate. For larger-scale preparations, a 30-mL Q-Sepharose High-Performance column (GE Healthcare) and a 30-column-volume gradient from 0.15 to 0.4 M NaCl was run. Eluted fractions were concentrated to 5–10 mg/mL using a Vivaspin cutoff 30-kDa centrifugal concentrator (GE Healthcare), dialyzed thrice into PBS, and stored at − 80 °C. All purification steps were performed using an ÄKTA purifier 10 (GE Healthcare). Yield from refolding 1 L of biotin-binding subunit mixed with 1 L of non-binding subunit was approximately 9 mg of the relevant divalent and 12 mg of the monovalent streptavidin.
Monovalent streptavidin (SAe1D3) can also be purified, if preferred, in a gravity-flow column. The mixed refold of SAe and D, prepared in 20 mM Tris–HCl, pH 8.0, as above, is loaded onto 1 mL Q-Sepharose High-Performance resin (GE Healthcare) in a Poly-Prep chromatography column (Bio-Rad) pre-equilibrated with binding buffer of 20 mM Tris–HCl, pH 8.0. The column is washed with 10 mL 20 mM Tris–HCl, pH 8.0, plus 0.15 M NaCl (to wash out D4 and impurities), followed by elution of SAe1D3 with 5 mL 20 mM Tris–HCl, pH 8.0, plus 0.25 M NaCl, collecting 1-mL fractions. The resin is regenerated by washing with 10 mL 20 mM Tris–HCl, pH 8.0, plus 2 M NaCl.
Glutathione S-transferase-BirA (the plasmid was a kind gift from Chris O'Callaghan, University of Oxford) was expressed in E. coli and purified using glutathione-Sepharose as described previously [71]. AP-Affibody was expressed in E. coli and purified using Ni-NTA (Qiagen) [68]. Enzyme-mediated biotinylation was performed as described previously [19].
Protein concentrations were determined from A280 via ProtParam. Concentrations of all streptavidin forms refer to the concentration of the monomer. pI was predicted using ProtParam.
SDS-PAGE
SDS-PAGE was performed on 10% or 14% polyacrylamide gels, using an XCell SureLock system (Life Technologies). Protein samples (10 μM) for denaturation were mixed with an equal volume of 2 × SDS loading buffer (20% glycerol, 100 mM Tris–HCl, 4% SDS, and 0.2% bromophenol blue, pH 6.8) and heated for 3 min at 95 °C. Non-denatured samples were loaded without boiling, in SDS-free loading buffer. Gels were typically run for 1 h at room temperature at 200 V in running buffer (25 mM Tris–HCl, 192 mM glycine, and 0.1% SDS, pH 8.2).
Biotinylated affibody and streptavidin were incubated at the indicated concentrations in PBS for 16 h at 25 °C, before mixing with an equal volume of 2 × loading buffer (20% glycerol, 100 mM Tris–HCl, and 0.2% bromophenol blue, pH 6.8). Native PAGE was performed on 10% polyacrylamide gels using standard running and loading buffers but without SDS. Gels were stained with InstantBlue (Expedeon) and imaged using a ChemiDoc XRS imager and QuantityOne (version 4.6) software (Bio-Rad).
Thermostability testing
Samples at 5 μM in PBS were heated at the indicated temperature for 3 min in a DNA Engine® Peltier Thermal Cycler (Bio-Rad). After cooling to room temperature, samples were mixed with an equal volume of 2 × loading buffer (20% glycerol, 100 mM Tris–HCl, and 0.2% bromophenol blue, pH 6.8). SDS-PAGE to discriminate monomers from tetramers was performed on 14% polyacrylamide gels as previously described [36]. Wild-type streptavidin samples heated for 3 min at 95 °C in the presence of SDS were included as controls.
Divalent streptavidin crystallization and data collection
Apo 1,3 trans-divalent streptavidin crystals (space group P1211; a = 47.04 Å, b = 81.59 Å, c = 65.12 Å, with two SAe and two D monomers in the asymmetric unit) were obtained from a 1-μL drop of a solution containing 0.5 μL of 740 μM 1,3 trans-divalent streptavidin in PBS mixed with 0.5 μL of reservoir solution, 65% v/v MPD, and 0.1 M 2-(N-morpholino)ethanesulfonic acid (Mes), pH 6.0. Crystals were obtained by the sitting-drop vapor-diffusion method at 291 K, reached a maximum size after ~ 10 days, and were harvested soon after.
For crystallization of 1,3 trans-divalent streptavidin with biotin-4-fluorescein, we added biotin-4-fluorescein to a final concentration of 100 μM along with 75 μM 1,3 trans-divalent streptavidin in PBS (0.5 mL final volume). This sample was then concentrated 10-fold using a Vivaspin cutoff 30-kDa centrifugal concentrator. Crystals (space group P3121; a = 64.41 Å, b = 64.41 Å, c = 103.13 Å, with one SAe and one D monomer in the asymmetric unit) were obtained from a 1-μL drop of a solution containing 0.5 μL 750 μM 1,3 trans-divalent streptavidin mixed with 0.5 μL reservoir solution: 65% v/v MPD and 0.1 M Mes, pH 4.0. Crystals were obtained by the sitting-drop vapor-diffusion method at 277 K, reached a maximum size after ~ 14 days, and were harvested soon after.
Apo cis-divalent streptavidin crystals (space group P1211; a = 46.22 Å, b = 84.24 Å, c = 58.25 Å, with two SA and two Dd monomers in the asymmetric unit) were obtained from a 0.1-μL drop of a solution containing 113 μM cis-divalent streptavidin and 28 μM ALAD-Streptag [11] and a reservoir solution of 2.5% w/v polyethylene glycol 1000, 12.5% w/v polyethylene glycol 3350, 12.5% v/v MPD, 30 mM of ethylene glycol mix (di-ethyleneglycol, tri-ethyleneglycol, tetra-ethyleneglycol, penta-ethyleneglycol), and 0.1 M Mes/imidazole, pH 6.5, corresponding to condition E4 of the Morpheus screen [34]. Crystals appeared after 2 days and were harvested soon after.
Prior to data collection, crystals were mounted and immersed into liquid nitrogen. Crystallographic data were collected at 100 K, using an Oxford Cryosystems 700 series Cryostream on an ADSC Quantum 315 charge-coupled device detector with an oscillation range of 0.5° at beamline iO4 at the Diamond Light Source, Didcot, UK.
Structure solution and refinement
Data were auto-indexed and integrated by the xia2 program upon collection [72]. The cis- and 1,3 trans-divalent streptavidin structures were phased by molecular replacement using PDB 3RY1 (apo streptavidin) [35] and further refined using the Phenix [73] suite of programs implemented through a Python graphical user interface [74]. The models were altered to better fit the electron density using Coot [75]. Throughout the refinement of apo 1,3 trans-divalent, all data were included from 24.98 Å resolution to the highest limit (1.59 Å), and anisotropic temperature factors were refined. For the 1,3 trans-divalent streptavidin with bound biotin-4-fluorescein, all data from 49.06 Å resolution to the highest limit (2.26 Å) and isotropic temperature factors were refined. Similarly, for the cis-divalent structure, all data were included from 42.12 Å resolution to the highest limit (1.43 Å), and anisotropic temperature factors were refined. All models were continually evaluated with MolProbity [76].
The refinement statistics and other structure factors for the two structures are shown in Table 1.
Structures were visualized, and images for figures were prepared using PyMOL (Schrödinger, LLC). Electron density maps were visualized and images were prepared (contoured at 1 rmsd) with the CCP4mg program [77]. Predicting the clash between cis-bound biotin-4-fluorescein molecules was done by overlaying the biotin-4-fluorescein-bound streptavidin monomer with the adjacent ligand-free monomer using PyMOL.
Biotin-4-fluorescein off-rate assay
The off-rate of biotin-4-fluorescein (Life Technologies) was measured from the fluorescence increase of biotin-4-fluorescein upon unbinding from streptavidin at 37 °C with competing free biotin (Sigma-Aldrich), as previously described [36]. Samples (180 μL) in a sealed clear 96-well microtiter plate containing 1 μM streptavidin variant in PBS were incubated with 12 nM biotin-4-fluorescein at 37 °C for 1 h. We then added 1 mM biotin (20 μL) and biotin-4-fluorescein release was monitored from the increase in fluorescence (λex = 480 nm, λem = 520 nm) using a SpectraFluor Plus plate reader (Tecan) over time at 37 °C. The percentage of dissociation was calculated as (signal with biotin − signal without biotin)/(signal without quenching − signal without biotin) × 100. The signal without quenching was taken as the biotin-4-fluorescein fluorescence in the absence of streptavidin. One-way ANOVA statistical analysis with Tukey's multiple comparison test for the dissociation rate curves was performed using the Prism program (GraphPad Software).
Biotin-4-fluorescein titration
To 20 nM streptavidin variant in PBS was added 0 to 15 nM biotin-4-fluorescein. Samples were incubated for 1 h at 37 °C in PBS. Subsequently, the fluorescence was measured (λex = 485 nm, λem = 523 nm) using a SpectraMax M5 plate reader (Molecular Devices).
Biotinylated DNA gel shift
A monobiotinylated 439-bp PCR product was prepared using Taq polymerase with primer Fts1 and terminally biotinylated primer bioFts3 (5′ biotin triethylene glycol linker), as previously described [36]. Streptavidin variant (100 nM) was incubated with 50 nM monobiotinylated PCR product in electrophoretic mobility shift assay buffer (20 mM Hepes, pH 7.9, 1 mM reduced dithiothreitol, 0.1 mM ethylenediaminetetraacetic acid, 50 mM KCl, 5% glycerol, and 0.2 mg/mL bovine serum albumin) for the indicated time at 37 °C, mixed with 2 × loading buffer [10% glycerol, 2 mg/mL Orange G (Sigma-Aldrich)], and run on a 1.5% agarose gel containing ethidium bromide with TAE (40 mM Tris–HCl, 20 mM acetic acid, and 1 mM ethylenediaminetetraacetic acid, pH 8.3) for 1 h at 130 V. Gels were imaged under UV using a ChemiDoc XRS imager and QuantityOne (version 4.6) software.
For Fig. 7b, the indicated concentration of the streptavidin variant was incubated with 50 nM of the monobiotinylated PCR product in electrophoretic mobility shift assay buffer for 16 h at 25 °C, before mixing with loading buffer and agarose electrophoresis as above.
Dye labeling
SAe1D3 was labeled with AlexaFluor 555 succinimidyl ester (Life Technologies) according to the manufacturer's instructions. After conjugation (10:1 dye:streptavidin monomer), excess dye was first removed by gel filtration on a PD10 column (GE Healthcare) and subsequently by dialysis three times against excess PBS. The number of dye molecules per streptavidin monomer was calculated, following manufacturer's instructions, from A280 and A555 to be 1.2.
Cell culture and receptor labeling
HeLa cells (American Type Culture Collection) were grown in high-glucose Dulbecco's modified Eagle's medium with 10% fetal bovine serum (GE Healthcare), 50 U/mL penicillin, and 50 μg/mL streptomycin (growth medium). HeLa cells were either co-transfected with AP-GFP-LDLR and BirA-ER plasmids or transfected with AP-GFP-LDLR plasmid alone using Lipofectamine 2000 and incubated overnight in growth medium supplemented with 10 μM biotin. Cells were then placed on ice, rinsed with cold PBS-Mg (PBS + 5 mM MgCl2), and labeled with 200 nM SAe1D3-AlexaFluor 555 in PBS-Mg with 1% dialyzed bovine serum albumin for 15 min on ice. After three washes with cold PBS-Mg, cells were imaged live.
Microscopy
Cells were imaged using a wide-field DeltaVision fluorescence microscope (Applied Precision) with a 40 × oil immersion objective and Optovar lens (1.6 ×), using softWoRx 5.0.0 software (Applied Precision). AlexaFluor 555 was imaged with 540DF40 excitation, 600DF50 emission, and a Chroma 84100bs polychroic filter set. Typical exposure times were 0.1–0.5 s. Samples shown in the same figure were imaged, analyzed, and displayed under identical conditions.
Accession numbers
Coordinates and structure factors have been deposited with PDB ID: 4BX5 for cis-divalent streptavidin, PDB ID: 4BX6 for 1,3 trans-divalent ligand-free streptavidin, and PDB ID: 4BX7 for 1,3 trans-divalent streptavidin bound to biotin-4-fluorescein.
The following are the supplementary data related to this article.
Supplementary material.
Supplementary material 1.
Supplementary material 2.
Supplementary material 3.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2013.09.016.
Acknowledgements
M.F. and M.H. were funded by the Biotechnology and Biological Sciences Research Council (BBSRC). D.K. and M.H. were funded by the Wellcome Trust. E.D.L. was funded by the Department of Biochemistry, University of Oxford. We would like to thank the Micron Oxford Advanced Bioimaging Unit and the Department of Biochemistry Biophysical Facility at the University of Oxford.
Conflict of interest: M.H. is an author on a patent application in 2006 for monovalent streptavidin compositions (USPTO 20070099248).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Sano T., Vajda S., Reznik G.O., Smith C.L., Cantor C.R. Molecular engineering of streptavidin. Ann N Y Acad Sci. 1996;799:383–390. doi: 10.1111/j.1749-6632.1996.tb33229.x. [DOI] [PubMed] [Google Scholar]
- 2.Green N.M. Avidin and streptavidin. Methods Enzymol. 1990;184:51–67. doi: 10.1016/0076-6879(90)84259-j. [DOI] [PubMed] [Google Scholar]
- 3.Laitinen O.H., Hytonen V.P., Nordlund H.R., Kulomaa M.S. Genetically engineered avidins and streptavidins. Cell Mol Life Sci. 2006;63:2992–3017. doi: 10.1007/s00018-006-6288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim M., Wang C.C., Benedetti F., Rabbi M., Bennett V., Marszalek P.E. Nanomechanics of streptavidin hubs for molecular materials. Adv Mater. 2011;23:5684–5688. doi: 10.1002/adma.201103316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niemeyer C.M., Adler M., Pignataro B., Lenhert S., Gao S., Chi L. Self-assembly of DNA-streptavidin nanostructures and their use as reagents in immuno-PCR. Nucleic Acids Res. 1999;27:4553–4561. doi: 10.1093/nar/27.23.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber C., Liu J., Egelseer E.M., Moll D., Knoll W., Sleytr U.B. Heterotetramers formed by an S-layer-streptavidin fusion protein and core-streptavidin as a nanoarrayed template for biochip development. Small. 2006;2:142–150. doi: 10.1002/smll.200500147. [DOI] [PubMed] [Google Scholar]
- 7.Sims S., Willberg C., Klenerman P. MHC-peptide tetramers for the analysis of antigen-specific T cells. Expert Rev Vaccines. 2010;9:765–774. doi: 10.1586/erv.10.66. [DOI] [PubMed] [Google Scholar]
- 8.Grumbach I.M., Veh R.W. The SA/rABC technique: a new ABC procedure for detection of antigens at increased sensitivity. J Histochem Cytochem. 1995;43:31–37. doi: 10.1177/43.1.7822761. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto D., Nagura N., Omote S., Taniguchi M., Ando T. Streptavidin 2D crystal substrates for visualizing biomolecular processes by atomic force microscopy. Biophys J. 2009;97:2358–2367. doi: 10.1016/j.bpj.2009.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L., Sigworth F.J. Liposomes on a streptavidin crystal: a system to study membrane proteins by cryo-EM. Methods Enzymol. 2010;481:147–164. doi: 10.1016/S0076-6879(10)81007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinclair J.C., Davies K.M., Venien-Bryan C., Noble M.E. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat Nanotechnol. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- 12.Yeates T.O., Padilla J.E. Designing supramolecular protein assemblies. Curr Opin Struct Biol. 2002;12:464–470. doi: 10.1016/s0959-440x(02)00350-0. [DOI] [PubMed] [Google Scholar]
- 13.Padilla J.E., Colovos C., Yeates T.O. Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci U S A. 2001;98:2217–2221. doi: 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sano T., Vajda S., Smith C.L., Cantor C.R. Engineering subunit association of multisubunit proteins: a dimeric streptavidin. Proc Natl Acad Sci U S A. 1997;94:6153–6158. doi: 10.1073/pnas.94.12.6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim K.H., Huang H., Pralle A., Park S. Stable, high-affinity streptavidin monomer for protein labeling and monovalent biotin detection. Biotechnol Bioeng. 2013;110:57–67. doi: 10.1002/bit.24605. [DOI] [PubMed] [Google Scholar]
- 16.Aslan F.M., Yu Y., Vajda S., Mohr S.C., Cantor C.R. Engineering a novel, stable dimeric streptavidin with lower isoelectric point. J Biotechnol. 2007;128:213–225. doi: 10.1016/j.jbiotec.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Wu S.C., Wong S.L. Engineering soluble monomeric streptavidin with reversible biotin binding capability. J Biol Chem. 2005;280:23225–23231. doi: 10.1074/jbc.M501733200. [DOI] [PubMed] [Google Scholar]
- 18.Stayton P.S., Freitag S., Klumb L.A., Chilkoti A., Chu V., Penzotti J.E. Streptavidin–biotin binding energetics. Biomol Eng. 1999;16:39–44. doi: 10.1016/s1050-3862(99)00042-x. [DOI] [PubMed] [Google Scholar]
- 19.Howarth M., Chinnapen D.J., Gerrow K., Dorrestein P.C., Grandy M.R., Kelleher N.L. A monovalent streptavidin with a single femtomolar biotin binding site. Nat Methods. 2006;3:267–273. doi: 10.1038/NMETHXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori Y., Wakabayashi R., Goto M., Kamiya N. Protein supramolecular complex formation by site-specific avidin–biotin interactions. Org Biomol Chem. 2013;11:914–922. doi: 10.1039/c2ob26625c. [DOI] [PubMed] [Google Scholar]
- 21.Oohora K., Burazerovic S., Onoda A., Wilson Y.M., Ward T.R., Hayashi T. Chemically programmed supramolecular assembly of hemoprotein and streptavidin with alternating alignment. Angew Chem Int Ed Engl. 2012;51:3818–3821. doi: 10.1002/anie.201107067. [DOI] [PubMed] [Google Scholar]
- 22.Nordlund H.R., Hytonen V.P., Horha J., Maatta J.A., White D.J., Halling K. Tetravalent single-chain avidin: from subunits to protein domains via circularly permuted avidins. Biochem J. 2005;392:485–491. doi: 10.1042/BJ20051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim K.H., Huang H., Pralle A., Park S. Engineered streptavidin monomer and dimer with improved stability and function. Biochemistry. 2011;50:8682–8691. doi: 10.1021/bi2010366. [DOI] [PubMed] [Google Scholar]
- 24.Loosli A., Rusbandi U.E., Gradinaru J., Bernauer K., Schlaepfer C.W., Meyer M. (Strept)avidin as host for biotinylated coordination complexes: stability, chiral discrimination, and cooperativity. Inorg Chem. 2006;45:660–668. doi: 10.1021/ic051405t. [DOI] [PubMed] [Google Scholar]
- 25.Sano T., Cantor C.R. Cooperative biotin binding by streptavidin. Electrophoretic behavior and subunit association of streptavidin in the presence of 6 M urea. J Biol Chem. 1990;265:3369–3373. [PubMed] [Google Scholar]
- 26.Green N.M. Avidin. 5. Quenching of fluorescence by dinitrophenyl groups. Biochem J. 1964;90:564–568. doi: 10.1042/bj0900564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones M.L., Kurzban G.P. Noncooperativity of biotin binding to tetrameric streptavidin. Biochemistry. 1995;34:11750–11756. doi: 10.1021/bi00037a012. [DOI] [PubMed] [Google Scholar]
- 28.Green N.M. Avidin. Adv Protein Chem. 1975;29:85–133. doi: 10.1016/s0065-3233(08)60411-8. [DOI] [PubMed] [Google Scholar]
- 29.Howarth M., Ting A.Y. Imaging proteins in live mammalian cells with biotin ligase and monovalent streptavidin. Nat Protoc. 2008;3:534–545. doi: 10.1038/nprot.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayer E.A., Ehrlich-Rogozinski S., Wilchek M. Sodium dodecyl sulfate-polyacrylamide gel electrophoretic method for assessing the quaternary state and comparative thermostability of avidin and streptavidin. Electrophoresis. 1996;17:1319–1324. doi: 10.1002/elps.1150170808. [DOI] [PubMed] [Google Scholar]
- 31.Kada G., Kaiser K., Falk H., Gruber H.J. Rapid estimation of avidin and streptavidin by fluorescence quenching or fluorescence polarization. Biochim Biophys Acta. 1999;1427:44–48. doi: 10.1016/s0304-4165(98)00177-9. [DOI] [PubMed] [Google Scholar]
- 32.Kada G., Falk H., Gruber H.J. Accurate measurement of avidin and streptavidin in crude biofluids with a new, optimized biotin-fluorescein conjugate. Biochim Biophys Acta. 1999;1427:33–43. doi: 10.1016/s0304-4165(98)00178-0. [DOI] [PubMed] [Google Scholar]
- 33.Chivers C.E., Koner A.L., Lowe E.D., Howarth M. How the biotin–streptavidin interaction was made even stronger: investigation via crystallography and a chimaeric tetramer. Biochem J. 2011;435:55–63. doi: 10.1042/BJ20101593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorrec F. The MORPHEUS protein crystallization screen. J Appl Crystallogr. 2009;42:1035–1042. doi: 10.1107/S0021889809042022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Trong I., Wang Z., Hyre D.E., Lybrand T.P., Stayton P.S., Stenkamp R.E. Streptavidin and its biotin complex at atomic resolution. Acta Crystallogr D. 2011;67:813–821. doi: 10.1107/S0907444911027806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chivers C.E., Crozat E., Chu C., Moy V.T., Sherratt D.J., Howarth M. A streptavidin variant with slower biotin dissociation and increased mechanostability. Nat Methods. 2010;7:391–393. doi: 10.1038/nmeth.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mittal R., Bruchez M.P. Biotin-4-fluorescein based fluorescence quenching assay for determination of biotin binding capacity of streptavidin conjugated quantum dots. Bioconjug Chem. 2011;22:362–368. doi: 10.1021/bc100321c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takakura Y., Tsunashima M., Suzuki J., Usami S., Kakuta Y., Okino N. Tamavidins—novel avidin-like biotin-binding proteins from the Tamogitake mushroom. FEBS J. 2009;276:1383–1397. doi: 10.1111/j.1742-4658.2009.06879.x. [DOI] [PubMed] [Google Scholar]
- 39.Gotz M., Hess S., Beste G., Skerra A., Michel-Beyerle M.E. Ultrafast electron transfer in the complex between fluorescein and a cognate engineered lipocalin protein, a so-called anticalin. Biochemistry. 2002;41:4156–4164. doi: 10.1021/bi015888y. [DOI] [PubMed] [Google Scholar]
- 40.Urano Y., Kamiya M., Kanda K., Ueno T., Hirose K., Nagano T. Evolution of fluorescein as a platform for finely tunable fluorescence probes. J Am Chem Soc. 2005;127:4888–4894. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- 41.Whitlow M., Howard A.J., Wood J.F., Voss E.W., Hardman K.D. 1.85-Angstrom structure of antifluorescein 4-4-20-Fab. Protein Eng. 1995;8:749–761. doi: 10.1093/protein/8.8.749. [DOI] [PubMed] [Google Scholar]
- 42.Li J., Lundberg E., Vernet E., Larsson B., Hoiden-Guthenberg I., Graslund T. Selection of affibody molecules to the ligand-binding site of the insulin-like growth factor-1 receptor. Biotechnol Appl Biochem. 2010;55:99–109. doi: 10.1042/BA20090226. [DOI] [PubMed] [Google Scholar]
- 43.Lau P.W., Potter C.S., Carragher B., MacRae I.J. DOLORS: versatile strategy for internal labeling and domain localization in electron microscopy. Structure. 2012;20:1995–2002. doi: 10.1016/j.str.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blois T.M., Hong H., Kim T.H., Bowie J.U. Protein unfolding with a steric trap. J Am Chem Soc. 2009;131:13914–13915. doi: 10.1021/ja905725n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kattah M.G., Coller J., Cheung R.K., Oshidary N., Utz P.J. HIT: a versatile proteomics platform for multianalyte phenotyping of cytokines, intracellular proteins and surface molecules. Nat Med. 2008;14:1284–1289. doi: 10.1038/nm.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sacca B., Meyer R., Erkelenz M., Kiko K., Arndt A., Schroeder H. Orthogonal protein decoration of DNA origami. Angew Chem Int Ed Engl. 2010;49:9378–9383. doi: 10.1002/anie.201005931. [DOI] [PubMed] [Google Scholar]
- 47.Sakash J.B., Kantrowitz E.R. The contribution of individual interchain interactions to the stabilization of the T and R states of Escherichia coli aspartate transcarbamoylase. J Biol Chem. 2000;275:28701–28707. doi: 10.1074/jbc.M005079200. [DOI] [PubMed] [Google Scholar]
- 48.Miles G., Bayley H., Cheley S. Properties of Bacillus cereus hemolysin II: a heptameric transmembrane pore. Protein Sci. 2002;11:1813–1824. doi: 10.1110/ps.0204002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hammarstrom P., Schneider F., Kelly J.W. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 50.You C., Wilmes S., Beutel O., Lochte S., Podoplelowa Y., Roder F. Self-controlled monofunctionalization of quantum dots for multiplexed protein tracking in live cells. Angew Chem Int Ed Engl. 2010;49:4108–4112. doi: 10.1002/anie.200907032. [DOI] [PubMed] [Google Scholar]
- 51.Huang J., Zarnitsyna V.I., Liu B., Edwards L.J., Jiang N., Evavold B.D. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helppolainen S.H., Nurminen K.P., Maatta J.A., Halling K.K., Slotte J.P., Huhtala T. Rhizavidin from Rhizobium etli: the first natural dimer in the avidin protein family. Biochem J. 2007;405:397–405. doi: 10.1042/BJ20070076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meir A., Bayer E.A., Livnah O. Structural adaptation of a thermostable biotin-binding protein in a psychrophilic environment. J Biol Chem. 2012;287:17951–17962. doi: 10.1074/jbc.M112.357186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nordlund H.R., Hytonen V.P., Laitinen O.H., Kulomaa M.S. Novel avidin-like protein from a root nodule symbiotic bacterium, Bradyrhizobium japonicum. J Biol Chem. 2005;280:13250–13255. doi: 10.1074/jbc.M414336200. [DOI] [PubMed] [Google Scholar]
- 55.Park S.I., Shenoi J., Frayo S.M., Hamlin D.K., Lin Y., Wilbur D.S. Pretargeted radioimmunotherapy using genetically engineered antibody-streptavidin fusion proteins for treatment of non-Hodgkin lymphoma. Clin Cancer Res. 2011;17:7373–7382. doi: 10.1158/1078-0432.CCR-11-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stayton P.S., Ding Z., Hoffman A.S. Smart polymer–streptavidin conjugates. Methods Mol Biol. 2004;283:37–43. doi: 10.1385/1-59259-813-7:037. [DOI] [PubMed] [Google Scholar]
- 57.Funabashi H., Ubukata M., Ebihara T., Aizawa M., Mie M., Kobatake E. Assessment of small ligand–protein interactions by electrophoretic mobility shift assay using DNA-modified ligand as a sensing probe. Biotechnol Lett. 2007;29:785–789. doi: 10.1007/s10529-006-9301-6. [DOI] [PubMed] [Google Scholar]
- 58.Sletten E.M., Bertozzi C.R. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinner M.J., Johnsson K. How to obtain labeled proteins and what to do with them. Curr Opin Biotechnol. 2010;21:766–776. doi: 10.1016/j.copbio.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 60.Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N., Zimprich C. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 61.Liu D.S., Phipps W.S., Loh K.H., Howarth M., Ting A.Y. Quantum dot targeting with lipoic acid ligase and HaloTag for single-molecule imaging on living cells. ACS Nano. 2012;6:11080–11087. doi: 10.1021/nn304793z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zakeri B., Fierer J.O., Celik E., Chittock E.C., Schwarz-Linek U., Moy V.T. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A. 2012;109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Woolfson D.N., Bartlett G.J., Bruning M., Thomson A.R. New currency for old rope: from coiled-coil assemblies to alpha-helical barrels. Curr Opin Struct Biol. 2012;22:432–441. doi: 10.1016/j.sbi.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 64.Keren K., Berman R.S., Buchstab E., Sivan U., Braun E. DNA-templated carbon nanotube field-effect transistor. Science. 2003;302:1380–1382. doi: 10.1126/science.1091022. [DOI] [PubMed] [Google Scholar]
- 65.Sano T., Pandori M.W., Chen X., Smith C.L., Cantor C.R. Recombinant core streptavidins. A minimum-sized core streptavidin has enhanced structural stability and higher accessibility to biotinylated macromolecules. J Biol Chem. 1995;270:28204–28209. doi: 10.1074/jbc.270.47.28204. [DOI] [PubMed] [Google Scholar]
- 66.Chiu J., March P.E., Lee R., Tillett D. Site-directed, Ligase-Independent Mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 2004;32:e174. doi: 10.1093/nar/gnh172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gama L., Breitwieser G.E. Generation of epitope-tagged proteins by inverse PCR mutagenesis. Biotechniques. 1999;26:814–816. doi: 10.2144/99265bm03. [DOI] [PubMed] [Google Scholar]
- 68.Holm L., Moody P., Howarth M. Electrophilic affibodies forming covalent bonds to protein targets. J Biol Chem. 2009;284:32906–32913. doi: 10.1074/jbc.M109.034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beckett D., Kovaleva E., Schatz P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howarth M., Liu W., Puthenveetil S., Zheng Y., Marshall L.F., Schmidt M.M. Monovalent, reduced-size quantum dots for imaging receptors on living cells. Nat Methods. 2008;5:397–399. doi: 10.1038/nmeth.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O'Callaghan C.A., Byford M.F., Wyer J.R., Willcox B.E., Jakobsen B.K. BirA enzyme: production and application in the study of membrane receptor–ligand interactions by site-specific biotinylation. McMichael A.J., Bell J.I., editors. Anal Biochem. 1999;266:9–15. doi: 10.1006/abio.1998.2930. [DOI] [PubMed] [Google Scholar]
- 72.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J Appl Crystallogr. 2010;43:186–190. [Google Scholar]
- 73.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Echols N., Grosse-Kunstleve R.W., Afonine P.V., Bunkoczi G., Chen V.B., Headd J.J. Graphical tools for macromolecular crystallography in PHENIX. J Appl Crystallogr. 2012;45:581–586. doi: 10.1107/S0021889812017293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 76.Chen V.B., Arendall W.B., III, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McNicholas S., Potterton E., Wilson K.S., Noble M.E. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.
Supplementary material 1.
Supplementary material 2.
Supplementary material 3.