

Abstract
While microRNAs (miRNAs) clearly regulate multiple pathways integral to disease development and progression, the lack of safe and reliable means for specific delivery of miRNAs to target tissues represents a major obstacle to their broad therapeutic application. Our objective was to explore the use of nucleic acid aptamers as carriers for cell-targeted delivery of a miRNA with tumor suppressor function, let-7g. Using an aptamer that binds to and antagonizes the oncogenic receptor tyrosine kinase Axl (GL21.T), here we describe the development of aptamer-miRNA conjugates as multifunctional molecules that inhibit the growth of Axl-expressing tumors. We conjugated the let-7g miRNA to GL21.T and demonstrate selective delivery to target cells, processing by the RNA interference machinery, and silencing of let-7g target genes. Importantly, the multifunctional conjugate reduced tumor growth in a xenograft model of lung adenocarcinoma. Therefore, our data establish aptamer-miRNA conjugates as a novel tool for targeted delivery of miRNAs with therapeutic potential.
Introduction
A growing body of literature implicates microRNAs (miRNAs) in regulation of a vast network of genes involved in development and disease. These short noncoding RNAs modulate gene expression by either preventing the translation of messenger RNAs and/or by targeting messenger RNAs for degradation. Deregulation of miRNA biogenesis has been implicated in many diseases, including cancer. For example, loss of miRNAs with tumor suppressor functions has been documented in several cancer types. In contrast, miRNAs that target tumor suppressor genes are often upregulated in oncogenic states,1,2 highlighting their potential as novel anticancer therapeutics. However, the fact that a single miRNA can regulate expression of multiple genes illustrates the need for specific delivery miRNAs to target cells. This is expected to minimize undesired effects in nontarget tissues. Therefore, the development of effective and safe tools for selective delivery of miRNAs into tumor cells is recognized as a key step toward adapting miRNA-based therapeutics for human diseases.
Synthetic RNA ligands, aptamers, have been used for targeted delivery of oligonucleotides to cells in vivo.3 Aptamers are evolved from a complex library through a process termed systematic evolution of ligands through exponential enrichment (SELEX). In order to identify aptamers that bind to cell-surface receptor targets, several variants of the cell-SELEX approach have been recently described.4,5,6,7,8,9,10 In addition to providing exquisite cell specificity in vivo, aptamers represent a powerful tool for delivery of therapeutic cargos.11,12 For example, direct conjugation of small interfering RNAs (siRNAs) to such aptamers has been established as an effective means to silence target genes in specific cell types in vitro and in vivo.13,14,15,16,17,18,19,20,21,22,23 This technology has been expanded to develop dual-functional reagents composed of inhibitory aptamers conjugated to therapeutic cargos.24
In previous studies, we employed the cell-SELEX methodology to identify the GL21.T aptamer, which we showed binds the receptor tyrosine kinase oncogene, Axl, with high affinity (low nmol/l range) and specificity.7 We demonstrated that GL21.T inhibits Axl tyrosine kinase activity and interferes with target cell growth and motility.7,25 Our objective in this study was to provide the first demonstration of a one-component multifunctional agent capable of in vivo targeted delivery of therapeutic miRNAs. Toward this end, we engineered an aptamer-miRNA conjugate by conjugating the tumor suppressor let-7g miRNA sequence26,27 to the GL21.T aptamer. We demonstrate specific delivery of the multifunctional conjugate to Axl-expressing cancer cells and silencing of let-7g target genes in vitro and in vivo. Treatment of Axl+ cancer cells with the conjugate resulted in pronounced inhibition of (i) cancer cell survival and migration in culture and (ii) tumor growth in a xenograft model of lung cancer. This study provides the first proof-of-concept for in vivo targeted delivery of miRNAs with therapeutic potential and the findings have broad applicability to pathologies with underlying miRNA deregulation.
Results
Rational design of multifunctional aptamer-miRNA conjugates
We designed a multifunctional conjugate, which we term “GL21.T-let,” containing the GL21.T aptamer,7 an antagonist of the Axl receptor, as delivery carrier and the human let-7g miRNA as a gene-silencing moiety (Figure 1a). The passenger strand of let-7g sequence (with UU-3′ overhang) is fused to the 3′ end of the aptamer sequence, and the complementary guide strand of let-7g is annealed to the template. The conjugate is modified with 2′-fluoro pyrimidines (2′F-Py) to protect against nuclease degradation, which is essential for in vivo applications.28
Figure 1.

GL21.T-let conjugate specificity and processing. (a) Scheme (left panel) and secondary structure predicted by RNA structure v4.5 (right panel, free energy: −44.5 J/mol) of GL21.T-let. Guide strand is in red. (b) Left panel: binding of 50 nmol/l radiolabeled GL21.T or GL21.T-let on A549 (Axl+) and MCF-7 (Axl−) cells. Right panel: binding isotherm of GL21.T-let to soluble extracellular domain of Axl (EC-Axl). (c) Time-dependent internalization of radiolabeled GL21.T, GL21.T-let, or a scrambled sequence of GL21.T (scraGL21.T) used as negative control. Results are expressed as percent of internalized RNA relative to total bound. (d) Left panel: the indicated RNAs were untreated or treated with recombinant Dicer, resolved on a nondenaturing polyacrylamide gel and stained with ethidium bromide. Right panel: GL21.T-let containing radiolabeled guide strand was incubated with Dicer, resolved on gel, and analyzed by autoradiography. Size of single strand (ss) let-7g guide and expected size of miRNA duplex (ds) are indicated. (e) A549 (Axl+) cells were transfected with let-7g miRNA mimic (let-7g-miR), control-microRNA (ctrl-miR), GL21.T, or GL21.T-let. HMGA2 protein was analyzed by immunoblotting (upper panel) or let-7g miRNA was measured by RT-qPCR (lower panel), 48 hours after transfection. Values below the blots indicate signal levels relative to mock-treated cells (indicated as “-”), arbitrarily set to 1 (with asterisk). Intensity of bands was calculated using ImageJ (v1.46r). Equal loading was confirmed by immunoblot with anti-α-tubulin (αTub) antibodies. (f) RNAs from A549 (Axl+) cells transfected with let-7g-miR, ctrl-miR, GL21.T-let, or GL21.T were immuprecipitated with anti-Ago2 antibody and processed for let-7g miRNA RT-qPCR. No let-7g miRNA was immunoprecipitated with negative control mouse IgG (data not shown). In (b, e, and f) error bars depict mean ± SD (n = 3). HMGA2, high mobility group AT-hook 2; miRNA, microRNA; RT-qPCR, reverse transcription quantitative polymerase chain reaction.
An important feature of the conjugate design is the let-7g mimetic. Although complementary miRNA sequences have been shown to be sufficient for targeted gene silencing,29,30,31 the thermodynamic stability at each end of the duplex impacts recruitment of the guide strand into the RNA-induced silencing complex.32,33,34,35,36 Several recent reports on the use of siRNA as therapeutics have shown that silencing efficacy and specificity can be improved by introducing internal partial complementarity and increased length extension to obtain a more effective Dicer substrate.15,23,34 Therefore, in order to encourage correct strand selection and thereby target specificity, we used the distal stem portion of the human let-7g pre-miRNA encompassing the first 27 bases of the 5′ strand and 26 of the 3′ strand, derived from the let-7g precursor, rather than a mature miRNA mimetic.35 The thermodynamic end stability properties of the resulting duplex miRNA should closely correspond to the native mature miRNA.
Initially, we confirmed that the predicted secondary structure of the GL21.T aptamer (Figure 1a), as well as binding of the aptamer to Axl-positive cells (Figure 1b), are preserved in the context of the conjugate. We next asked whether conjugation of the miRNA to the aptamer alters binding of the GL21.T to Axl. Specifically, as compared to the GL21.T aptamer, the GL21.T-let conjugate retains high binding efficiency to the target A549 (Axl+) cells with no detectable binding to the Michigan Cancer Foundation (MCF)-7 (Axl−) cells (Figure 1b, left panel). The apparent Kd of GL21.T-let (19 nmol/l; Figure 1b, right panel) and its internalization rate (Figure 1c) correlates with that of the unconjugated aptamer, whereas conjugate internalization into MCF-7 (Axl−) cells is less than 10% (data not shown). These data demonstrate that conjugation of the let-7g miRNA does not affect affinity of the GL21.T aptamer for its target or internalization into target cells.
We next determined whether the let-7g miRNA moiety is still recognized as a Dicer substrate when conjugated to the GL21.T aptamer. As demonstrated by nondenaturing gel electrophoresis, incubation of conjugates with recombinant human Dicer results in cleaved products corresponding to the expected size of the duplex let-7g miRNA (Figure 1d, left panel). To confirm that the cleaved Dicer product corresponds to the duplex miRNA portion, we 32P-end-labeled the guide strand and annealed it to the GL21.T-let conjugate. Following Dicer processing, the labeled strand migrates on nondenaturing gel as a dimer of ~22 nucleotides (Figure 1d, right panel), consistent with proper Dicer processing.
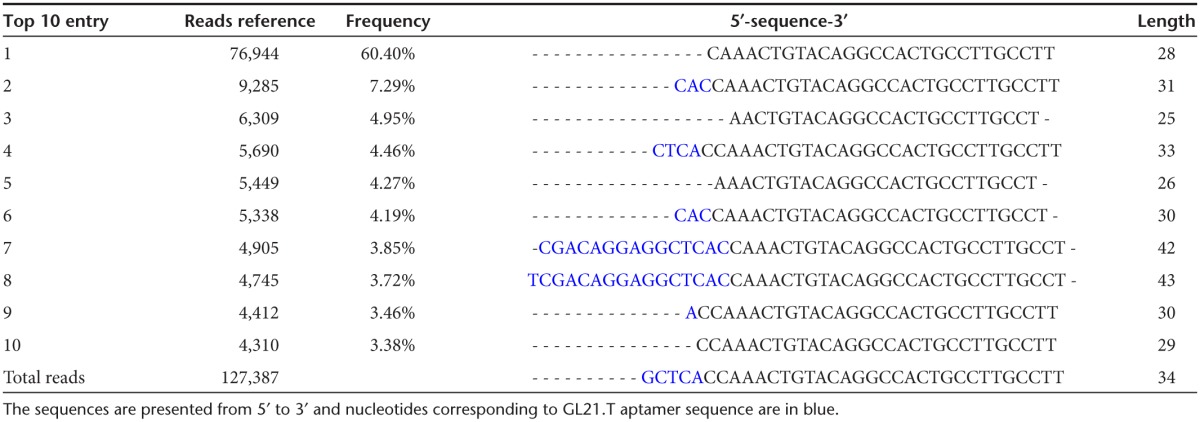
Next, we verified that the GL21.T-let conjugate was efficiently processed once inside the target cells to produce the mature let-7g miRNA duplex. The levels of the processed duplexes were determined by Illumina Deep sequencing analyses in A549 (Axl+) cells transfected with the conjugate. As shown, the let-7g guide strand is processed with variable lengths ranging between 24 and 27 bases (Table 1) while the passenger strand includes three bases coming from the sequence at the 3′ end of the aptamer (Table 2), and most importantly, sequences at the 5′ end of the aptamer are instead absent or present at less than 0.05% of total reads. These data indicate that while the mature guide strand is the major product produced in cells, base wobbling at the site of Dicer cleavage is also present. These results are not surprising and are in agreement with previous observations made by John Rossi's group regarding processing of aptamer-siRNA conjugate in cells.23
Table 1. Illumina Deep sequence analyses: total reads of the top 10 guide sequences.
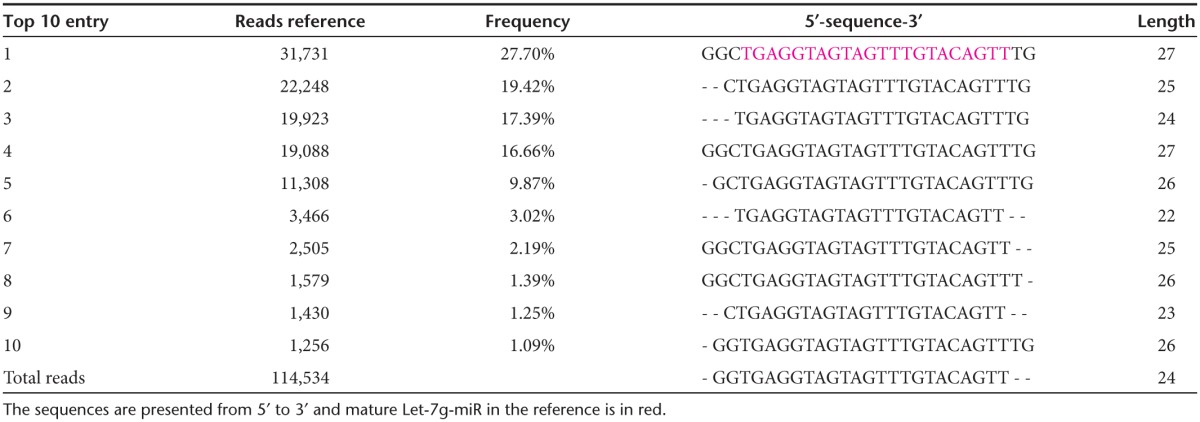
Table 2. Illumina Deep sequence analyses: total reads of the top 10 passenger sequences.
We next assessed whether transfecting the conjugate in A549 (Axl+) cells leads to let-7g target-specific silencing. Our data demonstrate that GL21.T-let acts as powerful inhibitor of the expression of high mobility group AT-hook 2 (HMGA2), a validated target of let-7g, involved in cell transformation37 (Figure 1e, upper panel). Both the amount of intracellular let-7g (Figure 1e, lower panel) and its HMGA2 silencing efficiency (Figure 1e, upper panel) are comparable to that achieved with transfection of a commercial let-7g miRNA mimic, also indicating that 2′F-Py modification of the conjugate do not compromise its silencing capability.38 Following GL21.T-let transfection, the let-7g miRNA is recruited to the Ago2 complex (Figure 1f); further indicating that processing of the conjugate by the silencing machinery is functional.39 Taken together, these data provide compelling evidence that the GL21.T-let conjugate selectively binds to Axl-expressing cells, is properly processed by the RNA interference (RNAi) machinery to produce functional double strand let-7g miRNA, and silences a validated let-7g target, HMGA2.
The GL21.T aptamer mediates delivery of functional miRNAs
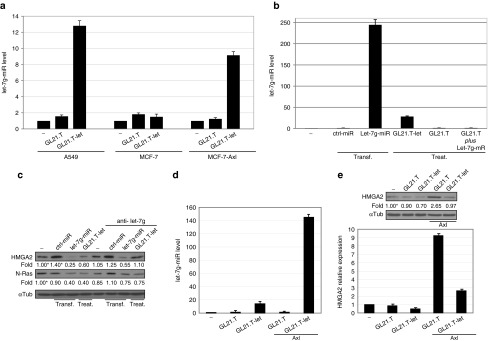
Since transfecting the GL21.T-let conjugate downregulates the expression of a known let-7g miRNA target, we asked whether the GL21.T sequence may act as selective delivery moiety for the conjugated miRNA. To this end, we first treated A549 (Axl+) cells for 48 hours with 400 nmol/l of GL21.T-let and determined by reverse transcription quantitative polymerase chain reaction (RT-qPCR) the levels of let-7g miRNA. As compared to treatment with GL21.T aptamer, treating cells with the aptamer-miRNA conjugate results in a significant increase of let-7g miRNA levels, though to a less extent as compared to let-7g mimic transfection (Figure 2a, left panel). Accordingly, HMGA2 expression levels are strongly reduced in cells treated with GL21.T-let (Figure 2a, right panel).
Figure 2.

GL21.T mediates delivery of let-7g miRNA. (a) A549 (Axl+) cells were transfected with 100 nmol/l let-7g-miR or ctrl-miR or treated with 400 nmol/l GL21.T-let or GL21.T. After 48 hours, let-7g miRNA was quantified by RT-qPCR (left panel) or cell lysates were immunoblotted with anti-HMGA2 and anti-αTub antibodies (right panel). (b, c) A549 (Axl+) cells were transfected with 100 nmol/l let-7g-miR or ctrl-miR or treated with GL21.T-let or GL21.T at (b) indicated concentration or at (c) 400 nmol/l for indicated times. (b) After 48 hours or (c) the indicated times, cell lysates were immunoblotted with anti-HMGA2, anti-N-Ras, and anti-αTub antibodies (upper panel in b and in left panel c). let-7g miRNA was quantified by RT-qPCR (lower panel in b and right panel in c). Integral for let-7g-miRNA (A1) or GL21.T-let (A2) at different time points was calculated (inset). (d) A549 (Axl+) cells were transfected with HMGA2-3′UTR-Lenti-reporter-GFP-vector alone or in combination with 50 nmol/l let-7g-miR-, ctrl-miR-, GL21.T-let-, or GL21.T-transfection or 400 nmol/l GL21.T-let- or GL21.T-treatment. Fluorescence was measured following 24 hours. No fluorescence changes were detected with Blank-3′UTR-GFP-control-vector ( data not shown). Statistics (compared to mock-treated cells) are indicated, ***P < 0.0005, *P < 0.01. (e) A549 (Axl+) cells were transfected with 100 nmol/l let-7g-miR or ctrl-miR or treated with 400 nmol/l GL21.T-let or GL21.T in absence or in presence of Dicer siRNA. After 48 hours, cell lysates were immunoblotted with anti-Dicer, anti-HMGA2, and αTub antibodies. In (a–c and e), band intensity was calculated as in Figure 1. In (a–d) error bars, mean ± SD (n = 3). In (a–e), transfection (transf) or treatment (treat) are indicated. GFP, green fluorescent protein; HMGA2, high mobility group AT-hook 2; miRNA, microRNA; UTR, untranslated region.
To further support the efficacy of silencing, we determined whether the GL21.T-let alters the expression of N-Ras, another predicted target of let-7g.40 In A549 (Axl+) cells, we detect a dose-dependent decrease in HMGA2 and N-Ras expression (Figure 2b, upper panel) that corresponds with intracellular let-7g levels (Figure 2b, lower panel) and reaches a plateau at 400 nmol/l GL21.T-let. These results suggest that the amounts of internalized conjugate are saturable and likely depend on the cell-surface availability of the aptamer target, Axl. In addition, we performed a time course of GL21.T-let conjugate-treatment. Silencing of HMGA2 is maximum at 24 hours and subsequently declines up to 72 hours, while silencing of N-Ras is more effective at 72 hours than at earlier times (Figure 2c, left panel), likely reflecting the different half-life of these two targets. In parallel studies, we determined the kinetics of let-7g intracellular accumulation. Cellular levels of let-7g reach a maximum at 24 hours following treatment of target cells with the conjugate (Figure 2c, right panel and inset). As expected, the levels of let-7g (A2) in treated cells are several folds lower compared to transfection of let-7g (A1) using a cationic lipid (Figure 2c, right panel and inset). Despite this difference, the inhibitory effect of the conjugate on let-7g targets (HMGA2 and N-Ras) is similar to that achieve with transfection of the let-7 miRNA (Figure 2c, left panel).
In order to confirm the specificity of let-7g targeting, we transfected A549 (Axl+) cells with a green fluorescent protein (GFP)-based reporter plasmid containing the 3′-untranslated region (UTR) of the human HMGA2 (HMGA2-3′-UTR-Lenti-reporter-GFP-vector) and showed that treatment with the GL21.T-let, but not with the GL21.T aptamer, reduces the GFP signal (Figure 2d). Furthermore, using a Dicer-specific siRNA, we show that downregulation of the HMGA2 protein by the GL21.T-let conjugate requires expression of Dicer (Figure 2e), which is in agreement with data in Figure 1d. Taken together, these experiments demonstrate that GL21.T-let delivers functional let-7g into target cells. Furthermore, as compared to transfected miRNA mimics, the amount of miRNA delivered with the conjugate is only a few times above the physiological levels, thus decreasing the likelihood of off-target effects.
The GL21.T aptamer as cell-specific delivery agent
As previously shown,7 the GL-21.T aptamer selectively binds to and is internalized in Axl-expressing cells. We thus determined whether appending a miRNA moiety alters cell-type specificity of the GL-21.T aptamer, by treating A549 (Axl+) and MCF-7 (Axl−) cells with GL21.T-let and determining the relative amounts of intracellular let-7g. As shown in Figure 3a, the let-7g miRNA levels increase in target cells, but remain unchanged in Axl-negative MCF-7 (Axl−) cells, indicative of receptor-dependent delivery and intracellular processing of the let-7g miRNA moiety of the conjugate. Consistently, treating the MCF-7 (Axl−) cells with the conjugate elicits the intracellular accumulation of let-7g, solely if cells were forced to express Axl (Figure 3a). Conversely, as shown in Figure 3b, treating A549 (Axl+) cells with a mixture of GL21.T aptamer and let-7g miRNA does not affect levels of intracellular let-7g, thus confirming that the effect depends on the presence of both the miRNA and the aptamer moieties within the conjugate. In addition, downregulation of the let-7g specific targets, HMGA2 and N-Ras, is rescued by a competitive-specific inhibitor of let-7g miRNA (anti-let-7g), thus demonstrating that target silencing depends on the presence of the functional let-7g miRNA moiety (Figure 3c). We next asked if the functional delivery of let-7g is dependent on the amount of Axl on the cell surface. We thus transiently overexpressed Axl in A549 (Axl+) cells (Supplementary Figure S1). We show that the intracellular levels of let-7g, following treatment with the conjugate, increase by 10-fold in A549 (Axl+) cells which overexpress Axl, compared to parental A549 (Axl+) cells (Figure 3d). In addition, the cellular levels of the let-7g target, HMGA2, were significantly reduced despite an observed increase in HMGA2 levels upon Axl overexpression (Figure 3e).41 Furthermore, we demonstrate that different conjugation strategies (e.g., stick-end annealing)16,22,23 and different aptamer sequences (e.g., prostrate specific membrane antigen aptamer)13,15,42 result in a functional conjugate (Supplementary Figure S2). Likewise, conjugation of different miRNA sequences (e.g., miR-16) to the GL21.T aptamer, results in a functional conjugate capable of suppressing the miRNA target genes (Supplementary Figure S3). Taken together, these data indicate that aptamer-miRNA conjugates can be broadly designed to selectively deliver a functional miRNA to any cell expressing the aptamer target on the cell surface.
Figure 3.
GL21.T aptamer specific delivery. (a) let-7g-miR levels were quantified by RT-qPCR in A549 (Axl+), MCF-7 (Axl−), or Axl TruClone-transfected MCF-7 cells (MCF-7-Axl) treated with 400 nmol/l of GL21.T-let or GL21.T. (b) A549 (Axl+) cells were transfected (transf) with 100 nmol/l let-7g-miR or ctrl-miR or were treated (treat) with 400 nmol/l GL21.T-let, GL21.T, or a mixture of GL21.T and let-7g-miR (GL21.T plus Let-7g-miR). let-7g miRNA levels were quantified by RT-qPCR. (c) A549 (Axl+) cells were transfected (transf) with 100 nmol/l let-7g-miR or ctrl-miR or were treated (treat.) with 400 nmol/l GL21.T-let or GL21.T in absence or in presence of let-7g competitive-specific inhibitor (anti-let-7g). After 48 hours, cell lysates were immunoblotted with anti-HMGA2 or anti-N-Ras antibodies, as indicated. (d, e) A549 (Axl+) cells, following 24-hour transfection with Axl TruClone (Axl), were treated with 400 nmol/l GL21.T-let or GL21.T for additional 24 hours. (d) let-7g miRNA levels were quantified by RT-qPCR. (e) HMGA2 levels were analyzed by immunoblotting (upper panel) or RT-qPCR (lower panel). In (c, e), values below the blots indicate signal levels relative to mock-treated cells, arbitrarily set to 1 (“-”, labeled with asterisk). Intensity of bands has been calculated as in Figure 1. In (a, b, d, and e), error bars depict mean ± SD (n = 3). HMGA2, high mobility group AT-hook 2; miRNA, microRNA; RT-qPCR, reverse transcription quantitative polymerase chain reaction.
GL21.T-let acts as a multifunctional inhibitor of cell survival and motility
Next, we investigated the activities of the aptamer and miRNA moieties within the context of the GL21.T-let conjugate. As shown in Figure 4a, inhibition of Axl tyrosine kinase activity is comparable between the GL21.T aptamer and the GL21.T-let conjugate further confirming that conjugation of the miRNA to the aptamer does not abrogate aptamer function. As predicted, GL21.T-let, but not the GL21.T aptamer alone, interferes with the expression of downstream let-7g targets: Bcl-XL (anti-apoptotic),43 and cyclin D1 (proproliferative)44 (Figure 4b). Using genome-wide expression analysis, we demonstrate that the changes in gene expression induced by the GL21.T-let conjugate were both qualitatively and quantitatively similar to those induced by the aptamer and miRNA moieties used separately (Figure 4c and Supplementary Figure S4). The majority (around 80%) of genes (down- or up-) regulated by treating cells with the GL21.T-let chimera were similarly regulated by either the let-7g mimic transfection or by the GL21.T treatment; genes regulated by both let-7g miRNA and GL21.T, were also regulated by the chimera, but at a greater degree (see selected examples indicated with a black arrow in Figure 4c). However, we also noted several genes that are uniquely regulated by either the aptamer or the miRNA (Supplementary Figure S4a). In addition, we performed gene ontology analyses of genes that are changed following GL21.T-let treatment showing that the conjugate predominantly regulates genes involved in cell cycle, death, and proliferation (Supplementary Figure S4b). Next, we determined whether the let-7g miRNA and GL21.T aptamer moieties produce additive or synergistic effects on cell growth and motility in the context of the GL21.T-let conjugate. As shown in Figure 4d, cells treated with the conjugate have reduced viability by ~30–40% (at 400 and 800 nmol/l), whereas cells treated with either the GL21.T aptamer or let-7g miRNA alone display a significantly lower reduction in viability by ~20–25%. In addition, treatment of cells with the GL21.T-let conjugate inhibits A549 (Axl+) cell migration by ~60%. In contrast, the aptamer and miRNA show ~40% reduction in cell migration when used separately (Figure 4e). Taken together, these results indicate that the effects of GL21.T-let on cell survival and migration are additive and are due to the combined effects on Axl signaling (mediated by the GL21.T aptamer) and on let-7g miRNA target genes (mediated by the let-7g miRNA).
Figure 4.

GL21.T-let in vitro function. (a) Serum-starved A549 (Axl+) cells were left untreated or treated for 3 hours with 200 nmol/l GL21.T, scraGL21.T, or GL21.T-let and then stimulated Gas 6 (15 minutes with 400 ng/ml) in presence of each RNA. Cell lysates were immunoblotted with anti-phospho-Axl (pAxl), anti-Axl, and αTub antibodies. (b) A549 (Axl+) cells were transfected with 100 nmol/l let-7g-miR or ctrl-miR or treated with 400 nmol/l GL21.T-let or GL21.T. Following 72 hours, cell lysates were immunoblotted with anticyclin D1 (cyc D1), anti-Bcl-XL, and anti-αtubulin antibodies. In (a, b), band intensity was calculated as in Figure 1. (c) RNAs from A549 (Axl+) cells transfected with let-7g-miR or ctrl-miR or treated with 400 nmol/l GL21.T-let or GL21.T were subjected to Illumina-HT12 v.4.0 array. Heat maps of genes that are significantly downregulated or upregulated (≥±1.5) following GL21.T-let-treatment are shown. The color code is indicated at the top. Distance trees represent the relative difference between genes. Arrows indicate representative examples. (d) A549 (Axl+) cells were transfected with 100 nmol/l let-7g-miR or ctrl-miR or treated GL21.T-let or GL21.T as indicated. After 72 hours, cell viability was measured. (e) Motility of A549 (Axl+) cells transfected with 100 nmol/l let-7g-miR or treated with 400 nmol/l of GL21.T-let or GL21.T was analyzed by Transwell Migration Assay using 10% FBS as migration inducer. In (d, e) the results are relative to untreated cells. Statistics were analyzed ***P < 0.0005; **P < 0.005; *P < 0.01 (n = 6). Error bars, mean ± SD (n = 3). In (b, d, e), transfection (transf) or treatment (treat) are indicated. FBS, fetal bovine serum.
In vivo targeting with GL21.T-let multifunctional conjugate
We next determined the target specificity of the GL21.T-let conjugate in vivo. To this end, immunodeficient (nu/nu) mice-bearing A549 (Axl+)-Luc tumors (right flank) and MCF-7 (Axl−)-Luc tumors (left flank) were treated with a single intravenous injection (1,600 pmol) of GL21.T-let, GL21.T aptamer or control, nontargeting aptamer (scraGL21.T). Mice were then sacrificed 24 hours after administration of the RNAs and tumors/organs (kidney, liver, and spleen) excised and analyzed by RT-qPCR. Following GL21.T-let treatment, the intracellular levels of let-7g increase in Axl-expressing A549 (Axl+) but not in MCF-7 (Axl−) tumors. Importantly, no let-7g was detected in liver or spleen, two well-known oligodeoxynucleotide target tissues.45 In contrast, we did see accumulation of let-7g miRNA in kidney, likely due to renal clearance and not uptake of the miRNA into renal cells (Figure 5a). Together, these data suggest that the conjugate retains cell/tissue-target specificity in vivo. Furthermore, the increase of let-7g levels in tumor-bearing mice treated with GL21.T-let (Figure 5b, left panels) results in downregulation HMGA2 (let-7g target) and a concomitant induction in active caspase-3, a measure of apoptosis46 (Figure 5b, middle and right panels).
Figure 5.
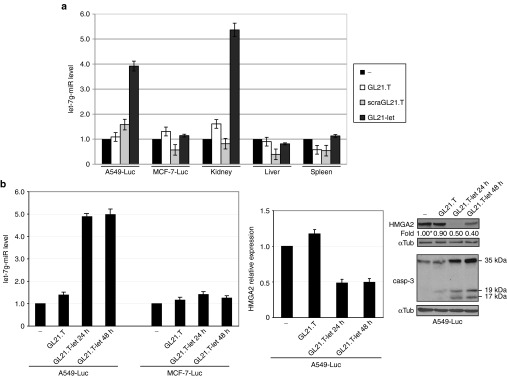
GL21.T-let in vivo selective targeting. (a, b) (nu/nu) Mice-bearing MCF-7 (Axl−)-Luc cells and A549 (Axl+)-Luc xenografts were injected intravenously either with 1,600 pmol of GL21.T or GL21.T-let conjugate. (a) After 24 hours tumors, kidney, liver, and spleen were excised, lysed for RNA extraction, and pooled for let-7g miRNA RT-qPCR. (b) After 24 or 48 hours A549 (Axl+)-Luc tumors were excised, lysed for protein or RNA extraction and pooled. Left panel: let-7g miRNA levels were quantified by RT-qPCR (middle panel), HMGA2 mRNA levels were quantified by RT-qPCR. Right panel: lysates were immunoblotted with anti-HMGA2, anti-caspase-3, and anti-αTub antibodies, as indicated. Band intensity was calculated as in Figure 1. Molecular weights of full-length and cleaved (active) caspase-3 are shown. In (a, b), error bars depict mean values of experimental replicates ± SD (n = 3). HMGA2, high mobility group AT-hook 2; mRNA, messenger RNA; miRNA, microRNA; RT-qPCR, reverse transcription quantitative polymerase chain reaction.
Next, we evaluated the ability of the conjugate to inhibit tumor growth in vivo. Treatment of GL21.T-let (1,600 pmol/injection three times a week for 2 weeks, administered via tail vein) resulted in a significant reduction in A549 (Axl+)-Luc, but not in MCF-7 (Axl−)-Luc tumors as assessed by caliper (Figure 6a left and right panel, respectively). The same results were obtained by bioluminescence by in vivo imaging system spectrum analyses (data not shown). At the end of the experiment (2 weeks following first treatment), mice were sacrificed, tumor masses excised and analyzed. We observed high levels of let-7g miRNA in A549 (Axl+)-Luc tumors (Figure 6b, left panel) and a concomitant downregulation of HMGA2 protein levels (Figure 6b, right panel) as compared to A549 (Axl+)-Luc tumors from mice treated with the GL21.T aptamer or the control aptamer (scraGL21.T). In addition, the antitumor activity was also confirmed by immunohistochemical staining for Ki-67 (Figure 6c). Finally, in order to exclude nonspecific immune activation due to dsRNA, we demonstrate that the expression levels of interferon-inducible IFIT1 (p56), 2′-5′ oligoadenylate synthetase 1, and interleukin-8 (IL-8) genes were not increased in liver and spleen of treated animals (Figure 6d and Supplementary Figure S5). Together, these data confirm the cell/tissue-target specificity and efficiency of the conjugate in vivo.
Figure 6.

GL21-let in vivo tumor growth inhibition. (a) (nu/nu) Mice-bearing A549 (Axl+)-Luc xenografts (left panel) or MCF-7 (Axl−)-Luc xenografts (right panel) were injected intravenously with 1,600 pmol of scraGL21.T, GL21.T, or GL21.T-let conjugate (three times a week for 2 weeks). Day 0 marks the start of treatments (23 days following cell injection). Tumor volumes were measured by caliper and experimental raw data were interpolated with curve fitting or regression analysis. Statistics on experimental data were analyzed (GL21.T-let compared to GL21.T), **P < 0.005 (n = 5). Error bars depict mean values of experimental replicates ± SD (n = 5). (b) Left panel, RNAs from A549 (Axl+)-Luc recovered tumors were extracted and pooled; let-7g miRNA levels were quantified by RT-qPCR. Statistics are indicated ***P < 0.0005. Right panel, immunoblot with anti-HMGA2 and anti-αTub antibodies of pooled lysates from A549 (Axl+)-Luc recovered tumors. Intensity of bands was calculated as in Figure 1. Error bars, mean values ± SD (n = 3). (c) Representative sections of A549 (Axl+)-Luc tumors from the scraGL21.T, GL21.T, and GL21.T-let groups stained with H&E and Ki-67 antibody. Note a reduction in Ki-67-positive cells and in density of cells stained with H&E in the GL21.T and GL21.T-let-treated sections. Scale bars are indicated. (d) To measure immune response, mice were injected intravenously with 1,600 pmol of scraGL21.T, GL21.T or GL21.T-let conjugate. After 6 hours, liver and spleen were recovered, lysed for RNA extraction, and pooled. PBS and Poly (I:C)-treated mice were used as a negative and positive control, respectively. P56, OAS1, and IL-8 mRNAs were analyzed and values expressed as relative to control arbitrarily set to 1. Variations are in line with previous report.23 Error bars depict mean values of experimental replicates ± SD (n = 3). H&E, hematoxylin and eosin stain; IL, interleukin; mRNA, messenger RNAs; miRNA, microRNA; OAS1, 2′-5′ oligoadenylate synthetase 1; PBS, phosphate-buffered saline; RT-qPCR, reverse transcription quantitative polymerase chain reaction.
Discussion
In this study, we demonstrate that cell-targeted aptamers can be used to deliver therapeutic miRNAs into specific cell types in vivo. An RNA aptamer that binds the Axl tyrosine kinase receptor with low nanomolar affinity and specificity7 was covalently linked to the let-7g miRNA. We designed an optimized aptamer-miRNA conjugate (GL21.T-let) that has improved recognition and processing by the RNAi machinery (Figure 1). We demonstrate that in vitro targeting and processing of the GL21.T-let conjugate results in cell-specific silencing of multiple let-7g targets, including HMGA2, N-Ras, Bcl-XL, and cyclin D1 (Figures 2–4). Uptake of let-7g in the target cells, correlated with reduction in cell viability and migration of target cells in vitro (Figure 4). Following systemic administration of the optimized GL21.T-let conjugate, we observed uptake into target tumors (A549) but not nontarget tumors, which do not express Axl (MCF-7). Furthermore, cell uptake, resulted in the downregulation of let-7g target genes leading to (i) caspase-3 activation (apoptosis), (ii) decreased cell proliferation, and (iii) a reduction in tumor volume (Figures 5, 6). Importantly, no nonspecific immunostimulatory effects were seen with these reagents (Figure 6d). These findings are the first example of targeted delivery of a miRNA with biologic activity to a specific cell type in vivo and serve as a proof-of-concept for future development of other targeted miRNA-based therapies.
This study is innovative on two levels. First, it is the first example of targeted miRNA delivery using RNA aptamers. Second, this is the first example of generation of a multifunctional aptamer conjugate. In contrast to siRNAs, which target a single gene, miRNAs can produce a global effect on many genes, gene regulatory networks, and thereby cellular processes, as evidenced by the numerous genes that were altered by introduction of a let-7g mimetic. Thus, miRNAs are an ideal single-component therapy with multiple functions. The ability to target miRNAs with a functional aptamer further increases the therapeutic potential of an all-RNA reagent. The Axl aptamer used in this study, GL21.T, has both antagonistic activity and cell-targeting capability.7 Whereas dual-functional conjugates have been previously described,24 the aptamer-miRNA conjugates described herein combine the multifunctional properties of the miRNA moiety with those of the GL21.T aptamer to thereby produce a multifunctional molecule. Given that single-agent targeted therapies have not fulfilled their original promise as anticancer strategies, this multifunctional strategy represents the shift toward the increasing use of combination therapies.
RNAi offers the attractive possibility of selectively knocking down disease-causing genes. Within the past year, phase 1 and 2 clinical trials based on RNAi have shown promising, durable gene knockdown, and clinical benefit in a handful of diverse diseases caused by aberrant liver gene expression.47 RNA delivery in these studies mostly used liposome-encapsulated RNAs that are selectively trapped in the liver.48 Indeed, direct conjugation of RNAi to a targeting agent is revealing a promising alternative to nanoparticle-based cargoes for selective delivery. Realizing the therapeutic potential of gene knockdown for treating cancer, however, requires a robust method to deliver RNAs into cancer cells.49 The aptamer delivery approach offers the possibility to target tissues other than the liver. Our study joins only a handful of other studies demonstrating targeted delivery of small noncoding RNAs to target tissues using RNA aptamers. These studies describe the use of cancer targeting aptamers for delivery of therapeutic siRNAs to cancer cells. The siRNAs were designed to either: kill the cancer cells,13,15 augment sensitivity to chemotherapy20 or radiation18, or elicit an antitumor immune response.50 The next iteration of conjugates, which have been applied to HIV therapy, is bifunctional through use of aptamers with antagonistic properties.24 Our development of an aptamer-miRNA conjugate represents the first description of a multifunctional conjugate that harnesses the broad function of let-7g to target gene networks.
Safety issues associated with RNA-based therapies. Targeting alleviates these concerns. A major concern with the use of interfering RNAs as cancer therapeutics is the frequent occurrence of undesired effects in nontarget tissues. Targeted delivery via RNA aptamers alleviates this issue. Indeed, we show that both the amounts of intracellular let-7g and the extent of target downregulation depend on the level of expression of Axl on the cell surface, suggesting that nontarget cells effects due to aptamer internalization into cells with moderate-to-low Axl levels will likely be negligible. The safety of the aptamer-miRNA conjugate is also enhanced by the use of chemically modified nucleotides, which minimize nonspecific immunostimulatory effects.
In conclusion, we provide the first description of aptamer-based conjugates that act as novel multifunctional molecules for the selective in vivo targeted delivery of miRNAs with therapeutic potential. Moreover, our data in which we use aptamer targeting a different receptor or append a different miRNA highlights the generality of this technology. While our findings were in the setting of cancer, this study makes an important contribution to the larger field of RNAi therapeutics by defining a safe, effective strategy to delivery functional miRNAs to target tissues.
Materials and Methods
Aptamer-miRNA conjugates. GL21.T: 5′ AUGAUCAAUCGCCUCAAUUCGACAGGAGGCUCAC 3′. scraGL21.T: 5′ GCACGCAAAUAGCGACUUCAGACGACUCUUCAGU 3′. GL21.T-let passenger strand: 5′ AUGAUCAAUCGCCUCAAUUCGACAGGAGGCUCACCAAACUGUACAGGCCACUGCCUUGCCUU 3′. Let guide strand: 5′GGCUGAGGUAGUAGUUUGUACAGUUUG3′. GL21.T-miR-16 passenger strand: 5′ AUGAUCAAUCGCCUCAAUUCGACAGGAGGCUCACACUCCAGUAUUAACUGUGCUGCUGAGGUU 3′. MiR-16 guide strand: 5′ CCUUAGCAGCACGUAAAUAUUGGCGU 3′. GL21.T-stick: 5′ AUGAUCAAUCGCCUCAAUUCGACAGGAGGCUCACXGUACAUUCUAGAUAGCC 3′. scraGL21.T-stick: 5′ GCACGCAAAUAGCGACUUCAGACGACUCUUCAGUXGUACAUUCUAGAUAGCC 3′. A10-stick: 5′ GGGAGGACGAUGCGGAUCAGCCAUGUUUACGUCACUCCUXGUACAUUCUAGAUAGCC 3′. Let passenger-stick: 5′ CAAACUGUACAGGCCACUGCCUUGCCGGCUAUCUAGAAUGUAC 3′.
All RNA were modified with 2′-F pyrimidines and synthesized by Synthetic and Biopolymer Chemistry Core at the Beckman Research Institute of City of Hope (Duarte, CA) Bold UU are 3′-overhang. Stick sequence, consisting of 2′-F-Py and 2′-OMe-Pu, is underlined. X indicated C3 carbon linker. Before each treatment, the aptamers were subjected to a short denaturation-renaturation step (5 minutes 85 °C, 2 minutes snap-cooling on ice, warming up to 37 °C).
To prepare GL21.T-let and GL21.T-miR-16 conjugates, 5 µmol/l aptamer-passenger RNA strand was combined with 5 µmol/l of the appropriate guide RNA in annealing buffer (20 mmol/l 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid pH 7.5, 150 mmol/l NaCl, 2 mmol/l CaCl2), heated to 95 °C for 15 minutes, incubated at 55 °C for 10 minutes, and then at 37 °C for 20 minutes.
For stick-based conjugates, stick aptamers were refolded and the passenger-stick strand was annealed to the complementary guide partner (ratio 1:1) by incubating at 95 °C for 15 minutes, 55 °C for 10 minutes, and 37 °C for 20 minutes to form the stick-miRNAs. The same amount of the refolded aptamer was added and incubated at 37 °C for 30 minutes.
Cell lines and transfection. NSCLC A549 (Axl+), human megakaryoblastic leukemia Meg-01 and human prostate carcinoma LNCaP cells (American Type Culture Collection, Manassas, VA) were grown in Roswell Park Memorial Institute medium (Sigma, St Louis, MO) supplemented with 10% fetal bovine serum (Sigma); human breast MCF-7 (Axl−) cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Growth condition for A549-Luc-C8 and MCF-7 (Axl−)-Luc-F5 (Caliper Life Sciences, Hopkinton, MA) were previously reported.7 All transfections were performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Cells were transfected with: miRNA precursor hsa-let-7g miRNA (let-7g-miR), hsa-miR-16 (miR-16), and Negative Control #1 (ctrl-miR) (Applied Biosystems, Foster City, CA); GL21.T or GL21.T-let, as indicated; locked nucleic acid anti-miR-let-7g (Exiqon, Woburn, MA); Axl TruClone (Origene, Rockville, MD); Dicer-specific siRNA (Cell Signalling Technology, Danvers, MA). Axl overexpression on A549 (Axl+) cell surface was measured by fluorescence activated cell sorting using anti-Axl primary antibodies (R&D Systems, Minneapolis, MN) and FITC-labeled secondary antibodies (Jackson ImmunoResearch, West Grove, PA).
Immunoblot and RT-qPCR. The day before conjugate treatment, cells were seeded in 3.5-cm plates (1.5 × 105 cells/plate). Cell extracts and immunostaining were performed as described.5 The primary antibodies used were: anti-HMGA2, anti-Dicer, anti-caspase-3, anti-phospho-Axl (tyr-702), anti-cycline D1, anti-Bcl-XL (Cell Signalling Technology); anti-N-Ras (Santa Cruz Biotechnology, CA); anti-Axl (R&D Systems); anti-αtubulin (DM 1A) (Sigma). Ligand-dependent Axl receptor activation was performed by using Gas 6 from R&D Systems.
For Real-time PCR, RNAs were extracted by TRiZol (Invitrogen), and 1 µg total RNA was reverse transcribed. To assess level of let-7g miRNA, miR-16, or U6 RNA as housekeeping gene, reverse transcription was done using miScript Reverse Transcription Kit and amplification was performed with the miScript-SYBR Green PCR Kit and specific miScript Primer Assay (QIAGEN, Milan, Italy). To analyze HMGA2 or Axl mRNA level, RNA was reverse transcribed with iScript cDNA Synthesis Kit and amplified by using IQ-SYBR Green supermix (Bio-Rad, Hercules, CA). Primers used were: HMGA2 Fw: 5′GCGCCTCAGAAGAGAGGAC3′, HMGA2 Rev: 5′GGTCTCTTAGGAGAGGGCTCA3′; Axl Fw: 5′GGTGGCTGTGAAGACGATGA3′, Axl Rev: 5′CTCAGATACTCCATGCCACT3′; β-actin Fw: 5′CAAGAGATGGCCACGGCTGCT3′, β-actin Rev: 5′TCCTTCTGCATCCTGTCGGCA3′; IL8 Fw: 5′ GCGCCCAGACAGAAGTCATAG3′; IL8 Rev: 5′GGCAAACTTTTTGACCGCC3′. P56 and 2′-5′ oligoadenylate synthetase 1 mRNAs were analyzed as described.23 The ΔΔCt method for relative quantization of gene expression was used.
Binding assays. Binding to cells, internalization and filter binding with the soluble extracellular domain of human Axl (EC-Axl, R&D Systems) were performed as described.7 To check the endocytosis rate, 100 nmol/l radiolabeled GL21.T, scraGL21.T or GL21.T-let conjugate been incubated on A549 (Axl+) cells for increasing incubation times (from 15 minutes up to 2 hours) and at desired times, cells have been left untreated (to measure total bound aptamer) or treated with 0.5 μg/μl proteinase K (Roche Diagnostics, Indianapolis, IN) for 30-minute at 37 °C (to measure internalized aptamer). RNA was recovered and the amount of labeled aptamer determined by 32P counting. For each aptamer, the percentage of internalization has been expressed as the amount of internalized aptamer relative to total bound aptamer.
Cell viability assays. A549 (Axl+) cells were transfected with let-7g-miR or treated for 24 hours GL21.T-let or GL21.T and then trypsinized, and seeded in 96-well plates (4 × 103 cells/well). Following additional 48-hour cell viability was assessed by CellTiter 96 Proliferation Assay (Promega, Madison, WI).
Transwell migration assays. A549 (Axl+) cells transfected with let-7g-miR or treated for 24 hours with 400 nmol/l GL21.T or GL21.T-let, were trypsinized and resuspended in Dulbecco's modified eagle's medium serum free. Migration was assessed as previously reported.7
In vitro Dicer assay. GL21.T or GL21.T-let (1 µg) were left undigested or digested using Recombinant Human Turbo Dicer Kit (Genlantis, San Diego, CA) according to the supplier's instructions. RNAs were then loaded on a 15% nondenaturing polyacrylamide gel electrophoresis gel, stained with ethidium bromide and visualized with GEL.DOC XR (BioRad) gel camera. Alternatively, 32P-end-labeled miRNA guide strand was annealed to aptamer-passenger strand, before digestion, loading on nondenaturing gel and autoradiography.
Ago2 immunoprecipitation. A549 (Axl+) cells were transfected with 50 nmol/l Ctrl-miR, Let-7g-miR, GL21.T aptamer or GL21.T-let and cell extracts were prepared with Polysome lysis buffer (100 mmol/l KCl, 5 mmol/l MgCl2, 10 mmol/l 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid pH 7.0, 0.5% NP40, 1 mmol/l dithiothreitol) supplemented with 100 units/ml RNase-inhibitor and Protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). RIPAb+ Ago2 - Antibody (Millipore, Billerica, MA) was incubated with Protein A/G PLUS-Agarose (Santa Cruz Biotechnology) over night at 4 °C. After three washes with NT2 buffer (50 mmol/l Tris–HCl pH 7.4, 150 mmol/l NaCl, 1 mmol/l MgCl2, 0.05% NP40), antibody-coated beads were resuspended in 850 μl of ice-cold NT2 buffer supplemented with 200 units of RNase inhibitor, 1 mmol/l dithiothreitol, 20 mmol/l ethylenediaminetetraacetic acid; 1 µg (100 μl) of cleared lysate was added and incubated for 5 hours at 4 °C. Beads were washed three times and RNA was extracted by TriZoL. The levels of let-7g miRNA were measured by RT-qPCR. Normal mouse IgG was used as a negative control.
Illumina Deep sequence and data analysis. A549 (Axl+) cells were seeded in six-well plates and transfected with 50 nmol/l GL21.T-let conjugate using Lipofectamine 2000. The total RNAs were isolated with TriZol reagent (Invitrogen) following 48 hours and prepared for Illumina Deep sequencing.
To identify the most frequent guide and passenger products, the raw sequences were matched with the corresponding mature sequences after removing the 3′-adapter using the cutadapt tool (http://code.google.com/p/cutadapt/). The matched sequences were then reduced to a set of unique sequences along with their number of occurrences. This set of sequences was then aligned with the guide and passenger reference sequences using the ClustalX2 multiple alignment tool not allowing gaps. The multiple aligned sequences were visualized and exported using JalView. The extra bases at end of the products were highlighted manually.
3′UTR-HMGA2 GFP assay. A549 (Axl+) cells were transfected with HMGA2-3′UTR-Lenti-reporter-GFP-vector (Abmgood, Richmond, BC) alone or in combination with 50 nmol/l let-7-miR-, GL21.T-, or GL21.T-let-transfection or 400 nmol/l GL21.T- or GL21.T-let- treatment. Following 24 hours, cells were recovered and GFP levels were measured by fluorescence activated cell sorting. P-Lenti-3′UTR-GFP-Blank was used as negative control.
Illumina array analyses. A549 (Axl+) cells were transfected with 100 nmol/l Ctrl-miR or let-7g-miR or were treated with 400 nmol/l GL21.T or GL21.T-let. RNA was extracted and subjected to Illumina HT12v.4.0 array. Data were analyzed by MeVsoftware v4.7. Venn diagram and gene ontology analyses were obtained by David-GO database and R-project (package “VennDiagram”) software, respectively.
In vivo experiments. Athymic CD-1 nude mice (nu/nu) were injected subcutaneously with 3 × 106 A549 (Axl+)-Luc cells (right flank) and 6 × 106 MCF-7 (Axl−)-Luc (left flank). Tumors (three mice/group) were injected with GL21.T or GL21.T-let by caudal vein (1,600 pmol in 100 µl/injection). Following 24 or 48 hours, mice were sacrificed. To follow tumor growth inhibition, mice were injected subcutaneously with 3 × 106 A549 (Axl+)-Luc cells (five mice for group) or 6 × 106 MCF-7 (Axl−)-Luc and were injected with scraGL21.T, GL21.T, or GL21.T-let by caudal vein (1,600 pmol/injection, three injections a week). Tumor growth was measured with calipers or bioluminescence by in vivo imaging system spectrum. Animals were sacrificed following 16 days. Tumors from sacrificed animals were processed for protein or RNA extraction and pooled for immunoblot or RT-qPCR analyses. Histological and immunohistochemical examinations were performed as described.7 To determine potential immune activation induction following treatment, livers and spleens of treated animals (three mice/group) at 6 hours and 16 days following treatment were excise and processed for total RNA and RT-qPCR. Poly (I:C) (10 µg in 100 µl, Sigma), and phosphate buffered saline were used as a positive and negative control, respectively.
Ethics statement. All the experimental procedures were approved by the Ethical Committee for the Animal Use (CESA) of the Istituto di Ricerche Genetiche Gaetano Salvatore (IRGS) and where communicated to the national authorities accordingly with national and European rules.
Statistical analysis. Statistics were performed with GraphPad Prism v.6.0 by t-test or two-way Analysis of variance for tumor size data. P value of 0.01 or less was considered to indicate a significant difference.
SUPPLEMENTARY MATERIAL Figure S1. Axl over-expression in A549 (Axl+) cells. Figure S2. Stick-based conjugates. Figure S3. GL21.T-miR-16 conjugate. Figure S4. Illumina array analyses. Figure S5. Immune response analyses 2-week posttreatment.
Acknowledgments
This work was supported by funds from: CNR, AICR No 11–0075 (LC), MIUR grant, MERIT RBNE08YFN3_001 (VdF), AIRC No 11781 (LC), No.14046 (GC) and No 13345 (VdF); from the Italian Ministry of Economy and Finance to the CNR for the Project FaReBio di Qualità (VdF); from The Mary Kay Foundation 033-12 (PHG), the NIH/NCI 1R01 CA138503 (PHG), Elsa U Pardee Foundation (PHG), from Fondazione Berlucchi (GC) and POR Campania FSE 2007–2013 Project “CRÈME” (VdF). We are grateful to J.J. Rossi for invaluable suggestions, comments and continuous encouragement. We wish to thank L. Baraldi for technical assistance; M. Iaboni, L. Insabato, and J.O. McNamara for advice; K. Thiel for critically reading the manuscript. The authors disclose no conflict of interest. C.L.E. designed and performed the majority of the experiments, interpreted results and assisted with manuscript preparation. S.C. and G.dV. performed and/or assisted with several experiments. P.S. provided useful reagents and advice. V.dF. provided intellectual input, coordinated the research, secured the funding and guided experimental design and the preparation of the manuscript. L.C., P.H.G., G.C., J.P.D., and G.S. provided expertise, analyzed data, and assisted with manuscript preparation.
Supplementary Material
Axl over-expression in A549 (Axl+) cells.
Stick-based conjugates.
GL21.T-miR-16 conjugate.
Illumina array analyses.
Immune response analyses 2-week posttreatment.
References
- Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143–159. doi: 10.1002/emmm.201100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo M, Condorelli GL, Croce CM, Condorelli G. MicroRNAs as regulators of death receptors signaling. Cell Death Differ. 2010;17:200–208. doi: 10.1038/cdd.2009.105. [DOI] [PubMed] [Google Scholar]
- Thiel KW, Giangrande PH. Intracellular delivery of RNA-based therapeutics using aptamers. Ther Deliv. 2010;1:849–861. doi: 10.4155/tde.10.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Hu J, Peng M, Liu J, Liu J, Liu H, et al. Generating Aptamers by Cell-SELEX for Applications in Molecular Medicine. Int J Mol Sci. 2012;13:3341–3353. doi: 10.3390/ijms13033341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito CL, Passaro D, Longobardo I, Condorelli G, Marotta P, Affuso A, et al. A neutralizing RNA aptamer against EGFR causes selective apoptotic cell death. PLoS ONE. 2011;6:e24071. doi: 10.1371/journal.pone.0024071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Nguyen HH, Byrom M, Ellington AD. Inhibition of cell proliferation by an anti-EGFR aptamer. PLoS ONE. 2011;6:e20299. doi: 10.1371/journal.pone.0020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchia L, Esposito CL, Camorani S, Rienzo A, Stasio L, Insabato L, et al. Targeting Axl with an high-affinity inhibitory aptamer. Mol Ther. 2012;20:2291–2303. doi: 10.1038/mt.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Duan JH, Song YM, Ma J, Wang FD, Lu X, et al. Novel HER2 aptamer selectively delivers cytotoxic drug to HER2-positive breast cancer cells in vitro. J Transl Med. 2012;10:148. doi: 10.1186/1479-5876-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel WH, Bair T, Peek AS, Liu X, Dassie J, Stockdale KR, et al. Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS ONE. 2012;7:e43836. doi: 10.1371/journal.pone.0043836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlknecht G, Maron R, Mancini M, Schechter B, Sela M, Yarden Y. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc Natl Acad Sci USA. 2013;110:8170–8175. doi: 10.1073/pnas.1302594110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchia L, de Franciscis V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol. 2010;28:517–525. doi: 10.1016/j.tibtech.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Zhou J, Rossi JJ. Cell-specific aptamer-mediated targeted drug delivery. Oligonucleotides. 2011;21:1–10. doi: 10.1089/oli.2010.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- Wullner U, Neef I, Eller A, Kleines M, Tur MK, Barth S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr Cancer Drug Targets. 2008;8:554–565. doi: 10.2174/156800908786241078. [DOI] [PubMed] [Google Scholar]
- Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Swiderski P, Li H, Zhang J, Neff CP, Akkina R, et al. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009;37:3094–3109. doi: 10.1093/nar/gkp185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H, et al. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci Transl Med. 2011;3:66ra6. doi: 10.1126/scitranslmed.3001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Zhang Y, Ribas J, Chowdhury WH, Castanares M, Zhang Z, et al. Prostate-targeted radiosensitization via aptamer-shRNA chimeras in human tumor xenografts. J Clin Invest. 2011;121:2383–2390. doi: 10.1172/JCI45109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler LA, Trifonova R, Vrbanac V, Basar E, McKernan S, Xu Z, et al. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J Clin Invest. 2011;121:2401–2412. doi: 10.1172/JCI45876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel KW, Hernandez LI, Dassie JP, Thiel WH, Liu X, Stockdale KR, et al. Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012;40:6319–6337. doi: 10.1093/nar/gks294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilner SE, Wengerter B, Maier K, de Lourdes Borba Magalhães M, Del Amo DS, Pai S, et al. An RNA alternative to human transferrin: a new tool for targeting human cells. Mol Ther Nucleic Acids. 2012;1:e21. doi: 10.1038/mtna.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Shibata T, Kabashima T, Kai M. Inhibition of HIV-1 protease expression in T cells owing to DNA aptamer-mediated specific delivery of siRNA. Eur J Med Chem. 2012;56:396–399. doi: 10.1016/j.ejmech.2012.07.045. [DOI] [PubMed] [Google Scholar]
- Zhou J, Neff CP, Swiderski P, Li H, Smith DD, Aboellail T, et al. Functional in vivo delivery of multiplexed anti-HIV-1 siRNAs via a chemically synthesized aptamer with a sticky bridge. Mol Ther. 2013;21:192–200. doi: 10.1038/mt.2012.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Li H, Li S, Zaia J, Rossi JJ. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol Ther. 2008;16:1481–1489. doi: 10.1038/mt.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, et al. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene. 2009;28:3442–3455. doi: 10.1038/onc.2009.212. [DOI] [PubMed] [Google Scholar]
- Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19–F36. doi: 10.1677/ERC-09-0184. [DOI] [PubMed] [Google Scholar]
- Esposito CL, Catuogno S, de Franciscis V, Cerchia L. New insight into clinical development of nucleic acid aptamers. Discov Med. 2011;11:487–496. [PubMed] [Google Scholar]
- Wu X, Ding B, Gao J, Wang H, Fan W, Wang X, et al. Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int J Nanomedicine. 2011;6:1747–1756. doi: 10.2147/IJN.S23747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dai F, Zhang Y, Zhu X, Shan N, Chen Y. Anticancer role of MUC1 aptamer-miR-29b chimera in epithelial ovarian carcinoma cells through regulation of PTEN methylation. Target Oncol. 2012;7:217–225. doi: 10.1007/s11523-012-0236-7. [DOI] [PubMed] [Google Scholar]
- Liu N, Zhou C, Zhao J, Chen Y. Reversal of paclitaxel resistance in epithelial ovarian carcinoma cells by a MUC1 aptamer-let-7i chimera. Cancer Invest. 2012;30:577–582. doi: 10.3109/07357907.2012.707265. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Amarzguioui M, Lundberg P, Cantin E, Hagstrom J, Behlke MA, Rossi JJ. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat Protoc. 2006;1:508–517. doi: 10.1038/nprot.2006.72. [DOI] [PubMed] [Google Scholar]
- Amarzguioui M, Rossi JJ. Principles of Dicer substrate (D-siRNA) design and function. Methods Mol Biol. 2008;442:3–10. doi: 10.1007/978-1-59745-191-8_1. [DOI] [PubMed] [Google Scholar]
- De N, Young L, Lau PW, Meisner NC, Morrissey DV, MacRae IJ. Highly complementary target RNAs promote release of guide RNAs from human Argonaute2. Mol Cell. 2013;50:344–355. doi: 10.1016/j.molcel.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezhnoy A, Brenneman R, Bajgelman M, Seales D, Gilboa E. Thermal Stability of siRNA Modulates Aptamer- conjugated siRNA Inhibition. Mol Ther Nucleic Acids. 2012;1:e51. doi: 10.1038/mtna.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff LA, Davila J, Swerdel MR, Moore JC, Cohen RI, Wu H, et al. Ago2 immunoprecipitation identifies predicted microRNAs in human embryonic stem cells and neural precursors. PLoS ONE. 2009;4:e7192. doi: 10.1371/journal.pone.0007192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Li D, Lin HH, McMahon M, Ma H, Ann DK. Oncogenic raf-1 induces the expression of non-histone chromosomal architectural protein HMGI-C via a p44/p42 mitogen-activated protein kinase-dependent pathway in salivary epithelial cells. J Biol Chem. 1997;272:25062–25070. doi: 10.1074/jbc.272.40.25062. [DOI] [PubMed] [Google Scholar]
- Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- Shimizu S, Takehara T, Hikita H, Kodama T, Miyagi T, Hosui A, et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J Hepatol. 2010;52:698–704. doi: 10.1016/j.jhep.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Schultz J, Lorenz P, Gross G, Ibrahim S, Kunz M. MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008;18:549–557. doi: 10.1038/cr.2008.45. [DOI] [PubMed] [Google Scholar]
- Juliano R, Alam MR, Dixit V, Kang H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008;36:4158–4171. doi: 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Yang C, Wang X, Xu C, Li S, Zhang B, et al. Overexpression of Smac promotes Cisplatin-induced apoptosis by activating caspase-3 and caspase-9 in lung cancer A549 cells. Cancer Biother Radiopharm. 2013;28:177–182. doi: 10.1089/cbr.2012.1261. [DOI] [PubMed] [Google Scholar]
- Zhou J, Shum KT, Burnett JC, Rossi JJ. Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges. Pharmaceuticals (Basel) 2013;6:85–107. doi: 10.3390/ph6010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedervall T, Lynch I, Lindman S, Berggård T, Thulin E, Nilsson H, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci USA. 2007;104:2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrocca F, Lieberman J. Promise and challenge of RNA interference-based therapy for cancer. J Clin Oncol. 2011;29:747–754. doi: 10.1200/JCO.2009.27.6287. [DOI] [PubMed] [Google Scholar]
- Pastor F, Kolonias D, Giangrande PH, Gilboa E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature. 2010;465:227–230. doi: 10.1038/nature08999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Axl over-expression in A549 (Axl+) cells.
Stick-based conjugates.
GL21.T-miR-16 conjugate.
Illumina array analyses.
Immune response analyses 2-week posttreatment.