

Abstract
Pregnancy-associated breast cancers (PABCs) are malignancies diagnosed during pregnancy or up to five years following parturition, and are usually aggressive, stroma-rich, and estrogen receptor/progesterone receptor-negative; but little is known about the cellular origin of PABCs or the mechanisms by which PABCs initiate. Using the RCAS retrovirus to deliver the ErbB2 oncogene into the mammary epithelium of our previous reported MMTV-tva transgenic mice, we detected human PABC-like tumors during pregnancy and lactation but not in involuted mice or in age-matched virgin mice. More importantly, by generating a WAP-tva transgenic line for expression of ErbB2 selectively in WAP+ mammary alveolar cells, we found that the resulting tumors exhibited the hallmarks of PABCs irrespective of the time since pregnancy and even in the absence of pregnancy. These data suggest that PABCs arise preferentially from an alveolar cell population that expands during pregnancy and lactation. This somatic mouse model may also be useful for preclinical testing of new prophylactic and therapeutic strategies against PABC.
Keywords: Pregnancy, breast cancer, alveolar cells, PABC
Background
Breast cancer arising either during pregnancy or up to 5 years postpartum is defined as pregnancy-associated breast cancer (PABC) (1). PABCs form a subtype of breast cancer characterized by increased tumor grade, Ki67 positivity, invasiveness, stromal involvement, estrogen receptor (ER)/progesterone receptor (PR) negativity, HER2 overexpression, and poor prognosis (2, 3). Over the last few decades there has been a gradual but consistent increase in the incidence of PABCs globally and correspondingly, in the number of PABC-associated deaths (4). It is, therefore, increasingly important to understand the mechanisms by which PABCs arise in order to identify novel risk factors and biomarkers, as well as more effective therapeutic and prophylactic strategies.
Xenograft studies in animal models have suggested that stromal accumulation during post-lactational involution in the breast is chiefly responsible for the poor prognosis associated with PABCs in humans (5, 6). A tumor-promoting stromal milieu associated with involution has been reported to underlie the especially aggressive nature of PABCs diagnosed after parturition but within 2 years postpartum (7–9). Moreover, the use of non-steroidal anti-inflammatory drugs (NSAIDs) to circumvent this stromal recruitment during involution is able to ameliorate the increased metastatic potential of PABCs in rats (10). Immature macrophages that accumulate in the breast during involution have also been reported to generate a pro-tumorigenic microenvironment promoting PABC growth (7).
The adult mammary gland has a well-structured cellular hierarchy that constitutes a ductal tree branching out from a main duct leading up to the nipple. Each duct in the ductal tree is comprised of two layers of cells: luminal epithelial cells that line the lumens of ducts, and basal (myoepithelial) cells that surround the luminal cells (11). Most ducts end in alveoli, whose luminal cells are also called alveolar epithelial cells and usually produce whey acidic protein (WAP) and casein (12). Alveoli expand rapidly in number during pregnancy as a result of stimulation by several growth factor and hormonal pathways that activate STAT5 and STAT6 (13, 14), and become highly secretory by late pregnancy and lactation (15). Scattered in these basal and luminal layers are various compartmental progenitors. These different cell populations can be transformed to form tumors representing different histopathological subtypes of human breast cancer (16–20). Besides impacting histopathological feature, the cell of origin of breast cancer also significantly influences cellular composition, gene expression profile, and cancer progression (16, 17, 21). Here we report the surprising finding that the cardinal features of PABC may also be encoded in a distinct subset of mammary cells from which the tumor originates.
Results
ErbB2-induced PABCs in mice are aggressive and proliferate rapidly
To study the contribution of the cell of origin on PABC phenotype, we used the RCAS-TVA mouse model system that allows an oncogene cloned into the RCAS retroviral vector to be delivered into a selected population of mammary epithelial cells (<0.3% of the mammary gland), more closely replicating human breast cancer initiation from a single cell (22). This virus is derived from avian leukosis virus sub-group A, and these mammary cells are made susceptible to RCAS infection using a mammary-selective promoter such as MMTV to drive transgenic expression of tva, which encodes the RCAS receptor (23). While the MMTV promoter is responsive to ovarian hormones, baseline TVA protein levels in this transgenic line are easily detected and are not significantly altered during estrus (Fig. S1a). As soon as the RCAS integrates into the host genome, any gene cloned in this retroviral vector is completely under the control of RCAS LTR, which is constitutive and not hormonally influenced (24). We used RCAS expressing a constitutively activated version of rat ErbB2 (HER2/Neu), RCAS-caErbB2, to initiate tumorigenesis as human HER2 amplification/overexpression has been associated with PABCs (25) and its protein activates an oncogenic pathway commonly activated in human breast cancer (26, 27).
We have reported that intraductal injection of RCAS-caErbB2 into MMTV-tva mice leads to tumors with a median latency of approximately six months (22, 24). To demonstrate that we could generate PABCs, we injected 6-week-old MMTV-tva mice intraductally, mated half of the injected mice one week later, and palpated them as well as the virgin control mice for tumor appearance. A subpopulation of the parous cohort acquired tumors during lactation and the first four days of involution (Fig. 1a). These tumors were collectively termed mouse PABCs (mPABCs) since they conformed to the most rigorous definition of postpartum human PABCs (hPABCs) as arising during lactation and early involution. More than half the mice in the parous cohort (12/22) acquired mPABCs, while only one of 17 (5.9%) virgin control mouse acquired a tumor during the same time frame (p=0.003; Fig. 1a). Parous mice that acquired tumors >50 days after parturition (after one month of involution) constituted the late onset parous group and, along with tumors arising in virgin mice, formed two control groups for tumor phenotype comparison with the PABC group (Fig. 1a). Of note, the decreased tumor latency in the mPABC group was not incidental to a pregnancy-associated increase in oncogene expression, a common concern with conventional transgenic models. The caErbB2 protein level as measured by the HA tag was confirmed to be similar in all three subsets of tumors (Fig. S1b+d), as predicted from the ubiquitous nature of the RCAS LTR.
Figure 1. Pregnancy-associated breast cancer (PABC) arises in MMTV-tva mice intraductally injected with RCAS-caErbB2.
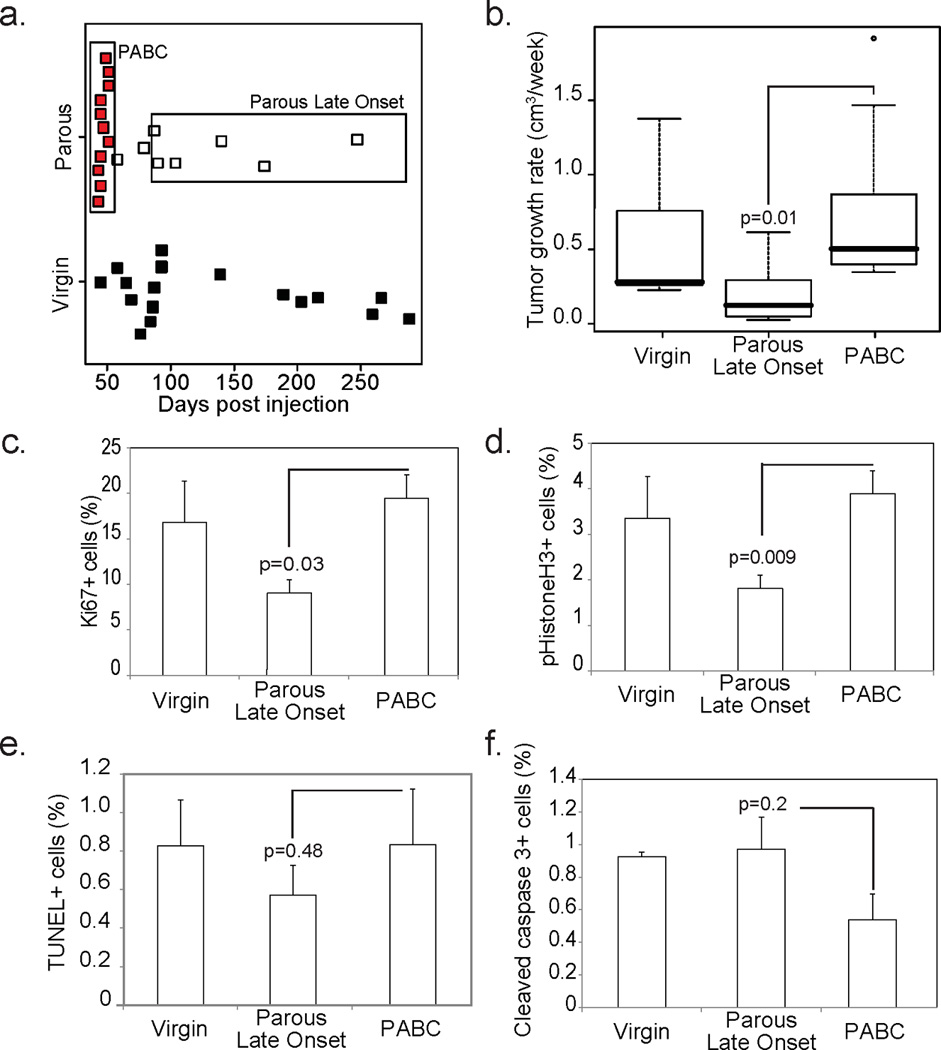
(a) Dot plot showing that PABCs (red) arose during lactation and the first 4 days of involution (≤25 days after parturition), while late onset tumors in parous mice arose more than 1 month after weaning (white) and tumors in virgin mice predominantly arose ≥80 days after intraductal injection with the oncogene (comparable to ≥50 days postpartum) (black). Boxes denote the tumors included in each parous group.
(b) Box plot showing tumor growth rate for all tumors calculated as number of weeks required for tumors detected at 0.5 cm diameter to reach 1.8 cm at euthanasia. Tumor diameter was measured using calipers twice-weekly. n=10 PABCs, n=7 parous late onset and n=8 virgin.
(c–f) Percentage of proliferating cells (c+d) and apoptotic cells (e+f) were determined based on the number of Ki67+ (c; n=3) and pHistone3+ (d; n=6) cells, and the number of TUNEL+ (e; n=3) and cleaved caspase 3+ (f; n=3) cells respectively as detected by immunofluorescence.
Student’s t-test of PABC vs Parous Late Onset detected all p-values. For all bar graphs, columns represent the mean and error bars represent the SEM.
Similar to the aggressive phenotype of hPABCs, mPABCs had a 4-fold increase in growth rate compared to late onset tumors in parous mice (Fig. 1b). In further concordance with hPABCs, mPABCs had a 3-fold higher Ki67 index than late onset parous tumors (Fig. 1c). The higher proliferation of mPABCs was also reflected in a 2.5-fold increase in the number of mitotic cells relative to late onset tumors in parous mice (Fig. 1d). Apoptosis, as detected both by TUNEL and cleaved caspase3 (CC3) levels, was not affected by pregnancy and remained comparable between mPABCs and late onset tumors in parous mice (Fig. 1e+f). Tumor histology in all three cohorts (nulliparous, PLO and mPABC) was complex, comprising two or more distinctive histological subtypes. Areas resembling squamous cell carcinoma, poorly differentiated adenocarcinomas, and EMT tumors appeared in all three tumor groups; however, the relative area of the total tumor occupied by these aggressive histological subtypes was least in the nulliparous tumors and greatest in the mPABCs (Fig 2a). Squamous cell carcinoma was particularly prominent in the mPABC group. The peripheries of these squamous-differentiated tumors were aggressively invasive with high cellular pleomorphism. Like hPABCs, mPABCs had increased collagen deposition indicating a significant increase in stromal involvement relative to the late onset tumors in both parous and virgin mice (Fig. 2b). Finally, similar to hPABCs, mPABCs were predominantly ER-negative, as were the late onset tumors in both parous and virgin animals (Fig. 4a). Of note, the intra-tumor heterogeneity in these tumors is unlikely to be due to polyclonality — the tumor latency was several months for most of the tumors especially for non-PABC tumors (Fig. 1), indicating the need for a rarely occurring spontaneous secondary somatic mutation and therefore lowering the chance of two or more neighboring infected cells gaining a key secondary mutation at a similar time point and then co-evolving into an overt tumor. Accordingly, using FISH to detect the RCAS provirus, we found that seven of the nine tumors analyzed were homogeneous for either one or two integration sites, suggesting that they likely evolved from a single infected cell although we could not exclude the possibility that some of them might have arisen from two neighboring cells that were both infected by the same number of viral particles. Two tumors comprised cells with variable integration events, suggesting that they were oligoclonal (Fig. S1c). Since the PABC group did not appear to have more FISH foci or a stronger indication of non-clonal origin (Fig. S1c), neither clonality nor oncogene levels would likely account for the observed histopathological differences between the PABC tumors and other tumors.
Figure 2. PABCs arising in mice are aggressive and have increased stromal involvement.
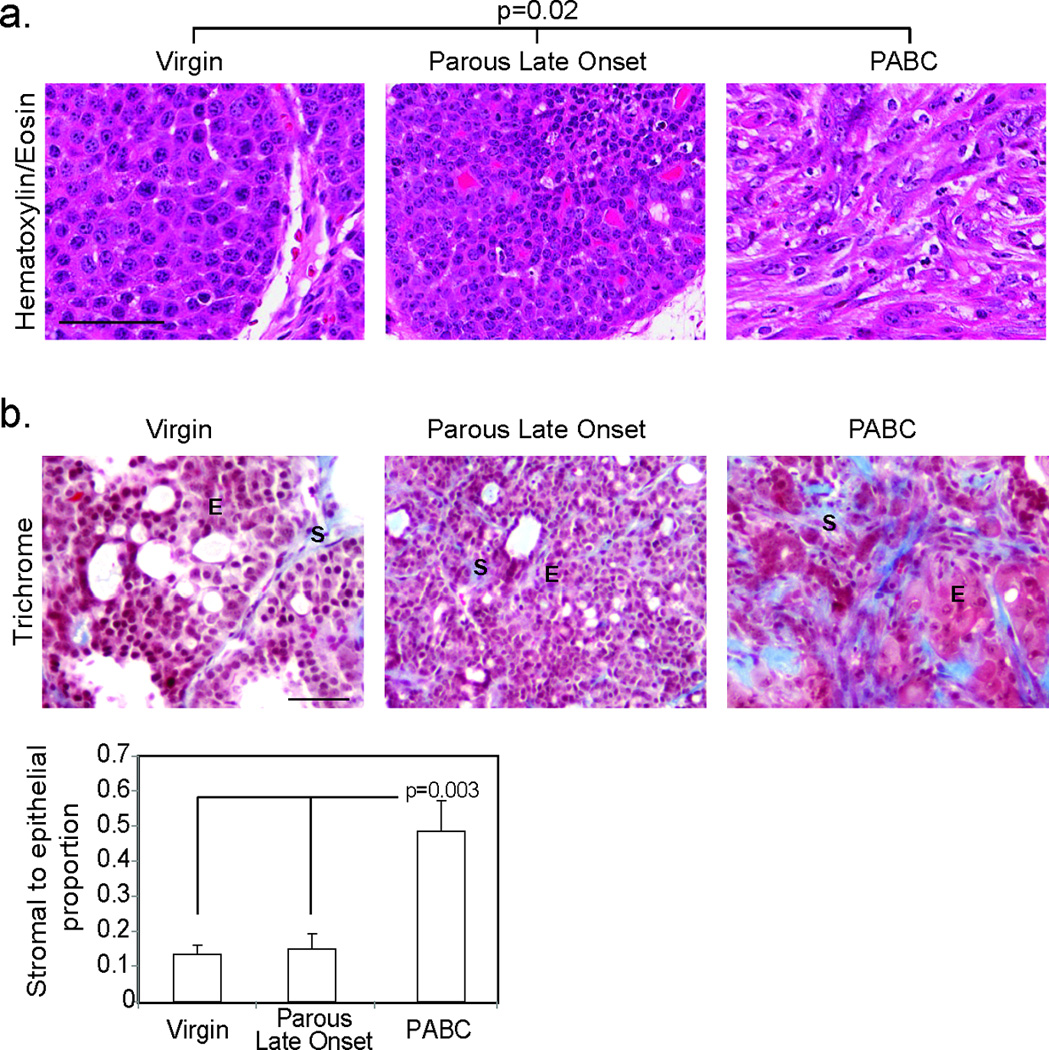
Representative photomicrographs depict Hematoxylin and Eosin staining (a; n=10 virgin, 6 parous late onset and 8 PABCs) of tumor morphology allowing characterization of tumors as solid or disorganized, and trichrome detection of collagen deposition with accompanying quantification of stromal involvement (b; n=4). Scale bars = 50 µm. For bar graph, columns represent the mean and error bars represent the SEM. ANOVA determined p-value. E, epithelia; S, stroma.
Figure 4. PABCs are ER− and enriched for alveolar markers.
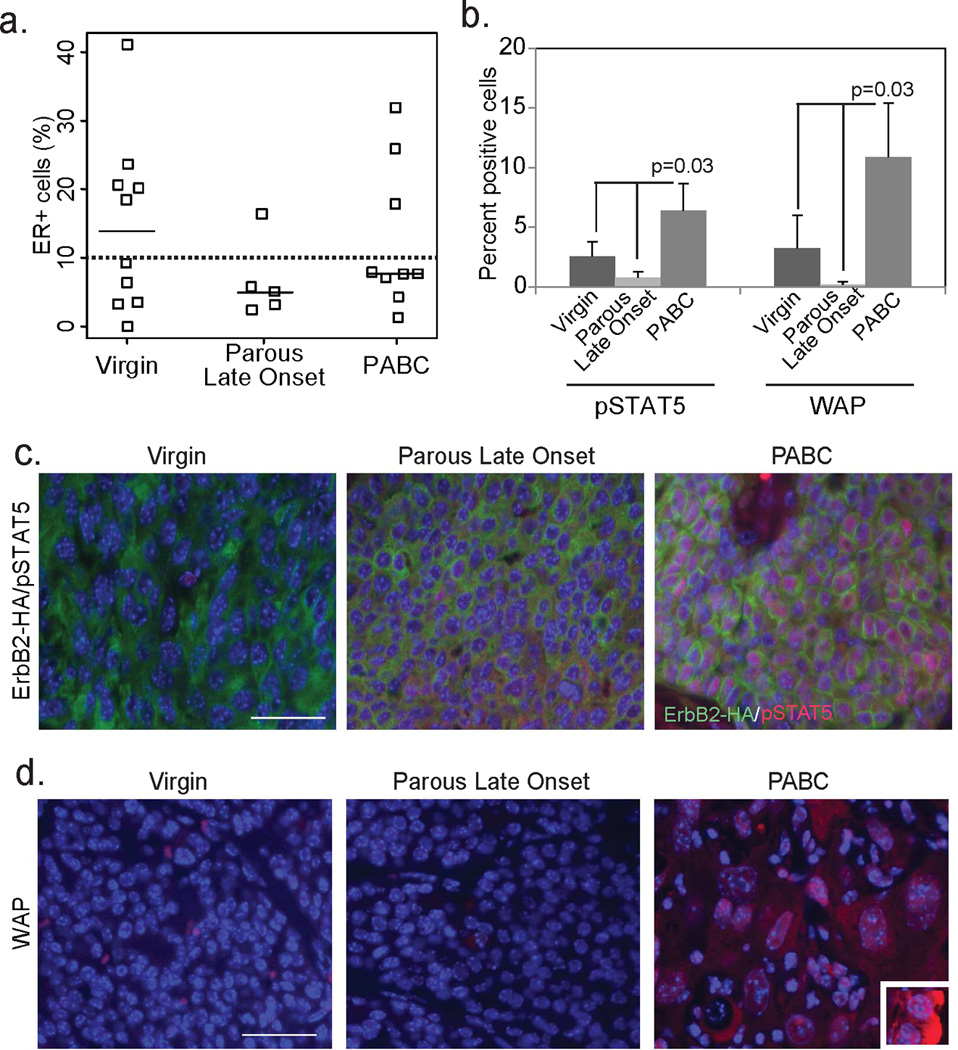
(a) Dot plot showing quantification of ER positivity and categorization of tumors as ER+ and ER− (10% cut-off indicated by dotted line) based on immunohistochemical detection of ER. Horizontal bar represents the median.
(b–d) Immunofluorescence for pSTAT5+ (c; n=4) and WAP+ cells (d; n=4) in the three groups of tumors (c+d) with accompanying quantification (b). ANOVA determined p-value. Co-immunofluorescence for the HA tag of RCAS-caErbB2 indicates cells originating from the tumor (green; c). Inset in (d) depicts WAP+ cells in a lactating mouse mammary gland, serving as a positive control. Columns represent the mean and error bars represent the SEM. Scale bars = 20µm.
ErbB2-induced PABCs have intratumor cellular heterogeneity and upregulated levels of alveolar markers
We next used cell lineage markers to confirm both the intra- and inter-tumor cellular heterogeneity indicated by the above histopathological studies. Both the well- and poorly-differentiated adenocarcinoma areas in all three groups comprised predominantly keratin 8 (K8, a luminal cell marker)-positive cells and only a few K5 (a basal cell marker)-positive cells (Fig. 3). The tumor areas resembling squamous cell carcinoma in every group were uniformly positive for K5 and weakly positive for K8, while the EMT-type tumor cells in every group stained for both vimentin and K8 and had occasional K5 positive cells (not shown). Important, the mPABCs had significantly higher K5 positivity than the late onset tumors (Fig. 3a+d), while the late onset tumors in both parous and virgin mice were predominantly K8+ (Fig. 3b+e). K5+ cells in the mPABCs were largely HA+ (Fig. 3c+f), indicating that they were progeny of cells infected by RCAS-caErbB2 and were not normal basal cells entrapped within the tumor. In contrast, the few cells that were K5+ in the late onset tumors of both parous and virgin mice were predominantly HA−, indicating that these cells were part of the normal mammary epithelium entrapped in tumors (Fig. 3c+f). These differences in cellular heterogeneity suggest that PABCs may have a different cell of origin—possibly a bipotential progenitor—from the late onset tumors in these parous mice.
Figure 3. PABCs encompass oncogene expressing cells of both luminal and basal lineage.
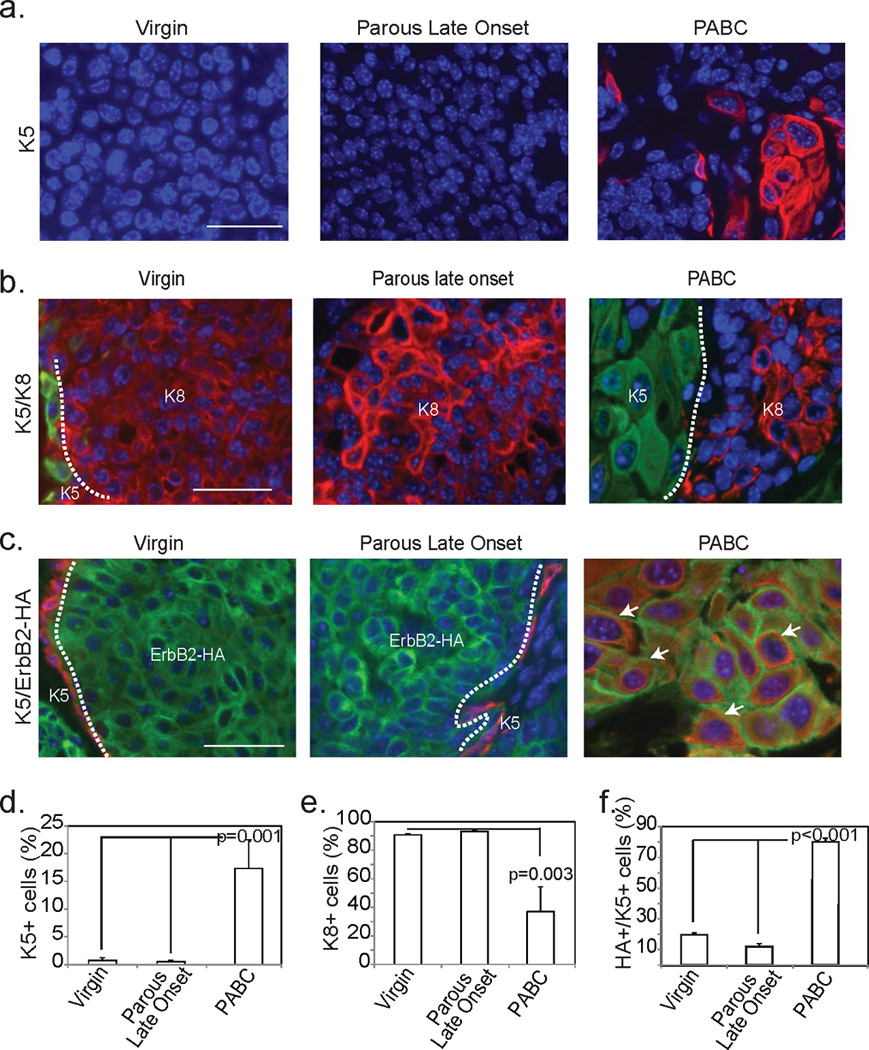
Representative photomicrographs depict immunofluorescent detection of K5 (a), K8 (b) and colocalization of K5 and the HA tag of the RCAS-caErbB2 construct (c) along with accompanying quantification of the number of basal cells (d; n=6), the number of luminal cells (e; n=3) and the number of basal cells expressing the oncogene (f; n=4). Scale bars = 20 µm. ANOVA determined all p-values. Columns represent the mean and error bars represent the SEM. White dotted lines demarcate regions of differential immunofluorescence and arrows indicate cells expressing both K5 and ErbB2.
We also found a >3-fold enrichment for two traditional alveolar markers, phosphorylated signal transducer and activator of transcription 5 (pSTAT5) and whey acidic protein (WAP) (12, 28, 29) in mPABCs relative to late onset tumors in both parous and virgin mice (Fig. 4b–d). The enrichment of alveolar markers in mPABCs suggests that these tumors potentially originate from an alveolar cell. Together, these observations suggest that mPABCs constitute a tumor subtype that is distinct from late onset tumors in parous mice as well as in virgin mice, is comparable to the hPABC tumor subtype, and may originate from alveolar cells possessing the potential to differentiate into both luminal and basal lineages upon oncogene activation.
WAP-tva transgenic mice enable selective expression of oncogene in alveolar cells
To test whether PABC phenotypes could indeed be recapitulated by alveolar cells undergoing transformation, we targeted caErbB2 selectively into the alveolar cell population. WAP is an integral component of breast milk and is generally restricted to cells of the alveolar lineage (12). WAP expression, in accordance with its function, remains at baseline levels in the virgin mammary gland where it is regulated by the estrus cycle, and is maximal during pregnancy and lactation (12). Therefore, we generated a transgenic mouse line transcribing the gene encoding the TVA receptor from the promoter of WAP (Fig. 5a). In virgin WAP-tva mice, the TVA protein level of individual mammary cells varied within the estrous cycle (Fig. S2a), as expected of this hormone-responsive promoter. Based on flow cytometry analysis of single cell suspensions of mammary cells that were stained for TVA, 13.6±3.7% of cells were TVA+ in MMTV-tva mice (n=18), but the TVA+ cell number dropped to 9.9±4.6% in WAP-tva mice (n=21) (p=0.007), consistent with a restricted population of cells expressing WAP in the mammary gland compared to the broader cell population that is responsive to MMTV LTR. Important, based on qPCR, TVA+ cells were enriched for WAP mRNA >8-fold (p=0.015) relative to TVA−/Lin− cells, demonstrating the specificity of TVA expression in WAP-expressing cells (Fig. 5c). During pregnancy and lactation, the TVA protein increased in intensity and could be detected on the membrane of many more cells (Fig. 5b), again in accord with the endogenous pattern of WAP expression. Together, these data indicate that similar to endogenous WAP, TVA in the WAP-tva mice is restricted to cells from an alveolar lineage in the mouse mammary gland.
Figure 5. WAP-tva mice produce TVA in alveolar WAP+ cells.
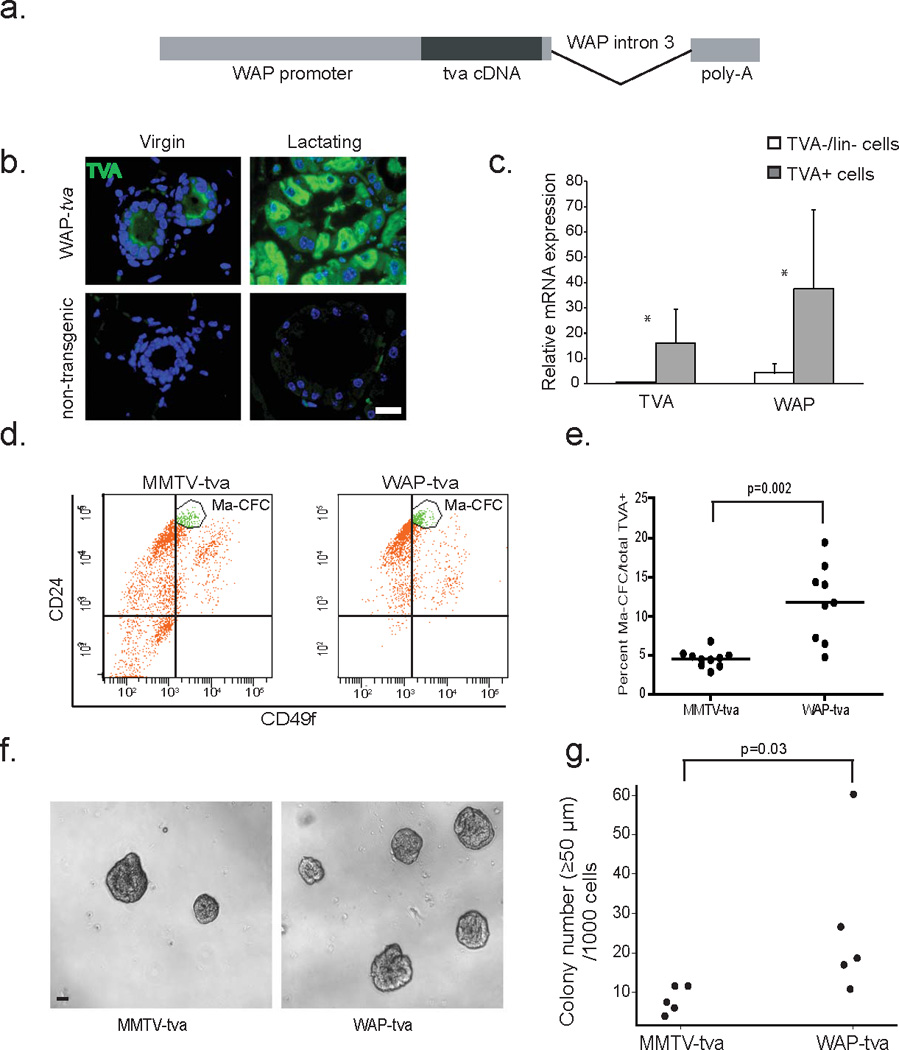
(a) Pictorial representation of construct used for the generation of the WAP-tva mouse line.
(b) Photomicrographs indicating TVA localization in 12 week-old virgin and lactation day 7.5 (L7.5) WAP-tva mice, when endogenous WAP expression is highest. Non-transgenic mice were included as a negative control.
(c) Bar graph depicting relative levels of WAP and TVA genes from a qRT-PCR assay in TVA+/Lin− and TVA−/Lin− cells isolated using FACS. n=7 mice. Asterisks indicate p<0.05.
(d+e) Flow cytometry for CD24 and CD49 cell population markers within TVA+ cells from virgin WAP-tva mice (n=9) relative to TVA+ cells in virgin MMTV-tva mice (n=10).
(f+g) Representative photomicrographs of colonies formed by infected WAP-tva and MMTV-tva cells (f) with accompanying quantification (g).
Student’s t-test determined all p-values. For dot plots, horizontal line depicts the mean. For bar graphs, columns represent the mean and error bars the SEM.
To further characterize the TVA+ cell population in this new WAP-tva mouse line, we used flow cytometry to analyze their mammary cell lineage markers, CD24 and CD49f. Mouse mammary luminal progenitor cells are enriched in the CD24hi/CD49flo mammary colony-forming cell (Ma-CFC) population (30). The Ma-CFC fraction was almost 3-fold larger in the TVA+ cell population in the WAP-tva line than in the MMTV-tva line (Fig. 5d+e). This result suggests that TVA+ cells in WAP-tva mice, while being alveolar, are enriched for cells with progenitor potential relative to TVA+ cells in MMTV-tva mice. In accord with this inference, in an in vitro colony forming assay, the TVA+ cell population from the WAP-tva line formed significantly more colonies than TVA+ cells from MMTV-tva mice (Fig. 5f+g).
An alveolar cell of origin leads to rapidly growing, aggressive, PABC-like tumors regardless of time since pregnancy or even in the absence of a pregnancy
Having established a mouse line where oncogenic signaling could be targeted to a luminal cell population largely restricted to the alveolar lineage, we next tested whether tumors generated from these cells preferentially formed a PABC-like tumor subtype. We first determined the infection rate to be approximately 0.3% of the mammary epithelial cells based on intraductal injection of 1 × 107 IUs of the RCAS-GFP reporter virus per gland of 12-week-old WAP-tva virgin mice (Fig. S2b). This rate is similar to what is observed in MMTV-tva mice, despite the higher frequency of TVA+ cells in MMTV-tva mice (Fig. S2b). Then, we injected WAP-tva mice with the RCAS-caErbB2 virus, and one week later allowed half the mice to undergo a full-term pregnancy and three weeks of lactation while maintaining the other half as virgin controls. As in the MMTV-tva mice, we detected mPABCs that arose during lactation and the first week of involution, but also detected an early onset subgroup of tumors that arose with comparable latency in the virgin mice (p=0.99; Fig. 6a). We confirmed that the ErbB2 protein level was unperturbed by pregnancy/lactation hormones in mPABCs from WAP-tva mice (Fig. S2c). We also validated that the caErbB2 protein level in tumors was comparable between MMTV-tva and WAP-tva mice regardless of parity or time at detection (Fig. S3).
Figure 6. Targeting oncogenic signaling to alveolar cells results in rapidly growing PABC-like tumors irrespective of time since pregnancy.
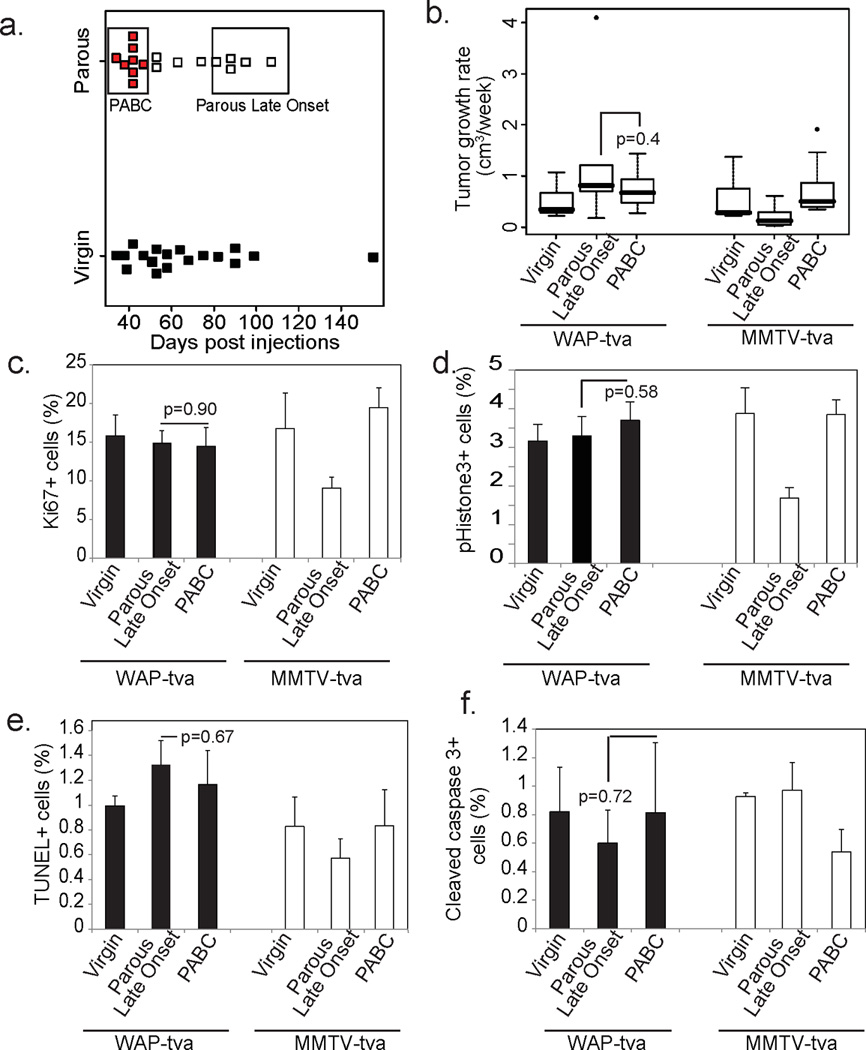
(a) Dot plot indicating the latency of PABCs (red), comparable early onset tumors in virgin WAP-tva mice (black), as well as late onset tumors. Tumors encompassed by the respective boxes designate PABC and parous late onset groups used for subsequent analyses.
(b) Graphical representation of tumor growth rate. n=10 Virgin, 8 PABC and 7 Parous Late Onset tumors.
(c–f) Percentage of proliferating (c+d) and apoptotic cells (e+f) was determined based on the number of Ki67+ (c; n=3) and pHistone3+ (d; n=4) cells, and the number of TUNEL+ (e; n=3) and cleaved caspase 3+ (f; n=3) cells, respectively, as detected by immunofluorescence.
Columns represent mean and error bars represent SEM for all bar graphs. Student’s t-test between PABC and Parous Late Onset determined all p-values. Black columns represent tumors from WAP-tva mice and white columns from MMTV-tva mice.
mPABCs and late onset tumors in parous mice in the WAP-tva mice grew at a similar rate, which was comparable to the aggressive growth rate of mPABCs in the MMTV-tva mice (Fig. 6b). Ki67 positivity was similarly high in mPABCs and late onset tumors from parous WAP-tva mice as well as from virgin WAP-tva mice (Fig. 6c). Also, comparable numbers of tumor cells were in mitosis (Fig. 6d) and there was no observable difference in apoptosis (Fig. 6e+f). Therefore, both PABCs and late onset tumors in parous WAP-tva mice grow equally fast with similarly high levels of proliferation. These data suggest that the recency of a pregnancy did not affect growth or proliferation of tumors arising from an alveolar cell population, unlike in the MMTV-tva line.
All three groups of WAP-tva tumors (mPABC, late onset in parous mice, and in virgin mice) also contained complex histopathological subtypes including squamous cell carcinomas, poorly differentiated adenocarcinomas, and EMT-like tumors, but they resembled mPABCs in MMTVtva mice more than the other tumor subsets in MMTV-tva mice (Fig. 7a). Stromal involvement was extensive in all three tumor groups of WAP-tva mice, as in mPABCs in the MMTV-tva mice (Fig. 7b). Moreover, all three groups had significant K5 positivity, comparable to the level in mPABCs from MMTV-tva mice and much higher than the level in late onset tumors from the MMTV-tva mice (Fig. 8a+d). Again, the K5+ cells also produced the HA tag of the RCAS-caErbB2 provirus (Fig. 8b+e), indicating that these cells originated from the initial virus-infected cell population and were not normal myoepithelial cells recruited to the tumor. Corresponding to the higher K5 positivity, tumors from all three groups of WAP-tva mice had lower K8 positivity than late onset tumors from the parous MMTV-tva mice (Fig. 8c+f).
Figure 7. Oncogenic signaling targeted to alveolar cells results in PABC-like tumor histopathology irrespective of parity status or time since pregnancy.
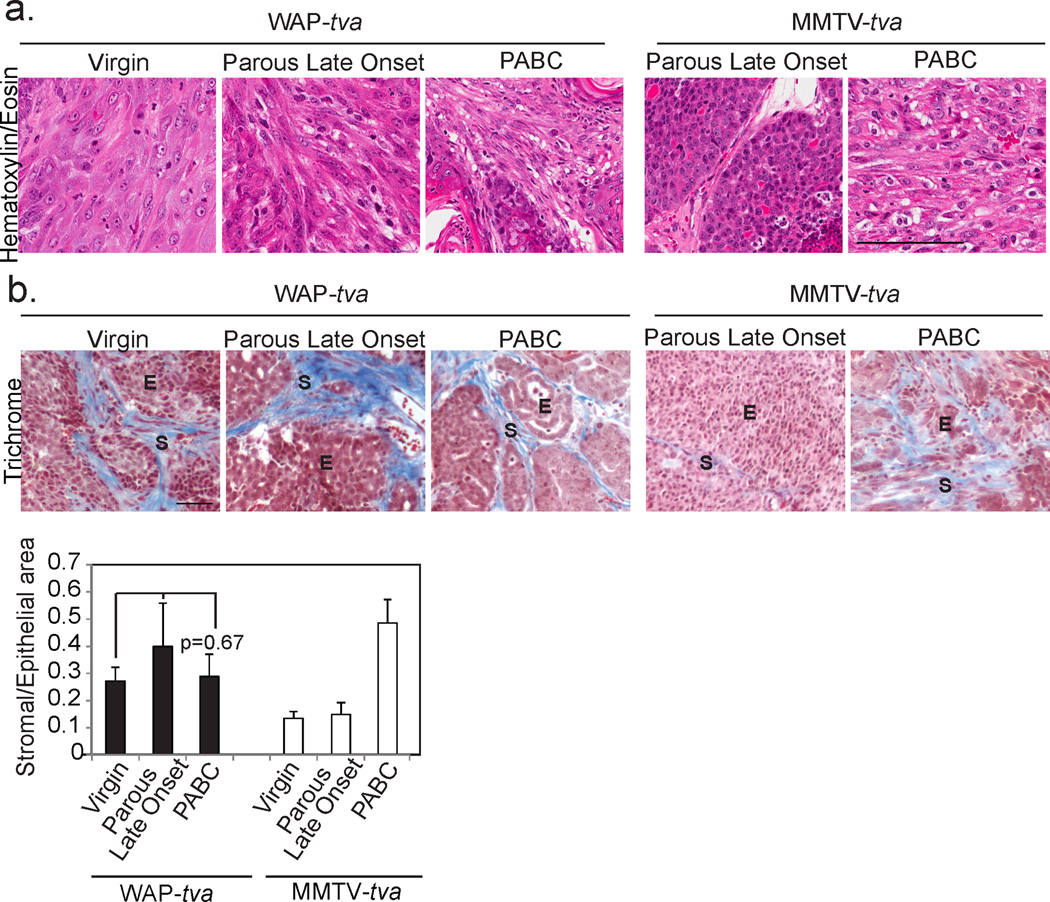
Representative photomicrographs depict Hematoxylin and Eosin staining (a; n=9 virgin, 5 parous late onset and 7 PABCs) of tumor morphology allowing characterization of tumors as solid or disorganized and trichrome detection of collagen deposition (b; n=4) with accompanying quantification of stromal area. ANOVA measured p-value. Dotted black lines demarcate epithelial and stromal regions in (b). E, epithelium and S, stroma. Columns represent mean and error bars represent SEM for all bar graphs. Black columns represent tumors from WAP-tva mice and white columns from MMTV-tva mice.
Figure 8. Tumors originating from alveolar cells have increased cellular heterogeneity irrespective of parity status.
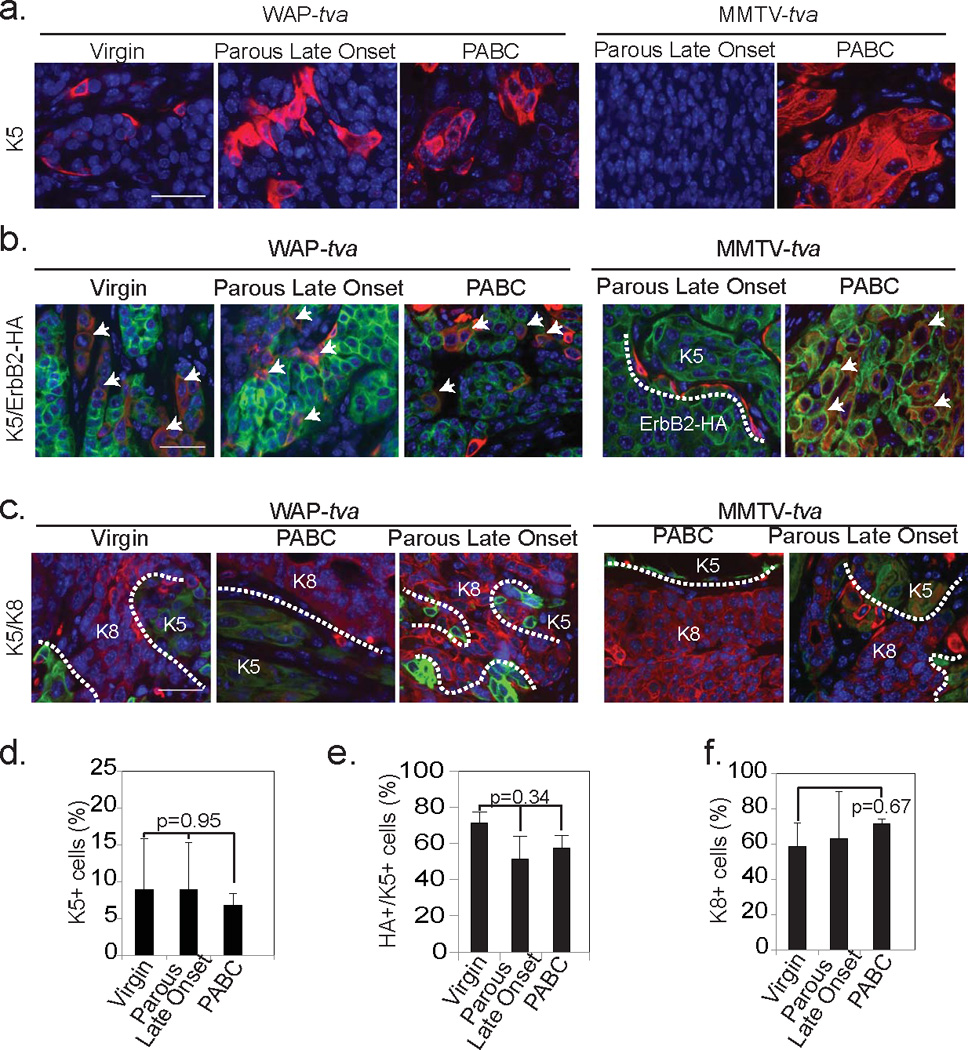
Representative photomicrographs indicating K5 positivity (a; n=5), co-localization of K5 (red) with oncogenic construct (green) (b; n=4) and lack of colocalization between basal (K5; green) and luminal (K8; red) cell markers (c; n=3) in all three tumor groups with accompanying quantifications (d–f). Arrows indicate colocalization (b). White dotted lines demarcate HA+ and K5+ regions (b) and K8+ and K5+ regions (c). Scale bars = 20µm. Bars represent mean and error bars the SEM. ANOVA determined all p-values.
As with the MMTV-tva mice, all three groups of tumors in WAP-tva mice were largely ER-negative (Fig. 9a). Tumors from WAP-tva virgin mice were more likely to be ER-negative than those from virgin MMTV-tva mice (p=0.001; Fig. 9b), indicating that ER status is also influenced by the tumor cell of origin. Taken together, these results suggest that an alveolar cell of origin can endow a tumor with the hallmarks of a PABC, irrespective of the time since pregnancy and even in the absence of pregnancy.
Figure 9. Tumors originating from alveolar cells are predominantly ER−.
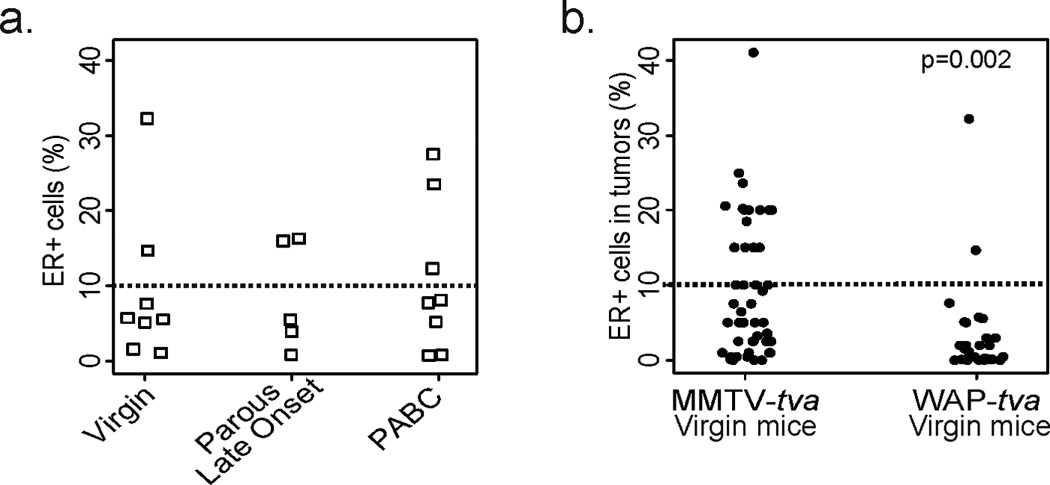
ER positivity was determined based on immunohistochemical detection of ER in tumor sections (cut-off set at 10% ER+ cells indicated by dotted line). Pearson’s chi-squared test determined p-value.
Discussion
Our study is the first to report an impact of cell of origin on PABC phenotype and our results indicate that the cellular origin of the tumor is a critical contributing factor to the hallmarks of PABCs. While late onset tumors in parous MMTV-tva mice lacked PABC characteristics including high basal cell involvement and collagen deposition, tumors from parous WAP-tva mice, irrespective of the time since pregnancy, had PABC features (Fig. 7+8). Since the late onset tumors in these parous mice are not exposed to involution stroma, our data indicate that an alveolar cell of origin can endow a tumor with PABC characteristics long after the pregnancy/involution stimuli have disappeared. In support of this interpretation, tumors arising in virgin mice of the WAP-tva line exhibited the cardinal features of hPABCs and were similar to both mPABC and late onset tumors in WAP-tva parous mice.
The dramatic phenotypic differences between PABCs and late onset tumors in parous mice of the MMTV-tva line are likely due to a difference in the cell of origin. In this transgenic line, tva expression, and therefore oncogene delivery, is not restricted to a specific subset of mammary cells but rather distributed to various subtypes of mammary cells; therefore tumors could arise from different subsets of RCAS-caErbB2-infected cells depending upon selective pressure and evolutionary advantage (such as a pregnancy). However, the microenvironment of pregnancy, lactation, and involution may also modulate tumor phenotypes in this transgenic line, and may convert the phenotype of tumors arising from non-alveolar cells into phenotypes that bear the cardinal features of PABC. Therefore, our results and data interpretation do not disprove previous reports regarding the impact of involuting stroma on phenotypes of PABC.
It is of interest to note that tumor proliferation and growth appeared dependent more on the cell of origin than on pregnancy association. Compared to parous late onset tumors in MMTV-tva virgin mice, the Ki67 index and the number of cells entering mitosis were high in the WAP-tva line irrespective of time since pregnancy or even in the absence of pregnancy (Fig. 6c+d). Similarly, time to tumor doubling was comparable among all three groups of WAP-tva tumors (Fig. 6b). These results also suggest the possibility that the cell of tumor origin can affect pregnancy protection against breast cancer. Epidemiological studies indicate that pregnancy in young women protects against breast cancer (31). Moreover, based on reduced mammary cell proliferation following a pregnancy in women and rodents, it has been postulated that pregnancy-induced protection against breast cancer occurs through the inhibition of proliferation (32). Our observation that the growth of tumors originating from WAP+ cells is not affected by a pregnancy suggests that tumors as well as premalignant lesions arising from alveolar cells might be outside the protective effects of pregnancy. However, it remains to be tested whether alveolar cell tumorigenesis initiated by a different oncogene may still be modulated by a pregnancy.
The risk of PABCs appears to increase with a woman’s age at pregnancy (1), but the reasons are unclear. This age-dependence might be due to the normal changes of breast epithelia and/or stroma during ageing; however, it is also possible that preexistent oncogenic insults (such as HER2 amplification) in the breast epithelium might appear with greater likelihood in older women (33–35). These preexistent insults may be preferentially responsive to the potent hormonal milieu induced by pregnancy and lactation, and therefore may be able to form tumors more rapidly under these stimuli. It is also possible that with pregnancy, mammary cells with oncogenic mutations in an ageing breast might be more readily forced into an alveolar lineage which might evolve into tumors more rapidly. There are few animal models that can recapitulate PABC initiation, and previous models largely utilize xenograft tissue (6). The RCAS-TVA mouse model system appears to comprehensively recapitulate the PABC phenotype, and the use of this somatic model system may help uncover the molecular and cellular mechanisms by which PABCs arise and progress.
The cell of origin has been reported to affect breast tumor ER status (19, 36), and the results of our study further support this connection. The preponderance of ER-negative PABC tumors is a contributory factor to poor prognosis and survival of PABC patients. In fact, several epidemiological studies suggest that the tumor subtype associated with PABCs, rather than the pregnancy itself, is responsible for the poor prognosis of PABCs (37). Our data support this epidemiological observation since the alveolar cell of origin in itself predicates an ER-negative breast cancer even in the absence of a pregnancy (Fig. 9b).
PABCs, being largely ER− and advanced at diagnosis, are difficult to treat. It is therefore crucial to identify new molecular targets for therapeutic and prophylactic strategies. The identification of a potential distinct alveolar cell of origin for PABCs suggests that proteins regulating alveolar differentiation, such as pSTAT5, might be important as targets for intervention strategies. Moreover, the identification of an alveolar gene expression signature or protein biomarker that can be estimated non-invasively might be invaluable for predicting women at higher risk of being diagnosed with postpartum PABC. In addition, our mouse model may also be valuable for preclinical testing of therapeutics that may be especially potent for PABC.
Taken together, the results of our study identify for the first time the critical effects of cell of tumor origin on PABC characteristics. Using a mouse model well suited for studying tumor formation in vivo, we recapitulated the hallmarks of PABCs as distinct from those of late onset tumors in parous mice. Furthermore, we showed that tumor origination in alveolar cells significantly increased PABC characteristics irrespective of time since pregnancy and even parity per se. Translationally, it may be illuminating to investigate whether human PABCs are associated with alveolar markers or pSTAT5 levels. Identification of an alveolar signature in human PABCs might provide therapeutic targets, as well as prophylactic strategies. Most importantly, identification of key dysregulated molecular pathways contributing to PABC initiation in humans may be valuable for the identification of risk factors and biomarkers, facilitating early detection of PABCs.
Materials and Methods
Experimental Animals
MMTV-tva (MA) mice in a FVB genetic background used in these studies have been previously reported (22). All animals were euthanized according to the NIH guidelines. The animal protocol was approved by the IACUC of Baylor College of Medicine, Houston, TX. The WAP-tva transgenic vector was constructed in pBS-WAP, which contains the 7.3 kb WAP gene (38). TVA was removed from pSP72(0.8) (39) using Asp718 and EcoRI (Klenow) and inserted into Asp718 and SalI (Klenow) sites in pBS-WAP (a gift of Dr. Lothar Hennighaussen), thus resulting in removal of the WAP gene between exon 1 and exon 3 respectively. The resulting transgenic construct contains the WAP promoter, the quail tva cDNA (850 bp) encoding a glycosylphosphatidylinositol-linked form of TVA, and the WAP poly-A signal. This 6.4-kb transgene fragment was removed from the transgenic vector by digestion with EcoRI and injected into pronuclei from FVB/N mice. Candidate founders were screened by PCR using primers specific to tva. MMTV-tva mice have been previously described (22).
Virus Preparation and Delivery
RCAS virus was prepared as described earlier (22). The lentiviral vector (FC-CGW) carrying either GFP alone or both GFP and caErbB2 was prepared as described earlier (40).
Tumor studies
One week after intraductal injection of RCAS, the experimental group consisting of approximately half the mice was mated. Pups were weaned at lactation day 21. All mice were palpated thrice-weekly for tumor incidence. Tumor-free mice were euthanized 12 months post-injection. Tumor growth rate was calculated based on two diametric measurements taken at initial detection (diameter≤0.5 cm) and at time of euthanasia (diameter=1.8 cm) and the intervening time interval.
FISH analysis
FISH was performed to identify the copy number or integration sites for the RCAS retrovirus on paraffin sections from MMTV-tva mPABC and nulliparous tumors (5569, 6213, and 7166). The plasmid clone containing RCAS sequences was labeled by nick translation with Spectrum Red (Abbott Laboratories, Des Plaines, IL). Hybridization and detection were performed according to the manufacturer's protocols. The slides were counterstained with 4, 6-diamidino-2-phenylindole (DAPI) and the images were captured using Nikon E800 microscope equipped with a cooled-charge coupled devices (CCD) camera. The cells were analyzed using Quips Pathvysion (Applied Imaging, Santa Clara, CA). A total of 50 interphase nuclei were analyzed to determine the amplification status.
Single cell suspension preparation from mammary glands and qRT-PCR
Mammary glands were collected with removal of lymph nodes. Single cell suspensions were prepared as previously described (30). For qRT-PCR of WAP and TVA genes, single-cell suspensions were prepared from mammary glands, and cells were stained using antibodies against CD45, CD31, and Ter119 (Lin) and TVA. Using FACS, TVA+ and TVA−/Lin− cells were sorted out, RNA was isolated, and reverse transcription was performed to generate cDNA.
Flow cytometry analysis and FACS
Flow cytometry analysis was performed on a BD LSRII (BD Bioscience). FACS was performed on an AriaI w/ UV (BD Bioscience) with a 130 µm nozzle. Antibodies used for flow cytometry include a PE-conjugated rat antibody against-CD24 (553262, BD Bioscience), PE-conjugated rat IgG2b κ isotype (553989, BD Bioscience), FITC-conjugated rat anti-CD49f (555735, BD Bioscience), and FITC-conjugated rat IG2b κ isotype (553988, BD Bioscience). A rabbit polyclonal antibody against TVA (gift from Andy Leavitt) and an APC-conjugated goat anti-rabbit IgG (A10931, Invitrogen) were used. Dead cells were gated out based on propidium iodide positivity.
Colony forming assay
Mammary glands from virgin MMTV-tva and WAP-tva mice were digested as described above and FACS was performed to sort out TVA+ cells. 1000 TVA+ cells from each mouse was plated in triplicate on reduced growth factor Matrigel (354230, BD Bioscience) on plastic with DME/F12 medium supplemented with 20µl/ml B27 (17504-044; Invitrogen), 20 ng/ml bFGF (13256-029; Invitrogen), 20 ng/ml EGF (13247-051; Invitrogen), and Antibiotic/Antimycotic (15240-062; Invitrogen), and 5% FBS. One week after plating, colonies (≥ 50 µm) were quantified.
Tissue Harvest
Tumors and mammary tissue were fixed in 4% paraformaldehyde overnight at 4°C, paraffin-embedded, and sliced into 3 µm sections. Sections were deparaffinized in xylene, rehydrated in graded alcohol, and used for histology and immunostaining.
Immunostaining and Microscopy
Immunohistochemistry (IHC) and immunofluorescence (IF) were performed as described earlier (22). Antigen retrieval was carried out by heating sections in 10 mM sodium citrate, pH6.0. MOM and VectaStain Elite ABC Rabbit kits (Vector Labs; cat.no. PK-2200 & PK-6101) were used according to manufacturer’s protocols. Primary antibodies used included mouse monoclonal antibody against HA (Covance; 1:500); rabbit IgG specific for cleaved caspase 3 (Cell Signaling; 1:200), K5 (1:200), ER (Santa Cruz; 1:200), pHistone3 (Millipore; 1:200) and Ki67 (Novocastra; 1:200); and rat IgG specific for K8 (1:200). Incubation with the primary antibody for IF staining was overnight at 4°C, while incubation with primary antibody for IHC was 1 hour at RT. Nuclei were counterstained with 4’-6-diamidino-2-phenylindole (DAPI)-containing mounting medium and hematoxylin, respectively, for IF and IHC. Bright field images were captured using a Leica DMLB microscope, and images were processed with Magnavision and Adobe Photoshop software. Fluorescent images were captured with a Zeiss Axiskop2 plus microscope. Images were processed with Axiovision and Adobe Photoshop software.
Terminal Deoxynucleotidyl Transferase dUTP Nick-end Labeling Assay (TUNEL)
Paraffin-embedded gland and tumor sections were treated in proteinase K and subjected to the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay using the ApopTag Red in situ TUNEL detection kit (Chemicon, S7165). Nuclei were counterstained with DAPI-containing mounting medium.
Trichrome staining
Trichrome staining was performed on paraffin-embedded sections using Accustain Trichrome Stain Kit (Sigma, catalog#HT15), Bouin’s solution (Sigma catalog#HT10132) and Weigert’s Iron Hematoxylin Set (Sigma catalog#HT10-79). Sections were deparaffinized as for other immunohistochemical procedures and then treated as per manufacturer’s instructions. Sections were allowed to clear in Xylene overnight before being mounted.
Quantification of Stained Sections
5 random fields were viewed and a total of at least 5000 cells were counted per sample. Image J software was used for counting, and either DAPI or hematoxylin nuclear staining was used to identify the total number of cells. For tumor histopathological quantification, tumor sections were viewed microscopically and scored by the investigators and independently by a pathologist.
Statistical Analysis
All numbers in the text were represented as mean ± standard error of the mean. Statistical analysis of quantification of stained sections was done using ANOVA or Student’s T-test for independent samples with Holm’s correction for multiple comparisons when distribution of data was judged to be normal. Where distribution was not normal (assessed using Q-Q plots with the Wilk-Shapiro test of normality), either Kruskal-Wallis or Wilcoxon’s Rank Sum test was used. Holm’s correction was also used where required when using non-parametric tests. For categorical data with <15 data points in each group, the Fisher’s Exact test was used. For categorical data with ≥15 data points in each group, the Pearson’s chi-square test was used. All graphs and statistical analyses were generated either in MS Excel or R.
Supplementary Material
(a) Representative photomicrographs depicting levels of TVA in the mammary gland of MMTV-tva mice at various stages of estrus. n=3 mice.
(b) Bar graph representing relative fluorescent intensity of the ErbB2 oncogene in tumors from MMTV-tva virgin, PABC, and parous late onset mice (n=4 each) based on ImageJ quantification of immunofluorescence. ANOVA determined p-value. Columns indicate mean and error bars, the SEM.
(c) Representative photomicrographs and table showing number of copies of RCAS-ErbB2 seen in mPABCs (n=5) and tumors from virgin MMTV-tva mice (n=4) based on FISH analysis using a probe for the RCAS virus (red).
(d) Western blot showing relative levels of RCAS-ErbB2 construct in PABCs and late onset tumors from MMTV-tva mice.
(a) Representative photomicrographs depicting immunohistochemical evaluation of TVA levels at various indicated stages of estrus. n=4 mice. Mammary glands from FVB mice included as negative controls.
(b) Dot plot comparing percentage of cells infected with the RCAS-GFP virus in MMTV-tva vs WAP-tva mammary glands. Student’s t-test identified p-value. Median represented by horizontal line.
(c) Bar graphs depicting mean relative ErbB2 intensity based on ImageJ quantification of immunofluorescence of tumors from indicated groups (n=4 each). ANOVA determined p-values. Columns represent mean and error bars, the SEM.
Bar graph depicting mean relative ErbB2 intensity based on ImageJ quantification of immunofluorescence of tumors from indicated groups (n=4 each). White columns represent MMTV-tva and black columns represent WAP-tva mice. ANOVA determined p-values. Columns represent mean and error bars, the SEM.
Acknowledgements
The authors acknowledge Jay Reddy and Zhijun Du for technical help with experiments. This work was supported in part by funds from NIH CA124820 (to Y.L) and U54CA149196 (to Y. L; PI: Stephan Wong); from CDMRP BC085050 (to Y. L) and BC073703 (to Y.L); and from the Nancy Owens Memorial Foundation (to Y. L); as well as by the resources from the Dan L. Duncan Cancer Center (P30CA125123) and the Sue & Lester Breast Center (P50-CA058183).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006 Apr;6(4):281–291. doi: 10.1038/nrc1839. PubMed PMID: 16557280. Epub 2006/03/25. eng. [DOI] [PubMed] [Google Scholar]
- 2.Middleton LP, Amin M, Gwyn K, Theriault R, Sahin A. Breast carcinoma in pregnant women: assessment of clinicopathologic and immunohistochemical features. Cancer. 2003 Sep 1;98(5):1055–1060. doi: 10.1002/cncr.11614. PubMed PMID: 12942575. Epub 2003/08/28. eng. [DOI] [PubMed] [Google Scholar]
- 3.Mathelin C, Annane K, Treisser A, Chenard MP, Tomasetto C, Bellocq JP, et al. Pregnancy and post-partum breast cancer: a prospective study. Anticancer research. 2008 Jul-Aug;28(4C):2447–2452. PubMed PMID: 18751433. Epub 2008/08/30. eng. [PubMed] [Google Scholar]
- 4.Lyons TR, Schedin PJ, Borges VF. Pregnancy and breast cancer: when they collide. Journal of mammary gland biology and neoplasia. 2009 Jun;14(2):87–98. doi: 10.1007/s10911-009-9119-7. PubMed PMID: 19381788. Pubmed Central PMCID: 2693784. Epub 2009/04/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fornetti J, Martinson H, Borges V, Schedin P. Emerging targets for the prevention of pregnancy-associated breast cancer. Cell Cycle. 2012 Feb 15;11(4):639–640. doi: 10.4161/cc.11.4.19358. PubMed PMID: 22374663. Pubmed Central PMCID: 3318100. Epub 2012/03/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyons TR, O'Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, et al. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nature medicine. 2011 Sep;17(9):1109–1115. doi: 10.1038/nm.2416. PubMed PMID: 21822285. Epub 2011/08/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Brien J, Lyons T, Monks J, Lucia MS, Wilson RS, Hines L, et al. Alternatively activated macrophages and collagen remodeling characterize the postpartum involuting mammary gland across species. The American journal of pathology. 2010 Mar;176(3):1241–1255. doi: 10.2353/ajpath.2010.090735. PubMed PMID: 20110414. Pubmed Central PMCID: 2832146. Epub 2010/01/30. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pilewskie M, Gorodinsky P, Fought A, Hansen N, Bethke K, Jeruss J, et al. Association between recency of last pregnancy and biologic subtype of breast cancer. Ann Surg Oncol. 2012 Apr;19(4):1167–1173. doi: 10.1245/s10434-011-2104-6. PubMed PMID: 21997350. Epub 2011/10/15. eng. [DOI] [PubMed] [Google Scholar]
- 9.Stensheim H, Moller B, van Dijk T, Fossa SD. Cause-specific survival for women diagnosed with cancer during pregnancy or lactation: a registry-based cohort study. J Clin Oncol. 2009 Jan 1;27(1):45–51. doi: 10.1200/JCO.2008.17.4110. PubMed PMID: 19029418. Epub 2008/11/26. eng. [DOI] [PubMed] [Google Scholar]
- 10.O'Brien J, Hansen K, Barkan D, Green J, Schedin P. Non-steroidal anti-inflammatory drugs target the pro-tumorigenic extracellular matrix of the postpartum mammary gland. Int J Dev Biol. 2011;55(7–9):745–755. doi: 10.1387/ijdb.113379jo. PubMed PMID: 22161831. Epub 2011/12/14. eng. [DOI] [PubMed] [Google Scholar]
- 11.Watson CJ, Khaled WT. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development. 2008 Mar;135(6):995–1003. doi: 10.1242/dev.005439. PubMed PMID: 18296651. Epub 2008/02/26. eng. [DOI] [PubMed] [Google Scholar]
- 12.Robinson GW, McKnight RA, Smith GH, Hennighausen L. Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development. 1995 Jul;121(7):2079–2090. doi: 10.1242/dev.121.7.2079. PubMed PMID: 7635053. Epub 1995/07/01. eng. [DOI] [PubMed] [Google Scholar]
- 13.Yamaji D, Na R, Feuermann Y, Pechhold S, Chen W, Robinson GW, et al. Development of mammary luminal progenitor cells is controlled by the transcription factor STAT5A. Genes & development. 2009 Oct 15;23(20):2382–2387. doi: 10.1101/gad.1840109. PubMed PMID: 19833766. Pubmed Central PMCID: 2764497. Epub 2009/10/17. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khaled WT, Read EK, Nicholson SE, Baxter FO, Brennan AJ, Came PJ, et al. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development. 2007 Aug;134(15):2739–2750. doi: 10.1242/dev.003194. PubMed PMID: 17611223. Epub 2007/07/06. eng. [DOI] [PubMed] [Google Scholar]
- 15.Smith GH, Boulanger CA. Mammary epithelial stem cells: transplantation and self-renewal analysis. Cell Prolif. 2003 Oct;36(Suppl 1):3–15. doi: 10.1046/j.1365-2184.36.s.1.2.x. PubMed PMID: 14521512. Epub 2003/10/03. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bu W, Chen J, Morrison GD, Huang S, Creighton CJ, Huang J, et al. Keratin 6a marks mammary bipotential progenitor cells that can give rise to a unique tumor model resembling human normal-like breast cancer. Oncogene. 2011 Oct 27;30(43):4399–4409. doi: 10.1038/onc.2011.147. PubMed PMID: 21532625. Pubmed Central PMCID: 3156856. Epub 2011/05/03. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell. 2007 Aug;12(2):160–170. doi: 10.1016/j.ccr.2007.06.013. PubMed PMID: 17692807. Epub 2007/08/19. eng. [DOI] [PubMed] [Google Scholar]
- 18.Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011 Feb 4;8(2):149–163. doi: 10.1016/j.stem.2010.12.007. PubMed PMID: 21295272. Pubmed Central PMCID: 3050563. Epub 2011/02/08. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci U S A. 2012 Feb 21;109(8):2772–2777. doi: 10.1073/pnas.1017626108. PubMed PMID: 21940501. Pubmed Central PMCID: 3286919. Epub 2011/09/24. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010 Sep 3;7(3):403–417. doi: 10.1016/j.stem.2010.07.010. PubMed PMID: 20804975. Epub 2010/09/02. eng. [DOI] [PubMed] [Google Scholar]
- 21.Bu W, Zhang X, Dai H, huang S, Li Y. Mammary cells with active Wnt signaling resist ErbB2-induced tumorigenesis. PLoS One. 2013 doi: 10.1371/journal.pone.0078720. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du Z, Podsypanina K, Huang S, McGrath A, Toneff MJ, Bogoslovskaia E, et al. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proc Natl Acad Sci U S A. 2006 Nov 14;103(46):17396–17401. doi: 10.1073/pnas.0608607103. PubMed PMID: 17090666. Pubmed Central PMCID: 1635021. Epub 2006/11/09. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy JP, Li Y. The RCAS-TVA system for introduction of oncogenes into selected somatic mammary epithelial cells in vivo. Journal of mammary gland biology and neoplasia. 2009 Dec;14(4):405–409. doi: 10.1007/s10911-009-9157-1. PubMed PMID: 19936988. Epub 2009/11/26. eng. [DOI] [PubMed] [Google Scholar]
- 24.Toneff MJ, Du Z, Dong J, Huang J, Sinai P, Forman J, et al. Somatic expression of PyMT or activated ErbB2 induces estrogen-independent mammary tumorigenesis. Neoplasia. Sep;12(9):718–726. doi: 10.1593/neo.10516. PubMed PMID: 20824048. Pubmed Central PMCID: 2933692. Epub 2010/09/09. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruz GI, Martinez ME, Natarajan L, Wertheim BC, Gago-Dominguez M, Bondy M, et al. Hypothesized role of pregnancy hormones on HER2+ breast tumor development. Breast cancer research and treatment. 2013 Jan;137(1):237–246. doi: 10.1007/s10549-012-2313-0. PubMed PMID: 23135573. Epub 2012/11/09. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nature reviews Cancer. 2009;9(7):463–475. doi: 10.1038/nrc2656. PubMed PMID: 19536107. [DOI] [PubMed] [Google Scholar]
- 27.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nature reviews Cancer. 2008;8(5):387–398. doi: 10.1038/nrc2389. PubMed PMID: 18432252. eng. [DOI] [PubMed] [Google Scholar]
- 28.Brisken C. Hormonal control of alveolar development and its implications for breast carcinogenesis. J Mammary Gland Biol Neoplasia. 2002 Jan;7(1):39–48. doi: 10.1023/a:1015718406329. PubMed PMID: 12160085. Epub 2002/08/06. eng. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997 Jan 15;11(2):179–186. doi: 10.1101/gad.11.2.179. PubMed PMID: 9009201. Epub 1997/01/15. eng. [DOI] [PubMed] [Google Scholar]
- 30.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006 Jan 5;439(7072):84–88. doi: 10.1038/nature04372. PubMed PMID: 16397499. Epub 2006/01/07. eng. [DOI] [PubMed] [Google Scholar]
- 31.MacMahon B, Cole P, Lin TM, Lowe CR, Mirra AP, Ravnihar B, et al. Age at first birth and breast cancer risk. Bull World Health Organ. 1970;43(2):209–221. PubMed PMID: 5312521. Pubmed Central PMCID: 2427645. Epub 1970/01/01. eng. [PMC free article] [PubMed] [Google Scholar]
- 32.Medina D. Breast cancer: the protective effect of pregnancy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004 Jan 1;10(1 Pt 2):380S–384S. doi: 10.1158/1078-0432.ccr-031211. PubMed PMID: 14734495. Epub 2004/01/22. eng. [DOI] [PubMed] [Google Scholar]
- 33.Nielsen M, Thomsen JL, Primdahl S, Dyreborg U, Andersen JA. Breast cancer and atypia among young and middle-aged women: a study of 110 medicolegal autopsies. Br J Cancer. 1987 Dec;56(6):814–819. doi: 10.1038/bjc.1987.296. PubMed PMID: 2829956. Pubmed Central PMCID: 2002422. Epub 1987/12/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welch HG, Black WC. Using autopsy series to estimate the disease "reservoir" for ductal carcinoma in situ of the breast: how much more breast cancer can we find? Ann Intern Med. 1997 Dec 1;127(11):1023–1028. doi: 10.7326/0003-4819-127-11-199712010-00014. PubMed PMID: 9412284. Epub 1997/12/31. eng. [DOI] [PubMed] [Google Scholar]
- 35.Bartow SA, Pathak DR, Black WC, Key CR, Teaf SR. Prevalence of benign, atypical, and malignant breast lesions in populations at different risk for breast cancer. A forensic autopsy study. Cancer. 1987 Dec 1;60(11):2751–2760. doi: 10.1002/1097-0142(19871201)60:11<2751::aid-cncr2820601127>3.0.co;2-m. PubMed PMID: 3677009. Epub 1987/12/01. eng. [DOI] [PubMed] [Google Scholar]
- 36.Foulkes WD, Metcalfe K, Sun P, Hanna WM, Lynch HT, Ghadirian P, et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004 Mar 15;10(6):2029–2034. doi: 10.1158/1078-0432.ccr-03-1061. PubMed PMID: 15041722. Epub 2004/03/26. eng. [DOI] [PubMed] [Google Scholar]
- 37.Murphy CG, Mallam D, Stein S, Patil S, Howard J, Sklarin N, et al. Current or recent pregnancy is associated with adverse pathologic features but not impaired survival in early breast cancer. Cancer. 2012 Jul 1;118(13):3254–3259. doi: 10.1002/cncr.26654. PubMed PMID: 22086863. Epub 2011/11/17. eng. [DOI] [PubMed] [Google Scholar]
- 38.Campbell SM, Rosen JM, Hennighausen LG, Strech-Jurk U, Sippel AE. Comparison of the whey acidic protein genes of the rat and mouse. Nucleic acids research. 1984 Nov 26;12(22):8685–8697. doi: 10.1093/nar/12.22.8685. PubMed PMID: 6095207. Pubmed Central PMCID: 320407. Epub 1984/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proceedings of the National Academy of Sciences of the United States of America. 1998 Feb 3;95(3):1218–1223. doi: 10.1073/pnas.95.3.1218. PubMed PMID: 9448312. Pubmed Central PMCID: 18724. Epub 1998/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bu W, Xin L, Toneff M, Li L, Li Y. Lentivirus vectors for stably introducing genes into mammary epithelial cells in vivo. J Mammary Gland Biol Neoplasia. 2009 Dec;14(4):401–404. doi: 10.1007/s10911-009-9154-4. PubMed PMID: 19936990. Epub 2009/11/26. eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) Representative photomicrographs depicting levels of TVA in the mammary gland of MMTV-tva mice at various stages of estrus. n=3 mice.
(b) Bar graph representing relative fluorescent intensity of the ErbB2 oncogene in tumors from MMTV-tva virgin, PABC, and parous late onset mice (n=4 each) based on ImageJ quantification of immunofluorescence. ANOVA determined p-value. Columns indicate mean and error bars, the SEM.
(c) Representative photomicrographs and table showing number of copies of RCAS-ErbB2 seen in mPABCs (n=5) and tumors from virgin MMTV-tva mice (n=4) based on FISH analysis using a probe for the RCAS virus (red).
(d) Western blot showing relative levels of RCAS-ErbB2 construct in PABCs and late onset tumors from MMTV-tva mice.
(a) Representative photomicrographs depicting immunohistochemical evaluation of TVA levels at various indicated stages of estrus. n=4 mice. Mammary glands from FVB mice included as negative controls.
(b) Dot plot comparing percentage of cells infected with the RCAS-GFP virus in MMTV-tva vs WAP-tva mammary glands. Student’s t-test identified p-value. Median represented by horizontal line.
(c) Bar graphs depicting mean relative ErbB2 intensity based on ImageJ quantification of immunofluorescence of tumors from indicated groups (n=4 each). ANOVA determined p-values. Columns represent mean and error bars, the SEM.
Bar graph depicting mean relative ErbB2 intensity based on ImageJ quantification of immunofluorescence of tumors from indicated groups (n=4 each). White columns represent MMTV-tva and black columns represent WAP-tva mice. ANOVA determined p-values. Columns represent mean and error bars, the SEM.