

Abstract
Chronic exposure to cold caused pulmonary arterial hypertension (CIPH) and increased phosphodiesterase-1C (PDE-1C) expression in pulmonary arteries (PAs) in rats. The purpose of this study is to investigate a hypothesis that inhibition of PDE-1 would decrease inflammatory infiltrates and superoxide production leading to attenuation of CIPH. Three groups of male rats were exposed to moderate cold (5±1°C) continuously while 3 groups were maintained at room temperature (23.5±1°C, warm) (6 rats/group). Following 8-week exposure to cold, 3 groups in each temperature condition received continuous i.v. infusion of 8-IBMX (PDE-1 inhibitor), apocynin (NADPH oxidase inhibitor) or vehicle, respectively, for one week. Cold exposure significantly increased right ventricular (RV) systolic pressure compared to warm groups (33.8±3.2 vs 18.6±0.3 mmHg), indicating that animals developed pulmonary arterial hypertension (CIPH). Notably, treatment with 8-IBMX significantly attenuated the cold-induced increase in RV pressure (23.5±1.8 mmHg). Cold exposure also caused RV hypertrophy while 8-IBMX reversed cold-induced RV hypertrophy. Cold exposure increased PDE-1C protein expression, macrophage infiltration, NADPH oxidase activity, and superoxide production in PAs and resulted in PA remodeling. 8-IBMX abolished cold-induced upregulation of PDE1C in PAs. Interestingly, inhibition of PDE1 eliminated cold-induced macrophage infiltration, NADPH oxidase activation, and superoxide production in PAs and reversed PA remodeling. Inhibition of NADPH oxidase by apocynin abolished cold-induced superoxide production and attenuated CIPH and PA remodeling.
Conclusions
Inhibition of PDE-1 attenuated CIPH and reversed cold-induced PA remodeling by suppressing macrophage infiltration and superoxide production, suggesting that upregulation of PDE-1C expression may be involved in the pathogenesis of CIPH.
Keywords: pulmonary artery, blood pressure, remodeling, macrophage infiltration, NADPH oxidase, superoxide, smooth muscle cell proliferation, right ventricle
Introduction
The development of pulmonary hypertension (PH) is multi-factorial with genetic background and environmental stress being two critical components. The adverse effects of cold temperatures on the human cardiovascular system are well documented.1–5 Lungs are open to the environment and are susceptible to cold air stimulation. Clinical studies suggest that inhaled cold air causes pathophysiological responses such as vasoconstriction in the respiratory tract mucosa which may contribute to cold-related respiratory diseases6. Exposure to cold temperatures is an important public health concern, particularly for those dying from cardiorespiratory diseases.7 In the US, cold weather is associated with increased mortality from pulmonary and cardiovascular diseases.8,9–10 Cold exposure causes pulmonary vasospasm in patients with Raynaud’s phenomenon.11 Cold exposure has been demonstrated to induce PH in several animal models including broilers, rats and bovine.12–15 However, the underlying mechanism of cold-induced PH remains poorly understood.
Phosphodiesterases (PDEs) are a family of enzymes that catalyze the breakdown of the second messengers cGMP and cAMP. Currently eleven different families of PDEs have been described, each with several different subtypes. Three different PDE-1 variants have been described, PDE-1A, PDE-1B, and PDE-1C. Previously these PDEs were given the term CaM-PDEs for their reliance on calcium-calmodulin for activity.16 PDE-1A and 1B hydrolyze cGMP more efficiently while PDE-1C hydrolyzes both cGMP and cAMP equally.17 The PDE family has been a focus of drug development in recent years, especially for cardiovascular diseases, because of the favorable effects second messengers have in the vasculature that include increasing vasodilation and decreasing SMC proliferation. Early investigations revealed that PDE-1 inhibition increased aortic vasodilation. Recently, Shermuly et al showed that PDE-1C protein expression is increased in proliferating pulmonary vasculature in human IPAH.18 For these reasons, PDE-1 has generated significant interest in PH research.
Preliminary studies in our lab indicated that cold exposure causes CIPH and PA remodeling and upregulates expression of PDE-1C. Increased PDE-1 activity decreases intracellular cGMP levels which may increase PA resistance and smooth muscle cell proliferation. Cold exposure also increased inflammation in the cardiovascular system.2 The aim of this study is to investigate a hypothesis that inhibition of PDE-1 would decrease inflammatory infiltrates and superoxide production and attenuate CIPH.
Methods
For details, see the expanded methods section in the Online Supplemental Methods and Data (http://hyper.ahajournals.org).
Animal study protocols
Six groups of male Sprague–Dawley rats were used (150–180g, 6 rats/group). Three groups of rats were exposed to a climate-controlled walk-in chamber maintained at moderate cold (5.0±1°C). The remaining groups were kept in an identical chamber maintained at room temperature (23.5±1°C, warm) and served as controls.
After eight weeks of exposure to cold, 3 groups in each temperature condition received continuous IV infusion of 8-IBMX (PDE-1 inhibitor, 8.5 mg/kg/day18), apocynin (NADPH oxidase inhibitor, 25 mg/kg/day19–20) and vehicle (dimethyl sulfoxide -DMSO, 50%), respectively. The doses of drugs have been validated for effective inhibition of PDE-1 and NADPH oxidase activity, respectively.18–20 Body weight was measured weekly. After one week of drug infusion, the animals’ right ventricular systolic blood pressure (RVBP) was measured under anesthesia. The RVP is a reliable indicator of pulmonary arterial blood pressure (PAP) and has been used by numerous investigators for evaluating PH.21–25 For details, refer to the Online Supplemental Methods and Data (http://hyper.ahajournals.org).
Western blot analysis of PDE-1 and PDE-5 protein expression in tissue
Protein expression of PDE-1A, PDE-1B, PDE-1C and PDE-5 was measured as we described previously.2, 26
Morphometric measurements and IHC analysis of CD-68 and SM α-actin expression
The histological and IHC analysis of macrophage infiltration and SM α-actin expression were performed as described in our recent studies.2, 26 For details, refer to the Online Supplemental Methods and Data (http://hyper.ahajournals.org).
Measurement of in situ superoxide production
The in situ PA superoxide production was assessed using DHE staining as we described recently.2, 27–28 For details, refer to the Online Supplemental Methods and Data (http://hyper.ahajournals.org).
Measurement of NADPH oxidase activity
NADPH oxidase activity was assessed using the lucigenin assay as we described recently.27–28 For details, refer to the Online Supplemental Methods and Data (http://hyper.ahajournals.org).
Measurement of pulmonary cGMP levels
The pulmonary cGMP levels were measured using a KGE 003 cGMP Parameter Assay Kit (R&D Systems) according to the manufacturer’s instruction. The data were normalized with protein for each animal.
Statistical analysis
Data were analyzed by one-way ANOVA. Tukey’s multiple comparison tests were used to assess the significance of differences between means. Significance was set at a 95% confidence limit.
Results
8-IBMX and apocynin reduced cold-induced pulmonary hypertension
The RV systolic blood pressure measured at 9 weeks after exposure to cold was elevated significantly in the Cold-DMSO group (33.8±3.2 mmHg) compared to the Warm-DMSO, Warm-Apocynin and Warm-IBMX groups (18.6±0.6, 20.7±0.5 and 19.6±0.5 mmHg, respectively). Thus, cold exposure caused pulmonary arterial hypertension (CIPH). Treatments with IBMX and apocynin significantly decreased cold-induced elevation of RV pressure (23.5±1.8 and 24.2±0.6 mmHg, respectively) although they did not decrease RV pressure to the warm control levels (Fig 1A).
Figure 1. 8-IBMX and apocynin reduced right ventricular blood pressure (RVP).

A, In vivo RVP. After one week of drug infusion, animals were anesthetized and RV systolic blood pressure was monitored for 20 min (1 reading/min) utilizing a telemetry system (DSI). B, RV weight. The free RV was separated from the left ventricle (LV) for calculating the ratio of RV/(LV+septum). C, Morphometric measurements of the RV wall thickness (For histological micrographs, refer to supplemental figure S1C). *p<0.05, **p<0.01, ***p<0.001 vs Warm-DMSO; ++p<0.01, +++p<0.001 vs Warm-Apocynin; ^^p<0.05, ^^p<0.01, ^^^p<0.001 vs Cold-DMSO; #p<0.05, ##p<0.01, ###p<0.001 vs Warm-IBMX.
Compared to the 3 warm control groups, the Cold-DMSO group showed a significant increase in the ratio of RV/(LV+S) after normalizing to body weight (Fig. 1B), suggesting that cold-exposed rats developed RV hypertrophy. IBMX attenuated cold-induced RVH while treatment with apocynin slightly but not significantly decreased RVH (Fig. 1B). The RV wall thickness was increased by cold exposure (Fig. 1C & Supplemental Figure S1C). IBMX prevented the cold-induced increase in RV wall thickness. Trichrome staining showed that there is no significant difference in collagen levels between groups (not shown).
IBMX and apocynin did not affect systemic blood pressure or body weight gain (Supplemental Figure S1A&B).
8-IBMX and apocynin reversed cold-induced remodeling of pulmonary arteries (PAs)
We examined small PAs with diameters of 60–80 μm. Cold exposure increased medial layer thickness of small PAs in the Cold-DMSO group (22.7 ± 1.3 μm) compared to Warm-DMSO, Warm-Apocynin, and Warm-IBMX groups (17.4 ± 0.8, 15.6 ± 0.6, and 17.7 ± 0.3 μm, respectively) (Fig. 2A,B). Cold exposure also caused narrowing of the PA lumen in the Cold-DMSO group (32.2 ± 3.0 μm) compared to Warm-DMSO, Warm-Apocynin, and Warm-IBMX groups (56.8 ± 4.6, 55.9 ± 3.5, and 62.1 ± 3.1 μm, respectively) (Fig. 2A,C). IBMX or apocynin significantly reduced medial layer thickness (19.0 ± 0.9, and 16.9 ± 0.8 μm, respectively) and increased lumen diameter (62.7 ± 4.2, and 59.5 ± 4.3 μm, respectively) of small PAs in cold-exposed rats. Cold exposure increased the ratio of the medial layer thickness to the lumen diameter which can be abolished by 8-IBMX or apocynin (Fig. 2D).
Figure 2. 8-IBMX and apocynin reversed cold-induced remodeling of pulmonary arteries (PAs).

A, Photomicrograph of lung and PAs (H&E staining). B, PA medial layer thickness. C, PA lumen diameters. D, Ratio of the medial layer to lumen diameter. PAs with a diameter of 60–80 μm were selected for morphological analysis. Sections were viewed at 200x. **p<0.01, ***p<0.001 vs Warm-DMSO; +p<0.05, +++p<0.001 vs Warm-Apocynin; ^p<0.05, ^^p<0.01, ^^^p<0.001 vs Cold-DMSO; ##p<0.01, ###p<0.001 vs Warm-IBMX.
8-IBMX and apocynin attenuated cold-induced PA SMC proliferation
Alpha smooth muscle actin (α-SMA) is a marker of proliferating SMCs. We used IHC with an α-SMA specific antibody to semi-quantify the expression of α-SMA in small resistance PAs (40–80 μm). The α-SMA expression in PA medial layer of the Cold-DMSO group was increased significantly compared to the 3 warm groups (Fig. 3A,B). IBMX and apocynin significantly reduced α-SMA expression in PAs of cold-exposed rats (Fig. 3A,B), suggesting decreased PA SMC proliferation.
Figure 3. 8-IBMX and apocynin attenuated cold-induced PA SMC proliferation.

A, The IHC analysis of α-SMA staining in the smooth muscle layer of small PAs of the lungs. The negative control (without primary antibody) showed no α-SMA staining indicating antibody specificity. Arrows indicate α-SMA-positive staining (brown color). Sections were viewed at 400x. B, Semi-quantitative analysis of the α-SMA staining in small PAs. **p<0.01 vs Warm-DMSO; ++p<0.01 vs Warm-Apocynin; #p<0.05, ##p<0.01 vs Warm-IBMX; ^p<0.05, ^^p<0.01 vs Cold-DMSO.
8-IBMX and apocynin attenuated cold-induced superoxide production in PAs
Cold exposure increased PA superoxide production (Fig. 4A, photomicrograph in Fig. S2) and NADPH oxidase activity (Fig. 4B) compared to warm controls. Treatment with apocynin reduced superoxide production and NADPH oxidase activity to the warm control levels (Fig. 4A&B), indicating a role of NADPH oxidase in the cold-induced increase in superoxide in PAs. Interestingly, IBMX abolished the cold-induced increases in NADPH oxidase activity and superoxide production (Fig. 4A&B), suggesting that upregulation of PDE-1 may increase NADPH oxidase activity and superoxide production in PAs of cold-exposed rats.
Figure 4. 8-IBMX and apocynin reduced superoxide production PAs.
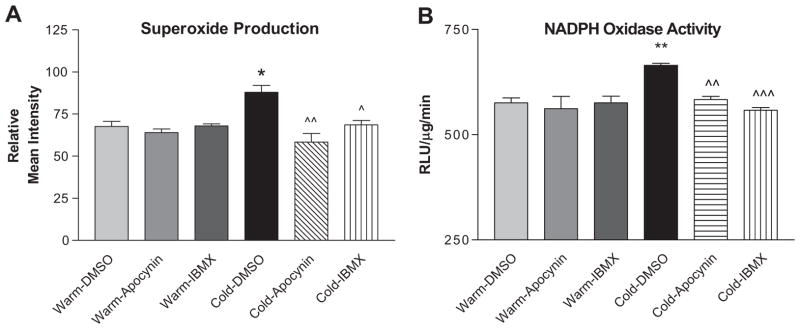
Superoxide production was viewed using the dye DHE (see photomicrograph in Supplemental Figure S2). A, Quantification of superoxide staining in PAs. B, NADPH oxidase activity in the PAs as measured by lucigenin assay. Sections were viewed at 200x. *p<0.05, **p<0.01 vs Warm-DMSO; ^p<0.05, ^^p<0.01, ^^^p<0.001 vs Cold-DMSO.
The Mn-SOD protein expression was not altered significantly by cold exposure or treatments with IBMX or apocynin (Fig. S3A&B), suggesting that the increases in PA superoxide levels may not be due to alteration of Mn-SOD protein expression.
8-IBMX abolished the cold-induced increase in PDE-1C protein expression in PAs
Cold exposure increased protein expression of PDE-1C in PAs compared to the 3 warm control groups (Fig. 5A,B). PDE-1A levels were unaffected by either cold exposure or drug treatments (Fig. 5A,C) while PDE-1B levels were not detectable by western blot (not shown). PDE-5 protein expression in the lungs was not altered significantly by cold exposure or drug treatments (Fig. S4). Thus, cold exposure selectively up-regulates PDE-1C expression in PAs. Treatment with IBMX or apocynin abolished cold-induced increases in PDE-1C expression on PAs (Fig. 5A,C). PDE1A and PDE1C protein expression in aortas and kidneys were not altered by cold exposure or by IBMX (Fig. S5&S6).
Figure 5. 8-IBMX abolished the cold-induced increase in PDE-1C protein expression in PAs.

Representative western blot bands of PDE-1C and PDE1A expression in PAs (A). Quantitative analysis of PDE-1C expression in PAs (B) and Quantitative analysis of PDE-1A expression in PAs (C). Pulmonary cGMP levels (D). *p<0.05, **p<0.01 vs Warm-DMSO; +p<0.05, ++p<0.01 vs Warm-Apocynin; ##p<0.01 vs Warm-IBMX; ^p<0.05 vs Cold-DMSO.
Cold exposure decreased the pulmonary cGMP levels which can be abolished by IBMX (Fig. 5D). These data indicate that the cold-induced decrease in pulmonary cGMP bio-availability can be rescued by PDE-1 inhibition.
8-IBMX decreased cold-induced macrophage infiltration
Chronic cold exposure remarkably increased infiltration of CD-68-positive macrophages around the small PAs of the lungs compared to the 3 warm groups (Fig. 6A,B,C). IBMX significantly attenuated cold-induced macrophage infiltration. Apocynin decreased macrophage infiltration although the difference was not significant when compared with the DMSO-Cold group (Fig. 6A,B). These findings indicate that cold exposure increased inflammation which can be abolished by PDE-1 inhibition.
Figure 6. 8-IBMX but not apocynin decreased cold-induced macrophage infiltration.

Semi-quantitative analysis of macrophage infiltration around the PAs in the lung as measured by density of CD-68 expression (A) and the average number of CD-68-positive cells (B). Photomicrograph of macrophage infiltration in the lung as measured by CD-68 staining (C). Arrows indicate CD-68-positive cells. Photos are shown at 400x. CD-68-Positive cells were examined in a series of 10–12 slices (84,000 μm2/slice). *p<0.05, **p<0.01 vs Warm-DMSO; ++p<0.01 vs Warm-Apocynin; ^p<0.05, ^^p<0.01 vs Cold-DMSO; ##p<0.01 vs Warm-IBMX.
Cold exposure did not increase IL-1β or IL-6 levels in the lungs (Fig. S7), suggesting that cold-induced macrophage infiltration may not be mediated by these cytokines. IBMX significantly decreased these cytokines (Fig. S7).
Discussion
Although the adverse effect of cold exposure on the systemic circulation is well documented,26, 29–35 its effects on the pulmonary circulation is poorly studied. The present study demonstrates that cold exposure caused pulmonary arterial hypertension (CIPH) and PA remodeling in rats which is supported by several previous reports.13–15 PDE-1C was up-regulated in PAs in response to cold exposure (Fig. 5). Interestingly, inhibition of PDE-1 effectively attenuated CIPH and RV hypertrophy and reversed PA remodeling in cold-exposed animals (Figs. 1 & 2), indicating that upregulation of PDE-1 may be involved in the pathogenesis of CIPH. These findings suggest that inhibition of PDE1 is an effective therapeutic strategy for cold-related impairment in the pulmonary circulation, which is important for people who live in cold regions or work outside during the winter months.
The PDE-1C enzyme regulates intracellular signaling mechanisms by degrading both cGMP and cAMP. Reduced cGMP levels promote vasoconstriction, proliferation of vascular smooth muscle cells and endothelial dysfunction, contributing to pulmonary hypertension.36–38 The present study demonstrated that cold exposure decreased pulmonary cGMP levels and that treatment with IBMX for one week restored cGMP to warm control levels and most importantly attenuated CIPH and PA remodeling (Figs. 2&6). It seems that BMX and apocynin mainly affected the pulmonary circulating system and lungs because PDE1A and PDE1C expression in aortas and kidneys were not altered by the treatments (Fig. S5 & S6). Therefore, following IV infusion via the jugular vein, the drugs were effectively taken up by the pulmonary circulation.
In addition to the regulation of cGMP levels, PDE-1C also has the ability to decompose cAMP. Decreased cAMP levels promote inflammation, including cytokine production39–40 that is known to activate NADPH oxidases (Fig. S9). It was recently shown that decreased cAMP levels promoted superoxide production via activating the Rac1-NADPH oxidase pathway.41 Superoxide derived from the NADPH oxidase is a major oxidant source in the vasculature and can promote the development of oxidative stress.43–45 While superoxide production is necessary for a variety of cellular processes (e.g., cell signaling, host defense), over-production of superoxide can lead to oxidative damage in the vasculature contributing to endothelial dysfunction, vascular remodeling and hypertension.42–43 The present study demonstrated that cold exposure increased NADPH oxidase activity and superoxide production in PAs (Fig. 4). Treatment with the NADPH oxidase inhibitor, apocynin, and the PDE-1 inhibitor, 8-IBMX, significantly reduced NADPH oxidase activity and superoxide production in PAs and improved CIPH and reversed PA remodeling (Figs. 2&6). While apocynin acts to directly inhibit NADPH oxidase, the mechanism by which 8-IBMX decreased cold-induced superoxide production is unclear at this time. Based on literatures, inhibition of PDE-1C could decrease NADPH activity via two pathways: 1) increasing cAMP levels and preventing low cAMP-related activation of NADPH oxidase41; and 2) eliminating low cAMP-associated inflammation39–40 and thus attenuating inflammation-related activation of NADPH oxidase (Fig. S9).
Inhibition of NADPH oxidase activity by apocynin abolished the cold-induced increase in PDE-1C protein expression (Fig. 5), suggesting that NADPH oxidase and superoxide may mediate cold-induced PDE-1C expression. On the other hand, inhibition of PDE-1 activity decreased NADPH oxidase activity and superoxide production (Fig. 4 B&C), which, in turn, decreased PDE-1C expression (Fig. 5). As depicted in Figure S9, the upregulation of PDE-1C may promote NADPH oxidase activity and superoxide production which then induce PDE-1C expression. Thus, this study revealed an important role of the NADPH oxidase and superoxide in the regulation of PDE-1C expression. Heumuller et al44 suggested that that in vascular cells, apocynin functions more as an antioxidant than as a specific inhibitor of NADPH oxidase. As the changes in NADPH oxidase activity are minor (≈ 15%) (Fig. 4C), the effects of apocynin may be partially mediated by its anti-oxidant action.
One of the major pathological changes in PH patients is the proliferation of SMCs in PAs, especially the small distal PAs. Proliferation of PA SMCs leads to extensive vascular remodeling and is a feature common to almost all forms of PH.45–47 Proliferating SMCs lead to narrowing of blood vessels and thus result in increased PA resistance and PA remodeling. In the present study, we showed that cold exposure increased PA SMC proliferation and medial thickness and decreased small PA lumen (Figs. 2&3). The improved PA remodeling by PDE1 inhibition is likely a cumulative effect of several results including: 1) Decreases in cold-induced macrophage infiltration and inflammation; 2) Reduction of cold-induced increases in NADPH oxidase activity and superoxide production; 3) Elimination of the cold-induced decrease in cGMP bioavailability; and 4) Reduction of PA pressure. While significant improvements in managing PA pressure and endothelial function have been made with recent therapeutics, treating PA remodeling has remained elusive. If PH is to be managed effectively, the PA remodeling must be addressed. To our knowledge, this is the first study demonstrating that 8-IBMX treatment reversed cold-induced PA remodeling.
An upregulation of PDEs has been suggested to play a role in human PH.48 However, the pulmonary PDE5 expression was not altered significantly by cold exposure or by IBMX (Fig. S4). PDE-5 is primarily expressed in the arterial walls of the lungs and penis and acts to decompose the second messenger cGMP. The first PDE-5 inhibitor, sildenafil citrate, was originally used for the treatment of erectile dysfunction but was approved by FDA in 2005 for the treatment of PH.36 It is marketed under the name of Revatio.36 Although PDE-1 is expressed in a relatively low level, a significant increase in expression is observed in proliferating pulmonary vasculature.48
It has been shown that lung inflammation precedes the development of PH in rodent models.49 Furthermore, the recruitment of monocytes/macrophages was demonstrated to be vital to vascular remodeling in hypoxia-induced PH.50 In this study, we found that cold exposure increased macrophage infiltration around PAs, indicating increased inflammation. Interestingly, treatment with 8-IBMX attenuated the infiltration of macrophages around the small PAs from the lungs of cold-exposed animals (Fig. 6). While examining macrophages in the lung tissue, we also found that the alveolar diameter was decreased but cell infiltrates were increased in cold-exposed rats, which can be abolished by IBMX (Fig. S8). Infiltration of macrophages in the lungs of cold exposed animals may promote a switch to a pro-inflammatory state that allows for leukocytes, cytokines and superoxide to initiate the proliferation and remodeling process of the pulmonary vasculature (Fig. S9). Significant remodeling of the lung vasculature, especially the small PAs, can lead to an increase in pulmonary vascular resistance and eventually the development of PH. Unexpectedly, the levels of IL-1β and IL-6 in the lungs were not altered significantly by cold exposure (Fig. S7). Additional studies are needed to explore the underlying mechanism of cold-induced inflammatory responses and its involvement in CIPH.
It is noted that both IBMX and apocynin decreased CIPH to a similar extent but only IBMX attenuated RV weight (Fig. 1), suggesting that cold-induced RVH may be independent of elevation of PAP. The anti-hypertrophic effect of IBMX is likely due to inhibition of PDE-1 in the heart. The NADPH oxidase may not contribute significantly to cold-induced RVH.
Perspectives
Chronic exposure to cold upregulated PDE-1C in small PAs and caused PH and PA remodeling. Inhibition of PDE-1 with 8-IBMX decreased cold-induced macrophage infiltration and superoxide production in PAs and attenuated CIPH and PA remodeling. Therefore, inhibition of PDE-1 represents an effective therapeutic strategy for CIPH and related cardiovascular dysfunctions. The findings have major implications for people who live in cold regions or in winter. Further studies are warranted to determine the mechanistic link of PDE-1C and NADPH oxidase in the context of inflammation in the pathogenesis of CIPH.
Supplementary Material
Novelty and Significance.
1. What is new?
It is new and interesting that continuous exposure to cold temperatures increased PDE1 C protein expression and macrophage infiltration in pulmonary arteries (PAs) which leads to pulmonary arterial hypertension and remodeling.
This study demonstrates, for the first time, that inhibition of PDE1 attenuated cold-induced increases in macrophage inflammation and NADPH oxidase activity in PAs which reveals a previously unidentified role of PDE1 in the regulation of inflammation and superoxide production.
2. What is relevant?
It is significant that inhibition of PDE1 attenuated cold-induced pulmonary arterial hypertension (CIPH) and PA remodeling which provides a new therapeutic approach for the management of CIPH and related cardiovascular disorders.
This study addresses an important role of PDE1 in CIPH which is a public health concern but remains poorly explored.
3. Summary
Inhibition of PDE-1 attenuated CIPH and reversed cold-induced PA remodeling by suppressing macrophage infiltration and superoxide production, which suggest that upregulation of PDE-1C expression may be involved in the pathogenesis of CIPH.
Acknowledgments
Sources of Funding
This work was supported by NIH R01 HL116863, HL105302 and HL102074 and AHA Predoctoral Fellowship 11PRE7830040. This publication was made possible by NIH Grant Number P20 RR 024215 from the COBRE Program of the National Center for Research Resources.
Footnotes
Disclosures
Nothing to disclose. The authors confirm that there are no conflicts of interest.
References
- 1.Chen GF, Sun Z. Effects of chronic cold exposure on the endothelin system. J Appl Physiol. 2006;100:1719–1726. doi: 10.1152/japplphysiol.01407.2005. [DOI] [PubMed] [Google Scholar]
- 2.Crosswhite P, Sun Z. Ribonucleic acid interference knockdown of interleukin 6 attenuates cold-induced hypertension. Hypertension. 2010;55:1484–1491. doi: 10.1161/HYPERTENSIONAHA.109.146902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giesbrecht GG. The respiratory system in a cold environment. Aviat Space Environ Med. 1995;66:890–902. [PubMed] [Google Scholar]
- 4.Wang X, Skelley L, Cade R, Sun Z. AAV delivery of mineralocorticoid receptor shRNA prevents progression of cold-induced hypertension and attenuates renal damage. Gene Ther. 2006;13:1097–1103. doi: 10.1038/sj.gt.3302768. [DOI] [PubMed] [Google Scholar]
- 5.Sun ZJ, Zhang ZE. Historic perspectives and recent advances in major animal models of hypertension. Acta Pharmacol Sin. 2005;26:295–301. doi: 10.1111/j.1745-7254.2005.00054.x. [DOI] [PubMed] [Google Scholar]
- 6.Mourtzoukou EG, Falagas ME. Exposure to cold and respiratory tract infections. Int J Tuberc Lung Dis. 2007;11:938–943. [PubMed] [Google Scholar]
- 7.Carder M, McNamee R, Beverland I, Elton R, Cohen GR, Boyd J, Agius RM. The lagged effect of cold temperature and wind chill on cardiorespiratory mortality in Scotland. Occup Environ Med. 2005;62:702–710. doi: 10.1136/oem.2004.016394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braga AL, Zanobetti A, Schwartz J. The effect of weather on respiratory and cardiovascular deaths in 12 U.S. cities. Environ Health Perspect. 2002;110:859–863. doi: 10.1289/ehp.02110859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keatinge WR, Donaldson GC, Bucher K, Jendritzky G, Cordioli E, Martinelli M, Katsouyanni K, Kunst AE, McDonald C, Nayha S, Vuori I. Winter mortality in relation to climate. Int J Circumpolar Health. 2000;59:154–159. [PubMed] [Google Scholar]
- 10.Keatinge WR. Winter mortality and its causes. Int J Circumpolar Health. 2002;61:292–299. doi: 10.3402/ijch.v61i4.17477. [DOI] [PubMed] [Google Scholar]
- 11.Barr WG, Fahey PJ. Reduction of pulmonary capillary blood volume following cold exposure in patients with Raynaud’s phenomenon. Chest. 1988;94:1195–1199. doi: 10.1378/chest.94.6.1195. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe K, Koizumi T, Ruan Z, Kubo K, Sakai A, Shibamoto T. Reduced pulmonary vascular reactivity after cold exposure to acute hypoxia: a role of nitric oxide (NO) High Alt Med Biol. 2007;8:43–49. doi: 10.1089/ham.2006.1015. [DOI] [PubMed] [Google Scholar]
- 13.Greenlees KJ, Tucker A, Robertshaw D, Vader CR. Pulmonary vascular responsiveness in cold-exposed calves. Can J Physiol Pharmacol. 1985;63:131–135. doi: 10.1139/y85-023. [DOI] [PubMed] [Google Scholar]
- 14.Kalmar ID, Cools A, Buyse J, Roose P, Janssens GP. Dietary N,N-dimethylglycine supplementation improves nutrient digestibility and attenuates pulmonary hypertension syndrome in broilers. J Anim Physiol Anim Nutr (Berl) 2010;94:e339–347. doi: 10.1111/j.1439-0396.2010.01018.x. [DOI] [PubMed] [Google Scholar]
- 15.Li JC, Pan JQ, Huang GQ, Tan X, Sun WD, Liu YJ, Wang XL. Expression of PDGF-beta receptor in broilers with pulmonary hypertension induced by cold temperature and its association with pulmonary vascular remodeling. Res Vet Sci. 2010;88:116–121. doi: 10.1016/j.rvsc.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 16.Rybalkin SD, Rybalkina I, Beavo JA, Bornfeldt KE. Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res. 2002;90:151–157. doi: 10.1161/hh0202.104108. [DOI] [PubMed] [Google Scholar]
- 17.Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 18.Schermuly RT, Pullamsetti SS, Kwapiszewska G, Dumitrascu R, Tian X, Weissmann N, Ghofrani HA, Kaulen C, Dunkern T, Schudt C, Voswinckel R, Zhou J, Samidurai A, Klepetko W, Paddenberg R, Kummer W, Seeger W, Grimminger F. Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: target for reverse-remodeling therapy. Circulation. 2007;115:2331–2339. doi: 10.1161/CIRCULATIONAHA.106.676809. [DOI] [PubMed] [Google Scholar]
- 19.Pechanova O, Jendekova L, Vrankova S. Effect of chronic apocynin treatment on nitric oxide and reactive oxygen species production in borderline and spontaneous hypertension. Pharmacol Rep. 2009;61:116–122. doi: 10.1016/s1734-1140(09)70013-1. [DOI] [PubMed] [Google Scholar]
- 20.Beswick RA, Dorrance AM, Leite R, Webb RC. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension. 2001;38:1107–1111. doi: 10.1161/hy1101.093423. [DOI] [PubMed] [Google Scholar]
- 21.Murata T, Kinoshita K, Hori M, Kuwahara M, Tsubone H, Karaki H, Ozaki H. Statin protects endothelial nitric oxide synthase activity in hypoxia-induced pulmonary hypertension. Arterioscler Thromb Va c Biol. 2005;25:2335–2342. doi: 10.1161/01.ATV.0000186184.33537.48. [DOI] [PubMed] [Google Scholar]
- 22.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tahara N, Kai H, Niiyama H, Mori T, Sugi Y, Takayama N, Yasukawa H, Numaguchi Y, Matsui H, Okumura K, Imaizumi T. Repeated gene transfer of naked prostacyclin synthase plasmid into skeletal muscles attenuates monocrotaline-induced pulmonary hypertension and prolongs survival in rats. Hum Gene Ther. 2004;15:1270–1278. doi: 10.1089/hum.2004.15.1270. [DOI] [PubMed] [Google Scholar]
- 24.van Albada ME, Schoemaker RG, Kemna MS, Cromme-Dijkhuis AH, van Veghel R, Berger RM. The role of increased pulmonary blood flow in pulmonary arterial hypertension. Eur Respir J. 2005;26:487–493. doi: 10.1183/09031936.05.00015405. [DOI] [PubMed] [Google Scholar]
- 25.Zhang TT, Cui B, Dai DZ, Tang XY. Pharmacological efficacy of CPU 86017 on hypoxic pulmonary hypertension in rats: mediated by direct inhibition of calcium channels and antioxidant action, but indirect effects on the ET-1 pathway. J Cardiovasc Pharmacol. 2005;46:727–734. doi: 10.1097/01.fjc.0000184470.58047.79. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Sun Z. RNAi silencing of brain klotho potentiates cold-induced elevation of blood pressure via the endothelin pathway. Physiol Genomics. 2010;41:120–126. doi: 10.1152/physiolgenomics.00192.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54:810–817. doi: 10.1161/HYPERTENSIONAHA.109.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dammanahalli JK, Wang X, Sun Z. Genetic interleukin-10 deficiency causes vascular remodeling via the upregulation of Nox1. J Hypertens. 2011;29:2116–2125. doi: 10.1097/HJH.0b013e32834b22a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bello Roufai M, Li H, Sun Z. Heart-specific inhibition of protooncogene c-myc attenuates cold-induced cardiac hypertrophy. Gene Ther. 2007;14:1406–1416. doi: 10.1038/sj.gt.3302995. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Sun Z, Cade R. Prolonged attenuation of cold-induced hypertension by adenoviral delivery of renin antisense. Kidney Int. 2005;68:680–687. doi: 10.1111/j.1523-1755.2005.00446.x. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Cade R, Sun Z. Human eNOS gene delivery attenuates cold-induced elevation of blood pressure in rats. Am J Physiol Heart Circ Physiol. 2005;289:H1161–1168. doi: 10.1152/ajpheart.01306.2004. [DOI] [PubMed] [Google Scholar]
- 32.Sun Z, Zhang Z, Cade R. Renal responses to chronic cold exposure. Can J Physiol Pharmacol. 2003;81:22–27. doi: 10.1139/y03-002. [DOI] [PubMed] [Google Scholar]
- 33.Sun Z, Cade R, Zhang Z, Alouidor J, Van H. Angiotensinogen gene knockout delays and attenuates cold-induced hypertension. Hypertension. 2003;41:322–327. doi: 10.1161/01.hyp.0000050964.96018.fa. [DOI] [PubMed] [Google Scholar]
- 34.Sun Z, Cade R, Morales C. Role of central angiotensin II receptors in cold-induced hypertension. Am J Hypertens. 2002;15:85–92. doi: 10.1016/s0895-7061(01)02230-0. [DOI] [PubMed] [Google Scholar]
- 35.Sun Z, Cade R, Katovich MJ, Fregly MJ. Body fluid distribution in rats with cold-induced hypertension. Physiol Behav. 1999;65:879–884. doi: 10.1016/s0031-9384(98)00250-9. [DOI] [PubMed] [Google Scholar]
- 36.Crosswhite P, Sun Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. J Hypertens. 2010;28:201–212. doi: 10.1097/HJH.0b013e328332bcdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arif SA, Poon H. Tadalafil: a long-acting phosphodiesterase-5 inhibitor for the treatment of pulmonary arterial hypertension. Clin Ther. 2011;33:993–1004. doi: 10.1016/j.clinthera.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Murray F, Maclean MR, Insel PA. Role of phosphodiesterases in adult-onset pulmonary arterial hypertension. Handb Exp Pharmacol. 2011;204:279–305. doi: 10.1007/978-3-642-17969-3_12. [DOI] [PubMed] [Google Scholar]
- 39.Oldenburger A, Roscioni SS, Jansen E, Menzen MH, Halayko AJ, Timens W, Meurs H, Maarsingh H, Schmidt M. Anti-Inflammatory Role of the cAMP Effectors Epac and PKA: Implications in Chronic Obstructive Pulmonary Disease. PLoS One. 2012;7:e31574. doi: 10.1371/journal.pone.0031574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schafer PH, Parton A, Gandhi AK, Capone L, Adams M, Wu L, Bartlett JB, Loveland MA, Gilhar A, Cheung YF, Baillie GS, Houslay MD, Man HW, Muller GW, Stirling DI. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–855. doi: 10.1111/j.1476-5381.2009.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diebold I, Djordjevic T, Petry A, Hatzelmann A, Tenor H, Hess J, Gorlach A. Phosphodiesterase 2 mediates redox-sensitive endothelial cell proliferation and angiogenesis by thrombin via Rac1 and NADPH oxidase 2. Circ Res. 2009;104:1169–1177. doi: 10.1161/CIRCRESAHA.109.196592. [DOI] [PubMed] [Google Scholar]
- 42.Sedeek M, Hebert RL, Kennedy CR, Burns KD, Touyz RM. Molecular mechanisms of hypertension: role of Nox family NADPH oxidases. Current Opin Nephrol Hypertens. 2009;18:122–127. doi: 10.1097/MNH.0b013e32832923c3. [DOI] [PubMed] [Google Scholar]
- 43.Sedeek MH, Llinas MT, Drummond H, Fortepiani L, Abram SR, Alexander BT, Reckelhoff JF, Granger JP. Role of reactive oxygen species in endothelin-induced hypertension. Hypertension. 2003;42:806–810. doi: 10.1161/01.HYP.0000084372.91932.BA. [DOI] [PubMed] [Google Scholar]
- 44.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 45.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 46.Agarwal R, Gomberg-Maitland M. Current therapeutics and practical management strategies for pulmonary arterial hypertension. Am Heart J. 2011;162:201–213. doi: 10.1016/j.ahj.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 47.Durmowicz AG, Stenmark KR. Mechanisms of structural remodeling in chronic pulmonary hypertension. Pediatr Rev. 1999;20:e91–e102. [PubMed] [Google Scholar]
- 48.Wilkins MR, Wharton J, Grimminger F, Ghofrani HA. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur Respir J. 2008;32:198–209. doi: 10.1183/09031936.00124007. [DOI] [PubMed] [Google Scholar]
- 49.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. 2011;123:1986–1995. doi: 10.1161/CIRCULATIONAHA.110.978627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.