

Abstract
To avoid genome instability, DNA repair nucleases must precisely target the correct damaged substrate before they are licensed to incise. Damage identification is a challenge for all DNA damage response proteins, but especially for nucleases that cut the DNA and necessarily create a cleaved DNA repair intermediate, likely more toxic than the initial damage. How do these enzymes achieve exquisite specificity without specific sequence recognition or, in some cases, without a non-canonical DNA nucleotide? Combined structural, biochemical, and biological analyses of repair nucleases are revealing their molecular tools for damage verification and safeguarding against inadvertent incision. Surprisingly, these enzymes also often act on RNA, which deserves more attention. Here, we review protein-DNA structures for nucleases involved in replication, base excision repair, mismatch repair, double strand break repair (DSBR), and telomere maintenance: apurinic/apyrimidinic endonuclease 1 (APE1), Endonuclease IV (Nfo), tyrosyl DNA phosphodiesterase (TDP2), UV Damage endonuclease (UVDE), very short patch repair endonuclease (Vsr), Endonuclease V (Nfi), Flap endonuclease 1 (FEN1), exonuclease 1 (Exo1), RNase T and Meiotic recombination 11 (Mre11). DNA and RNA structure-sensing nucleases are essential to life with roles in DNA replication, repair, and transcription. Increasingly these enzymes are employed as advanced tools for synthetic biology and as targets for cancer prognosis and interventions. Currently their structural biology is most fully illuminated for DNA repair, which is also essential to life. How DNA repair enzymes maintain genome fidelity is one of the DNA double helix secrets missed by Watson-Crick, that is only now being illuminated though structural biology and mutational analyses. Structures reveal motifs for repair nucleases and mechanisms whereby these enzymes follow the old carpenter adage: measure twice, cut once. Furthermore, to measure twice these nucleases act as molecular level transformers that typically reshape the DNA and sometimes themselves to achieve extraordinary specificity and efficiency.
Keywords: Exonuclease, Enzyme–DNA complex, Genome maintenance, Crystallography, Structure-specific nuclease, DNase, RNase, Nucleases, DNA, RNA, Endonucleases, Metals, Magnesium, Zinc, Manganese, DNA repair, Base excision repair, Mismatch repair, Double strand break repair, Nucleotide Incision Repair, Telomere, APE1, Nfo, EndoIV, TDP2, UVDE, Vsr, Nfi, EndoV, Mre11, FEN1, Exo1
1. Introduction
The discovery of the double helix transformed biology and opened the doors for molecular biology and the field of genetics. However, DNA repair was not considered. Francis Crick wrote in 1974, “We totally missed the possible role of enzymes in repair although due to Claud Rupert’s early very elegant work on photoreactivation, I later came to realize that DNA is so precious that probably many distinct repair mechanisms would exist.” [1]. DNA nucleases are essential players in DNA repair. For DNA, nucleases are a necessary evil. DNA damage needs to be trimmed off or removed, and this removal needs to be done both efficiently and accurately. Small errors in the substrate recognition or location of the incision can be deleterious to the cell and cause genomic instability. This review examines how nucleases ensure not only they have bound the correct substrate, but also that they do not bind and cut the wrong substrate. Here, we focus on DNA repair phosphoesterases that leave a 5′ phosphate and a 3′ hydroxyl suitable for polymerase extension and ligation. In particular, we analyze those whose structures have been determined with substrate and/or product DNA: apurinic/apyrimidinic endonuclease 1 (APE1), Endonuclease IV (Nfo), tyrosyl DNA phosphodiesterase (TDP2), UV Damage endonuclease (UVDE), very short patch repair endonuclease (Vsr), Endonuclease V (Nfi), Flap endonuclease 1 (FEN1), exonuclease 1 (Exo1), RNase T and Meiotic recombination 11 (Mre11). There is now a sufficient number of enzymes meeting this criteria that useful insights emerge, and these insights have general importance. For the eukaryotic enzymes, we also include an examination of motifs that can be used to identify mechanistically similar nucleases. These enzymes are central to cell biology: they act in replication, base excision repair (BER), mismatch repair (MMR) double strand break repair (DSBR), and telomere maintenance. Furthermore they are increasingly found to act on RNA as well as DNA, and these activities may well be important as well.
Some of these nucleases are endonucleases that make a single cut within the DNA and some are exonucleases that processively cut from a DNA end, but some fall into both categories. The “restriction nuclease” discovered by Stuart Linn and Werner Arber [2, 3] provided breakthroughs in genetics because they provided enzymatic tools needed to “cut and paste” DNA molecules. Their specificity was based upon methylation or specific sequences, and thus they are site-specific nucleases. For damaged DNA, the discoveries of nucleotide excision repair and transcription-coupled repair pioneered by Phil Hanawalt and others sparked a dramatic evolution in our understanding of DNA and molecular biology by revealing the intriguing systems of DNA repair essential to life plus sets of nucleases needed for the cut-and-patch repair that are specific to DNA structure rather than sequence [4-8]. Thus, DNA damage repair nucleases have a different challenge than restriction nucleases with targeted sequences for incision. Although some recognize a modified base or phosphate backbone, others must recognize their substrates containing canonical nucleotides in an aberrant structure. The structure-specific nucleases in this review therefore provide a paradox of both extreme specificity and the lack of any sequence dependence with broad implications. For biotechnology, they can provide powerful tools to probe and modify DNA structure, as seen for FEN1 [9, 10]. Biochemically, if misregulated, they would destroy the integrity of genomic information. Biologically, they are necessary to preserving genome integrity and life itself.
How are these nucleases regulated? What is the basis for their exquisite specificity? Nuclease cutting is a committed step and thus tightly regulated. Structural biology provides key knowledge to address specificity questions and to contribute to a more complete and detailed understanding of their activities and biological functions. Particularly for these nucleases, structures furthermore provide detailed and rigorous information with which all other data should be reconciled and that often allows the integration of biochemical and genetic results. Examining the existing structures provides a basis to design mutants and inhibitors for separation of functions as seen for Mre11 [11, 12]. Yet, structures provide key knowledge not only to design mutations and inhibitors but also to interpret the impact of disease-causing mutations, as seen for XPD helicase [13], and the likelihood that polymorphisms may impact risks. As we come to understand DNA repair networks as more accurate than classical linear pathway concepts, we wish to control pathway choice and network crosstalk and interactions for biology and medicine. A detailed structural and mechanistic understanding of structure-specific nucleases, which is the focus of this review, is key to this goal. Increasingly we are finding that repair nuclease function requires changes in protein and DNA architecture that impacts binding, activity, and partner recruitment. Furthermore, flexible components (intrinsically unstructured regions) reshape or fold themselves in the presence of target DNA, as shown for FEN1 and its family members such as XPG [14-17]. In essence these nucleases behave like molecular level transformers that can rebuild themselves by sometimes altering their protein conformations and typically sculpting the DNA to control both their specificity and efficiency functions. This knowledge suggests we need to re-think our understanding and the classic lock and key concept of how interactions, specificity, and activity are regulated with implications for inhibitor design.
2. Cell biology of DNA repair nucleases and increasing role as therapeutic targets
DNA repair nucleases permeate every DNA repair and processing pathway and are essential to the cell (Fig. 1). Damaged DNA can form spontaneously from endogenous metabolic sources, exogenously by DNA damaging agents (chemicals, radiation), or are intermediates from other repair or DNA processing enzymes. Damaged DNA must be incised from the DNA strand to prevent errors in coding or regulatory regions, to prevent mutations during replication, and to maintain genomic stability. Additionally, damaged DNA can often arrest RNA polymerase, setting the cell on a path towards apoptosis [18]. Thus, nucleases play a crucial role in removing the damaged DNA.
Fig. 1.

The ten nucleases discussed in this review, highlighting their biological roles and differences in substrate specificity and features. Under diseases, not applicable (N/A) and unknown (?) are denoted. Relative incision position(s) on substrate is shown in red.
Many DNA repair nucleases are essential for the cell. Homozygous null mutations are often cellular or embryonic lethal. Single site mutations are associated with increased risk for cancer, ageing, and neurological diseases. Once cancer has occurred, these enzymes may become upregulated and provide cancer cells resistance to DNA damaging treatments such as chemotherapy and radiation treatments. Thus, many of these nucleases have become targets for developing inhibitors that can lead to sensitizing cancer cells to DNA damaging treatments. Three DNA repair nucleases, APE1, TDP2, and FEN1, which have been particularly well-studied, will be reviewed as typifying examples.
Apurinic/apyrimidinic endonuclease 1 (APE1) acts on abasic sites that form spontaneously or are repair intermediates from BER glycosylases [19-24]. It is estimated that as many as 10,000 abasic sites are formed in one cell, each day in humans [25]. APE1 null mice are embroynic lethal [22, 26, 27], and heterozygous mice showed increased tumor susceptibility [28]. In humans, some APE1 mutations have been associated with amyotrophic lateral sclerosis (ALS) [29, 30] and endometrial cancers [31]. On the flip side, APE1 activity gives cells increased survival after radiation, oxidative stress, and chemotherapy, making it a drug target; down-regulation of APE1 can lead to increased sensitivity of tumor cells to various cancer treatments, reviewed in [19, 23, 32-34].
The duality of nucleases both preventing cancer, but also sustaining cancer once it has started is also true for FEN1. FEN1 incises 5′ flaps during lagging strand replication. Theoretically, there are as many as 50 million Okazaki fragments during each S1 phase in each cell. FEN1 must be efficient and absolutely precise in its incision. FEN1 can modulate CAG repeat expansion, and nuclease-deficient FEN1 blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability [35, 36]. FEN1 has been linked to genetic diseases, such as myotonic dystrophy, Huntington’s disease, several ataxias, and fragile X syndrome [37-40]. FEN1 mutants are also associated with liver, lung, gastrointestinal, melanoma and esophageal cancer [41-43]. A nuclease-defective FEN1 mouse model exhibited spontaneous mutations, chronic inflammation, autoimmune issues, and cancer [44, 45]. FEN1 is normally silenced in differentiated cells [46, 47], but FEN1 overexpression is associated with breast, uterine, kidney, ovarian, and colon cancers as well as pancreatic adenocarcinoma, glioblastoma and astrocystoma tumors [48-52]. As reducing FEN1 by RNAi kills tumor cells in vitro, FEN1 is a priority pharmaceutical target.
Related structurally to APE1, TDP2 is an interesting nuclease among DNA repair nucleases. It incises peptides covalently bound to the 5′ phosphate of DNA through a tyrosyl linkage. These DNA adducts are formed as failed topoisomerase type IA and IIA intermediates where the topoisomerase is covalently linked through an active site tyrosine and was unable to release itself during the religation step. These failed intermediates can form spontaneously or as a consequence of cancer therapeutic inhibitors of Top2, a type IIA topoisomerase, recently reviewed in [53]. Top2 inhibitors are often used in chemotherapy against a wide variety of cancers, including lung cancer, non-Hodgkin’s lymphomas, leukemias, Kaposi’s sarcoma, neuroblastoma, and soft-tissue sarcomas, thus making failed topoisomerase DNA intermediates a medically significant topic. TDP2 acts to resolve these failed topoisomerase intermediates, which is good normally but will antagonize Top2-targeted chemotherapy treatments. Inhibitors of TDP2 are likely to sensitize cells to Top2 inhibitors. Although this alone merits identification of TDP2 inhibitors, TDP2 recently was identified as THE human host protein that is required for picornaviral development [54]. For many years, researchers were searching for aVPg unlinkase that cleaves a protein-RNA covalent linkage. Picornaviruses use a protein, VPg, as a primer for RNA synthesis, and VPg must be cut from the RNA. TDP2 was found to be that human host aVPg unlinkase, with the linkage to the RNA formed by a tyrosyl group. This finding also showed that TDP2 could act on both RNA and DNA with 5′ tyrosyl adducts. These results are medically important, as picornaviruses cause a multitude of human illnesses, ranging from the common cold and hand and foot disease to polio and encephalitis. A TDP2 inhibitor could thus be a potential major medical breakthrough.
Developing inhibitors of DNA repair nucleases as tools and leads for therapeutic intervention is in process, in particular for treatment of cancers where nucleases may be upregulated. It is thus important to consider their detailed mechanisms, since it may lead to the development of specific inhibitors. The major problem with many nuclease inhibitors that are DNA analogs is their lack of specificity. Understanding essential steps in the damage recognition mechanism at the structural level will provide targets for development of specific, structure-based inhibitors.
3. Overview of DNA Repair Nuclease Structures and Mechanisms
Phosphodiesters are highly resistant to hydrolysis, with t1/2 of 30 million years at 25°C [55]. Nucleases, such as FEN1, can accelerate that reaction 1017 fold [56]. Nucleases achieve this acceleration through a multistep acid-base reaction: 1) orientation of the attacking water for a linear attack on the phosphodiester bond; 2) activation of the attacking water through acid deprotonation; 3) stabilization of the electronegative pentacovalent intermediate, and 4) base protonation of the leaving ribose oxygen. The nicked DNA has a 5′ phosphate and a 3′ hydroxyl on one strand. The 3′ hydroxyl provides a ready-substrate for DNA polymerases, which is particularly important for DNA repair.
The nucleases enzymatic reaction is achieved either through side chain coordination or through metal coordination. These details of the mechanism are not common to all repair nucleases. Even repair enzymes that perform functionally identical reactions can have distinct mechanisms [57, 58]. All nucleases that directly cleave the phosphodiester backbone, excluding bifunctional glycosylases, are metal-dependent [59,60]. There can be one to three metals. APE1 (apurinic endonuclease) uses Asp210 and Asn212 to orient and activate the attacking water and requires one Mg2+ ion, to stabilize the pentacovalent intermediate [57, 58]. VSR uses two Mg2+ ions [61]. Nfo, which acts on the same substrate as APE1, requires three Zn2+ ions that are involved in both activation of the attacking water and stabilization of the pentacovalent intermediate [62-64]; it is the only known endonuclease that uses Zn2+ for its catalytic metal.
However, what makes DNA repair nucleases distinct from other nucleases? When we examine DNA repair enzymes, we find that these phosphodiesterases share common elements, despite their differences. 1) Recognition is through a DNA contortion or sculpting that provides specificity for the DNA damage. 2) Although there is limited change in the endonuclease core structure, flexible regions of the protein often compact and trap the DNA. 3) Damaged DNA sculpting places the phosphodiester bond to be incised in the active site pocket to license the nucleases to incise. Normal DNA typically can not be similarly contorted and their phosphodiester backbones cannot reach the active site. 4) The nuclease active sites are generally open, with multiple waters available for protonation of the leaving ribose oxygen. 5) Repair endonucleases, particularly eukaryotic ones with regulatory regions, are product-inhibited, often mediated by clamping of the DNA through side chains or helices that form around the DNA. 6) Product release is often the rate limiting and a regulated step.
In our survey of structures of DNA repair nucleases in complex with substrate and/or product DNA, we have observed protein structures that sculpt and distort the DNA (or RNA) that in turn, license incision to occur and promote the fidelity needed for DNA repair and for life to continue. We find that the rule of measure twice, cut once works not only for carpenters but also for nucleases and is enabled in part by enzymes that can sometimes reshape themselves and typically sculpt their target DNA.
4. Structurally-related family members: APE1, Nfo, TDP2 and UVDE
Abasic residues occur spontaneously or as intermediates in BER. Acting in the BER pathway, Endonuclease IV (EndoIV or Nfo) and apurinic/apyrimidinic endonuclease 1 (APE1) are two endonucleases that recognize abasic sites in the context of intact duplex DNA. Abasic sites can occur spontaneously or as repair intermediates from mono-functional or bi-functional glycosylases. Bi-functional glycosylases leave a 3′ deoxyribose, and APE1 and Nfo act as 3′ deoxyribonucleases to clean the 3′ end of these byproducts. Intriguingly, APE1 and Nfo have been implicated in incising the phosphodiesterase backbone 5′ to certain types of base damage [65-70], indicating that these nucleases can also bind and recognize bases in their active site. Recently, APE1 has been shown to have activity on RNA as well [71, 72].
APE1 and Nfo have the same AP endonuclease activity, but their tertiary structure and their divalent metal requirements are completely different. APE1 is part of the exonuclease III (Xth) family, has a two-layered β-sheet core flanked by helices, and has a single Mg2+ ion in its active site [73]. Nfo has a TIM β barrel core, surrounded by helices with not one but three metal ions – either three Zn2+ or two Zn2+ and one Mn2+ [62, 63]. Additionally, although APE1 and Nfo flip out the abasic site, Nfo flips out the base on the opposite strand as well. Despite these differences, the steric manner in which they contort the abasic site in their recognition mechanism is surprisingly similar [57]. The DNA is bent severely by both proteins, approximately 35° for APE1 and 90° for Nfo. Intriguingly, the two abasic nucleotides and surrounding nucleotides overlay well, despite the fact that Nfo double flips the abasic site and the base on the opposite strand (Fig. 2A). One could think that this similarity is explained by the fact that the nucleotide-flipped abasic site adopts its lowest energy geometry. However, key features including the position of the scissile phosphate are not identical to an abasic site flipped out by uracil N-glycosylase (Fig. 2A) [74]. The abasic site in duplex DNA has two major characteristics that the AP endonucleases select for: no Watson-Crick pairing and no base. Both endonucleases flip out the sugar moiety into a small pocket that would not be able to accommodate a base (Fig. 2B). In both proteins, there is little change in protein structure between unbound and bound, a feature shared by many proteins that contort or sculpt DNA as part of their recognition mechanism. We postulate that the similarity of the AP site configuration is key to the hydrolysis step, as catalytic metals and residues overlay between the two proteins [57]. Phosphodiester incision is mediated by one or more metals in APE1 and three metals in Nfo. In both enzymes, residues most important to the catalytic activity or metals are positioned to activate the water for an in-line attack [57, 62-64, 75]. A significant feature of human APE1 is the trapping of the product by Arg177, which lies over the AP site (Fig. 2B). Mutation to alanine results in a 25% higher kcat/Km compared to WT due to an increase in the off rate.
Fig. 2.

Nfo and APE1 share a common DNA sculpting to select for AP sites. A) Overlay of the AP site in substrate structures highlights the common DNA structure, despite the differences in protein structure and catalytic metals. The closeup highlights that this is not a default flipped out AP nucleotide conformation, as the abasic site in UNG shows small but significant shifts in phosphate and sugar positions (cyan arrows). Below each enzyme, a DNA schematic shows nucleotide flip and incision position (*). B) The shallow binding pocket (protein shown as a surface) selects for abasic sites. For clarity, Arg177 was shown in cyan. C) After overlay of the protein in APE1 and TDP2 DNA-bound structures, the scissile phosphate (orange arrow) matches, but the nucleotide is oriented inversely.
Given the small binding pocket, the mechanism for how Nfo and APE1 can accommodate a damaged base in nucleotide incision repair is still unclear, although some suggestion is given by a structure with a complete nucleotide at the active site of APE1 [76]. Another clue is given by another enzyme that structurally resembles APE1, TDP2 that incise 5′ tyrosyl groups from ssDNA and RNA. Both enzymes share specific amino acid motifs bearing some of the catalytic residues (Table I). Moreover the scissile phosphate in TDP2 is located identically to that in APE1, but the sugar and base are on the opposite side of the phosphodiester (Fig. 2C) [77, 78]. In other words, TDP2 does not flip out the nucleotide, unlike APE1. TDP2 also has a narrower groove that forces the terminal nucleotide into a twist that would disrupt Watson-Crick basepairing. Another feature of TDP2 regulation that may be a important for other nucleases is an apparent DNA mimicry by its flexible terminus, and here as in uracil-DNA glycosylase the DNA mimicry would be of the sculpted DNA [79, 80. Given that TDP2 requires ssDNA for its activity, it is possible that melting of the DNA around the damage would allow APE1 and Nfo to work on substrates with DNA base damage. That the NIR substrate, α anomeric adenosine, disrupts the dsDNA, and that flanking sequences exacerbate this feature are better Nfo substrates [Aramini, 2004 #534, 81] is consistent with this melting hypothesis.
Table I.
Nuclease groups have specific amino acid motifs that can be searched against protein sequence databases to find members of each group in different organisms. In brackets [ ], different amino acid possibilities at one position. Any amino acid is abbreviated by x and the number in parentheses defines the number of times it is repeated. l represents any aliphatic amino acid [IVL]. Catalytic residues are highlighted in red.
| Protein | Motifs | |||
|---|---|---|---|---|
| APE-1, TDP-2 | [ST]WNlDGL | PDll[FC]LQE | GDxNlxxxE | SDHxxl |
| Nfi (Endonuclease V) | Ylxxx[LF]x[FM]RE | aG[HQN]GxxH | lAKxxL | |
| 5′ nucleases (FEN-1, EXO-1) | [DE]x(6)Kx(6)R | EA[DE]Ax(17)DxD! | ||
| Mre11 | DxH[lC]G | GDl[FY][HDEN] | G[NSDE]H[DE] | G[HD]xH |
Similar in topology to Nfo, UVDE recognizes and incises DNA 5′ to UV damage, such as pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PP) [82]. UVDE can also incise much smaller damage such as abasic sites, nicks, and gaps [83], which is surprising given the size of CPDs and 6-4PPs. UVDE has a similar Tim barrel structure as Nfo [84], and both enzymes flip out nucleotides from both strands [85] (Fig. 3). However, unlike Nfo, UVDE flips out two nucleotides from each strand in a quadruple flip, compared to only one nucleotide from each strand in the Nfo complex structures (double flip). Both the 6-4PP and the opposing adenines were rotated out of the helix and base stacking with the rest of the DNA was disrupted. A wedge, formed by Gln103 and Tyr104, stacks against the basepairs 5′ to the damage. Unlike all other repair nucleases in this review, a strikingly large space is formed in the DNA that remains unoccupied by the protein. However, given that the flipped out 6-4PP is exposed in this gap, it is intriguing to consider that this gap allows the next enzyme in the pathway spatial access to the damage, promoting handoff
Fig. 3.

UVDE, which incises the phosphodiester backbone 5′ to the 6-4 PP, quadruple nucleotide flips the covalently linked base damage and the two nucleotides opposite. A) Side and B) top views of UVDE, inserts a wedge (Gln103 and Tyr104), as it double flips two nucleotides on both strands. C) Schematic of DNA and relative position of wedge residues. It is notable how accessible is the bound 6-4 PP.
5. Very Short Patch Repair (Vsr) endonuclease
Microbial Vsr endonuclease recognizes and incises on the 5′ side of thymidine in TG mismatches in a very short patch repair process. Unlike the other enzymes discussed in this review, Vsr recognizes the damage within a specific sequence, CC*(A/T)GG that is the target for DNA-cytosine methyltransferase (Dcm). Spontaneous deamination of the second C (*) methylated by Dcm leads to the TG mismatch to be repaired. The overall structure of Vsr resembles type II restriction enzymes [86]. The product-bound structure showed specific hydrogen bonding to mediate recognition of the TG mismatch within the canonical methylation sequence [87]. Surprisingly, the DNA is distorted on the 3′ side of the thymidine in the TG mismatch, while still maintaining all basepairing (Fig. 4). From the major groove side, Phe67, Trp68, and Trp86 stack between the basepairs, with Met14 and Ile17 from the N-terminal helix coming in from the minor groove side. The basepair to basepair distance increased by a remarkable 6 Å compared to canonical B-DNA. We postulate that this intercalation helps mediate recognition of the TG mismatch. In the structure of DNA containing a TG mismatch, the 3′ side to the mismatched T shows disrupted stacking with the next basepair in the helix [88]. It is also notable that MutS, a major mismatch recognition protein, also inserts on the same side in its binding to TG mismatches [89]. Incision is catalyzed by two Mg2+ ions. Vsr also binds a Zn2+ ion, but it serves only a structural role. Analogous to APE1, the N-terminal helix of Vsr appears to trap product DNA. The DNA-free structure is of a N-terminally truncated construct, so we do not know the exact position of the N-terminus in the DNA-free enzyme. However, it is clear from the structure that the N-terminus would need to clamp down on the DNA in response to DNA binding. It is the only major change between DNA-free and DNA-bound Vsr structures. Product inhibition is suggested from the retention of a three nucleotide product in the crystallization that took several weeks [87]. Kinetic studies of an N-terminally deleted mutant show a decrease in kcat [90]. Unfortunately, the rate of product release has not been measured. Interestingly, the N-terminal helix appears not to clamp down until the product has been formed, as incubation with Mg2+ but not Ca2+ protected it from limited proteolysis [91]. This suggests that protection of the product DNA was engineered into the Vsr N-terminus.
Fig. 4.

Vsr breaks the helical stacking of its substrate as part of its recognition mechanism. A) DNA schematic shows how Vsr endonuclease incises 5′ to a TG mismatch. B) The structure unexpectedly reveals that Vsr inserts three residues (Phe67, Trp68, and Trp8) to break the stacking on the 3′ side of T in a T/G mismatch. Incision occurs on the 5′ side of the T. Product DNA is shown as surface, colored by chain. The N-terminal helix (N) clamps the product DNA down onto the main catalytic core.
6. Endonuclease V (Nfi)
Nfi recognizes a surprisingly wide range of base damage in BER, including hypoxanthine, xanthine, oxanine, uracil, base mismatches, abasic sites, insertion/deletion loops, hairpins, and other aberrant DNA structures [92-98]. Nfi also incises RNA [99, 100]. This wide range of DNA damage recognized by Nfi means that it not only recognizes both damaged purines and pyrimidines, already distinct in size, but it also must recognize aberrant DNA structures with undamaged bases. To add to the complexity, Nfi cleaves the second phosphodiester 3′ to the damage. This polarity is unlike other damage recognition nucleases that cleave the DNA 5′ to the damage. As a result of the 3′ incision, Nfi in itself cannot remove the damage with the standard polymerase strand displacement. Instead, Nfi incision likely acts to signal a repair pathway for 3′ strands.
As damage recognition is physically distinct from incision on the DNA strand, the steric location for both functions are also physically separated in Nfi enzyme. Crystal structures of Thermotoga maritima (Tma) Nfi with DNA-containing hypoxanthine show one pocket for damage recognition and a second pocket for incision (Fig. 5). Like the other nucleases above, there is little change between the DNA-free and DNA-bound enzymes, except for one small set of shifts discussed below. The hypoxanthine is rotated 90° into a deep pocket, which is different from the full 180° flip like many base damage recognition proteins. Incision occurs in a second pocket containing a single Mg2+ ion. A helical and hydrophobic wedge, formed by Tyr80, Ile81, and Pro82, is inserted into the DNA opposite to the hypoxanthine and breaks the duplex DNA basepair-to-basepair stacking, analogous to Vsr endonuclease. The residues of this wedge are part of one of the motifs specific to Nfi (Table I). It is not clear how Nfi is licenced to incise. Does a signal need to pass from the recognition pocket to the incision pocket? Comparison with the Tma DNA-free structure reveals small shifts leading to one of the residues coordinating the catalytic metal, although whether these shifts convey the signal needs to be experimentally tested.
Fig. 5.
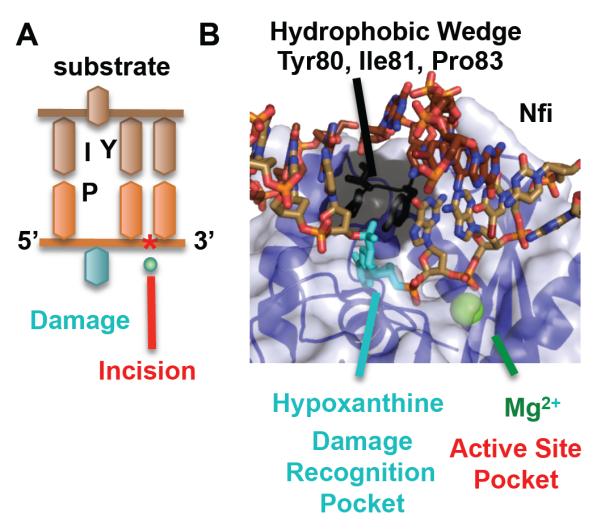
Two pockets, one for recognition and one for incision, underlie Nfi mechanism. A) DNA schematic shows the relative position of the damage and the phosphodiester incision 3′ to the damage. B) The substrate structure (2w36.pdb) overlaid with the Mg2+ from the product structure (2w35.pdb) shows two pockets, one for damage recognition (shown for hypoxanthine) and one for incision with a single Mg2+. A helical wedge separates the two pockets.
Pertinent to the broad substrate specificity of Nfi, a second, more recent crystal structure of Nfi bound to a one nucleotide loop showed the wedge pushing out two thymidines on the non-complementary strand and one adenine was rotated into the recognition site [101]. This is notable for two reasons. 1) Nfi broke the Watson-Crick basepairing of the A-T basepair next to the one nucleotide loop, forming a two nucleotide loop. 2) A normal adenine base was rotated into the recognition pocket. Hypoxanthine is deaminated adenine, and it was thought that rotation was stabilized by specific recognition of the hypoxanthine. However, it is clear that adenine can also be stably rotated into the damage recognition pocket. It is unexpected why Nfi does not put the one nucleotide loop into the “damage” recognition pocket but instead disrupted the loop further.
We postulate the following. 1) The helical wedge needs to be inserted into the DNA. It is located away from the scissile strand and inserts more on the side of the complementary strand, thus sterically forcing the loop to be positioned away from the incision site. 2) One nucleotide must be rotated out, in order to position the phosphodiester close to the active site. Nfi thus disrupts the A-T base pair, in order to rotate a nucleotide to go into the “damage” recognition pocket. Thus, this structure suggests a new structural basis for how Nfi handles its wide range of substrates, from single base damages that are still paired in the duplex to aberrant DNA structures with undamaged bases. It recognizes DNA where the bases can be rotated out of the helix. In the cases of loops, the stacking is disrupted, facilitating invasion in the DNA duplex by the helical wedge. In the case of specific base damage, it could be from disrupted hydrogen-bonding or disrupted basepair-to-basepair stacking. For example, although there are two hydrogen-bonds formed in the basepairing between inosine and thymidine, the base pair would be shifted in the DNA helix and the basepair-to-basepair stacking would be similarly disrupted. Thus, like Vsr, Nfi is recognizing disrupted DNA helical structures.
7. 5′ nuclease family: Flap Endonuclease 1 (FEN1) and Exonuclease 1 (Exo1)
FEN1 and Exo1 are part of the structure-specific 5′ nuclease superfamily that recognize ss-dsDNA junctions and cleave one nucleotide into the dsDNA. The members of this superfamily share two active site motifs in their sequences (Table I). FEN1 incises 5′ flaps formed during Okazaki fragment maturation and during long patch BER. Exo1 is a processive 5′-3′ exonuclease that acts in MMR, DSBR, and telomere maintenance. The challenge of these 5′ nucleases in recognition of their structure-specific damage is that they must distinguish their substrate from ssDNA and RNA and from dsDNA, or in other words, a bent hay stick in a hay stack problem. FEN1 incises 5′ flaps formed during Okazaki fragment maturation and during long patch BER. The crystal structures for FEN1 have been solved in a DNA-free state (in complex with PCNA), with a one nucleotide flap substrate and a product structure [16, 17] (Fig. 6). For Exo1, substrate and product bound structures have been determined [102].
Fig. 6.

EN1 and Exo1 use multiple mechanisms for substrate specificity. A) FEN1 and Exo1 use a hydrophobic wedge to block the path of the duplex DNA. B) FEN1 and Exo1 bind primarily to the complementary strand in two sections, the active site and the K+/H2/3TH. These regions are separated by 24 Å or a helical turn apart. C) The active sites, with seven invariantly conserved carboxylates and two catalytic metals, are protected by a helical gateway and a helical cap. The helical gateway could select for ssDNA or ssRNA. The terminal nucleotide stacks against an aromatic residue and is contacted by two conserved basic residues. D) A double base unpairing in the mechanisms is suggested by the observation of +1 and −1 paired nucleotides in the substrate and a −1 unpaired nucleotide in the product. The scissile phosphate is distant (5 Å) from the catalytic metals when the nucleotides are paired in the substrate structure, but is within catalytic distance (2.2 Å) in the product structure.
The key to ss/dsDNA junction recognition by the 5′ nucleases is recognition of duplex DNA with one end abruptly interrupted by a hydrophobic, helical wedge, analogous to Nfi. Only duplex DNA with a nick of ssDNA can bend around the hydrophobic wedge. This recognition is manifested in several ways. 1) Recognition of the helical features of duplex DNA. In both FEN1 and Exo1, the main DNA binding interfaces are not continuous, but is separated into two regions a DNA helical turn apart, one at the active site and one at a K+ binding site formed at a helix-two or three turn-helix (H2/3TH) motif. The distance between DNA binding sites bridges the major groove, enforcing specificity for 5′ flaps and not duplex RNA or 3′ flaps. This divided binding mode is distinct from most endonucleases which bind generally in a single connected interface that extends from the active site. 2) Binding to the complementary strand enforces specificity for dsDNA and selects against ssDNA or RNA. We found unexpectedly that over half of the protein-interaction interface was to the complementary strand. Excluding the terminal nucleotide, only one sixth was to the scissile strand. 3) Bent dsDNA binding path selects for 5′ flap. A helical hydrophobic wedge sits in the direct path of the DNA, forcing the remainder of the DNA (ss for Exo1, dsDNA for FEN1) to bend 90°. Only DNA that has a break in the ds DNA, such as found in a 5′ flap substrate, can do so. One notable advantage for FEN1 of focusing protein-DNA interactions to the complementary strand is that RNA in the incision strand would not disrupt interaction, allowing FEN1 its non-specificity toward RNA vs DNA.
In the case of FEN1, a disorder-to-order transition enforces substrate recognition for a one nucleotide 3′ flaps. To obtain a ligatable product, FEN1 forms and binds a one nucleotide 3′ flap. As FEN1 cuts one nucleotide into the dsDNA substrate, this flap can basepair back down to form a ligatable product [14, 56]. In the DNA-free structures [103], the protein tertiary region between the 3′ flap and the active site residues are disordered but this same region is ordered in the DNA-bound structure. This difference implies a disorder-to-order transition initiated by binding to a 3′ flap. Indeed, mutation of a lysine (R47A) that is in position to help order this region is as deleterious to the incision activity as a residue in the active site (Y40A) that helps position the substrate [16]. For Exo1, XPG, and GEN, there is no 3′ flap. It is possible that regulatory proteins or domains may induce a similar disorder-to-order transition to activate the nuclease catalytic residues [102, 104]. Also as seen for XPG and TFIIH, partner machines may couple DNA sculpting to licensing for nuclease cleavage [104, 105].
Another key and still cryptic mechanism in 5′ nuclease incision is the unpairing or fraying of the dsDNA around the scissile phosphate. For both Exo1 and FEN1, the substrate is paired around the scissile phosphate (+1 and −1 refer to bases on 5′ and 3′ side of scissile phosphate) with the scissile phosphate 5 Å distant from the two catalytic metals, while the product is unpaired at the −1 nucleotide with the scissile phosphate within catalytic distance ( 2.2 Å) of the metals. Presumably, not one but two nucleotides must have unpaired to move the scissile phosphate within catalytic distance. Why do these 5′ nucleases require unpairing for incision? Restriction enzymes routinely cleave the exterior-facing phosphodiesterases while in duplex DNA. So why are 5′ nucleases different?
We postulate that this double nucleotide unpairing only occurs after the 3′ flap has induced ordering of the active site and ordering of residues that promote unpairing [106]. A two helix gateway over the active site is wide enough only for ssDNA. A cap over the gateway, specific to FEN1 and Exo1 5′ nucleases, selects for ssDNA with free 5′ termini. Only if the helical gateway can form around the ssDNA are the two nucleotides unpaired. Alternatively, the ss nature of the substrate could be assessed by forcing the ssDNA portion of the substrate to bend severely back away from the active site. Duplex DNA would not be able to bend, disrupting the helical gateway and the unpairing residues on one of those two helices. Assessing the ss nature of their 5′ flaps by either of these two methods, the 5′ nucleases use multiple recognition steps before allowing incision to occur. Unlike other repair nucleases, FEN1 does show a significant shifting of conformation upon binding DNA, including both the gateway and cap, but the main core of the protein also shows significant shifts. A β hairpin sticking up from the catalytic core shifts back upon DNA binding. Also, 5′ nucleases show a notable crisscrossing of secondary structure elements across the long axis of the proteins. We hypothesize that this topology allow 5′ nucleases to be regulated by other proteins that can remotely “pull strings” to impact DNA binding and incision.
Like a Rube Goldberg machine, 5′ nucleases use multiple checks before incision, ensuring that these nucleases not incise other RNA or DNA substrates, but only their structure-specific substrate – a “measure twice” type of procedure. This recognition is accomplished purely through DNA structure, with no sequence specificity. Binding to broken duplex DNA (two binding sites to the complementary strand and space a helical turn apart, hydrophobic wedge in duplex DNA path) and 3′ flap-induced order-to-disorder (for FEN) leads to a double base unpairing of 5′ flap DNA and then, and only then incision. We postulate that this dsDNA binding – unpairing – ssDNA incision is a hallmark of the 5′ nuclease superfamily and may also be a mechanism for many structure-specific nucleases that recognize ss-dsDNA junctions, like XPF, which also cleaves in the dsDNA region, albeit 6-8 nucleotides from the junction [107].
8. RNaseT
A structure-specific nuclease that shares a similar steric wedge mechanism as the 5′ nucleases is RNase T. Although this DNA repair nuclease review has formally excluded RNases in its focus, Rnase T, despite its name, has been shown to have significant 3′-5′ exonuclease activity on ssDNA or 3′ overhangs with a 300-fold lower Km compared to RNA [108]. It has an unusual specificity, with its activity 100-fold reduced by a single C residue in the 3′ end [109]. Crystal structures with ss and 3′ overhang dsDNA revealed an active site deep in a narrow channel that would select against dsDNA (Fig. 7). Additionally, dsDNA is stopped from entering the active site by a phenylalanine in a helical wedge, ensuring that only the overhang as ssDNA or RNA enters the narrow channel to the active site. The enzyme does not change shape upon binding DNA, suggesting it acts as a rigid surface for DNA interaction. There are two metals, Mg2+, in the active site.
Fig. 7.

Rnase T dimer selects for ssDNA or ssRNA or 3′ overhangs by blocking the complementary strand (brown) with a steric helical wedge and Phe29. A) Global and B) closeup of the duplex structure (3va3.pdb) with Mg2+ atoms from an overlaid RnaseT/ssDNA structure (3v9w.pdb) to show relative position of active site metals and scissile phosphate (*). C) DNA schematic showing how the 3′ overhang is then selected through intercalation by Phe146, Phe124, and Phe77 and positioning of the phosphodiester bond for catalysis. For terminal cytosines with reduced incision activity, Glu73 will flip and pull on the cytosine, disrupting the positioning of the phosphodiester backbone and one of the two Mg2+ ions.
Comparison to the DNA-free structure showed little DNA-induced conformational changes of the catalytic core. Selectivity against incision past C is manifested by a fascinating chain reaction of flipped and shifted side chains. This chain reaction is initiated by a 180° rotation of Glu73 that hydrogen bonds to the 3′ terminal C. The C is pulled up and away from the active site, and only one metal is observed in the active site. This loss of a catalytic metal and shifting of the phosphodiester from an ideal catalytic position would explain the loss of activity on substrates with a terminal C.
9. Mre11
The dsDNA exonuclease and ssDNA endonuclease Mre11 recognizes, and processes double strand break (DSB) DNA ends. It also initiates the activation of the DNA-damage response through ATM. Furthermore, Mre11 degrades stalled replication forks [11, 110] where it also removes covalently bound topoisomerase [111]. Finally, Mre11 promotes micro-homology end joining at breaks during transcription [112]. With all these cellular functions, it is not surprising that Mre11 mutations are associated with some cancers [113-116]. Moreover, hypomorphic mutations in Mre11 that cause Ataxia telangiectasia-like disorder (ATLD) [117] may block DSB repair [118]. None of these mutations are in the specific amino acid motifs bearing the catalytic residues (Table I). Instead, the mutations disturb protein-protein interactions [118, 119]. In vitro, Mre11 is known to cleave various types of substrates: blunt DNA ends, branched DNA ends, 3′ recessed DNA ends, hairpins and ssDNA [120, 121]. In vivo, Mre11 licenses with an endonuclease cut, and then resects broken DNA with its exonuclease activity to create a suitable ssDNA template for homology search in homologous recombination [12]. Thus both its ssDNA endonuclease activity and its 3′ to 5′ exonuclease activity have key functions at forks and breaks.
Although the DNA does not enter the active site, the structure of Mre11 from Pyrococcus furiosus bound to a branched DNA does suggest a model involving an exonuclease mechanism in three steps [11]. First, both subunits of the Mre11 dimer interact with the minor groove and the phosphate backbone of the dsDNA (Fig. 8A). This part of the DNA is far from the active site and retains the ideal B-DNA geometry. Second, Mre11 undergoes conformational changes that promote the ATP-independent melting of the dsDNA. The angle between the subunits of the dimer changes, a feature called the capping domain rotates and a helical wedge interacts with the DNA minor groove. The precise mechanism by which those structural events are melting the DNA is unknown, but Mre11 must first sculpt the scissile strand towards the capping domain in order to redirect it towards the active site (Fig. 8B). This is the most important step for the recognition of the dsDNA end, since Mre11 likely does not recognize a precise structure, but uses the malleability of the DNA end, and the possibility to melt it without ATP, for substrate identification. The third and last step of the mechanism requires the sculpting of the scissile strand towards the active site containing two catalytic Mn2+ (Fig. 8C). This is done by phosphate rotation using a histidine (His52 in P. furiosus). A mutation of this residue specifically reduces the exonuclease activity of Mre11, but not its endonuclease activity. After those three steps, Mre11 cuts exonucleatically, from the 3′ end towards the 5′ end, in a processive manner.
Fig. 8.

The Mre11 exonuclease mechanism involves sculpting the DNA end to direct the scissile strand to the active site. A) Both subunits of the Mre11 dimer interact with a branched DNA end (3dsd.pdb). In this structure, the substrate was not cleaved because the catalytic His85 is mutated into a serine. B) Closeup of the active site (shown by purple Mn2+ ions) and its surroundings. The complementary strand (brown) is unmodified while the scissile strand (orange) is sculpted by the helical wedge (blue). The postulated phosphate rearrangement residues involved in the rotation of the 3′ strand into the active site are shown in red (includes His52). On the other side of the active site, a pocket (green) is hypothesized to bind ssDNA for the endonuclease activity of Mre11. C) Schematic of the phosphate rearrangement mechanism (red) in order to sculpt the scissile strand into the active site. The scissile bond is shown with a red star (*).
The endonuclease mechanism is even less understood, due to the lack of informative structures bound to ssDNA. But computational modeling identified a ssDNA-binding pocket on the other side of the active site from the dsDNA-binding interface [11]. Compared to dsDNA, ssDNA is more flexible and there is no need for substrate rotation. This explains why the mutation of His52 does not impact the endonuclease activity. Those differences in mechanisms between endo- and exonuclease activities were exploited for the development of inhibitors specific for each nuclease activities [12]. With these inhibitors as tools, the roles of the Mre11 nuclease activities in directing DNA double stands breaks into the NHEJ and HR pathways can be assessed in vivo for the first time.
Another structural determinant of Mre11 nuclease activities is the conformational state of its partner, the ATPase Rad50 [122]. In the presence of ADP, the heterotetramer composed of two Mre11 and two Rad50, is in an “open” complex, promoting the exonuclease activity. On the other hand, ATP closes the complex, occludes the active site of Mre11 and stimulates the endonuclease activity. In the closed Mre11-Rad50 structures, only ssDNA would be able to access the active site [123-125]. Employing SAXS, which can accurately define conformational states [126], experiments show that Rad50 conversion between closed and flexible states controls the Mre11 nuclease and end tethering versus end processing [127]. In future research, it will be exciting to see how the assembled Mre11-Rad50 machinery acts in concert to control replication forks and DSBs.
10. Integrating nuclease structures, chemistry, and biology
As a group, what do these structures inform us about the chemistry of the reaction and about the biology?
a) Nucleases sculpt their substrate DNA to physically validate their substrates
Nucleases cannot see. Although this statement is obvious, we and other FEN1 researchers focused on the ssDNA, the most obvious visual difference between 5′ flaps and dsDNA. Instead, the structure revealed that FEN1 recognized that 5′ flap DNA had a break in the dsDNA and could bend >90° over a single phosphodiester, a physical and tangible characteristic of 5′ flaps. As nucleases are blind, the nucleases in this review test their substrates through an obstacle course of wedges and narrow grooves. These wedges often are hydrophobic and almost always include an aromatic residue that stacks against the DNA, often against the strand complementary to the strand incision. In Nfi, RnaseT, FEN1, Exo1 and Mre11, these wedges are helical, suggesting that the secondary structure acts to gird the wedge. The role of basepair-to-basepair stacking in damaged DNA plays a recognition role in Vsr and Nfi. Narrow grooves or gateways that enclose the active site are found by structure-specific nucleases such as TDP2, FEN1, Exo1, RNase T, and Mre11 with specificity for substrates with ssDNA overhangs. These obstacle courses ensure that only true substrates that can contort or fit their phosphodiesters, can reach the active site. Only when the substrates finish the obstacle course are the DNA repair nucleases licensed to cut. This obstacle course testing of the substrates is how nucleases measure twice before cutting.
b) DNA repair nucleases need, as a rule, rigid DNA-binding platforms for that obstacle course to sculpt and select for their substrate DNA
We observe the lack of DNA-induced conformational changes in most of the nucleases in this review: APE1, Nfo, TDP2, Nfi, UVDE, Vsr, and RnaseT. 5′ nucleases (shown for FEN1) and Mre11 are exceptions to this rule, which likely points to facets in their regulation. The rigidness of the DNA binding interface with little DNA-induced conformational change is not uncommon for DNA repair enzymes; the conformation of MutS, a major mismatch recognition protein, was more dependent on small nucleotides than on its much larger DNA substrate [126].
c) The manner in which DNA repair nucleases bind DNA reflect not only how they bind their target DNA but also how they exclude particular DNA substrates
Many repair nucleases bind primarily to one strand, along the phosphate backbone. However, other nucleases interact with both strands, such as UVDE, FEN1, Exo1, and RnaseT. Indeed, the 5′ nucleases go so far as to have more interaction with the strand complementary to the scissile strand and use that interaction to place the scissile strand close to the active site, a feature that we believe provides specificity for dsDNA for nucleases that cleave ssDNA overhangs. If the primary interaction of 5′ nucleases were to the ssDNA, the cells risks having its RNA inadvertently incised.
d) Product release from repair nucleases is a key biological mechanism
As they have induced a nick in the DNA, it would be terrible for the cell if they simply released it to wreak havoc on other DNA repair processes, like strand invasion, double strand breaks, etc. Structurally, DNA repair enzymes do this through clamping down on the product DNA, through helices or long side chains. It is probably no coincidence that the crystal structures of product complexes are generally higher resolution than substrate complexes.
e) Product inhibition implies a needed handoff from one enzyme to the next enzyme in the repair pathway
The major DNA binding interface for most DNA repair nucleases is to one side of the DNA, enabling exchange with the next enzyme in the pathway. The interchange of the nuclease’s product to the next enzyme in the pathway has been referred to as passing the baton or molecular choreography [128, 129]. The structures of sequential pathway enzymes show a common bend in the DNA, with base stacking and/or base pairing disrupted in the product being handed over. Is stimulation of DNA binding of the subsequent enzyme through a direct protein-protein interaction or is it because the basepairing or base stacking of the dsDNA has already been destabilized?
Another major question in the field that remains unanswered is how the exchange occurs. Despite the valiant efforts of many laboratories, no one has been able to get a structural snapshot of an exchange. Is it truly an ordered passing of the baton or a spring-loaded product release mechanism? The latter may make sense since most proteins will be acting on the same strand close to the damage. The one exception may be FEN1 to the Ligase 1 (Lig1) DNA binding domain (DBD). Structural models suggest that they are able to bind to the opposite sides of the same DNA, FEN1 facing the major groove and Lig1 facing the minor groove [16, 17].
The broad question remains as to how these nuclease-DNA structures look in the context of chromosomes. These enzymes have DNA interactions between 6-14 nucleotides in general, approximately one helical turn of the DNA. For the most part, one can envision complexes where the nucleases are binding the DNA loosely lying on the histone core. How do these nucleases find their substrates? FEN1 is thought to work in a PCNA-scaffold complex with the replicative polymerase and ligases [44, 103, 130-138]. However, how are the other repair nucleases recruited to sites of damage or are they always scanning the DNA? Going forward, an important consideration is how these nucleases act on RNA substrates. Most of these enzymes have been found to be active on RNA, including APE1, TDP2, Nfi, FEN1, and RnaseT. Also, what is their role in RNA incision – as regulatory nucleases and/or RNA repair nucleases? Will their substrate specificity be identically mediated by RNA distortion?
Integrated analyses of current structures are providing connections between the structural chemistry and the biology. For example, in some cases the active site does not fully assume its activated conformation until the target substrate is properly bound. The active site conformations (inactive vs active) are controlling the chemistry to provide DNA substrate specificity, as seen for the FEN1 superfamily in the ordering of the helical arch and basic residues in the gateway above the active site. Inhibitors that provide chemical knock-downs, as seen for Mre11 exonuclease and endonuclease activities [12], are a powerful tool to understand distinct functions in a multifunctional nuclease in cells and examine enzymes for which knock-outs are cellular lethal, such as Mre11 and FEN1 [38, 139]. Chemical knock-downs are thus valuable tools that are potential leads for possible therapeutic interventions. Furthermore, although the enzymes tend to bind about one turn of DNA, the tight binding regions are often smaller and the active sites have pocket-like features that appear suitable to inhibitor design.
11. Synopsis and perspectives
This unified analysis of DNA repair nucleases reveals conserved themes in their mechanisms. The primary theme is the sculpting of the DNA with distortions, disruption of basepair stacking, flipped out nucleotides. These distortions are mediated near the active site through steric wedges that stick up in the path of the duplex DNA. Surprisingly, the structures of multiple nucleases with distinct mechanisms are teaching us that substrate specificity does not necessarily conform to the classic rigid lock-and-key paradigm. Instead these enzymes sculpt their nucleotide targets in ways that prevent normal DNA from binding appropriately in the active site. For some of these nucleases, DNA binding may induce a reshaping of the protein, although this is not as common as one would think. Nucleases thus achieve their exquisite specificities and efficiencies by forming protein-DNA complexes that are more like molecular versions of the science-fiction transformer robots that reshape DNA to achieve their activities than they are like the classical lock and key enzyme specificity hypothesis.
Acknowledgements
Work for this review and the authors’ efforts were funded by NIH RO1CA081967, RO1GM46312, and P01CA092584. J.L.V. is recipient of a fellowship from the Canadian Research Institutes of Health Research (CIHR).
Glossary
Abbreviations used in this Review
- (BER)
Base excision repair
- MMR
Mismatch repair
- DSBR
Double strand break repair
- Ss
single stranded
- Ds
double stranded
- APE1
apurinic/apyrimidinic endonuclease 1
- Nfo
Endonuclease IV
- TDP2
tyrosyl DNA phosphodiesterase 2
- UVDE
UV Damage endonuclease
- Vsr
very short patch repair endonuclease
- Nfi
Endonuclease V
- Mre11
Meiotic REcombination 11
- FEN1
Flap endonuclease 1
- Exo1
Exonuclease 1
- Dcm
DNA-cytosine methyltransferase
- 6-4PP
(6-4) photoproduct or (6,4) pyrimidine-pyrimidones
- CPD
cyclobutane pyrimidine dimer
- NIR
Nucleotide incision repair
- Lig1
Ligase 1
- DBD
DNA binding domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Crick F. The double helix: a personal view. Nature. 1974;248:766–769. doi: 10.1038/248766a0. [DOI] [PubMed] [Google Scholar]
- [2].Arber W, Linn S. DNA modification and restriction. Annual Review of Biochemistry. 1969;38:467–500. doi: 10.1146/annurev.bi.38.070169.002343. [DOI] [PubMed] [Google Scholar]
- [3].Linn S, Arber W. Host specificity of DNA produced by Escherichia coli, X. In vitro restriction of phage fd replicative form. Proceedings of the National Academy of Sciences of the United States of America. 1968;59:1300–1306. doi: 10.1073/pnas.59.4.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hanawalt PC. The Awakening of DNA Repair at Yale. Yale J Biol Med. 2013;86:517–523. [PMC free article] [PubMed] [Google Scholar]
- [5].Harrington JJ, Lieber MR. Functional domains within FEN-1 and RAD2 define a family of structure-specific endonucleases: implications for nucleotide excision repair. Genes Dev. 1994;8:1344–1355. doi: 10.1101/gad.8.11.1344. [DOI] [PubMed] [Google Scholar]
- [6].Harrington JJ, Lieber MR. DNA structural elements required for FEN-1 binding. J Biol Chem. 1995;270:4503–4508. doi: 10.1074/jbc.270.9.4503. [DOI] [PubMed] [Google Scholar]
- [7].Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- [8].Bennett RJ, Dunderdale HJ, West SC. Resolution of Holliday junctions by RuvC resolvase: cleavage specificity and DNA distortion. Cell. 1993;74:1021–1031. doi: 10.1016/0092-8674(93)90724-5. [DOI] [PubMed] [Google Scholar]
- [9].Mein CA, Barratt BJ, Dunn MG, Siegmund T, Smith AN, Esposito L, Nutland S, Stevens HE, Wilson AJ, Phillips MS, Jarvis N, Law S, de Arruda M, Todd JA. Evaluation of single nucleotide polymorphism typing with invader on PCR amplicons and its automation. Genome Res. 2000;10:330–343. doi: 10.1101/gr.10.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lyamichev VI, Kaiser MW, Lyamicheva NE, Vologodskii AV, Hall JG, Ma WP, Allawi HT, Neri BP. Experimental and theoretical analysis of the invasive signal amplification reaction. Biochemistry. 2000;39:9523–9532. doi: 10.1021/bi0007829. [DOI] [PubMed] [Google Scholar]
- [11].Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, Rossum-Fikkert S. van, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Molecular Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Finger LD, Atack JM, Tsutakawa S, Classen S, Tainer J, Grasby J, Shen B. The wonders of flap endonucleases: structure, function, mechanism and regulation. Subcell Biochem. 2012;62:301–326. doi: 10.1007/978-94-007-4572-8_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Grasby JA, Finger LD, Tsutakawa SE, Atack JM, Tainer JA. Unpairing and gating: sequence-independent substrate recognition by FEN superfamily nucleases. Trends Biochem Sci. 2012;37:74–84. doi: 10.1016/j.tibs.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, Cooper PK, Grasby JA, Tainer JA. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tsutakawa SE, Tainer JA. Double strand binding-single strand incision mechanism for human flap endonuclease: implications for the superfamily. Mechanisms of ageing and development. 2012;133:195–202. doi: 10.1016/j.mad.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Belotserkovskii BP, Mirkin SM, Hanawalt PC. DNA sequences that interfere with transcription: implications for genome function and stability. Chem Rev. 2013;113:8620–8637. doi: 10.1021/cr400078y. [DOI] [PubMed] [Google Scholar]
- [19].Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36:425–435. doi: 10.1016/j.ctrv.2009.12.006. [DOI] [PubMed] [Google Scholar]
- [20].Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA repair. 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Izumi T, Brown DB, Naidu CV, Bhakat KK, Macinnes MA, Saito H, Chen DJ, Mitra S. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci U S A. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li M, Wilson DM., 3rd Human Apurinic/Apyrimidinic Endonuclease 1. Antioxidants & redox signaling. 2013;20:678–707. doi: 10.1089/ars.2013.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA repair. 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- [25].Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- [26].Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, Meneses J, Pedersen RA, Chen DJ. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutation research. 1998;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- [28].Meira LB, Devaraj S, Kisby GE, Burns DK, Daniel RL, Hammer RE, Grundy S, Jialal I, Friedberg EC. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer research. 2001;61:5552–5557. [PubMed] [Google Scholar]
- [29].Hadi MZ, Coleman MA, Fidelis K, Mohrenweiser HW, Wilson DM., 3rd Functional characterization of Ape1 variants identified in the human population. Nucleic acids research. 2000;28:3871–3879. doi: 10.1093/nar/28.20.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kisby GE, Milne J, Sweatt C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport. 1997;8:1337–1340. doi: 10.1097/00001756-199704140-00004. [DOI] [PubMed] [Google Scholar]
- [31].Pieretti M, Khattar NH, Smith SA. Common polymorphisms and somatic mutations in human base excision repair genes in ovarian and endometrial cancers. Mutation research. 2001;432:53–59. doi: 10.1016/s1383-5726(00)00002-9. [DOI] [PubMed] [Google Scholar]
- [32].Wilson DM, 3rd, Simeonov A. Small molecule inhibitors of DNA repair nuclease activities of APE1. Cellular and molecular life sciences : CMLS. 2010;67:3621–3631. doi: 10.1007/s00018-010-0488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- [34].Tell G, Fantini D, Quadrifoglio F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cellular and molecular life sciences : CMLS. 2010;67:3589–3608. doi: 10.1007/s00018-010-0486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu Y, Prasad R, Beard WA, Hou EW, Horton JK, McMurray CT, Wilson SH. Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion. J Biol Chem. 2009;284:28352–28366. doi: 10.1074/jbc.M109.050286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Spiro C, McMurray CT. Nuclease-deficient FEN-1 blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability. Mol Cell Biol. 2003;23:6063–6074. doi: 10.1128/MCB.23.17.6063-6074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279:853–856. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- [38].Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, Heyer J, Kane MF, Fan K, Russell R, Brown AM, Kneitz B, Edelmann W, Kolodner RD, Lipkin M, Kucherlapati R. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc Natl Acad Sci U S A. 2002;99:9924–9929. doi: 10.1073/pnas.152321699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Henneke G, Friedrich-Heineken E, Hubscher U. Flap endonuclease 1: a novel tumour suppresser protein. Trends Biochem Sci. 2003;28:384–390. doi: 10.1016/S0968-0004(03)00138-5. [DOI] [PubMed] [Google Scholar]
- [40].Stoimenov I, Helleday T. PCNA on the crossroad of cancer. Biochem Soc Trans. 2009;37:605–613. doi: 10.1042/BST0370605. [DOI] [PubMed] [Google Scholar]
- [41].Liu L, Zhou C, Zhou L, Peng L, Li D, Zhang X, Zhou M, Kuang P, Yuan Q, Song X, Yang M. Functional FEN1 genetic variants contribute to risk of hepatocellular carcinoma, esophageal cancer, gastric cancer and colorectal cancer. Carcinogenesis. 2012;33:119–123. doi: 10.1093/carcin/bgr250. [DOI] [PubMed] [Google Scholar]
- [42].Yang M, Guo H, Wu C, He Y, Yu D, Zhou L, Wang F, Xu J, Tan W, Wang G, Shen B, Yuan J, Wu T, Lin D. Functional FEN1 polymorphisms are associated with DNA damage levels and lung cancer risk. Hum Mutat. 2009;30:1320–1328. doi: 10.1002/humu.21060. [DOI] [PubMed] [Google Scholar]
- [43].Kim CJ, Shin JW, Jung SW, Park BR, Cha HJ, Park NH. Mutational analysis of FEN1 Gene in hepatocellular carcinomas. Apmis. 2013 doi: 10.1111/apm.12202. [DOI] [PubMed] [Google Scholar]
- [44].Zheng L, Dai H, Hegde ML, Zhou M, Guo Z, Wu X, Wu J, Su L, Zhong X, Mitra S, Huang Q, Kernstine KH, Pfeifer GP, Shen B. Fen1 mutations that specifically disrupt its interaction with PCNA cause aneuploidy-associated cancer. Cell Research. 2011;21:1052–1067. doi: 10.1038/cr.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, Tsark W, Huang Q, Kernstine K, Zhang X, Lin D, Shen B. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med. 2007;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- [46].Kim IS, Lee MY, Lee IH, Shin SL, Lee SY. Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim Biophys Acta. 2000;1496:333–340. doi: 10.1016/s0167-4889(00)00029-x. [DOI] [PubMed] [Google Scholar]
- [47].Warbrick E, Coates PJ, Hall PA. Fen1 expression: a novel marker for cell proliferation. J Pathol. 1998;186:319–324. doi: 10.1002/(SICI)1096-9896(1998110)186:3<319::AID-PATH184>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- [48].Nikolova T, Christmann M, Kaina B. FEN1 is overexpressed in testis, lung and brain tumors. Anticancer Res. 2009;29:2453–2459. [PubMed] [Google Scholar]
- [49].Sato M, Girard L, Sekine I, Sunaga N, Ramirez RD, Kamibayashi C, Minna JD. Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer. Oncogene. 2003;22:7243–7246. doi: 10.1038/sj.onc.1206977. [DOI] [PubMed] [Google Scholar]
- [50].Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, Kernstine KH, Lin D, Shen B. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol Cancer Res. 2008;6:1710–1717. doi: 10.1158/1541-7786.MCR-08-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Iacobuzio-Donahue CA, Ashfaq R, Maitra A, Adsay NV, Shen-Ong GL, Berg K, Hollingsworth MA, Cameron JL, Yeo CJ, Kern SE, Goggins M, Hruban RH. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63:8614–8622. [PubMed] [Google Scholar]
- [52].Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, Song KS, Rho SM, Yoo HS, Kim YS, Kim JG, Kim NS. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin Cancer Res. 2005;11:473–482. [PubMed] [Google Scholar]
- [53].Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Virgen-Slane R, Rozovics JM, Fitzgerald KD, Ngo T, Chou W, van der Heden van Noort GJ, Filippov DV, Gershon PD, Semler BL. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc Natl Acad Sci U S A. 2012;109:14634–14639. doi: 10.1073/pnas.1208096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schroeder GK, Lad C, Wyman P, Williams NH, Wolfenden R. The time required for water attack at the phosphorus atom of simple phosphodiesters and of DNA. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4052–4055. doi: 10.1073/pnas.0510879103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Finger LD, Blanchard MS, Theimer CA, Sengerova B, Singh P, Chavez V, Liu F, Grasby JA, Shen B. The 3′-flap pocket of human flap endonuclease 1 is critical for substrate binding and catalysis. J Biol Chem. 2009;284:22184–22194. doi: 10.1074/jbc.M109.015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tsutakawa SE, Shin DS, Mol CD, Izumi T, Arvai AS, Mantha AK, Szczesny B, Ivanov IN, Hosfield DJ, Maiti B, Pique ME, Frankel KA, Hitomi K, Cunningham RP, Mitra S, Tainer JA. Conserved Structural Chemistry for Incision Activity in Structurally Non-homologous Apurinic/Apyrimidinic Endonuclease APE1 and Endonuclease IV DNA Repair Enzymes. Journal of Biological Chemistry. 2013;288:8445–8455. doi: 10.1074/jbc.M112.422774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mol CD, Hosfield DJ, Tainer JA. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat Res. 2000;460:211–229. doi: 10.1016/s0921-8777(00)00028-8. [DOI] [PubMed] [Google Scholar]
- [59].Yang W. Nucleases: diversity of structure, function and mechanism. Quarterly reviews of biophysics. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dupureur CM. Roles of metal ions in nucleases. Curr Opin Chem Biol. 2008;12:250–255. doi: 10.1016/j.cbpa.2008.01.012. [DOI] [PubMed] [Google Scholar]
- [61].Tsutakawa SE, Morikawa K. New recognition mode for a TG mismatch: the atomic structure of a very short patch repair endonuclease-DNA complex. Cold Spring Harb Symp Quant Biol. 2000;65:233–239. doi: 10.1101/sqb.2000.65.233. [DOI] [PubMed] [Google Scholar]
- [62].Garcin ED, Hosfield DJ, Desai SA, Haas BJ, Bjoras M, Cunningham RP, Tainer JA. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nature structural & molecular biology. 2008;15:515–522. doi: 10.1038/nsmb.1414. [DOI] [PubMed] [Google Scholar]
- [63].Hosfield DJ, Guan Y, Haas BJ, Cunningham RP, Tainer JA. Structure of the DNA repair enzyme endonuclease IV and its DNA complex: double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell. 1999;98:397–408. doi: 10.1016/s0092-8674(00)81968-6. [DOI] [PubMed] [Google Scholar]
- [64].Ivanov I, Tainer JA, McCammon JA. Unraveling the three-metal-ion catalytic mechanism of the DNA repair enzyme endonuclease IV. Proc Natl Acad Sci U S A. 2007;104:1465–1470. doi: 10.1073/pnas.0603468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chou KM, Cheng YC. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature. 2002;415:655–659. doi: 10.1038/415655a. [DOI] [PubMed] [Google Scholar]
- [66].Ishchenko AA, Sanz G, Privezentzev CV, Maksimenko AV, Saparbaev M. Characterisation of new substrate specificities of Escherichia coli and Saccharomyces cerevisiae AP endonucleases. Nucleic acids research. 2003;31:6344–6353. doi: 10.1093/nar/gkg812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ishchenko AA, Yang X, Ramotar D, Saparbaev M. The 3′->5′ exonuclease of Apn1 provides an alternative pathway to repair 7,8-dihydro-8-oxodeoxyguanosine in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:6380–6390. doi: 10.1128/MCB.25.15.6380-6390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature. 2002;415:183–187. doi: 10.1038/415183a. [DOI] [PubMed] [Google Scholar]
- [69].Kerins SM, Collins R, McCarthy TV. Characterization of an endonuclease IV 3′-5′ exonuclease activity. The Journal of biological chemistry. 2003;278:3048–3054. doi: 10.1074/jbc.M210750200. [DOI] [PubMed] [Google Scholar]
- [70].Demple B, Johnson A, Fung D. Exonuclease III and endonuclease IV remove 3′ blocks from DNA synthesis primers in H2O2-damaged Escherichia coli. Proc Natl Acad Sci U S A. 1986;83:7731–7735. doi: 10.1073/pnas.83.20.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A, Radicella JP, Kelley MR, D’Ambrosio C, Scaloni A, Quadrifoglio F, Tell G. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Molecular and cellular biology. 2009;29:1834–1854. doi: 10.1128/MCB.01337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Berquist BR, McNeill DR, Wilson DM., 3rd Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. Journal of molecular biology. 2008;379:17–27. doi: 10.1016/j.jmb.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mol CD, Kuo CF, Thayer MM, Cunningham RP, Tainer JA. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature. 1995;374:381–386. doi: 10.1038/374381a0. [DOI] [PubMed] [Google Scholar]
- [74].Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- [76].Mazouzi A, Vigouroux A, Aikeshev B, Brooks PJ, Saparbaev MK, Morera S, Ishchenko AA. Insight into mechanisms of 3′-5′ exonuclease activity and removal of bulky 8,5′-cyclopurine adducts by apurinic/apyrimidinic endonucleases. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E3071–3080. doi: 10.1073/pnas.1305281110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Schellenberg MJ, Appel CD, Adhikari S, Robertson PD, Ramsden DA, Williams RS. Mechanism of repair of 5′-topoisomerase II-DNA adducts by mammalian tyrosyl-DNA phosphodiesterase 2. Nature structural & molecular biology. 2012;19:1363–1371. doi: 10.1038/nsmb.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Shi K, Kurahashi K, Gao R, Tsutakawa SE, Tainer JA, Pommier Y, Aihara H. Structural basis for recognition of 5′-phosphotyrosine adducts by Tdp2. Nature structural & molecular biology. 2012;19:1372–1377. doi: 10.1038/nsmb.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Putnam CD, Shroyer MJ, Lundquist AJ, Mol CD, Arvai AS, Mosbaugh DW, Tainer JA. Protein mimicry of DNA from crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with Escherichia coli uracil-DNA glycosylase. J Mol Biol. 1999;287:331–346. doi: 10.1006/jmbi.1999.2605. [DOI] [PubMed] [Google Scholar]
- [80].Putnam CD, Tainer JA. Protein mimicry of DNA and pathway regulation. DNA Repair. 2005;4:1410–1420. doi: 10.1016/j.dnarep.2005.08.007. [DOI] [PubMed] [Google Scholar]
- [81].Johnson CN, Spring AM, Desai S, Cunningham RP, Germann MW. DNA sequence context conceals alpha-anomeric lesions. Journal of molecular biology. 2012;416:425–437. doi: 10.1016/j.jmb.2011.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bowman KK, Sidik K, Smith CA, Taylor JS, Doetsch PW, Freyer GA. A new ATP-independent DNA endonuclease from Schizosaccharomyces pombe that recognizes cyclobutane pyrimidine dimers and 6-4 photoproducts. Nucleic Acids Res. 1994;22:3026–3032. doi: 10.1093/nar/22.15.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Avery AM, Kaur B, Taylor JS, Mello JA, Essigmann JM, Doetsch PW. Substrate specificity of ultraviolet DNA endonuclease (UVDE/Uve1p) from Schizosaccharomyces pombe. Nucleic Acids Res. 1999;27:2256–2264. doi: 10.1093/nar/27.11.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Paspaleva K, Thomassen E, Pannu NS, Iwai S, Moolenaar GF, Goosen N, Abrahams JP. Crystal structure of the DNA repair enzyme ultraviolet damage endonuclease. Structure. 2007;15:1316–1324. doi: 10.1016/j.str.2007.05.010. [DOI] [PubMed] [Google Scholar]
- [85].Meulenbroek EM, Cane C. Peron, Jala I, Iwai S, Moolenaar GF, Goosen N, Pannu NS. UV damage endonuclease employs a novel dual-dinucleotide flipping mechanism to recognize different DNA lesions. Nucleic Acids Res. 2013;41:1363–1371. doi: 10.1093/nar/gks1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tsutakawa SE, Muto T, Kawate T, Jingami H, Kunishima N, Ariyoshi M, Kohda D, Nakagawa M, Morikawa K. Crystallographic and functional studies of very short patch repair endonuclease. Mol Cell. 1999;3:621–628. doi: 10.1016/s1097-2765(00)80355-x. [DOI] [PubMed] [Google Scholar]
- [87].Tsutakawa SE, Jingami H, Morikawa K. Recognition of a TG mismatch: the crystal structure of very short patch repair endonuclease in complex with a DNA duplex. Cell. 1999;99:615–623. doi: 10.1016/s0092-8674(00)81550-0. [DOI] [PubMed] [Google Scholar]
- [88].Hunter WN, Brown T, Kneale G, Anand NN, Rabinovich D, Kennard O. The structure of guanosine-thymidine mismatches in B-DNA at 2.5-A resolution. The Journal of biological chemistry. 1987;262:9962–9970. doi: 10.2210/pdb113d/pdb. [DOI] [PubMed] [Google Scholar]
- [89].Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, Wind N. de, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- [90].Monastiriakos SK, Doiron KM, Siponen MI, Cupples CG. Functional interactions between the MutL and Vsr proteins of Escherichia coli are dependent on the N-terminus of Vsr. DNA repair. 2004;3:639–647. doi: 10.1016/j.dnarep.2004.02.008. [DOI] [PubMed] [Google Scholar]
- [91].Polosina YY, Cupples CG. Changes in the conformation of the Vsr endonuclease amino-terminal domain accompany DNA cleavage. J Biochem. 2009;146:523–526. doi: 10.1093/jb/mvp095. [DOI] [PubMed] [Google Scholar]
- [92].Yao M, Kow YW. Strand-specific cleavage of mismatch-containing DNA by deoxyinosine 3′-endonuclease from Escherichia coli. The Journal of biological chemistry. 1994;269:31390–31396. [PubMed] [Google Scholar]
- [93].Yao M, Kow YW. Interaction of deoxyinosine 3′-endonuclease from Escherichia coli with DNA containing deoxyinosine. The Journal of biological chemistry. 1995;270:28609–28616. doi: 10.1074/jbc.270.48.28609. [DOI] [PubMed] [Google Scholar]
- [94].Yao M, Kow YW. Cleavage of insertion/deletion mismatches, flap and pseudo-Y DNA structures by deoxyinosine 3′-endonuclease from Escherichia coli. The Journal of biological chemistry. 1996;271:30672–30676. doi: 10.1074/jbc.271.48.30672. [DOI] [PubMed] [Google Scholar]
- [95].Yao M, Kow YW. Further characterization of Escherichia coli endonuclease V. Mechanism of recognition for deoxyinosine, deoxyuridine, and base mismatches in DNA. The Journal of biological chemistry. 1997;272:30774–30779. doi: 10.1074/jbc.272.49.30774. [DOI] [PubMed] [Google Scholar]
- [96].Huang J, Lu J, Barany F, Cao W. Multiple cleavage activities of endonuclease V from Thermotoga maritima: recognition and strand nicking mechanism. Biochemistry. 2001;40:8738–8748. doi: 10.1021/bi010183h. [DOI] [PubMed] [Google Scholar]
- [97].Fladeby C, Vik ES, Laerdahl JK, Gran Neurauter C, Heggelund JE, Thorgaard E, Strom-Andersen P, Bjoras M, Dalhus B, Alseth I. The human homolog of Escherichia coli endonuclease V is a nucleolar protein with affinity for branched DNA structures. PLoS One. 2012;7:e47466. doi: 10.1371/journal.pone.0047466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hitchcock TM, Gao H, Cao W. Cleavage of deoxyoxanosine-containing oligodeoxyribonucleotides by bacterial endonuclease V. Nucleic acids research. 2004;32:4071–4080. doi: 10.1093/nar/gkh747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Morita Y, Shibutani T, Nakanishi N, Nishikura K, Iwai S, Kuraoka I. Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nat Commun. 2013;4:2273. doi: 10.1038/ncomms3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Vik ES, Nawaz MS, Andersen P. Strom, Fladeby C, Bjoras M, Dalhus B, Alseth I. Endonuclease V cleaves at inosines in RNA. Nat Commun. 2013;4:2271. doi: 10.1038/ncomms3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Rosnes I, Rowe AD, Vik ES, Forstrom RJ, Alseth I, Bjoras M, Dalhus B. Structural basis of DNA loop recognition by endonuclease V. Structure. 2013;21:257–265. doi: 10.1016/j.str.2012.12.007. [DOI] [PubMed] [Google Scholar]
- [102].Orans J, McSweeney EA, Iyer RR, Hast MA, Hellinga HW, Modrich P, Beese LS. Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell. 2011;145:212–223. doi: 10.1016/j.cell.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, Uchida M, Ohtsuka E, Morioka H, Hakoshima T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005;24:683–693. doi: 10.1038/sj.emboj.7600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–1120. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair. 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Finger LD, Patel N, Beddows A, Ma L, Exell JC, Jardine E, Jones AC, Grasby JA. Observation of unpaired substrate DNA in the flap endonuclease-1 active site. Nucleic Acids Res. 2013;41:9839–9847. doi: 10.1093/nar/gkt737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].de Laat WL, Appeldoorn E, Jaspers NG, Hoeijmakers JH. DNA structural elements required for ERCC1-XPF endonuclease activity. The Journal of biological chemistry. 1998;273:7835–7842. doi: 10.1074/jbc.273.14.7835. [DOI] [PubMed] [Google Scholar]
- [108].Viswanathan M, Dower KW, Lovett ST. Identification of a potent DNase activity associated with RNase T of Escherichia coli. The Journal of biological chemistry. 1998;273:35126–35131. doi: 10.1074/jbc.273.52.35126. [DOI] [PubMed] [Google Scholar]
- [109].Zuo Y, Deutscher MP. The physiological role of RNase T can be explained by its unusual substrate specificity. The Journal of biological chemistry. 2002;277:29654–29661. doi: 10.1074/jbc.M204252200. [DOI] [PubMed] [Google Scholar]
- [110].Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lee KC, Padget K, Curtis H, Cowell IG, Moiani D, Sondka Z, Morris NJ, Jackson GH, Cockell SJ, Tainer JA, Austin CA. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol Open. 2012;1:863–873. doi: 10.1242/bio.20121834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rahal EA, Henricksen LA, Li Y, Williams RS, Tainer JA, Dixon K. ATM regulates Mre11-dependent DNA end-degradation and microhomology-mediated end joining. Cell Cycle. 2010;9:2866–2877. doi: 10.4161/cc.9.14.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Fukuda T, Sumiyoshi T, Takahashi M, Kataoka T, Asahara T, Inui H, Watatani M, Yasutomi M, Kamada N, Miyagawa K. Alterations of the double-strand break repair gene MRE11 in cancer. Cancer Res. 2001;61:23–26. [PubMed] [Google Scholar]
- [114].Giannini G, Ristori E, Cerignoli F, Rinaldi C, Zani M, Viel A, Ottini L, Crescenzi M, Martinotti S, Bignami M, Frati L, Screpanti I, Gulino A. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002;3:248–254. doi: 10.1093/embo-reports/kvf044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- [116].Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, Mistrik M, Aittomaki K, Blomqvist C, Heikkila P, Lukas J, Nevanlinna H, Bartek J. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- [118].Limbo O, Moiani D, Kertokalio A, Wyman C, Tainer JA, Russell P. Mre11 ATLD17/18 mutation retains Tel1/ATM activity but blocks DNA double-strand break repair. Nucleic Acids Res. 2012;40:11435–11449. doi: 10.1093/nar/gks954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Park YB, Chae J, Kim YC, Cho Y. Crystal structure of human Mre11: understanding tumorigenic mutations. Structure. 2011;19:1591–1602. doi: 10.1016/j.str.2011.09.010. [DOI] [PubMed] [Google Scholar]
- [120].Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- [121].Trujillo KM, Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J Biol Chem. 2001;276:35458–35464. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- [122].Majka J, Alford B, Ausio J, Finn RM, McMurray CT. ATP hydrolysis by RAD50 protein switches MRE11 enzyme from endonuclease to exonuclease. J Biol Chem. 2012;287:2328–2341. doi: 10.1074/jbc.M111.307041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Lim HS, Kim JS, Park YB, Gwon GH, Cho Y. Crystal structure of the Mre11-Rad50-ATPgammaS complex: understanding the interplay between Mre11 and Rad50. Genes Dev. 2011;25:1091–1104. doi: 10.1101/gad.2037811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, Guenther G, SilDas S, Hammel M, Russell P, Tainer JA. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol. 2011;18:423–431. doi: 10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Mockel C, Lammens K, Schele A, Hopfner KP. ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 2012;40:914–927. doi: 10.1093/nar/gkr749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, Tainer JA. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nat Methods. 2013;10:453–454. doi: 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Deshpande RA, Williams GJ, Limbo O, Williams RS, Kunhlein J, Lee JH, Classen S, Guenther G, Russell P, Tainer JA, Paull TT. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO Journal. 2014;33 doi: 10.1002/embj.201386100. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- [129].Parikh SS, Mol CD, Hosfield DJ, Tainer JA. Envisioning the molecular choreography of DNA base excision repair. Curr Opin Struct Biol. 1999;9:37–47. doi: 10.1016/s0959-440x(99)80006-2. [DOI] [PubMed] [Google Scholar]
- [130].Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- [131].Frank G, Qiu J, Zheng L, Shen B. Stimulation of eukaryotic flap endonuclease-1 activities by proliferating cell nuclear antigen (PCNA) is independent of its in vitro interaction via a consensus PCNA binding region. J Biol Chem. 2001;276:36295–36302. doi: 10.1074/jbc.M103397200. [DOI] [PubMed] [Google Scholar]
- [132].Friedrich-Heineken E, Toueille M, Tannler B, Burki C, Ferrari E, Hottiger MO, Hubscher U. The two DNA clamps Rad9/Rad1/Hus1 complex and proliferating cell nuclear antigen differentially regulate flap endonuclease 1 activity. J Mol Biol. 2005;353:980–989. doi: 10.1016/j.jmb.2005.09.018. [DOI] [PubMed] [Google Scholar]
- [133].Gomes XV, Burgers PM. Two modes of FEN1 binding to PCNA regulated by DNA. EMBO J. 2000;19:3811–3821. doi: 10.1093/emboj/19.14.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Hosfield DJ, Mol CD, Shen B, Tainer JA. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell. 1998;95:135–146. doi: 10.1016/s0092-8674(00)81789-4. [DOI] [PubMed] [Google Scholar]
- [135].Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- [136].Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- [137].Querol-Audi J, Yan C, Xu X, Tsutakawa SE, Tsai MS, Tainer JA, Cooper PK, Nogales E, Ivanov I. Repair complexes of FEN1 endonuclease, DNA, and Rad9-Hus1-Rad1 are distinguished from their PCNA counterparts by functionally important stability. Proc Natl Acad Sci U S A. 2012;109:8528–8533. doi: 10.1073/pnas.1121116109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Zheng L, Dai H, Qiu J, Huang Q, Shen B. Disruption of the FEN-1/PCNA interaction results in DNA replication defects, pulmonary hypoplasia, pancytopenia, and newborn lethality in mice. Mol Cell Biol. 2007;27:3176–3186. doi: 10.1128/MCB.01652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic acids research. 1997;25:2985–2991. doi: 10.1093/nar/25.15.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]