

Abstract
We used four mutants having albino or pale green phenotypes with disrupted nuclear-encoded chloroplast proteins to analyze the regulatory system of metabolites in chloroplast. We performed an integrated analyses of transcriptomes and metabolomes of the four mutants. Transcriptome analysis was carried out using the Agilent Arabidopsis 2 Oligo Microarray, and metabolome analysis with two mass spectrometers; a direct-infusion Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR/MS) and a gas chromatograph-time of flight mass spectrometer. Among approximately 200 known metabolites detected by the FT-ICR/MS, 71 metabolites showed significant changes in the mutants when compared with controls (Ds donor plants). Significant accumulation of several amino acids (glutamine, glutamate and asparagine) was observed in the albino and pale green mutants. Transcriptome analysis revealed altered expressions of genes in several metabolic pathways. For example, genes involved in the tricarboxylic acid cycle, the oxidative pentose phosphate pathway, and the de novo purine nucleotide biosynthetic pathway were up-regulated. These results suggest that nitrogen assimilation is constitutively promoted in the albino and pale green mutants. The accumulation of ammonium ions in the albino and pale green mutants was consistently higher than in Ds donor lines. Furthermore, genes related to pyridoxin accumulation and the de novo purine nucleotide biosynthetic pathway were up-regulated, which may have occurred as a result of the accumulation of glutamine in the albino and pale green mutants. The difference in metabolic profiles seems to be correlated with the disruption of chloroplast internal membrane structures in the mutants. In albino mutants, the alteration of metabolites accumulation and genes expression is stronger than pale green mutants.
Electronic supplementary material
The online version of this article (doi:10.1007/s11103-014-0194-9) contains supplementary material, which is available to authorized users.
Keywords: Albino or pale-green, Arabidopsis thaliana, Chloroplast, Metabolome, Transcriptome, Nitrogen assimilation
Introduction
In the post genome-sequencing era, functional analyses of identified genes have been performed systematically by using various mutant collections. Transcriptome analyses have provided us systematic characterization of gene expression profiles not only in various tissues and organs but also under various growth conditions. Proteome analyses have revealed various novel proteins not only in various tissues and organelle but also under various environmental conditions. Recently, technology development in mass spectrometry has improved its reliability, sensitivity of detection and measurement speed in comprehensive analyses of whole metabolites (Sumner et al. 2003; Villas-Bôas et al. 2005; Hall 2006) and whole proteins (proteome) (Kersten et al. 2002; Baginsky and Gruissem 2006; Giacomelli et al. 2006). More recently there have been reports which include the integrated analysis of transcriptome and metabolome data (Caldana et al. 2011; Fortes et al. 2011) as well as research regarding the transcriptomic, proteomic and metabolomic analysis of maize response to UV-B (Casati et al. 2011a, b). Tools have also been developed for the joined visualization of transcriptomics and metabolomics (Wägele et al. 2012).
An organism has the ability to acquire tolerance to external stresses such as light, nutrient starvation and cold. Internal biological networks including gene expression and cellular signal transduction are regulated in response to these external environmental changes, which results in an accumulation of stress-specific metabolites. Such accumulations of metabolites may cause phenotypic changes. An abundance of transcripts measured by microarray does not always correlate with an abundance of proteins (Gygi et al. 1999) and their enzymatic activities (Sumner et al. 2003). Thus expression profiles (of transcriptome and proteome) in mutants are not always consistent with biochemical and metabolic phenotypes. However, metabolite distribution in the organism is the final product of gene expression, which is directly connected to biochemical phenotype. Therefore, metabolome analyses can provide insights into novel gene functions that cannot be obtained with a conventional morphological analysis of various mutants. In particular, metabolome analyses have revealed silent mutant phenotypes that are analogous to those of wild-type (Weckwerth et al. 2004), and have predicted gene functions from classification of mutants with the same phenotype, a process known as metabolic fingerprinting.
We previously isolated albino or pale-green (apg) mutants of Arabidopsis thaliana. The mutations resulted from disruption of nuclear-encoded chloroplast proteins by the insertion of the Ds transposon. Since there were various phenotypes of these apg mutants, metabolic regulation in chloroplast contained several metabolic cascades. Active biosynthesis of various metabolites is carried out in chloroplast. Most of the gene products involved in metabolic pathways are encoded in cell nuclei and transported into chloroplast to perform various functions. As the first step to identify regulatory systems of metabolites in chloroplast, we thought it important to show how metabolite profiles are altered in the apg mutants. This study was designed to use integrated analyses of transcriptome and metabolome of four mutants: apg2 (albino or pale green 2) (Motohashi et al. 2001); apg3 (albino or pale green 3) (Motohashi et al. 2007); cla1 (cloroplastos alterados1) (Mandel et al. 1996; Estévez et al. 2000); and ch42 (chlorina locus 42) (Rissler et al. 2002).
We performed transcriptome analysis by using the Agilent Arabidopsis 2 Oligo Microarray (V2), and metabolome analysis by using two mass spectrometers; Apex-q70e with Apollo II ESI (electron spray ionization) source FT-ICR/MS (Fourier transform ion cyclotron resonance mass spectrometer) and GC-TOF/MS (gas chromatograph-time of flight mass spectrometer). Plant metabolome analyses were performed using various mass spectrometers, namely GC/MS (Kaplan et al. 2004; Kolbe et al. 2006), and FT-ICR/MS (Aharoni et al. 2002; Mungur et al. 2005; Tohge et al. 2005; Oikawa et al. 2006; Nakamura et al. 2007). Mass spectrometers have different advantages depending on their type. The GC/MS is a traditional mass spectrometer, the GC device of which can separate complex biological mixtures. It is effective for the analysis of semi-volatile organic compounds such as sugars, organic acids, amino acids, sugar alcohols, amines and amides. Unlike other mass spectrometers, the advantages of FT-ICR/MS are its high-resolution and ability to determine exact mass in order to predict chemical formulae. We used direct infusion FT-ICR/MS with ESI, which is known as a soft ionization method. In this study we report an integrated analyses of the transcriptomes and metabolomes of four mutants to compare their biochemical properties, phenotypes, metabolic profiles and related gene expressions.
Results
Characteristics of Albino and pale green Arabidopsis mutants
As mentioned in the introduction, we selected four albino and pale green mutants for this study: apg2 (Motohashi et al. 2001); apg3 (Motohashi et al. 2007); cla1 (Mandel et al. 1996; Estévez et al. 2000); and ch42 (Rissler et al. 2002). Descriptions of the mutations in the four mutants are as follows. The apg2 mutant is disrupted in a TatC homologue of the Escherichia coli delta-pH dependent protein transporter (Motohashi et al. 2001). The apg3 mutant is disrupted in a gene homologous to a eukaryotic ribosome release factor (RF1) and thus APG3 operates as a ribosome release factor in chloroplast (Motohashi et al. 2007). The gene disrupted in the cla1 mutant encodes the 1-deoxy-d-xylulose-5-phosphate (DXP) synthase, which is an enzyme upstream of isoprenoid biosynthesis (Mandel et al. 1996; Estévez et al. 2000). The mutation in the ch42 mutant is in the protoporphyrin IX chelatase subunit CHLI1 that functions in chlorophyll biosynthesis (Rissler et al. 2002). The gene loci references according to TAIR (version 10) are as follows: APG2 gene, AT2G01110; APG3 gene, AT3G62910; CLA1 gene, AT4G15560; CHLI1 gene, AT4G18480. The phenotypes of the apg2 and cla1 mutants are albino, whereas the apg3 mutant is a pale-green, and the ch42 mutant is a yellowish pale-green (Fig. 1a). In previous research, it has been reported that the chloroplast internal membrane structures have been deficient in all 4 of the mutants (Fig. 1b) (Motohashi et al. 2001; 2007, Mandel et al. 1996; Estévez et al. 2000; Rissler et al. 2002). The plastids of the apg2 and cla1 mutants did not contain thylakoid membranes, while those of apg3 and ch42 mutants contained immature thylakoid membranes. The Fv/Fm value measured by pulse amplitude modulated (PAM) fluorometer showed a potential quantum yield of photosystem II (Krause et al. 1988). That value in a 3-week old Ds donor plant is approximately 0.72; however the values in the three mutants apg2, cla1, and apg3 were nearly 0 (Fig. 1c). In contrast, the Fv/Fm value in the mutant ch42 was approximately 0.71 (Fig. 1c). HPLC analyses showed that amounts of various pigments (chlorophyll a, chlorophyll b, cis-neoxanthin, trans-violaxanthin, lutein, and beta-carotene) in each of the mutants were reduced to 0–20 % of that of Ds donor plants (Fig. 1d).
Fig. 1.
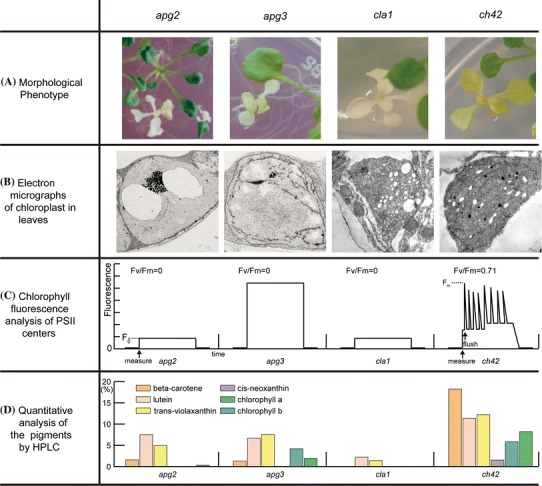
Summary of four apg mutants’ phenotypes that have been reported previously. a Morphological phenotype of the mutants that were grown for 21 days on GM medium containing 1 % sucrose. b Electron micrographs of leaf chloroplasts. c Schemes of chlorophyll fluorescence profiles measured by PAM. The values in each left top panel show Fv/Fm (=FmFo/Fm) indicating the state of PSII. d Bar plots show the relative abundance of five pigments compared with that of wild-type measured by HPLC
Analysis of the metabolic phenotypes of albino and pale green mutants versus Ds donor plants using direct-infusion ESI FT-ICR/MS
Using direct-infusion ESI FT-ICR/MS, we detected more than 2,500 independent ion peaks in every spectral scan of each sample, irrespective of experimental setting with different extraction solvents and two ionization operation modes (positive and negative). The negative operation mode was usually characterized by larger numbers of detectable peaks compared with the positive mode, and total peak numbers from Ds donor plants were larger than those from the mutants.
Almost 40 % of those 2500 peaks were detected at very low intensities (close to background levels), and such peaks were not consistently reproduced in subsequent experiments. We also evaluated the performance characteristics of direct-infusion ESI FT-ICR/MS for detection and quantification of the four mutants. We made the following four kinds of comparisons of peak intensity values found in spectral scan data. First, we checked the consistency in results of MS analysis by extracting one sample and analyzing it twice. Second, to examine the difference that occurs during extraction, we extracted the same sample twice and analyzed it. Third, we made the same comparison as the second but this time using methanol and acetone extraction. Fourth and finally, we extracted and analyzed completely different samples. Data spread was greater in the third and fourth comparisons than in those of the first and second. After performing these various tests we decided that direct-infusion ESI FT-ICR/MS was a suitable method to use for the metabolic phenotyping of the four mutant plants in our study. Four scatter plots and detailed descriptions of each are shown in Supplemental Figure 1.
Metabolomics profiling using principal component analysis and t test
Principal components analysis (PCA) is a technique used to reduce the dimensionality of multivariate data. We projected our data to two principal components. The first principal component (PC1) shows a clear difference between the mutants and the Ds donor plants (Fig. 2). The contribution ratio of PC1 was approximately 40–45 % in each matrix, which may reflect a vast difference of metabolites between the mutants and the Ds donor plants. PC2 separates the mutants, which had similar data patterns under different extraction solvent conditions (Fig. 2a, c), but differing data patterns across positive and negative ionization operation modes (Fig. 2c, d). For example the cla1 mutant was plotted far from the other mutants under positive mode, and the apg2 mutant was plotted far from the other mutants under negative mode.
Fig. 2.
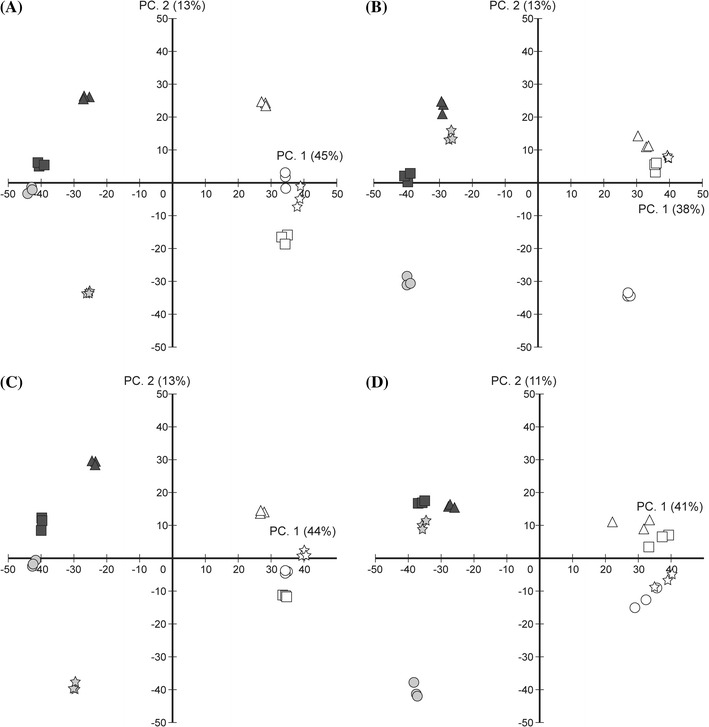
Biplot of Principal Component Analysis (PCA) applied for ESI-FT-ICR/MS. X-axis and Y-axis are principal component 1 (PC1) and principal component 2 (PC2), respectively. Solid circle is the apg2 mutant, unfilled circle is the apg2 Ds donor line, solid square is the apg3 mutant, unfilled square is the apg3 Ds donor line, solid star is the cla1 mutant, unfilled star is cla1 Ds donor line, solid triangle is the ch42 mutant, and unfilled triangle is the ch42 Ds donor line. Gray colored symbol is albino phenotype, black is pale-green phenotype. a Methanol extraction, positive charge mode measurement. b Methanol extraction, negative charge mode measurement. c Acetone extraction, positive charge mode measurement. d Acetone extraction, negative charge mode measurement.
To estimate the volume of the metabolites from the obtained ion peaks, we used Welch’s t-tests on the mutant values and the Ds donor line quantitative values as described in “Materials and methods”. When we searched manually for candidate compounds based on empirical formulas, we found approximately 150–200 peaks corresponding to known metabolites. Peaks that showed significant statistical difference between mutants and Ds donor lines (FDR < 0.05) were selected, corresponding to a total of 71 compounds (Supplemental Table 1). As follows, a short list of the compounds and their difference in relative abundance between mutants and their Ds donor lines: in the mutants there was an increase in five amino acids (asparagine, glutamine, glutamate, arginine, and histidine) and two organic acids (citrulline and glucarate), an increase in vitamin B6 and its derivatives, and a decrease in ascorbate and monodehydroascorbate (Supplemental Table 1). Phenylpropanoids were fewer for the most part, but levels of sinapoyl-glucose and bis-sinapoyl-glucoside were found to be increased in the cla1 mutant. There was also a reduction in nine terpenoids including carotenoids, but a relative increase was found in monoterpene (6-endo-Hydroxycineole) and sesquiterpene (copaene) in the apg2 and cla1 mutants. It is noteworthy that intermediate metabolites of de novo purine biosynthesis were affected differently among the mutants (Supplemental Table 1; Fig. 5; Supplemental Fig. 3). Nine flavonoids were detected. In particular, levels of flavonol glycosides such as cyanidin-rhamnosylglucoside, kaempferol-galactoside-rhamnoside and kaempferol-sophoroside were greater in the mutants. Among the sugars there were fewer pentoses and septuloses (sedoheptulose) and an increased number of hexoses, but flavonol and sinapoyl-glucose levels were distinctly different among the mutants. For example there were very high levels of flavonol and sinapoyl-glucose accumulated in the cla1 mutant in positive ion mode (Fig. 2a, c). Although the apg2 mutant differed from the other mutants on the PC2 axis in negative ion mode (Fig. 2b, d), we could not detect obvious apg2-specific metabolites, due to similar profiles between the apg2 mutant and its Ds donor control plant (Ds389-13). It might be because in Ds389-13, the Ds transposon has been inserted into the gene locus AT5G66210 [which encodes a calcium-dependent protein kinase (Ito et al. 2002)] before the APG2 gene was inserted.
Fig. 5.
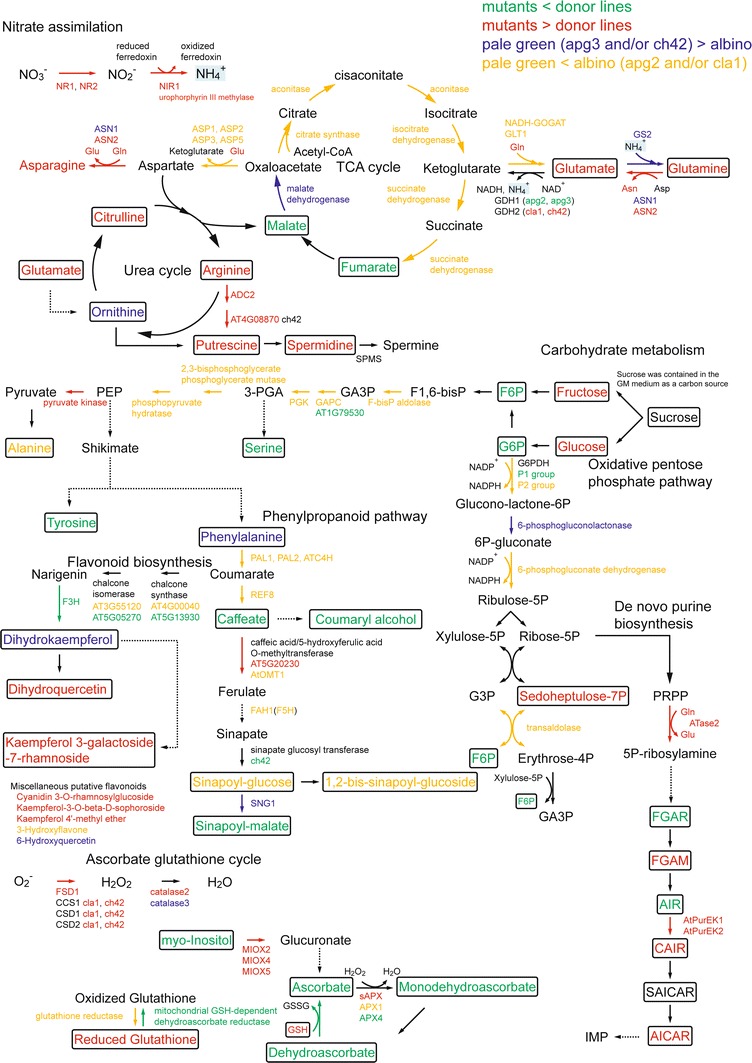
The scheme of metabolic pathways determined using metabolome analysis and transcriptome analysis. Boxed compounds indicate metabolites detected by the FT-ICR/MS or GC-TOF/MS. Solid arrows connected by compounds indicate an enzyme reaction. Dashed lines indicate abbreviation of several enzyme reactions. Colors of compounds and enzyme reactions indicate the relative abundance as indicated by mass spectrometers and gene expression by microarray, respectively. The following color codes are used: green, reduction of transcripts and metabolites in the mutants rather than Ds donor lines; red, accumulation of transcripts and metabolites in the mutants rather than Ds donor lines; blue, accumulation of transcripts and metabolites in the pale-green mutants (apg3, ch42) rather than albino mutants (apg2, cla1) or accumulation only in the ch42 mutant; yellow, accumulation of transcripts and metabolites in the albino mutants (apg2, cla1) rather than pale-green mutants (apg3, ch42) or accumulation only in the cla1 mutant
Parallel analysis of amino acids and sugar metabolites using GC-TOF/MS
It is difficult to identify compounds that have the same mass when using direct-infusion FT-ICR/MS. For this reason we also used GC-TOF/MS to analyze compounds. Six sample pools were created from each mutant and Ds donor plant. In a manner similar to FT-ICR/MS, log ratios were calculated and Welch’s t-tests were performed. As a result, the 68 compounds that were statistically significant (FDR < 0.05) were identified in at least one out of four experiments. Fourteen of those compounds, tyrosine, tryptophan, asparagine, arginine, glutamate, histidine, pyroglutamate, glutamine, fructose, glucose, glycerol 3-phosphate, alpha-tocopherol, linoleate and stearate were also detected in FT-ICR/MS, and their relative abundances were similar. The polyamines putrescine and spermidine were also accumulated in the mutants (Supplemental Table 2). The hexoses that accumulated in the mutants (shown by FT-ICR/MS) were fructose and glucose, whereas myo-inositol levels were decreased.
When performing metabolomics profiling analysis using either FT-ICR/MS or GC-TOF/MS, we observe an increase of several amino acids. Moreover, results of previously performed proteome analyses suggest the accumulation of proteins related to amino acid metabolism is a common characteristic in albino mutants (Motohashi et al. 2012). Activation of amino acid synthesis or metabolism is considered to be a feature common to heterotrophic metabolism caused by an albino mutation.
Transcriptomics profiling of microarrays using hierarchical clustering and functional categorization based on Gene Ontology Slim terms
A two-color microarray was used for transcriptome analysis. Intensity log ratio was calculated from mutant/Ds donor (see Materials and methods). The 2,812 significantly altered genes were chosen where FDR was <10−4 (Supplemental Table 3).
By hierarchical cluster analysis, we obtained eight clusters that showed similar gene expression profiles among the four mutants (Fig. 3). We then annotated these clusters by biological classification using Gene Ontology and GO Slim.
Fig. 3.
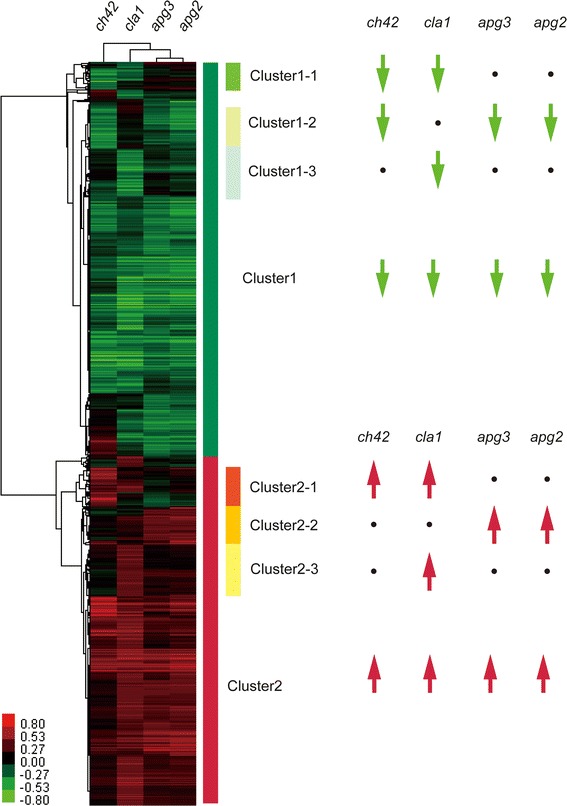
GeneTree of Hierarchical Cluster Analysis applied for microarray gene expression profiles. The GeneTree was calculated from 2,812 significantly altered genes (FDR < 1−4). Gene expression in the mutants higher and lower than those of the Ds donor lines are shown in red and green, respectively. Eight clusters were chosen based on their expression and their profiles were displayed as arrow symbols on the right panel. The altered genes were divided between 1,490 up-regulated genes as cluster1, and 1,322 down-regulated genes as cluster2. We extracted three subclusters from each of cluster1 and cluster2. Cluster1-1 (ch42 down-regulated, cla1 down-regulated, apg3 not changed, apg2 not changed), cluster1-2 (ch42 down-regulated, cla1 not changed, apg3 down-regulated, apg2 down-regulated), cluster1-3 (ch42 not changed, cla1 down-regulated, apg3 not changed, apg2 not changed) and cluster2-1 (ch42 up-regulated, cla1 up-regulated, apg3 not changed, apg2 not changed), cluster2-2 (ch42 not changed, cla1 not changed, apg3 up-regulated, apg2 up-regulated), cluster2-3 (ch42 not changed, cla1 up-regulated, apg3 not changed, apg2 not changed) were extracted from the two clusters
To determine whether consensus features of genes were contained in each cluster, we first annotated them by biological classification based on Gene Ontology (GO) provided by the Gene Ontology Consortium (http://www.geneontology.org/). We totaled the corresponding genes for each GO term, and then using Fisher’s exact test, screened to find GO terms within each cluster that had statistically higher relative enrichment factors in respect to Ds donor plants (Supplemental Table 3). In addition, we also referenced GO Slim provided by TAIR (Rhee et al. 2003), as GO terms alone are in many cases too specific to grasp an appropriate overview of a mutants’ characteristics (Fig. 4).
Fig. 4.
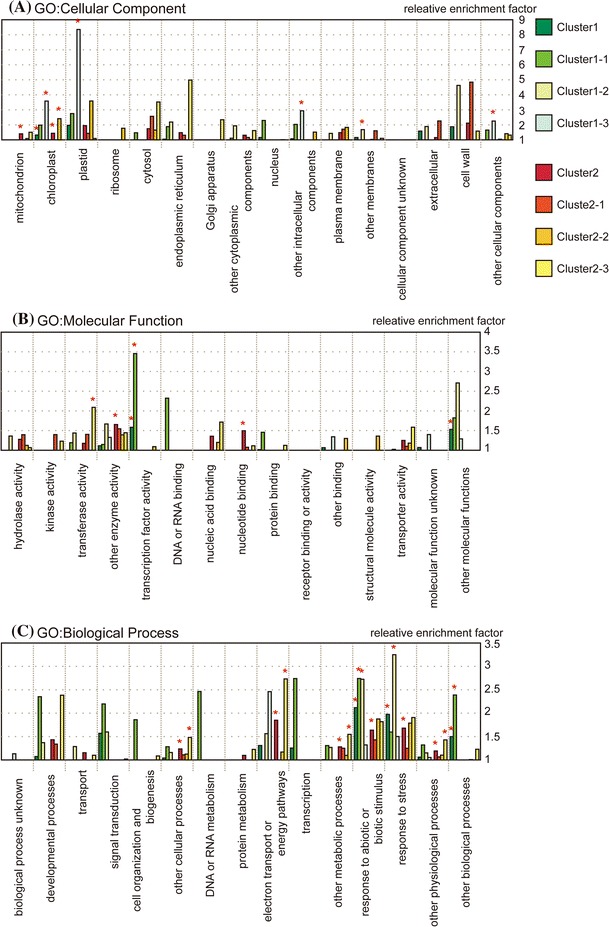
Functional classification of gene clusters based on GO Slim. Bar plot represents the relative enrichment factor that is the odds ratio of GO class corresponding genes in a cluster, compared with those in overall Arabidopsis. Asterisked GO classes were statistically significant (FDR < 10−4) in each of the three GO categories a Cellular Component, b Molecular Function, c Biological Process
‘Chloroplast’ appeared to be the most enriched class among almost all clusters. This suggests that the expression of nuclear-encoded chloroplast protein genes was affected greatly. An enrichment analysis of Cluster2 (composed of up-regulated genes over each of the four mutants) indicated the activation of genes related to nucleoside monophosphate metabolism (including purine nucleoside), and of genes related to the metabolism of nitrogen compounds, such as amino acids and amine. This is in agreement with metabolome results gained via FT-ICR/MS described above.
Under the classification for ‘Cellular Component’ we can see that genes related to plastids were highly active in Cluster1-3. In Cluster1-3 the cla1 mutant down-regulated the most, which suggests that genes related to plastids that were not as repressed in the other mutants were more strongly repressed in the cla1 mutant. Detailed classification with GO terms revealed that Cluster1-3 contained genes related to ‘chlorophyll biosynthesis’, ‘porphyrin biosynthesis’ and ‘photosynthesis, light reaction’ (Supplemental Table 4). From this we can deduce that the cla1 mutant specifically repressed genes that are considered to be photosynthesis-related genes, such as genes encoding chlorophyll biosynthesis.
Under “Molecular Function” the following genes were enriched: ‘other enzyme activity’ and ‘nucleotide binding’ in Cluster2, ‘transferase activity’ in Cluster2-3, and ‘transcription factor activity’ in Cluster1 and Cluster1-2 (Fig. 4b). Detailed classification of genes using GO terms in Cluster2-3 revealed up-regulated genes mainly in the cla1 mutant, related to glycosyl- or acyl-transfer enzymes. It should be noted that Cluster2-3 contained six UDP-glucosyl transferase genes. UDP-glucosyl transferase belongs to glycosyltransferase family 1, which uses phenylpropanoids such as flavonol and sinapate as substrates (Rohde et al. 2004) (http://afmb.cnrs-mrs.fr/CAZY/). Not coincidentally our data shows that sinapoyl-glucose, bis-sinapoyl-glucoside and flavonol were all accumulated in the cla1 mutant (Supplemental Table 1).
Finally under ‘Biological Process’, two GO classes ‘response to abiotic or biotic stimulus’ and ‘response to stress’ were enriched (Fig. 4c). The genes belonging to these classes are redundant, however it is worthy to note that ‘response to abiotic or biotic stimulus’ contains genes related to endogenous stimuli, such as sugars and hormones. These two classes were enriched not only in Cluster1 but also in Cluster2, which indicates that the mutants have suffered from various stimuli and stresses. The same accumulation could be found via proteome analysis as well (Motohashi et al. 2012). It is clear that responses to stress are active in albino mutants, likely due to their being weak from various stresses. In these GO classes, up-regulated genes were glutathione S-transferase, LEA (late embryogenesis abundant) proteins, peroxidase, catalase, heat shock proteins and superoxide dismutase, all of which are induced by oxidative stress. In contrast, down-regulated genes were those affected by a variety of stresses, including oxidative stress. In particular, the repressed genes were auxin-responsive proteins and cold responsive proteins such as cor47, cor15a and cor15b. The GO class ‘electron transport or energy pathway’ was enriched in Cluster2 and Cluster2-3. This class represents genes involved in reduction and energy liberation pathways. Typically, these genes are related to production of energy compounds such as ATP and NADPH. Detailed classification with GO terms revealed the GO term ‘main pathways of carbohydrate metabolism’ and its children terms ‘hexose catabolism’ and ‘glycolysis’ were enriched in Cluster2-3 (Supplemental Table 4). Gene expression related to metabolic pathways, such as the TCA cycle, glycolysis, and the oxidative pentose phosphate pathway were up-regulated among the mutants, in particular the cla1 mutant.
Discussion
An overview of the metabolome and transcriptome in albino and pale green mutants
In this study we observed that nitrogen assimilation was promoted in all four mutants, but this was achieved in different ways. One was the up-regulation of several genes related to the nitrate assimilation pathway. The other was accumulation of glutamine and glutamate synthesized from ketoglutarate via the GS/GOGAT cycle, and asparagine synthesized by asparagine synthetase. These three amino acids are compounds that play a role in nitrogen storage and transport (Lea and Mifflin 2003). TCA (Tricarboxylic acid) cycle and OPPP (Oxidative pentose phosphate pathway) genes were highly expressed in the mutants; the former is for ketoglutarate synthesis and the latter is for providing reducing power to nitrate assimilation (Emes and Neuhaus 1997). In addition, the gene expressions of enzymes associated with glycolysis and phenylpropanoid biosynthesis pathways were affected. This correlated with metabolomic profiles and the degree of disruption of the chloroplast inner membrane. We also detected the alteration of intermediate metabolites in the de novo purine nucleotide biosynthesis pathway, and the accumulation of pyridoxine and its derivatives. As both of these phenomena require glutamine amides for initiation, it is suggested that the increase in glutamine accumulation activates the down-stream events of these pathways.
There is a correlation between the degree of disruption of the chloroplast inner membrane and the degree of accumulation of the metabolomics profiles mentioned above. In albino mutants, the alteration of metabolite accumulation and gene expression is stronger than in pale green mutants. We also detected the alteration of intermediate metabolites in de novo purine nucleotide biosynthesis pathways. Because this pathway requires glutamine amides for initiation, it is suggested that strongly accumulated glutamine activates the down-stream events of these pathways.
In plants, there are relationships between carbohydrates such as sugars and the regulation of glutamine synthesis (Oliveira and Coruzzi 1999). Endogenous accumulation of glutamine represses the transcription of the nitrate and nitrite reductase genes (Vincentz et al. 1993; Hoff et al. 1994). In contrast, when sugars are fed to starved leaves, transcripts of nitrate reductase increase (Vincentz et al. 1993). In this study we found that glutamine was accumulated in the mutants (Supplemental Table 1; Supplemental Table 2). However, the transcripts of nitrate reductase were also up-regulated (Supplemental Table 5; Fig. 3). The reason may be that the accumulation of sugars such as glucose and fructose overrides the regulation of nitrate reductase genes (Klein et al. 2000).
The reason for the activation of nitrogen assimilation in the albino and pale green mutants is uncertain, but it is possibly associated with increasing endogenous ammonium. We measured endogenous ammonium levels in the mutants. The accumulation of ammonium ions in the mutants was consistently higher than in their Ds donor lines (Supplemental Figure 2). We thought that the reason for the degradation of photosynthetic proteins such as ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) and the chlorophyll a/b binding protein (Lhcp) was because the mediums of grown albino and pale green mutants and Ds donor lines had similar ammonium ion content (data not shown).
Extensive protein degradation leads to a drastic increase of internal ammonium content (Feng and Barker 1992). Even though the photosynthetic genes of the mutants tend to be repressed, expressions of Rubisco and Lhcp were higher than that of the other genes, thus some translated proteins of these genes have been imported into chloroplasts. Lhcp binds pigments in chloroplasts, which are then protected against protein degradation (White and Green 1987). However, chlorophyll content was low in the mutants (Fig. 1), thus almost all Lhcp could be degraded due to it being unable to bind to chlorophyll. It has been reported that the degradation of Rubisco proteins is triggered by ROS (Ishida et al. 1998). The ascorbate–glutathione cycle has the ability to remove ROS that are generated in tissue, but the cycle was not activated in this case because of the reduction of ascorbate (del Ri´o et al. 1998). For this reason, a large amount of ROS induced Rubisco degradation in the mutants. It is possible also that in order to compensate for the degradation of Lhcp and Rubisco proteins, their expressions were higher than that of the other genes.
In the following sections, we discuss in greater detail individual metabolic pathways that are significantly affected by albino and pale green mutations, focusing on the roles of the genes responsible for the respective mutations as proteins in chloroplast.
Promotion of nitrogen assimilation
Microarray analysis of the albino and pale green mutants revealed the most affected genes in metabolic pathways were those associated with nitrate assimilation. Genes encoding enzymes of nitrate assimilation have been reported (Wang et al. 2003). Of the 2,812 altered genes, six of these genes (Supplemental Table 4) were strongly up-regulated. These genes included one in the pathway leading from nitrate to ammonium, a nitrate transporter, a nitrate reductase (NR1, NR2), a ferredoxin-nitrite reductase (NiR1) (Fig. 5, top left red arrows; Supplemental Fig. 3) and an uroporphyrin III methylase that works as coenzyme for NiR1. The gene expressions of ferredoxin required for NiR1 and ferredoxin- NADP reductase were up-regulated. As previously reported (Wang et al. 2003), nitrate induced the expression of nicotianamine synthase. The up-regulation of the nicotianamine synthase gene was also observed in the mutants. Therefore, the mutants appear to assimilate nitrate into ammonium.
The assimilated ammonium is incorporated into the GS/GOGAT cycle (glutamine synthetase/glutamine-oxoglutamate aminotransferase) and synthesised into glutamine and glutamate. Asparagine is synthesised by asparagine synthetase (ASN) transforming glutamine into glutamate (Oliveira and Coruzzi 1999; Weber and Flügge 2002). GS2 was up-regulated in the cla1 and apg2 mutants, and GLT1 (NADH-GOGAT) was slightly up-regulated in the cla1 mutant. Among asparagine synthesis enzymes, ASN1 was up-regulated only in ch42 and ASN2 was up-regulated in all mutants. ASN2 may play an important role in the re-assimilation of ammonium derived from protein degradation under stress conditions (Wong et al. 2004). The gene expressions of four aspartate aminotransferases involved in aspartate synthesis, ASP1, ASP2, ASP3 and ASP5, were strongly up-regulated in the cla1 mutants and slightly up-regulated in the apg2 and apg3 mutants, but were unchanged in the ch42 mutants. In the mutants, accumulation of glutamate, glutamine, and asparagine was confirmed by metabolome analysis using FT-ICR/MS and GC-TOF/MS. Therefore, it is possible that the pathways in which ammonium is synthesized into amino acids via nitrate assimilation are activated in the mutants.
TCA cycle and glycolysis pathway
The most affected mutant in the expression of TCA cycle genes was cla1 (Supplemental Table 5). The expression levels of genes in the pathway from citrate to fumarate were highly up-regulated in the cla1 mutant (Fig. 5, top TCA cycle, yellow arrows; Supplemental Fig. 3). In contrast, malate dehydrogenase was down-regulated in the cla1 mutant (Fig. 5, top TCA cycle, green arrows; Supplemental Fig. 3). GC-TOF/MS analysis revealed that amounts of fumarate and malate were reduced in the mutants, particularly in the apg2 and cla1 mutants that had an albino phenotype (Supplemental Table 2). It has been reported that in nitrogen-starved tobacco plants, the addition of either nitrate or ammonium increased the gene expression of isocitrate dehydrogenase, aconitase and citrate synthase in the TCA cycle, to transform assimilated nitrogen into amino acids. In the GS/GOGAT cycle, two glutamates are synthesized from 2-oxoglutarat via transamidation. One of the glutamates becomes glutamine, while the other is used for other biosynthetic reactions (Lancien et al. 1999). Therefore, it is possible that the reason the TCA cycle is activated in the cla1 mutant is to provide the GS/GOGAT cycle with 2-oxoglutarate for re-assimilated ammonium.
Expressions of glycolysis genes were up-regulated in the cla1 mutant and slightly up-regulated in the apg2 and apg3 mutants, but were unchanged in the ch42 mutant (Supplemental Table 5; Fig. 5, center of carbohydrate metabolism; Supplemental Fig. 3). These gene expression profiles correlate with the position of the mutants on the PC2 axis in the PCA results of FT-ICR/MS (positive mode) (Fig. 2), and also with the degree of disrupted chloroplast inner membranes in the mutants (Fig. 1). Thus it is suggested that the more chloroplast inner membranes are disrupted, the more glycolysis gene expression is activated. These results should be reflected in the amount of metabolites; GC-TOF/MS analysis revealed that fructose and glucose were accumulated in the mutants. It is noted that soluble sugars such as fructose, glucose and sucrose are associated with oxidative stress (Couée et al. 2006). It is surprising that these sugars were accumulated in mutants that cannot fix carbon dioxide due to the repressed expression of photosynthesis genes. This phenomenon may have occurred as a result of oxidative stress, such as the accumulation of reactive oxygen species (ROS).
Oxidative pentose phosphate pathway
The oxidative pentose phosphate pathway (OPPP) is a major pathway that generates reducing power like that of NADPH in non-photosynthetic tissues and photosynthetic tissues in the dark (Emes and Neuhaus 1997). The main regulatory enzymes of OPPP are glucose 6-phosphate dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PDGH). The OPPP acquires two molecules of NADPH upon transforming from glucose 6-phosphate into ribulose 5-phosphate (Fig. 5, center right; Supplemental Fig. 3). The generated reducing power is used as a major cofactor of ROS scavenging pathways such as ascorbate–glutathione cycles (Couée et al. 2006), and used for nitrate assimilation by ferredoxin- NADP reductase. The G6PDH enzymes are divided into a chloroplastic isoform (P1 group) and a plastidic isoform (P2 group) based on their physiological roles (Wakao and Benning 2005). The P1 group of G6PDH is expressed in the developing tissues except for roots, and is tightly regulated by the redox potential (NADPH/NADP+ ratio) in photosynthesis. On the other hand, those of the P2 group are highly expressed in the root and are more resistant to NADPH levels. The reducing power in the root is linked to nitrogen assimilation (Neuhaus and Emes 2000; Esposito et al. 2005). Of the three G6PDH enzymes, the expression of the P1 group enzyme AT5G35790.1 was down-regulated in all mutants, however the expressions of the P2 group enzymes AT5G13110.1 and AT1G24280.1 were up-regulated all but the ch42 mutant (Supplemental Table 5). The reason for the down-regulation of P1-G6PDH could be the lowering of the NADPH/NADP+ ratio due to lost photosynthetic ability (Fig. 1). It is thought that heterotrophic albino mutants using the nitrate in the GM medium are actively performing nitrate assimilation. The reason for the up-regulation of P2-G6PDH could be the production of the reducing power required for nitrate assimilation.
De novo biosynthesis of purine nucleotides
AMP and GMP (de novo biosynthetic products of the purine nucleotide pathway) are fundamental to the structure of DNA and RNA, as well as for a number of essential coenzymes such as NAD, NADP, and FAD. The amounts of intermediate metabolites in this pathway were strongly affected in the mutants as indicated by FT-ICR/MS analysis (Fig. 5, bottom right). Furthermore, the expression of PRPP amidotransferase 2 (ATase2), a rate-limiting step in this pathway, was confirmed by microarray analysis to be up-regulated in the mutants (Supplemental Table 5). These results suggested that the synthesis of purine nucleotides was activated in the mutants. One possibility is that the strong accumulation of glutamine in the mutants could activate this pathway. In nodules of N-fixing tropical legumes, such as soybean (Glycine max) and cowpea (Vigna unguiculata), fixed N is assimilated initially through the amide group of glutamine, and the majority of it is subsequently incorporated through this pathway to form IMP (Smith and Atkins 2002). Thus, strong accumulation of glutamine in the mutants might be regulating the activity of this pathway. Another possibility is that this pathway is involved in chloroplast biogenesis in some way. The de novo biosynthesis of purine nucleotides, starting from glutamine and PRPP (5-phosphoribosyl diphosphate) that are synthesized from ribose-5P, could be considered to be localized at chloroplast, because sequences of their related enzymes are predicted to encode N-terminal plastid-transit peptides (Zrenner et al. 2006). It has been reported that the cia1 mutant that is disrupted in the PRPP amidotransferase 2 gene committing step in de novo purine synthesis shows albino pale-green mosaic leaves, and reduced import efficiency of proteins into chloroplasts (Hung et al. 2004). Because the cia1 mutant plants have very low levels of ATP and GTP, a decrease in energy may be altering growth and chloroplast protein import significantly. A third possibility is that its role in recycling and remobilization and purine catabolism might be involved in plant response and adaptation to environmental stress. 2-azahypoxanthine (AHX) is a compound responsible for the fairy-ring phenomenon caused by fungus. AHX chemically synthesizes from 5-aminoimidazole-4-carboxamide (AICA), and AICA is one of the members of the purine metabolic pathway. AHX and its metabolite (2-aza-8-oxohypoxanthine) are thought to be produced by plants themselves through a pathway similar to chemical synthesis (Choi et al. 2010; Choi et al. 2014). They stimulate plant growth (Choi et al. 2014; Tobina et al. 2014) and give environmental tolerance (unpublished data). Moreover, the purine metabolite allantoin enhances abiotic stress tolerance (Watanabe et al. 2014). For the same reason, the albino and pale green mutants may be trying to activate this pathway in order to gain energy, reclaim chloroplast functions and protect against environmental stress.
Ascorbate–glutathione cycle and excess energy removal
Expressions of several genes related to the removal of excess energy changed between the four mutants and their controls. There were eight genes affected in the ascorbate–glutathione cycle (Chew et al. 2003), six genes among superoxide dismutase (SOD) and catalase, and three genes encoding myo-inositol oxygenase that are related to the synthesis of ascorbate (Lorence et al. 2004) (Supplemental Table 5; Fig. 5, bottom left; Supplemental Fig. 3). In particular, clearly up-regulated genes in all mutants were the chloroplast Fe superoxide dismutase (AT4G25100) gene, a stromal ascorbate peroxidase (AT4G08390) gene, and genes encoding three myo-inositol oxygenases, MIOX2 (AT2G19800), MIOX4 (AT4G26260), and MIOX5 (AT5G56640). These results suggested that the albino and pale green mutants have been required to scavenge generated ROS. Nevertheless, the metabolome analysis showed the strong reduction in amounts of ascorbate, monodehydroascorbate, and dehydroascorbate; compounds related to xanthophyll cycle such as zeaxanthin, violaxanthin (Gilmore and Yamamoto 1993); and alpha-tocopherol, a compound which functions as the antioxidant tocopherol (Fryer 1992) (Supplemental Table 1). Because terpenoids including xanthophyll are synthesized in chloroplast, the albino and pale green mutants that repressed photosynthesis genes were assumed to decrease the synthesis rates of these compounds. Therefore, we suggest that the functions of the ascorbate–glutathione cycle and xanthophyll cycle are not sufficient to remove ROS in the albino and pale green mutants.
Accumulation of pyridoxine (Vitamin B6) and its derivatives, glucosylpyridoxine and pyridoxate were detected by FT-ICR/MS (Supplemental Table 1). Pyridoxine has been reported to play a role in protecting membranes from lipid peroxidation (Chen and Xiong 2005). Synthesis of pyridoxine and its active form pyridoxal 5-phosphate start from glutamine and ribose 5-phosphate. Expressions of two genes related to this pathway PDX1.1 (AT2G38230) and PDX1.3 (AT5G01410) (Wagner et al. 2006) were slightly up-regulated (Supplemental Table 5). It is possible that the accumulation of glutamine activates the down-stream events of this pathway in the mutants, similar to the activation of the de novo purine biosynthesis pathway.
Phenylpropanoid biosynthesis pathways
The expression profiles of genes related to phenylpropanoid biosynthesis correlated with the position of the mutants along the PC2 axis in the PCA results of FT-ICR/MS (positive mode), which is the same as that of OPPP and glycolysis (Supplemental Table 5; Fig. 3 center). These results suggested that the disruption of the chloroplast inner membrane influenced the synthesis of aromatic secondary metabolites via the phenylpropanoid biosynthesis pathway. FT-ICR/MS analysis showed that five phenylpropanoids were detected to have different relative abundance when compared between the mutants and their Ds donor lines. Sinapoyl-glucose and bis sinapoyl-glucoside were accumulated in the cla1 and apg2 mutants whereas they were reduced in the ch42 mutant. Sinapoyl-malate, caffeate and coumaryl alcohol were reduced in all of the mutants (Supplemental Table 1). Three sinapoylesters, sinapoyl-malate, sinapoyl-glucose and sinapoyl-choline have been found predominantly in plants in the Brassicaceae family (Lorenzen et al. 1996), and each sinapoylester is utilized at different times in the course of plant development (Ruegger et al. 1999). Sinapoyl-malate plays a role in UV-B protection (Booij-James et al. 2000) and is synthesized from sinapoyl-glucose by a sinapoyl-glucose:malate sinapoyltransferase; however it is not synthesized in seedlings in the dark. Thus, the accumulation of sinapoyl-malate requires light factors (Ruegger et al. 1999). Sinapoyl-glucose and bis sinapoyl-glucoside had a greater accumulation in the cla1 mutants without chloroplast inner membranes. In contrast, the smallest level of reduction of sinapoyl-malate was in the ch42 mutant. It is possible that the reason sinapoyl-glucose accumulates is due to the fact that cla1, not having pigment and not being able to detect light, cannot use sinapoyl-glucose:malate sinapoyltransferase to synthesize Sinapoyl-glucose to sinapoyl-malate.
Inconsistencies in metabolome and transcriptome states of each albino and pale green mutant
Our results show that there are partial inconsistencies between metabolic and transcriptomic states in albino and pale green mutants. The apg2 and cla1 mutants have the same albino appearance but have different metabolic phenotypes, and the apg2 mutant is more similar to the apg3 mutant which has a pale-green appearance, than to the cla1 mutant. The cla1 mutant is the most unique among the four mutants, because it lacks chlorophylls and carotenoids. The effect of the ch42 mutation was relatively light compared with the other mutants. The reason for this is possibly that the gene mutated in ch42 is one of two genes that make up the Mg-chelatase subunit CHLI, therefore the only direct effect is a reduction in chlorophyll content. Between the apg2 and apg3 mutants, the profiles of gene expression and metabolic distribution were similar. The reason for this similarity is that the biochemical changes of these mutants caused by their mutations are equivalent to the down-regulation of photosynthesis-related genes. The APG2 gene encodes a component of the delta pH-dependent protein transporter in plastids, and the mutation in the gene causes a reduction of thylakoid membrane proteins such as D1, light-harvesting complex and OE23 (Motohashi et al. 2001). The APG3 gene is thought to be associated with the termination of plastid protein translation (Motohashi et al. 2007). From these results it is thought that the changes in transcriptome and metabolome data in apg2 and apg3 may be caused by the decrease of plastid proteins.
Although phenotype is similar in cla1 and apg2 mutants (neither has thylakoid at all in their plastids), the results of their transcriptome and metabolome analysis are not identical. The results of these omics analyses reflects the influence of the defect each present in gene.
The usefulness of integrated analysis of metabolome and transcriptome in order to understand chloroplast functions
We used two types of mass spectrometry systems, FT-ICR/MS and GC/MS. GC/MS is a standard system to analyze various metabolites in plants. By using Infusion ESI FT-ICR/MS, we can discriminate among mutants that show albino or pale-green phenotypes based on the relative abundance of metabolites, which concludes that FT-ICR/MS is an appropriate method for metabolome analysis. The ChloroP software that predicts chloroplast transit peptides indicated that approximately 2,100 proteins may be localized in plastids (Richly and Leister 2004). By comprehensively analyzing the mutants of those genes, we will be able to better understand the relationship between phenotypes (albino, pale-green, and silent phenotypes like wild-type) and the genes responsible, which may offer clues to understanding novel chloroplast systematic mechanisms. The analysis of albino mutants using one- and two- dimensional nuclear magnetic resonance spectroscopy has been reported (Tian et al. 2007). NMR can be used to analyze metabolome in planta. However, mass spectrometry systems detect metabolites at a much higher sensitivity than NMR.
Conclusion
The comparative transcriptome and metabolome analysis of the four albino mutants in comparison to control Ds donor plants revealed a common molecular and metabolite phenotype for photosynthesis-impaired mutants. The high number of similar gene expressions and metabolites is surprising because we used albino mutants that have a lack of function of different genes. Because of their similar phenotypes (phenome), we presumed correctly that their metabolomes would be similar to their phenomes, but what was interesting was that the transcriptomes of each mutant were also quite similar.
At same time we were also able to find characteristics specific to each mutant. In the future, when metabolome analysis techniques have advanced and we can quantitatively detect more metabolites in plants, we can obtain more useful mutation-specific data to further analyze gene functions.
Materials and methods
Plant material and growth conditions
Four apg mutants were isolated from transposon-tagged lines that were crosses between transgenic lines expressing Ac transposase in the female parent and Ds-GUS-T-DNA lines (Ds54: Ds389-13, Ds52: Ds391-20, Ds53: Ds392-13, Ds13: Ds3-390-1) in pollen parents (Ito et al. 2002, 2005; Kuromori et al. 2004; Myouga et al. 2010). The plants were grown on germination medium (GM) containing Murashige and Skoog salts and 1 % sucrose (Wako, Osaka, Japan), for 3 weeks in a growth chamber maintained at 22 °C, with 16 h light and 8 h dark cycles. In order to equalize the biological bias, the mutants and their Ds donor lines as controls were sown in the same plates and rotated every 3 days. Pooled samples of equally volume were gathered in the same manner from more than 15 dishes. Samples used for microarray analysis were whole tissues. For metabolome analysis, after removing roots, samples were frozen in liquid nitrogen immediately and stored at −80 °C until use.
Electron microscopic analysis
Leaves were fixed, ultrathin sections were cut and stained, and electron microscope (Jeol, Tokyo, Japan) observations were made according to protocol in Motohashi et al. (2001).
Chlorophyll fluorescence measurements
Chlorophyll fluorescence of leaves was measured at room temperature using a pulse-amplitude-modulated (PAM) fluorometer (TEACHING-PAM, Walz, Effeltrich, Germany) and a photosynthesis yield analyzer (MINI- PAM, Walz, Effeltrich, Germany). Before the chlorophyll fluorescence measurements, plants grown normally for 3 weeks were dark-adapted for 20 min. The results represent the mean values of at least four measurements.
Pigment analysis
Pigment extraction, analysis, and quantification were performed according to protocol in Iuchi et al. (2001).
Metabolome analysis using infusion ESI FT-ICR/MS
After the samples were homogenized with liquid nitrogen, the powdered samples were dissolved in extraction solvents to 20 % fresh weight/volume. Two extraction solvents, acetone and methanol, were used to elute various polar compounds. Extracted sample solutions were filtered through 0.4 µm filters and dissolved in 10 % (v/v) H2O: methanol:acetone (4:4:2). Mass analysis using FT-ICR/MS (Apollo II ESI Apex-Q70e, Bruker Daltonics, Billerica, MA, USA) was performed in positive and negative ionization operation modes. For positive and negative mode, acetic acid was added to extracted sample solutions at a final concentration of 2 % (v/v) and ammonium acetate added at 0.1 % (v/v). Mass spectra were acquired over the 100–1,000 m/z range and accumulated to improve the S/N ratio. We measured the product ion mass spectra of each sample three times. When ion peaks were detected at least twice out of three successive spectral scans, they were subjected to further data processing as ion signals from actual analytes. A total of four mass spectral peaks from two different extraction solvents (methanol and acetone) and two ionization operation modes (positive and negative) were aligned with our original Java program.
Following this, we applied global normalization to our data. To avoid zero division, missing values were filled with 105 as a background signal of FT-ICR/MS. Peak intensities were transformed using a logarithmic scale with a factor of 10. Four data matrices were used to apply global normalization. Global normalization calculation methods are as follows: (1) Average intensity was calculated by dividing the total signal by the number of detected peaks in each spectrum. (2) Average signal was calculated for all spectra in each elution and charge pair. (3) Normalization factor was calculated for each spectrum by dividing the average intensity for each spectrum by total average intensity. (4) Normalized intensity was calculated by multiplying the raw intensity in each spectrum by the previously calculated normalization factor. Empirical formulas were inferred by the accuracy of the FT-ICR/MS. Because the sample ions became adduct ions to attach proton and sodium ion, etc. on the ESI source, we assumed the following were involved in the detected peaks [M+H]+, [M+Na]+, [M+K]+, [M+H+methanol]+, [M+ammonium]+ in the positive ionization mode and [M−H]− in the negative ionization mode. We searched for candidate compounds using KEGG (http://www.genome.jp/kegg/), NIST (http://webbook.nist.gov/chemistry/) and KNApSAck (Shinbo et al. 2006).
Metabolome analysis using GC-TOF/MS
Six replicate samples, mutants and their Ds donor lines were obtained from unique dishes. Metabolites were extracted from 50 mg of frozen plant material with CHCl3:MeOH:H2O (2:6:2) that included 10 stable isotope reference compounds. Metabolites were derivatized by methoxyamination for 22 h, using a 20 mg/ml solution of MeONH3 in pyridine. The derivatives were subsequently trimethylsilated with N-methyl-N-trimethylsilyl-trifluoacetamide for 1 h. An n-heptane mixture (30 μl) was used for the determination of retention time indices. Samples were injected in splitless mode (2 μl/sample) and analyzed using GC-TOF/MS (Pegasus 4D; Leco, St. Joseph, MI, USA). Artifact peaks were manually identified and removed from the obtained peaks. Remaining peaks in each mass spectrum were transformed using × 1,000 and + 1 in order to form a normal distribution. Peak intensities were transformed using a logarithmic scale with a factor of 10.
Principal component analysis and t-test
Statistical analyses, including principal component analysis (PCA), t test and regression plots were performed in an R software environment for statistical computing version 2.0.1; http://www.r-project.org/ (Ihaka and Gentleman 1996). To calculate eigenvalue, the eigenvector for PCA, prcomp in R function was implemented with a correlation matrix. The contribution ratio (%) in each component was the eigenvalue divided by total number of peaks in a matrix, ×100. The component loadings in each peak were the square root of the eigenvalue multiplied by the eigenvector. Principal component scores were obtained from prcomp, the variance of which was adjusted to be the eigenvalue.
LogRatios for each peak were calculated by the weighted mean of the mutant log transformed intensities minus that of the Ds donor line. One-step Tukey’s Biweight Estimate was adopted as a weighted mean method, which is known as a robust method against outliers in microarray intensity calculation. P value was calculated by Welch’s t-test and adjusted to controlled probability using Q-VALUE software (Storey and Tibshirani 2003) in consideration of the multiple comparisons problem.
Transcriptome analysis using Agilent Arabidopsis2 Oligo Microarray
Total RNA was isolated using Trizol according to the supplier’s instructions (Life Technologies, Rockville, MD, USA) and purified with an RNeasy Plant Mini Kit (Qiagen). cDNA was synthesized with 700 ng of total RNA using MMLV-RT and T7 promoter primers (Agilent Technologies, Inc., Palo Alto, CA, USA). This cDNA was used as a template to synthesize cRNA. The cRNA was labeled with Cyanine-3 (Cy3) CTP or Cyanine-5 (Cy5) CTP (Perkin Elmer/NEN Life Sciences, Tokyo, Japan). Cy3-labeled cRNA (700 ng) was mixed with the same amount of Cy5-labeled cRNA and used for subsequent hybridization. After hybridization for 17 h at 60 °C, slides were washed and scanned with an Agilent Microarray Scanner. Scanned images were transformed into quantified figures by the Agilent Features extraction software (version 7.1). We attempted dye swap experiments to estimate for human error and the consistency of the dye labeling.
Data mining of microarray results for transcriptome analysis
Unreliable spots at four filters (IsSaturated, IsWellAboveBG, IsFeatureNonUnifOL, IsPosAndSignif) were excluded from subsequent analyses. We attempted to detect the spots that have statistically significant differences in signal intensity, based on the PValueLogRatio calculated from the Feature Extraction software; this indicates the level of significance in the differential expression of a gene as measured through the log ratio. Then the PValueLogRatios, including color swap replicates, were corrected to the q-value (Storey and Tibshirani 2003) and chosen when FDR < 10−4. The list of significant genes in all experiments was partitioned into clusters based on expression similarity using Cluster software (Eisen et al. 1998). The similarity metric was determined using Correlation (uncentered), calculation of centroids was carried out using Average Linkage, and we used Java TreeView to create the GeneTree (Saldanha 2004). Eight distinctive clusters were isolated. Functional categories of the genes in these clusters were determined on the basis of Gene Ontology and GO Slim (ATH_GO_GOSLIM.20060422) in TAIR (Rhee et al. 2003). As reported by Zeeberg et al. (2003), one-sided Fisher’s exact tests were performed. The null hypothesis was that the GO category is not enriched in cluster genes compared to what would have been expected by chance alone.
Ammonium analysis
NH4 + content was determined in whole 3-week plants crushed in liquid nitrogen. Ammonium extraction, purification and quantification were performed according to protocol in Bräutigam et al. (2007). We used a kit (modified Fujii-Okuda method, Wako) as the Berthelot reaction for the quantification of ammonium. NH4 + concentration was determined using an NH4Cl standard curve, normalized to gram fresh weight of the sample.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Hitoshi Sakakibara for valuable discussions on the nitrogen assimilation aspects of this study. The operation of FT-ICR/MS was supported by Bruker Daltonics, K.K. We also thank our colleagues at RIKEN Plant Genomic Network Research Team and Makoto Kobayashi, Naomi Hayashi at Metabolomics Research Group for GC-TOF/MS analyses. This work was supported by the Ministry of Education, Culture, Sports, Science and Technology (Japan) [Grants-in-Aid for Scientific Research (No. 17681022 to R.M.)].
Abbreviation
- WT
Wild type
References
- Aharoni A, Ric de Vos CH, Verhoeven HA, Maliepaard CA, Kruppa G, Bino R, Goodenowe DB. Nontargeted metabolome analysis by use of Fourier Transform Ion Cyclotron Mass Spectrometry. OMICS. 2002;6:217–234. doi: 10.1089/15362310260256882. [DOI] [PubMed] [Google Scholar]
- Baginsky S, Gruissem W. Arabidopsis thaliana proteomics: from proteome to genome. J Exp Bot. 2006;57:1485–1491. doi: 10.1093/jxb/erj130. [DOI] [PubMed] [Google Scholar]
- Booij-James IS, Dube SK, Jansen MA, Edelman M, Mattoo AK. Ultraviolet-B radiation impacts light-mediated turnover of the photosystem II reaction center heterodimer in Arabidopsis mutants altered in phenolic metabolism. Plant Physiol. 2000;124:1275–1284. doi: 10.1104/pp.124.3.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräutigam A, Gagneul D, Weber APM. High-throughput colorimetric method for the parallel assay of glyoxylic acid and ammonium in a single extract. Anal Biochem. 2007;362:151–153. doi: 10.1016/j.ab.2006.12.033. [DOI] [PubMed] [Google Scholar]
- Caldana C, Degenkolbe T, Cuadros-Inostroza A, Klie S, Sulpice R, Leisse A, et al. High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 2011;67:869–884. doi: 10.1111/j.1365-313X.2011.04640.x. [DOI] [PubMed] [Google Scholar]
- Casati P, Campi M, Morrow DJ, Fernandes JF, Walbot V. Transcriptomic, proteomic and metabolomic analysis of maize responses to UV-B. Plant Sign Behav. 2011;6:1146–1153. doi: 10.4161/psb.6.8.15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casati P, Campi M, Morrow DJ, Fernandes JF, Walbot V. Transcriptomic, proteomic and metabolomic analysis of UV-B signaling in maize. BMC Genomic. 2011;12:321–338. doi: 10.1186/1471-2164-12-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Xiong L. Pyridoxine is required for post-embryonic root development and tolerance to osmotic and oxidative stresses. Plant J. 2005;44:396–408. doi: 10.1111/j.1365-313X.2005.02538.x. [DOI] [PubMed] [Google Scholar]
- Chew O, Whelan J, Millar AH. Molecular definition of the ascorbate-glutathione cycle in Arabidopsis mitochondria reveals dual targeting of antioxidant defenses in plants. J Biol Chem. 2003;278:46869–46877. doi: 10.1074/jbc.M307525200. [DOI] [PubMed] [Google Scholar]
- Choi JH, Fushimi K, Abe N, Tanaka H, Maeda S, Morita A, Hara M, Motohashi R, Matsunaga J, Eguchi Y, Ishigaki N, Hashizume D, Koshino H, Kawagishi H. Disclosure of the “fairy” of fairy-ring-forming fungus Lepista sordida. ChemBioChem. 2010;11:1373–1377. doi: 10.1002/cbic.201000112. [DOI] [PubMed] [Google Scholar]
- Choi JH, Ohnishi T, Yamakawa Y, Takeda S, Sekiguchi S, Maruyama W, Yamashita K, Suzuki T, Morita A, Ikka T, Motohashi R, Kiriiwa Y, Tobina H, Asai T, Tokuyama S, Hirai H, Yasuda N, Noguchi K, Asakawa T, Sugiyama S, Kan T, Kawagishi H. The source of “fairy rings”: 2-azahypoxanthine and its metabolite found in a novel purine metabolic pathway in plants. Angew Chem Int Ed Engl. 2014;53:1552–1555. doi: 10.1002/anie.201308109. [DOI] [PubMed] [Google Scholar]
- Couée I, Sulmon C, Gouesbet G, El Amrani A. Involvement of soluble sugars in reactive oxygen species balance and responses to oxidative stress in plants. J Exp Bot. 2006;57:449–459. doi: 10.1093/jxb/erj027. [DOI] [PubMed] [Google Scholar]
- del Rı´o LA, Pastori GM, Palma JM, Sandalio LM, Sevilla F, Corpas FJ, et al. The activated oxygen role of peroxisomes in senescence. Plant Physiol. 1998;116:1195–1200. doi: 10.1104/pp.116.4.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emes MJ, Neuhaus HE. Metabolism and transport in non-photosynthetic plastids. J Exp Bot. 1997;48:1995–2005. [Google Scholar]
- Esposito S, Guerriero G, Vona V, Di Martino RV, Carfagna S, Rigano C. Glutamate synthase activities and protein changes in relation to nitrogen nutrition in barley: the dependence on different plastidic glucose-6P dehydrogenase isoforms. J Exp Bot. 2005;56:55–64. doi: 10.1093/jxb/eri006. [DOI] [PubMed] [Google Scholar]
- Estévez JM, Cantero A, Romero C, Kawaide H, Jiménez LF, Kuzuyama T, et al. Analysis of the expression of CLA1, a gene that encodes the 1-Deoxyxylulose 5-Phosphate Synthase of the 2-C-Methyl-D-Erythritol-4-phosphate pathway in Arabidopsis. Plant Physiol. 2000;124:95–104. doi: 10.1104/pp.124.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Barker AV. Ethylene evolution and ammonium accumulation by tomato plants with various nitrogen forms and regimes of acidity: Part I. J Plant Nutr. 1992;15:2457–2469. doi: 10.1080/01904169209364487. [DOI] [Google Scholar]
- Fortes AM, Agudelo-Romero P, Silva MS, Ali K, Sousa L, Maltese F, et al. Transcript and metabolite analysis in Trincadeira cultivar reveals novel information regarding the dynamics of grape ripening. BMC Plant Bio. 2011;11:149–183. doi: 10.1186/1471-2229-11-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer MJ. The antioxidant effects of thylakoid Vitamin E (a-tocopherol) Plant Cell Environ. 1992;15:381–392. doi: 10.1111/j.1365-3040.1992.tb00988.x. [DOI] [Google Scholar]
- Giacomelli L, Rudella A, van Wijk KJ. High light response of the thylakoid proteome in Arabidopsis thaliana wild type and the ascorbate deficient mutant vtc2-2. A comparative proteomics study. Plant Physiol. 2006;141:685–701. doi: 10.1104/pp.106.080150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AM, Yamamoto HY. Linear models relating xanthophylls and lumen acidity to non-photochemical fluorescence quenching. Evidence that antheraxanthin explains zeaxanthin-independent quenching. Photosynth Res. 1993;35:67–78. doi: 10.1007/BF02185412. [DOI] [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RD. Plant metabolomics: from holistic hope, to hype, to hot topic. New Phyto. 2006;169:453–468. doi: 10.1111/j.1469-8137.2005.01632.x. [DOI] [PubMed] [Google Scholar]
- Hoff T, Truong H-N, Caboche M. The use of mutants and transgenic plants to study nitrate assimilation. Plant Cell Environ. 1994;17:489–506. doi: 10.1111/j.1365-3040.1994.tb00145.x. [DOI] [Google Scholar]
- Hung WF, Chen LJ, Boldt R, Sun CW, Li HM. Characterization of Arabidopsis glutamine phosphoribosyl pyrophosphate amidotransferase-deficient mutants. Plant Physiol. 2004;135:1314–1323. doi: 10.1104/pp.104.040956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comp Graph Stat. 1996;5:299–314. [Google Scholar]
- Ishida H, Shimizu S, Makino A, Mae T. Light-dependent fragmentation of the large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase in chloroplasts isolated from wheat leaves. Planta. 1998;204:305–309. doi: 10.1007/s004250050260. [DOI] [PubMed] [Google Scholar]
- Ito T, Motohashi R, Kuromori T, Mizukado S, Sakurai T, Kanahara H, et al. A new resource of locally transposed dissociation elements for screening gene-knockout lines in silico on the Arabidopsis genome. Plant Physiol. 2002;129:1695–1699. doi: 10.1104/pp.002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Motohashi R, Kuromori T, Noutoshi Y, Seki M, Kamiya A, et al. A resource of 5,814 dissociation transposon-tagged and sequence-indexed lines of Arabidopsis transposed from start loci on chromosome 5. Plant Cell Physiol. 2005;46:1149–1153. doi: 10.1093/pcp/pci112. [DOI] [PubMed] [Google Scholar]
- Iuchi S, Kobayashi M, Taji T, Naramoto M, Seki M, Kato T, Tabata S, Kakubari Y, Yamaguchi-Shinozaki K, Shinozaki K. Regulation of drought tolerance by gene mani- pulation of 9-cis epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis. Plant J. 2001;27:325–333. doi: 10.1046/j.1365-313x.2001.01096.x. [DOI] [PubMed] [Google Scholar]
- Kaplan F, Kopka J, Haskell DW, Zhao W, Schiller KC, Gatzke N, et al. Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol. 2004;136:4159–4168. doi: 10.1104/pp.104.052142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten B, Bürkle L, Kuhn EJ, Giavalisco P, Konthur Z, Lueking A, et al. Large-scale plant proteomics. Plant Mol Biol. 2002;48:133–141. doi: 10.1023/A:1013784205292. [DOI] [PubMed] [Google Scholar]
- Klein D, Morcuende R, Stitt M, Krapp A. Regulation of nitrate reductase expression in leaves by nitrate and nitrogen metabolism is completely overridden when sugars fall below a critical level. Plant Cell Environ. 2000;23:863–871. doi: 10.1046/j.1365-3040.2000.00593.x. [DOI] [Google Scholar]
- Kolbe A, Oliver SN, Fernie AR, Stitt M, van Dongen JT, Geigenberger P. Combined transcript and metabolite profiling of Arabidopsis leaves reveals fundamental effects of the thiol-disulfide status on plant metabolism. Plant Physiol. 2006;141:412–422. doi: 10.1104/pp.106.081208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause GH, Grafflage S, Rumich-Bayer S, Somersalo S. Effects of freezing on plant mesophyll cells. Symp Soc Exp Biol. 1988;42:311–327. [PubMed] [Google Scholar]
- Kuromori T, Hirayama T, Kiyosue Y, Takabe H, Mizukado S, Sakurai T, et al. A collection of 11 800 single-copy Ds transposon insertion lines in Arabidopsis. Plant J. 2004;37:897–905. doi: 10.1111/j.1365.313X.2004.02009.x. [DOI] [PubMed] [Google Scholar]
- Lancien M, Ferrario-Mery S, Roux Y, Bismuth E, Masclaux C, Hirel B, et al. Simultaneous expression of NAD-dependent isocitrate dehydrogenase and other krebs cycle genes after nitrate resupply to short-term nitrogen-starved tobacco. Plant Physiol. 1999;120:717–726. doi: 10.1104/pp.120.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea PJ, Mifflin BJ. Glutamate synthase and the synthesis of glutamate in plants. Plant Physiol Biochem. 2003;41:555–564. doi: 10.1016/S0981-9428(03)00060-3. [DOI] [Google Scholar]
- Lorence A, Chevone BI, Mendes P, Nessler CL. Myo-inositol oxygenase offers a possible entry point into plant ascorbate biosynthesis. Plant Physiol. 2004;134:1200–1205. doi: 10.1104/pp.103.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen M, Racicot V, Strack D, Chapple C. Sinapic acid ester metabolism in wild type and a sinapoylglucose-accumulating mutant of Arabidopsis. Plant Physiol. 1996;112:1625–1630. doi: 10.1104/pp.112.4.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel MA, Feldmann KA, Herrera-Estrella L, Rocha-Sosa M, León P. CLA1, a novel gene required for chloroplast development, is highly conserved in evolution. Plant J. 1996;9:649–658. doi: 10.1046/j.1365-313X.1996.9050649.x. [DOI] [PubMed] [Google Scholar]
- Motohashi R, Nagata N, Ito T, Takahashi S, Hobo T, Yoshida S, Shinozaki K. An essential role of a TatC homologue of a ∆pH dependent protein transporter in thylakoid membrane formation during chloroplast development in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2001;98:10499–10504. doi: 10.1073/pnas.181304598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi R, Yamazaki T, Myouga F, Ito T, Ito K, Satou M, et al. Chloroplast ribosome release factor 1 (AtcpRF1) is essential for chloroplast development. Plant Mol Biol. 2007;64:481–497. doi: 10.1007/s11103-007-9166-7. [DOI] [PubMed] [Google Scholar]
- Motohashi R, Rödiger A, Agne B, Baerenfaller K, Baginsky S. Common and specific protein accumulation patterns in different albino/pale green (apg) mutants reveals regulon organization at the proteome level. Plant Physiol. 2012;160:2189–2201. doi: 10.1104/pp.112.204032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungur R, Glass AD, Goodenow DB, Lightfoot DA. Metabolite fingerprinting in transgenic Nicotiana tabacum altered by the Escherichia coli glutamate dehydrogenase gene. J Biomed Biotechnol. 2005;30:198–214. doi: 10.1155/JBB.2005.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myouga F, Akiyama K, Motohashi R, Kuromori T, Ito T, Iizumi H, et al. The chloroplast function database: a large-scale collection of Arabidopsis Ds/Spm- or T-DNA-tagged homozygous lines for nuclear-encoded chloroplast proteins, and their systematic phenotype analysis. Plant J. 2010;61:529–542. doi: 10.1111/j.1365-313X.2009.04074.x. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kimura A, Saga H, Oikawa A, Shinbo Y, Kai K, et al. Differential metabolomics unraveling light/dark regulation of metabolic activities in Arabidopsis cell culture. Planta. 2007;227:57–66. doi: 10.1007/s00425-007-0594-z. [DOI] [PubMed] [Google Scholar]
- Neuhaus HE, Emes MJ. Nonphotosynthetic metabolism in plastids. Annu Rev Plant Physiol Plant Mol Biol. 2000;51:111–140. doi: 10.1146/annurev.arplant.51.1.111. [DOI] [PubMed] [Google Scholar]
- Oikawa A, Nakamura Y, Ogura T, Kimura A, Suzuki H, Sakurai N, et al. Clarification of pathway-specific inhibition by Fourier transform ion cyclotron resonance/mass spectrometry-based metabolic phenotyping studies. Plant Phyiol. 2006;142:398–413. doi: 10.1104/pp.106.080317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira IC, Coruzzi GM. Carbon and amino acids reciprocally modulate the expression of glutamine synthetase in Arabidopsis. Plant Physiol. 1999;121:301–310. doi: 10.1104/pp.121.1.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, et al. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003;31:224–228. doi: 10.1093/nar/gkg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly E, Leister D. An improved prediction of chloroplast proteins reveals diversities and commonalities in the chloroplast proteomes of Arabidopsis and rice. Gene. 2004;329:11–16. doi: 10.1016/j.gene.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Rissler HM, Collakova E, DellaPenna D, Whelan J, Pogson BJ. Chlorophyll biosynthesis. Expression of a second chl I gene of magnesium chelatase in Arabidopsis supports only limited chlorophyll synthesis. Plant Physiol. 2002;128:770–779. doi: 10.1104/pp.010625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde A, Morreel K, Ralph J, Goeminne G, Hostyn V, De Rycke R, et al. Molecular phenotyping of the pal1 and pal2 mutants of Arabidopsis thaliana reveals far-reaching consequences on phenylpropanoid, amino acid, and carbohydrate metabolism. Plant Cell. 2004;16:2749–2771. doi: 10.1105/tpc.104.023705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegger M, Meyer K, Cusumano JC, Chapple C. Regulation of ferulate-5-hydroxylase expression in Arabidopsis in the context of sinapate ester biosynthesis. Plant Physiol. 1999;119:101–110. doi: 10.1104/pp.119.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Shinbo Y, Nakamura Y, Altaf-Ul-Amin M, Asahi H, Kurokawa K, Arita M, et al. KNApSAcK: a comprehensive species-metabolite relationship database. Biotech Agric Forest. 2006;57:165–181. doi: 10.1007/3-540-29782-0_13. [DOI] [Google Scholar]
- Smith PMC, Atkins CA. Purine biosynthesis. Big in cell division, even bigger in nitrogen assimilation. Plant Physiol. 2002;128:793–802. doi: 10.1104/pp.010912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genome-wide experiments. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner LW, Mendes P, Dixon RA. Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry. 2003;62:817–836. doi: 10.1016/S0031-9422(02)00708-2. [DOI] [PubMed] [Google Scholar]
- Tian C, Chikayama E, Tsuboi Y, Kuromori T, Shinozaki K, Kikuchi J, et al. Top-down phenomics of Arabidopsis thaliana-metabolic profiling by one- and two-dimensional nuclear magnetic resonance spectroscopy and transcriptome analysis of albino mutants. J Biol Chem. 2007;282:18532–18541. doi: 10.1074/jbc.M700549200. [DOI] [PubMed] [Google Scholar]
- Tobina H, Choi JH, Asai T, Kiriiwa Y, Asakawa T, Kan T, Morita A, Kawagishi H. 2-Azahypoxanthine and imidazole-4-carboxamide produced by the fairy-ring-forming fungus increase wheat yield. Field Crops Res. 2014;162:6–11. doi: 10.1016/j.fcr.2014.03.008. [DOI] [Google Scholar]
- Tohge T, Nishiyama Y, Hirai MY, Yano M, Nakajima J, Awazuhara M, et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005;42:218–235. doi: 10.1111/j.1365-313X.2005.02371.x. [DOI] [PubMed] [Google Scholar]
- Villas-Bôas SG, Mas S, Åkesson M, Smedsgaard J, Nielsen J. Mass spectrometry in metabolome analysis. Mass Spectrom Rev. 2005;24:613–646. doi: 10.1002/mas.20032. [DOI] [PubMed] [Google Scholar]
- Vincentz M, Moureaux T, Leydecker MT, Vaucheret H, Caboche M. Regulation of nitrate and nitrite reductase expression in Nicotiana plumbaginifolia leaves by nitrogen and carbon metabolites. Plant J. 1993;3:315–324. doi: 10.1111/j.1365-313X.1993.tb00183.x. [DOI] [PubMed] [Google Scholar]
- Wägele B, Witting M, Schmitt-Kopplin P, Suhre K. MassTRIX Reloaded: combined analysis and visualization of transcriptome and metabolome data. PLoS ONE. 2012;7:e39860. doi: 10.1371/journal.pone.0039860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Bernhardt A, Leuendorf JE, Drewke C, Lytovchenko A, Mujahed N, et al. Analysis of the Arabidopsis rsr4-1/pd1–3 mutant reveals the critical function of the PDX1 protein family in metabolism, development, and vitamin B6 biosynthesis. Plant Cell. 2006;18:1722–1735. doi: 10.1105/tpc.105.036269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao S, Benning C. Genome-wide analysis of glucose-6-phosphate dehydrogenases in Arabidopsis. Plant J. 2005;41:243–256. doi: 10.1111/j.1365-313X.2004.02293.x. [DOI] [PubMed] [Google Scholar]
- Wang R, Okamoto M, Xing X, Crawford NM. Microarray analysis of the nitrate response in Arabidopsis roots and shoots reveals over 1,000 rapidly responding genes and new linkages to glucose, trehalose-6-phosphate, iron, and sulfate metabolism. Plant Physiol. 2003;132:556–567. doi: 10.1104/pp.103.021253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Matsumoto M, Hakomori Y, Takagi H, Shimada H, Sakamoto A. The purine metabolite allantoin enhances abiotic stress tolerance through synergistic activation of abscisic acid metabolism. Plant Cell Environ. 2014;37:1022–1036. doi: 10.1111/pce.12218. [DOI] [PubMed] [Google Scholar]
- Weber A, Flügge UI. Interaction of cytosolic and plastidic nitrogen metabolism in plants. J Exp Bot. 2002;370:865–874. doi: 10.1093/jexbot/53.370.865. [DOI] [PubMed] [Google Scholar]
- Weckwerth W, Loureiro ME, Wenzel K, Fiehn O. Differential metabolic networks unravel the effects of silent plant phenotypes. Proc Natl Acad Sci USA. 2004;101:7809–7814. doi: 10.1073/pnas.0303415101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, Green BR. Polypeptides belongings to each of the three major chlorophyll a + b protein complexes are present in a chlorophyll-b-less barley mutant. Eur J Biochem. 1987;165:531–535. doi: 10.1111/j.1432-1033.1987.tb11471.x. [DOI] [PubMed] [Google Scholar]
- Wong HK, Chan HK, Coruzzi GM, Lam HM. Correlation of ASN2 gene expression with ammonium metabolism in Arabidopsis. Plant Physiol. 2004;134:332–338. doi: 10.1104/pp.103.033126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrenner R, Stitt M, Sonnewald U, Boldt R. Pyrimidine and purine biosynthesis and degradation in plants. Annu Rev Plant Biol. 2006;57:805–836. doi: 10.1146/annurev.arplant.57.032905.105421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.