

Abstract
Background
It is currently accepted that the perturbation of complex intracellular networks, rather than the dysregulation of a single gene, is the basis for phenotypical diversity. High-throughput gene expression data allow to investigate changes in gene expression profiles among different conditions. Recently, many efforts have been made to individuate which biological pathways are perturbed, given a list of differentially expressed genes (DEGs). In order to understand these mechanisms, it is necessary to unveil the variation of genes in relation to each other, considering the different phenotypes. In this paper, we illustrate a pipeline, based on Structural Equation Modeling (SEM) that allowed to investigate pathway modules, considering not only deregulated genes but also the connections between the perturbed ones.
Results
The procedure was tested on microarray experiments relative to two neurological diseases: frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) and multiple sclerosis (MS). Starting from DEGs and dysregulated biological pathways, a model for each pathway was generated using databases information biological databases, in order to design how DEGs were connected in a causal structure. Successively, SEM analysis proved if pathways differ globally, between groups, and for specific path relationships. The results confirmed the importance of certain genes in the analyzed diseases, and unveiled which connections are modified among them.
Conclusions
We propose a framework to perform differential gene expression analysis on microarray data based on SEM, which is able to: 1) find relevant genes and perturbed biological pathways, investigating putative sub-pathway models based on the concept of disease module; 2) test and improve the generated models; 3) detect a differential expression level of one gene, and differential connection between two genes. This could shed light, not only on the mechanisms affecting variations in gene expression, but also on the causes of gene-gene relationship modifications in diseased phenotypes.
Keywords: Structural equation modeling (SEM), Model generation, Pathway analysis, Perturbed models, Perturbed edges
Background
Most of known diseases are complex diseases. This means that they are caused by the combination of genetic and environmental factors. The introduction of the concept of network biology [1] allowed the application of network based approaches for studying this type of diseases. These approaches rely on the possibility to represent molecules, as proteins or genes, as interaction networks. Microarray experiments of gene expression represent a useful tool to examine the change of gene expression profile in diseases. Many efforts were performed to build, starting from gene expression, molecular networks. This activity is sometimes referred to as reverse engineering of gene regulatory networks [2]. One type of method applied for this goal, relies on the Structural Equation Modeling (SEM), a general methodology used to address questions about complex systems [3]. SEM finds a number of applications in biological networks, for example in the inference of causal phenotype networks (see [4] for a review), genome-wide association studies (GWAS) and gene-environment interactions [5,6], as well as to measure effects of quantitative trait loci (QTLs) in linkage analyses [7-9]. The use of SEM in the analysis of microarray is not new. One of the first applications is shown by [10], who demonstrated that covariance structure analysis is a useful statistical method to find common transcriptional factors for a set of genes and to specify and evaluate hypothesized biological pathways. [11] applied SEM systematically for gene network reconstruction using gene expression data pre-processed with genetic algorithms. In most of the applications of SEM the aim is to infer networks starting from data. Thus, we can define these approaches as exploratory approaches. More recently, [12] described a confirmatory approach in microarray analysis.
In this paper we propose a SEM pipeline that, from initial and revised a priori network models, obtained by pathway analysis [13], is able to compare the path strengths between several groups and to determine the effect of factors analyzed on the paths. Our framework takes into consideration the generation of pathway models based on the principles of network theory such as the small network phenomena and the detection of modules [14]. Firstly considering how differentially expressed genes (DEGs) are connected by other genes in the microarray, we try to bring out which modifications in the gene network could be responsible of the differences observed between groups considered. Our approach relies on: 1) curated biological pathway databases, 2) the principles that characterize disease genes in biological networks, 3) grouping genes in Protein Information Resource (PIR) super-families [15] for facilitating the interpretation of the model. The proposed SEM pipeline is a combination of data-driven and knowledge-driven approaches. In fact, in order to generate perturbed pathway modules, we used curated biological pathways, representing the a priori biological knowledge about genes and their connections. The hardest part is to highlight which portion of the pathway is actually distinctive of the phenomenon being analyzed. We consider, as initial model, the one obtained from the shortest paths between every couple of DEGs. This process preserves the biological knowledge completely, as the connections in the new model are not inferred, but already present in the original pathway. The model is then fitted with SEM and improved by balancing between data-driven and knowledge-driven evidences obtained by the combination of SEM with the knowledge enclosed in public databases, relative to real and putative connections among genes. Finally, SEM with multiple group analyses supplies useful information to clinicians and biologists about experimental group differences, unveiling which connections and genes are statistically significant in the perturbed pathway models.
Methods
Illustration of the proposed pipeline
The pipeline, similar to that described by [12], is illustrated in Figure 1. In step-1, DEGs were obtained by Significance Analysis of Microarray (SAM) [16] and the perturbed pathways by Signaling Pathway Impact Analysis (SPIA) [17] using KEGG database [18]; in step-2, the pathway models were generated by network analysis and evaluated with SEM in step-3 for: 1) improving the models generated by the biological pathways found; 2) testing if the pathway models differ across groups by multiple group analysis; 3) screening of single differences in expression (gene nodes) and in regulation (gene-gene edges) across groups. We used the implementation provided by the R packages samr [19] and SPIA [20] for SAM/SPIA procedures.
Figure 1.
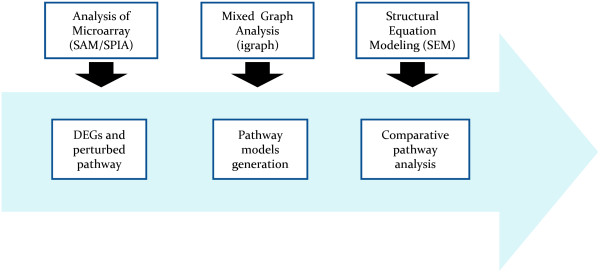
Pipeline proposed to generate and evaluate genetic pathway models.
Structural equations models (SEM)
SEM is a statistical procedure for confirmatory causal inference originated from path analysis proposed in 1921 by the American geneticist Sewall Wright [21]. It is based on multivariate linear regression equations, where the response variable in one regression equation may appear as a predictor in another equation. Indeed, variables may influence one-another reciprocally, either directly or through other variables as intermediaries. Additionally, correlated or uncorrelated unmeasured variables may indicate the presence of unobserved factors that influences observed variables.
In general, a SEM consists of a structural model describing (causal) relationships among latent (hidden) variables and a measurement model describing the relationships between the observed measurements and the underlying latent variables [22]. Here we consider SEM with observed variables only, and therefore no measurement models have been used [23]. Specifically, let V to be the index set of the Y observed variables, represented as the “parent” set {pa(i)|i ∈ V}, i.e. the explanatory variables of Y i , or as the “siblings” set {sib(i)|i ∈ V}, i.e. the unmeasured linked variables with Y i , respectively. These sets determine a system of linear equations:
and a covariance structure:
The system of linear equations affirms that every node is characterized by the relationships with his parents, while the covariance structure describes the relationships between unobserved nodes.
They encode two distinct causal assumptions: (1) a “weak” assumption on the possible existence of (direct) casual influences of explanatory variables on Y i , and (bi-directed) correlated unmeasured variables U i , quantified by the regression (path) coefficients β ij , and the covariances ψ ij , respectively; and 2) a “strong” assumption based on the absence of (direct) causal influences or (bi-directed) correlations of any observed/unobserved variables neither in the “parents” set pa(i) nor in the “siblings” set sib(i). In other terms, a weak assumption excludes some values for a parameter (the null value zero), but permits a range of other values; while, strong assumptions assume that parameters take specific values (null value zero or a fixed a priori value). The linear equations and the covariance structure can be encoded and visualized in a “path diagram”, that is, a mixed graph G = (V,E) featuring both directed (→) and bi-directed (↔) edges. The vertex set V includes the genes and the edge set E represents relations or reactions among vertexes. The “activity” of a given gene is embedded in a path diagram: the actions performed by a gene on downstream molecules, and the signals that it receives from upstream regulators. “Directed edges” between two genes (j→i, if and only if j ∈ pa(i)), measured by path coefficients (ranging usually from -1 to 1, if genes are standardized), represent expected change in the activity of the downstream gene, given a unit of change in the upstream gene while the values of the other genes remain constant. Considering that paths reflect a direct influence of one gene on another, negative path coefficients indicate ensemble inhibition (negative control) and positive paths measure net activation (positive control). “Bi-directed edges” between two genes (j↔i if and only if j ∈ sib(i), or equivalently, if and only if i ∈ sib(j)) encode a hidden common cause that may be interpreted as latent or unobserved measurement of upstream regulators that could account for the observed covariances (correlations) between the two genes.
One important feature of SEM is that direct and indirect effects can be computed and compared. “Directed paths” between two genes are the sequence of all the directed edges (j → k1,… → …, km → i) from genes Yj to Yi. Each directed path is a channel along which information (gene’s activities) can flow, and so a “total effect” (TE) of gene Yj on gene Yi is defined as the total sum of the products of the sequence of arrows (edges) along all directed paths from Yj to Yi. Accordingly, a “indirect effect” (IE) of gene Yj on gene Yi represents the portion of the total effects not considering the directed edge effect (DE), i.e. TE = DE + IE.
The well-known SEM analysis consists of four steps [22]: a) definition and identification of an initial path model, b) estimation of parameters, c) evaluation of the fitting, and d) model modification.
Initial model building
Specification of initial pathway models (step a) was obtained taking the perturbed pathways and converting them in directed graphs or gene networks. Generalizing, each pathway can be seen as a mixed graph. The idea is to understand how DEGs are connected in the perturbed pathways by other microarray genes. A natural way to solve this problem is to identify the shortest paths (geodesic distance) between DEGs. The geodesic distance dgeo(yi, yj) between two DEGs, yi and yj, is defined as the minimum distance between these two genes. The function get.shortest.paths( ) of the R package igraph was used to compute all the shortest paths [24]. Define the microarray genes, DEGs, and not DEGs in the following way: MG = {mg 1 , mg 2 , …, mg m }; DEG = {deg 1 , deg 2 ,…, deg n } and NDEG = {ndeg 1 , ndeg 2 ,…,ndeg m-n }, where MG = DEG ∪ NDEG and DEG ∩ NDEG = {∅}. Each shortest path could be represented as a list of nodes Y k = (y i , y i+1 ,..., y j-1, y j ) and a list of the corresponding edges Ek = (ei(i+1), ..., e(j-1)j) where (y i , y j ) ∈ DEG; (y i+1 ,…, y j-1 ) ∈ (DEG ∨ NDEG); Yk ⊆ Y and Ek ⊆ E. The shortest paths for each pathway constitute k (k = 1, …, K) subgraphs Gk = {Yk,Ek} of the original pathway, G = {Y, E}. Not all DEGs and NDEGs will be included in the shortest paths. Therefore, we define two new sets: DEG(s) and NDEG(s) respectively, the sets of DEGs and the set of not DEGs that include all genes in shortest paths, where DEG(s) ⊆ DEG and NDEG(s) ⊆ NDEG.
To reach the final model the NDEG(s) that connect DEG(s) are grouped in basis to their PIR superfamily (PIRSF). Based on the evolutionary relationships of whole proteins, this classification system allows annotation of both specific biological and generic biochemical functions. The PIRSF can be represented as SUPF = {supf 1 , supf 2 ,…, supf g }, where ∀ supf i ⊆ NDEG(s) and ∀i≠j supf i ∩ supf j = {∅}. Using this information, each original shorthest path Gk = {Yk,Ek} is transformed in a new shortest paths, G*k = {Y*k = (y*i, y*i+1,…, y*j-1, y*j), E*k = (e*i(i+1),…, e*(j-1)j) }, where (y*i+1,…, y*j-1) ∈ (DEG(s) ∨ SUPF ∨ NDEG(s)) and E* ⊆ E. The function to obtain the final graph, G* = (Y*,E*), is described in the following pseudo-code:
 |
The graph G* = (Y*,E*) is the fusion of all shortest paths found, where each node and each edge cannot be present more than once, the self-loops are not considered but the feed-backs and cycles were preserved. To ensure the identification of the initial models, the “block-recursive” criterion of Rigdon [25] and the “bow free” criterion of Brito and Pearl [26] were applied. The first affirms that reciprocal relationships, feedback loops, or covariances are segregated into groups, or blocks, with no more than two equations per block. The second affirms that a model is ensured if variables standing in direct causal relationships (directed edges) do not have correlated errors (bi-directed edges). So a new graph is attained in which the DEGs are connected by other DEGs, PIRSFs or NDEGs. In this way a model was created for each significant pathway found.
Successively, PIRSFs composite variables are defined considering not DEGs, present in shortest paths, as causal indicators of latent (hidden) constructs [27]. To generate the PIRSFs, a principal component analysis (PCA) was performed on genes belonging to a PIRSF and the principal component scores of the first principal component (PC1) were considered as the values that characterize the PIRSF. Only PIRSFs for which the PC1 represents 50% or more of the total variance are considered. At the end of process we have the initial SEM model.
The pathway graph conversion, the graph analysis, and the PC1 scores are obtained by graphite [28], igraph [24] and stats [29] R packages, respectively, while R functions for network analysis are implemented ad hoc, and are available Additional file 1.
SEM fitting
For parameter estimation (step-b), the classic derivation of the Maximum Likelihood estimation (MLE) is used, that assumes all observed variables are jointly Gaussian. The system of structural equations and covariance structure of unmeasured variables can be written compactly in a matrix form as: Y = BY + U, and Cov(U) = Ψ. This specification induces a structure on the covariance matrix of the joint distribution of the genes Y as:
where θ=(β; ψ) is the list of the free parameters in the model of dimension t. The unknown parameters are estimated so that the implied covariance matrix Σ(θ) is close to the observed sample covariance matrix S.
The assessment of the model (step-c) involves the Likelihood Ratio test (LRT) converted to a Chi-square test of the fitted model. Specifically, let Σ0 = E(S) to be the true population covariance matrix, and Σ(θ) the model-implied covariance matrix. The hypothesis to be tested is:
The chi-square test is then χ2 = -2logLRT = -2[logL(Σ(θ)) - logL(Σ0)] with d = p(p + 1)/2-t degree of freedom (d.f.). logL( ) represents the log-likelihood of the model, p the number of genes, t the number of parameters of the fitted model. Not-significant P-values (P >0.05) indicate that the model provides a good fit to the data. The P-values are derived by using the χ2(d) distribution or a resampling bootstrap distribution [30].
An alternative procedure [31] assumes that in the population, a model-implied covariance matrix Σ(θ0), which is approximately correct, is in the neighborhood of Σ0. So the null hypothesis of “exact fit” is replaced by the null hypothesis of “close fit”:
and the Root Mean Square Error of Approximation (RMSEA) measures the discrepancy ε for the fitted model:
P-values for RMSEA are set up from the non-central χ2(λ,d) distribution with non-centrality parameter, λ = (n-1) × d × 0.052 or from a resampling bootstrap distribution. The null hypothesis of close fit is not rejected if P > 0.05.
We also consider the Standardized Root-Mean-square Residual (SRMR), one of the most used SEM fit indices. SRMR is a measure based on the differences between observed (s) values and the ones obtained from the model (σ) of the covariance matrix:
SRMR values <0.10 are assumed as an adequate fitting measure, whereas values <0.05 may be considered as a good fit [32].
Finally, the model refinement (step-d) is obtained adding new directed or bi-directed edges to the initial model. This modification was needed considering that the initial model is only a simplified representation of the whole pathway. The criteria used for the refinement are based on the combination of three elements. First, the modification indexes (MI), that is an estimate of the decrease in the χ2-score statistic that would result by freeing each fixed (=0) parameter in the model; second, z-tests (=parameter estimate/standard error) of the MLE; and finally, biological evidences obtained by STRING database [33] and by the existence of a direct path between the nodes that MI proposes to connect. The following heuristic stepwise strategy was used:
Heuristic stepwise procedure:
Input: list of the fixed (=0) parameters (paths and/or covariances) in the model.
Output: new free parameters (paths and/or covariances) in the model.
Steps:
1. freeing just a single parameter (path coefficient or covariance) at a time, and these in turn are sorted in descending order of magnitude using MI;
2. verify if the edge (path coefficient or covariance) to add is present in STRING or when the edge is a path coefficient, if it represents a direct path that connects the nodes in the pathway selected, and then add this new edge in the model;
3. fit the model and if the new edge is statistically not significant (P > 0.05, one-sided), using a z value (z<|1.64|), remove it and repeat step 1-2;
4. STOP the selection procedure if the model achieves a non significant LRT (P > 0.05) or RMSEA (P of “close” fit > 0.05) or SRMR < 0.1, otherwise repeat step 1-3
Multiple-group analysis
When data are observed from multiple subsamples, the representation of groups with “indicator variables”, considered as nodes, allows to recognize DEGs. Instead, “multiple-group analysis” allows to identify differentially regulated genes (DRGs) across groups.
Specifically, define μ1(θ) and Σ1(θ) as the model-implied mean vector and covariance matrix of group 1 (experimental group) respectively and μ2(θ) and Σ2(θ) as the corresponding moments of group 2 (control group). For each models, two omnibus tests are performed considering the two experimental conditions (groups), one for the differential expression genes (nodes) and the other for the strength of the edges. In the first case, the hypothesis to be tested is:
while, in the second case, is:
In the “null” model (H0), the mean or covariance estimates are constrained to be equal across groups; in the “alternative” model (H1), they are allowed to differ across groups. The statistical significance is determined by comparison of LRT chi-square (χ2diff) values at a given degree of freedom (d.f. diff). If there is a significant difference (P < 0.05) in the chi-squared goodness-of-fit index, the groups differ significantly for one or more specific gene expression (nodes) and/or gene-relationships (edges). Finally, three path-coefficient differences are screened: 1) “up/down” expression (gene nodes), testing the “zero value” for the group indicator variable (C = experimental =1, and C = control = 0) path coefficients; 2) “up/down” regulation (gene edges), testing the “zero value” for the differences of path coefficients across groups; 3) “on/off” regulation (gene edges) with respect to a priori KEGG gene regulation target, testing the “zero value” of the edge coefficients across groups.
Specifically, assume C to be the path coefficient matrix of the group indicator variables and c i be an element of the matrix C. Let B1 and B2 to be the corresponding path coefficient matrices in the experimental and control groups; D = B1 – B2 and d ij be an element of the matrix D. We consider the test statistics:
where SE( ) is the estimated standard error of the parameters. The statistic tC can be used to test the conditional “up/down” expression level difference of one gene between groups, given the parents of the gene in the network. Similarly, t D checks the conditional “up/down” expression regulatory differences of one gene on another between groups. Moreover, t1 and t2 check the “on/off” regulatory differences compared to a priori KEGG pathway. The P-values of these statistics (two-sided, for tC and tD and one-sided, for t1 and t2) are derived either asymptotically from the N(0,1)-distribution or empirically from the nonparametric-based or using model-based bootstrap distribution with B bootstrap samples (usually, B = 100, or 1000).
Note that the marginal bivariate test of DEGs with SAM approach can be regarded as the special case of the conditional test with tC, when the pathway graph is G = (Y, ∅), so pa(y) = ∅ for all genes ∈Y.
We use the implementation provided by the lavaan [34] R package for estimation, evaluation, and modification of SEM data analysis, and R codes is available in Additional file 1.
Results
The above described method was applied to two gene expression microarrays datasets, one from a study on FTLD-U and the other on MS.
FTLD-U analysis
The data were obtained from a microarray experiment that analyzes various brain regions of patients affected by FTLD-U in presence of the mutation in the progranulin gene. Two groups were selected: one affected by FTLD with mutation in the progranulin gene (15 samples) and the other constituted by the control (17 samples). Data are freely available at Gene Expression Omnibus (GEO) database with ID GSE13162. For our analysis, we used normalized expression values submitted in the database. In the first step, SAM was performed using a delta value of 1.03 and a minimum fold-change of 2. The number of genes up-expressed was 207 while the number of gene down-expressed 244. Using this list of DEGs, the SPIA analysis found seven important pathways for the explanation of the role of the progranulin mutation on the FTLD-U, as showed in the Table 1.
Table 1.
Perturbed pathways obtained by SPIA on FTLD-U data
| Name pathway | pSize | NDE | pNDE | tA | pPERT | pGFdr | Status |
|---|---|---|---|---|---|---|---|
| Glutamatergic synapse |
77 |
11 |
0.000 |
-6.557 |
0.064 |
0.006 |
Inhibited |
| GABAergic synapse |
60 |
10 |
0.000 |
0.632 |
0.804 |
0.017 |
Activated |
| Calcium signaling pathway |
166 |
17 |
0.000 |
0.072 |
0.993 |
0.021 |
Activated |
| Amphetamine addiction |
55 |
8 |
0.001 |
-2.685 |
0.457 |
0.047 |
Inhibited |
| Gap junction |
85 |
10 |
0.001 |
5.216 |
0.454 |
0.047 |
Activated |
| MAPK signaling pathway |
235 |
18 |
0.001 |
-5.802 |
0.253 |
0.047 |
Inhibited |
| ECM-receptor interaction | 82 | 7 | 0.022 | 6.150 | 0.015 | 0.047 | Activated |
pSize = number of genes in the pathway; NDE = number of DEGs in the pathway; pNDE = p-value of the enrichment; tA = total perturbation; pPERT = p-value of the perturbation; Status = direction of the perturbation.
The most of the dysregulated pathways, as the MPAK signalling pathway, the calcium signalling pathway, the gap junction and the ECM-receptor interaction, confirm the analysis of [35]. The dysregulated pathways with a significant p-PERT were the glutamatergic synapse and GABAergic synapse. The role of the glutamate in the acute and neurodegenerative processes were well described in literature [35-38]. Meldrum [36] illustrated three different pathological mechanisms of action of the glutamate in the neurodegeneration. Glutamate can be neurotoxic through an agonist effect on the N-methyl-D-aspartate (NMDA), α-amino-hydroxy-5-methyl-4-isoaxaleproprionicacid (AMPA), kainate or Group I metabotropic receptors. The relative contribution of these different classes of receptors vary according to the neurons involved and a variety of other circumstances. Selective neuronal death subsequent to the epileptics status appears to be highly dependent on NMDA receptor activation. Acute neuronal degeneration after transient global or focal cerebral ischemia seems to be dependent on both NMDA and AMPA receptors. Regarding the GABAergic pathway, a loss of glutamatergic pyramidal cells and calbindin-D28k-immunoreactive GABAergic neurons in the frontal and temporal cortices of patients with FTLD [39] and FTLD with motor neuron disease [40] was reported.
SEM analysis of glutamatergic synapse KEGG pathway
For sake of brevity, model specification was obtained only for the glutamatergic synapses pathway, but the same procedure could be used for all significant pathways. We started from the original KEGG pathway and then, passing through the shortest paths model, we reached the PIRSF model. Four composite variables (PCA1) were generated: 1) PLC-beta (PIRSF000956) by entrez ID genes 5330, 5331, 23236: variance explained 66%; 2) adenylate cyclase (PIRSF001445) by entrez ID genes 113, 114, 155: variance explained 68%; 3) GTP-binding regulatory protein Gs alpha chain (PIRSF002400) by entrez ID genes 2776, 2778: variance explained 53%; 4) ionotropic glutamate receptor (PIRSF002437) by entrez ID genes 2902, 2903, 2905: variance explained 54%. The graph reduction of the model specification process was the following: from KEGG (mean degree = 11.431, number nodes(edges) = 92 (526)) to DEGs shortest paths model (mean degree = 3.769 , number nodes(edges) = 26 (49)), and to PIRSF model (mean degree = 2.947, number nodes(edges) = 19 (28)). The initial fitting indices were poor (χ2 (df) = 600.1 (25), P < 0.001, RMSEA (P-close) =0.320 (<0.001), SRMR = 0.450). This is likely because the existing pathway databases do not even contain all pathway information presented in the public literature [41], and because the model generated was a simplification of the real connections between nodes. Twenty-two directed edges were added, six using STRING database and sixteen using graph information. The final pathway model was an adequate approximation of the observed covariance matrix, as demonstrated by the SRMR index (0.092). Two-group analysis of the final pathway model revealed a significant global mean (χ2 diff(df) = 48.5 (19), P < 0.001 of H0: μ1 = μ2 subject to Σ1 = Σ2) and covariance differences (χ2 diff (df) = 110.9 (51), P < 0.001 of H0: Σ1 = Σ2 subject to μ1≠ μ2). The specific tests that consider the effect of the progranulin mutation on every gene and every edge are summarized in Tables 2 and 3.
Table 2.
Single node and edge differences found between FTLD-U with progranulin mutation and control groups
| Path | Type | FTLD-U Progranulin (P) | Control (C) | Difference (95% CI) | P-value | up/down |
|---|---|---|---|---|---|---|
| 1742 ← group |
Group on node |
-6.024 |
-5.540 |
-0.484(-0.87; -0.10) |
0.014 |
P down-expressed |
| 5532 ← group |
Group on node |
9.606 |
10.706 |
-1.101 (-1.64; -0.56) |
0.000 |
P down-expressed |
| 2785 ← group |
Group on node |
8.453 |
9.734 |
-1.281 (-1.95; -0.62) |
0.000 |
P down-expressed |
| 5534 ← group |
Group on node |
7.233 |
8.308 |
-1.075 (-1.65; -0.51) |
0.000 |
P down-expressed |
| gtp_bind ← group |
Group on node |
2.374 |
2.626 |
-0.252 (-0.44; -0.06) |
0.000 |
P down-expressed |
| 2911 < - > 9454 |
Binding/association |
0.178 |
0.813 |
-0.635 (-1.27; -0.00) |
0.049 |
P down-regulated |
| 3708 < -9456 |
Binding/association |
0.457 |
-0.205 |
0.662 (0.34; 0.98) |
0.000 |
P up-regulated |
| ade_cycl < -gtp_bind |
activation |
-2.044 |
-0.840 |
-1.204 (-2.28; -0.13) |
0.028 |
P down-regulated |
| 5613 < -107 |
Indirect |
-0.012 |
-0.324 |
0.312 (0.02; 0.60) |
0.034 |
P up-regulated |
| 5613 < -ade_cycl |
Indirect |
0.446 |
-0.413 |
0.858(0.43; 1.20) |
0.000 |
P up-regulated |
| 5579 < -plc_b |
Indirect |
3.677 |
1.110 |
2.567(0.86; 4.28) |
0.003 |
P up-regulated |
| 22941 < -1742 |
String |
1.306 |
0.570 |
0.736(0.12; 1.35) |
0.002 |
P up-regulated |
| plc_b < -9229 |
Directed path |
0.262 |
0.558 |
-0.297(-0.56; -0.03) |
0.028 |
P down-regulated |
| ade_cycl < -5532 |
Directed path |
-0.463 |
0.208 |
-0.671(-1.16; -0.18) |
0.007 |
P down-regulated |
| ade_cycl < -5534 |
Directed path |
-0.713 |
-0.168 |
-0.545(-1.05 -0.05) |
0.032 |
P down-regulated |
| 5613 < -22941 |
Directed path |
-0.581 |
-0.039 |
-0.542(-0.95; -0.13) |
0.010 |
P down-regulated |
| 22941 < -glutam_recp |
Directed path |
0.044 |
-0.278 |
0.322(0.09; 0.55) |
0.006 |
P up-regulated |
| 9455 < -glutam_recp | Directed path | -0.128 | -0.462 | 0.334(0.04; 0.92) | 0.024 | P up-regulated |
glutam_recp = PIRSF ionotropic glutamate receptor; ade_cycl = PIRSF adenylate cyclise; plc_b = PIRSF PLC-beta; gtp_bind = PIRSF GTP-binding regulatory protein Gs alpha chain.
Table 3.
Not significant (null edge) in FTLD-U with progranulin mutation and in control groups
| |
|
Progranulin (P) |
Control (C) |
|
||
|---|---|---|---|---|---|---|
| Paths | Type | Estimate | P-value | Estimate | P-value | P/C |
| 2911 < -5534 |
Activation |
-0.024 |
0.238 |
0.232 |
0.927 |
OFF/OFF |
| gtp_bind < -2911 |
Activation |
-0.049 |
0.167 |
-0.138 |
0.500 |
OFF/OFF |
| 1742 < -glutam_recp |
Binding/association |
-0.192 |
0.008 |
-0.376 |
0.200 |
ON/OFF |
| 50944 < -9229 |
Binding/association |
0.131 |
0.138 |
0.160 |
0.282 |
OFF/OFF |
| 22941 < -9229 |
Binding/association |
-0.245 |
0.811 |
-0.034 |
0.276 |
OFF/OFF |
| 9456 < -22941 |
Binding/association |
-0.114 |
0.632 |
0.130 |
0.553 |
OFF/OFF |
| 9456 < -50944 |
Binding/association |
0.742 |
0.133 |
0.931 |
0.098 |
OFF/OFF |
| 9455 < -22941 |
Binding/association |
0.142 |
0.224 |
-0.407 |
0.042 |
OFF/ON |
| 9454 < -50944 |
Binding/association |
0.519 |
0.466 |
0.226 |
0.358 |
OFF/OFF |
| 3708 < -9455 |
Binding/association |
-0.322 |
0.067 |
-0.137 |
0.432 |
OFF/OFF |
| 3708 < -9454 |
Binding/association |
0.462 |
0.055 |
0.216 |
0.001 |
OFF/ON |
| 107 < -gtp_bind |
Activation |
0.198 |
0.525 |
0.136 |
0.743 |
OFF/OFF |
| plc_b < -gtp_bind |
Activation |
-0.275 |
0.225 |
-0.234 |
0.127 |
OFF/OFF |
| 5613 < -107 |
Indirect |
-0.012 |
0.000 |
-0.324 |
0.926 |
ON/OFF |
| 1742 < -50944 |
String |
0.971 |
0.013 |
1.166 |
0.062 |
ON/OFF |
| 3708 < -2911 |
String |
-0.068 |
0.054 |
0.143 |
0.501 |
OFF/OFF |
| 9229 < -50944 |
String |
-0.490 |
0.116 |
-1.755 |
0.502 |
OFF/OFF |
| ade_cycl < -5532 |
Directed path |
-0.463 |
0.169 |
0.208 |
0.022 |
OFF/ON |
| ade_cycl < -5534 |
Directed path |
-0.713 |
0.107 |
-0.168 |
0.003 |
OFF/ON |
| glutam_recp < -5532 |
Directed path |
-0.531 |
0.000 |
-1.017 |
0.061 |
ON/OFF |
| 5579 < -9456 |
Directed path |
0.228 |
0.000 |
0.654 |
0.470 |
ON/OFF |
| 5579 < -5534 |
Directed path |
0.295 |
0.000 |
0.986 |
0.388 |
ON/OFF |
| 5613 < -22941 |
Directed path |
-0.581 |
0.741 |
-0.039 |
0.001 |
OFF/ON |
| 22941 < -glutam_recp |
Directed path |
0.044 |
0.000 |
-0.278 |
0.696 |
ON/OFF |
| 3708 < -22941 |
Directed path |
0.264 |
0.000 |
0.443 |
0.072 |
ON/OFF |
| 1742 < -9229 | String | -0.626 | 0.027 | -1.399 | 0.126 | ON/OFF |
glutam_recp = PIRSF ionotropic glutamate receptor; ade_cycl = PIRSF adenylate cyclise; plc_b = PIRSF PLC-beta; gtp_bind = PIRSF GTP-binding regulatory protein Gs alpha chain.
Four genes and one PIRSF of the glutamatergic model resulted influenced by the group: genes 1742 or PSD-95, 5532 or PPP3CB, 2785 or GNG3, 5534 or Ppp3r1 and the PIRSF of the glutamate receptor. An important role could be played by PSD-95 gene that is believed to be involved in the synapse maturation, in the induction of a network of neurotransmitter receptors, scaffolding proteins and ionotropic glutamate receptors [42]. The gene PPP3CB and the gene GNG3 are associated to the Wnt signalling correlated to the dysregulation in the case of progranulin deficiency [43]. To note that four of the nodes influenced by the group were involved in the perturbed edges described in the Table 1. Giving a look to the significant edges found, the relationships 22941 <-1742 and 22941 <-PIRSF “glutamate receptors” are well note in the literature. In fact, the gene 229141 or SHANK2 plays a critical role both in the integration of the various postsynaptic membrane proteins, cell-adhesion molecules, signal components, scaffolding proteins, and actin-based cytoskeleton, part of the PSD protein network (activated by the PSD-95) [44], and in the organization of the glutamate receptors [45]. The edges 3708 <-9456 that involves the gene Itpr1 and the gene HOMER1 were very interesting. The relationships are involved in the spinocerebellar ataxia in human as described by [46]. Also other links that include the PIRSF adenylate cyclase, GTP-binding regulatory protein and the PLC-beta could be useful to interpret the role of mutation in the progranulin gene group.
The comparison with KEGG edges revealed that 17 edges were not active (off) in the “mutant” group and 21 in the “control”. In contrast the edges activated (on) was 9 and 5 in mutant and control, respectively (cf. Table 3). The Figure 2 illustrates the model and the links found statistically significant between the yes/no mutant groups.
Figure 2.
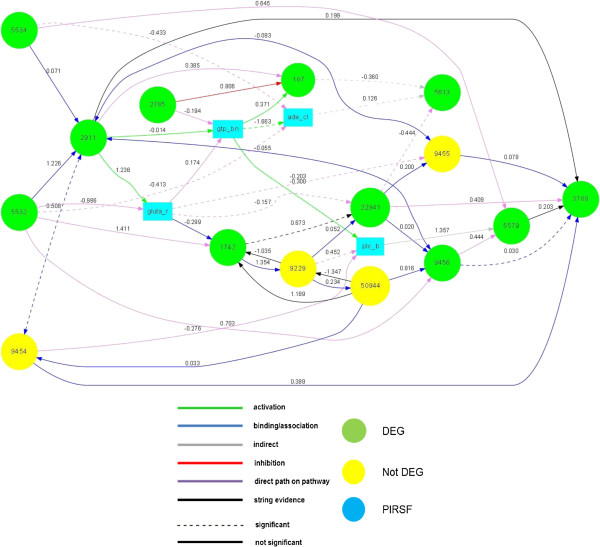
Final model of the Glutamatergic synapse pathway. The graph displays which nodes and edges are different between the yes/no mutant groups. The nodes (with “Entrez” name) or edges are mapped with different colours according to up/down expression or regulation, respectively.
Multiple sclerosis
MS is a neurodegenerative disease with a presumed autoimmune component. The genome-wide expression study in peripheral blood mononuclear cells (PBMC), from 12 MS patients and 15 controls, was performed in order to identify DEGs and dysregulated pathways. Data are freely available at GEO database with ID GSE21942. For the analysis, we used expression values submitted in the database. The SAM was performed using a delta value of 0.95 and a minimum fold-change of 2. The number of genes up-expressed was of 133 while the number of gene down-expressed of 92. Using this list of DEGs, the SPIA analysis found three important pathways that could be involved in the mechanism of the development of the MS, as showed in the Table 4.
Table 4.
Perturbed pathways obtained by SPIA on MS data
| Name pathway | pSize | NDE | pNDE | tA | pPERT | pGFdr | Status |
|---|---|---|---|---|---|---|---|
| B cell receptor signaling pathway |
73 |
8 |
0.000 |
6.822 |
0.372 |
0.000 |
Activated |
| Fc gamma R-mediated phagocytosis |
89 |
4 |
0.015 |
-13.66 |
0.006 |
0.036 |
Inhibited |
| Salmonella infection | 71 | 5 | 0.001 | -4.901 | 0.174 | 0.041 | Inhibited |
pSize = number of genes in the pathway; NDE = number of DEGs in the pathway; pNDE = p-value of the enrichment; tA = total perturbation; pPERT = p-value of the perturbation; Status = direction of the perturbation.
The first, B cell receptor (BCR) signaling pathway, is an important component of adaptive immunity. B cells produce and secrete millions of different antibody molecules, each of which recognizes a different antigen. This signalling ultimately results in the expression of immediate early genes that further activate the expression of other genes involved in B cell proliferation, differentiation and immunoglobulin (Ig) production as well as other processes. The role of B cells is well known in MS [47,48]. The second one, Fc gamma R-mediated phagocytosis pathway, includes specialized cell types as macrophages, neutrophils, and monocytes that take part in host-defence mechanisms. Expression of the inhibitory Fc gamma receptor IIB (FcγRIIB) plays an important role during peripheral B cell development, which prevents memory B cells with low affinity or self-reactive receptors from entering the germinal center and becoming IgG positive plasma cells [49]. Furthermore, the decreased expression of Fc gamma RIIB or non-functional Fc gamma RIIB variants are consistently associated with the development of autoimmune tissue inflammation [49]– [51]. Considering the connection between the Fc gamma R-mediated phagocytosis and B cells, and the association of this pathway with autoimmune inflammation, we could conclude that also this pathway could be implicated in the MS phenotype.
Salmonella infection, the third pathway, may appear less interesting, nevertheless it is connected with response to infections that, as showed for the two previous pathways, plays an important role in MS.
SEM analysis of Fc gamma R-mediated phagocytosis KEGG
As an example, we analyzed the Fc gamma R-mediated phagocytosis pathway. The model was obtained starting from the KEGG pathway and finding the shortest paths between DEGs (no PIRSF reduction was performed). The graph reduction of the model specification was from KEGG pathway (mean degree = 10.404, number nodes(edges) = 94 (489)) to DEGs shortest paths model (mean degree = 3.647, number nodes(edges) = 17(31)). The initial model had a poor fit (χ2 (df) = 339.410 (105), P < 0.001, RMSEA (P-close) =0.288 (<0.001), SRMR = 0.321) and the final model was adequate considering the SRMR index (0.098) and RMSEA (P-close) 0.111 (0.059). To reach the final model, twenty edges were added: fifteen using as driving criteria the presence of a directed path between the nodes in the original pathway, and five using STRING database information.
Two-group analysis of the final pathway-model revealed a significant global mean (χ2 diff(df) = 40.3 (16), P < 0.001 of H0: μ1 = μ2 subject to Σ1 = Σ2) and covariance differences (χ2 diff (df) = 124.4 (48), P < 0.001 of H0: Σ1 = Σ2 subject to μ1≠ μ2).
The specific tests, that analyze the effect of the disease-model on genes and edges, are reported in Tables 5 and 6. Five genes resulted group-sensitive: the genes 382 or ARF6, 10093 or ARPC4, 5580 or PRKCD, 5321 or plag2g4a, 1399 or CRKL. The gene ARF6, as showed by [52], could be implicated in the disruptive effects of IL-1b, a gene recently associated with MS [53]. In addition, other group sensitive genes could be associated with MS [54,55]. Considering the edges perturbed, the links between the gene 382 or ARF6 and the gene 1399 or CRKL resulted very interesting. The IL-1 beta, as reported [52], has an effect on the endothelial stability by the cascade MYD88 – ARNO and ARF6, a known regulator of adherents protein localization. In turn the gene CRKL, an adapter protein required for the spreading of epithelial colonies and breakdown of epithelial colonies and the breakdown of adherents junctions in response to hepatocyte growth factor, modulates the gene ARF6 [56]. So the genes IL-1b, ARF6 and CRKL could be involved in the same pathological mechanism. Other interesting significant edges were the connections between gene 23396 or PIP5K1C with gene 8612 or Ppap2c and with gene 8613 or Ppap2b. The gene PIP5K1C catalyzes the synthesis of phosphatidylinositol 4,5-bisphosphate, an essential lipid molecule in various cellular processes. As described by [57] the phosphatidylinositol-4, 5-bisphosphate synthesis has a critical role in the regulation of multiple steps of the synaptic vesicle cycle. This gene is connected with Ppap2c and Ppap2b, genes that have a role in metabolic pathways controlling the synthesis of glycerophospholipids and triacylglycerols, and in receptor-activated signal transduction mediated by phospholipase D, considered a susceptibility factor in sclerosis [58].
Table 5.
Single node and edge differences found between MS and control groups
| Path | Type | Multiple sclerosis ( MS ) | Control (C) | Difference (95% CI) | P-value | Up/down |
|---|---|---|---|---|---|---|
| 382 ← group |
Group on node |
-9.002 |
-7.963 |
-1.04 (-1.65; -0.43) |
0.001 |
MS down-expressed |
| 10093 ← group |
Group on node |
-0.452 |
0.032 |
-0.48 (-0.93; -0.04) |
0.033 |
MS down-expressed |
| 5580 ← group |
Group on node |
-3.054 |
-2.648 |
-0.41 (-0.70; -0.11) |
0.007 |
MS down-expressed |
| 5321 ← group |
Group on node |
0.694 |
1.240 |
-0.55 (-1.00;-0.09) |
0.020 |
MS down-expressed |
| 1399 ← group |
Group on node |
4.688 |
6.126 |
-1.44 (-1.82;-1.05) |
0.000 |
MS down-expressed |
| 8613 ↔ 8612 |
Indirect |
0.001 |
-0.002 |
0.003 (0.001;0.004) |
0.038 |
MS up-associated |
| 23396 ← 8613 |
Indirect |
0.643 |
-4.981 |
5.62 (2.44; 8.81) |
0.001 |
MS up-regulated |
| 23396 ← 5880 |
Activation |
0.079 |
1.207 |
-1.13 (-1.91;-0.35) |
0.005 |
MS down-regulated |
| 23396 ← 8612 |
Indirect |
-6.219 |
-0.945 |
-5.27 (-6.62; -3.92) |
0.000 |
MS down-regulated |
| 10093 ← 8936 |
Activation |
0.576 |
0.267 |
0.31 (0.08; 0.53) |
0.007 |
MS up-regulated |
| 5581 ← 8613 |
Indirect |
0.002 |
0.953 |
-0.95 (-1.41; -0.49) |
0.000 |
MS down-regulated |
| 5581 ← 8612 |
Indirect |
1.343 |
0.104 |
1.24 (0.78; 1.70) |
0.000 |
MS up-regulated |
| 382 ← 1399 |
Directed path |
0.970 |
0.144 |
0.83 (0.19; 1.46) |
0.011 |
MS up-regulated |
| 7454 ← 5338 |
Directed path |
1.070 |
0.111 |
0.96 (0.28; 1.64) |
0.006 |
MS up-regulated |
| 5604 ← 8613 |
Directed path |
0.618 |
3.133 |
-2.52 (-4.64; -0.39) |
0.020 |
MS down-regulated |
| 23396 ← 1399 | Directed path | -0.323 | -0.755 | 0.43 (0.24; 0.62) | 0.000 | MS up-regulated |
Table 6.
Not significant (null edge) in MS and in control groups
| |
|
Multiple sclerosis (MS) |
Control (C) |
|
||
|---|---|---|---|---|---|---|
| Paths | Type | Estimate | P-value | Estimate | P-value | MS/C |
| 23396 ← 382 |
Activation |
0.114 |
0.657 |
0.082 |
0.324 |
OFF/OFF |
| 10093 ← 7454 |
Activation |
0.419 |
0.199 |
0.065 |
0.726 |
OFF/OFF |
| 5580 ← 8612 |
Indirect |
0.326 |
0.388 |
5.119 |
0.144 |
OFF/OFF |
| 5894 ← 5580 |
Activation |
0.338 |
0.207 |
-0.103 |
0.657 |
OFF/OFF |
| 5894 ← 5581 |
Activation |
1.399 |
0.436 |
-2.815 |
0.349 |
OFF/OFF |
| 5595 ← 5604 |
Phoshorylation |
-0.107 |
0.808 |
-0.963 |
0.408 |
OFF/OFF |
| 1794 ← 1399 |
Binding/association |
0.056 |
0.399 |
-0.007 |
0.747 |
OFF/OFF |
| 8936 ← 5880 |
Activation |
0.015 |
0.972 |
-0.410 |
0.240 |
OFF/OFF |
| 7454 ← 382 |
Directed path |
0.057 |
0.753 |
-0.076 |
0.696 |
OFF/OFF |
| 5604 ← 5338 |
Directed path |
-0.024 |
0.810 |
-0.170 |
0.454 |
OFF/OFF |
| 5595 ← 5338 |
Directed path |
0.124 |
0.404 |
0.590 |
0.065 |
OFF/OFF |
| 5338 ← 382 |
Activation |
0.164 |
0.612 |
0.216 |
0.007 |
OFF/ON |
| 5580 ← 8613 |
Indirect |
3.021 |
0.133 |
-0.819 |
0.019 |
OFF/ON |
| 5604 ← 5894 |
Activation |
0.160 |
0.183 |
0.273 |
0.041 |
OFF/ON |
| 5321 ← 5595 |
Activation |
0.356 |
0.535 |
-0.285 |
0.052 |
OFF/ON |
| 382 ← 5880 |
Activation |
0.302 |
0.455 |
1.314 |
0.002 |
OFF/ON |
| 5580 ← 382 |
Directed path |
-0.073 |
0.688 |
0.156 |
0.005 |
OFF/ON |
| 382 ← 1399 |
Directed path |
0.144 |
0.414 |
0.970 |
0.001 |
OFF/ON |
| 7454 ← 5338 |
Directed path |
0.111 |
0.358 |
1.070 |
0.002 |
OFF/ON |
| 10093 ← 382 |
Directed path |
0.269 |
0.252 |
0.498 |
0.001 |
OFF/ON |
| 7454 ← 5880 |
String evidence |
0.109 |
0.621 |
0.864 |
0.077 |
OFF/OFF |
| 5581 ← 5595 |
String evidence |
0.001 |
0.978 |
0.049 |
0.001 |
OFF/ON |
| 23396 ← 5880 |
Activation |
1.207 |
0.000 |
0.079 |
0.708 |
ON/OFF |
| 5581 ← 8613 |
Indirect |
0.953 |
0.000 |
0.002 |
0.921 |
ON/OFF |
| 10093 ← 5880 |
Directed path |
1.044 |
0.000 |
0.365 |
0.397 |
ON/OFF |
| 8613 ← 1399 |
Directed path |
-0.024 |
0.027 |
-0.093 |
0.225 |
ON/OFF |
| 8936 ← 1794 |
Directed path |
1.819 |
0.008 |
-1.627 |
0.608 |
ON/OFF |
| 5580 ← 5338 | String evidence | 0.417 | 0.004 | 0.258 | 0.222 | ON/OFF |
The comparison with KEGG edges showed that in the model pathway, 22 edges were not active (off) in the MS patients and 18 in controls. In contrast, the edges activated (on) were 6 and 10 in MS patients and control subjects respectively (cf. Table 6). The Figure 3 illustrates the pathway model and the links found statistically significant between the MS/control groups.
Figure 3.
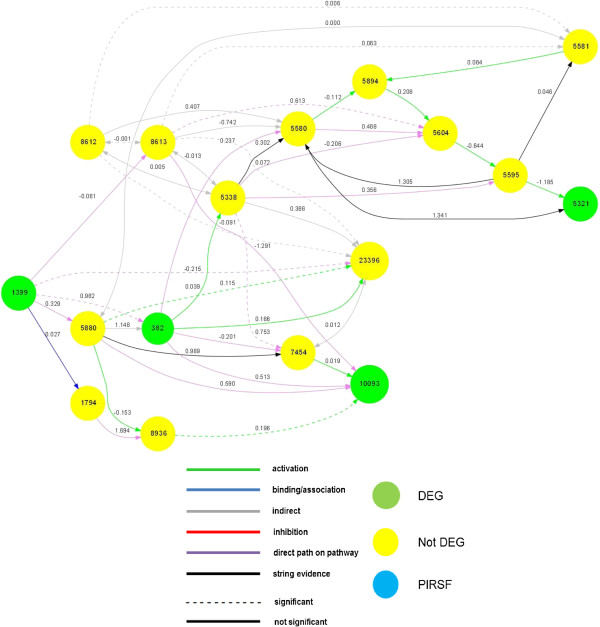
Final model of Fc gamma R-mediated phagocytosis pathway. The graph displays which nodes and edges are different between the disease/control groups. The nodes (with “Entrez” name) or edges are mapped with different colours according to up/down expression or regulation, respectively.
Discussion
This work illustrated a new procedure based on Structural Equation Modeling (SEM) to discover and compare perturbed pathway-modules, similar to that proposed by [12]. Unlike these authors, new pathway analysis (SPIA) and new model generation, based on mixed graph theory and principles of network biology, were added. Starting from the significant dysregulated pathways, a model for each pathway was generated, that allowed both to verify how DEGs were connected between them in a causal structure and to improve the model interpretation by grouping not DEGs in PIR superfamilies. To note that the initial model is not inferred by data, because connections among genes represent the biological knowledge enclosed in the pathway and in PIRSFs. The use of SEM proved to be very versatile in the downstream analysis of microarray data. It was used to: 1) test and improve the generated models (this could be a useful way to overcome the limitation of accuracy relative to the public pathway database); 2) verify the overall differences between groups, and 3) individuate differential expression level of one gene and differential connection between two genes.
Our procedure was tested on two experiments of gene expression microarray data finalized to unravel the biological mechanisms that allowed us to explain the differences between yes/no mutant groups and disease/healthy groups. Starting from the output of SPIA, the model generation procedure, illustrated in this paper, was applied. The first step was to obtain a subgraph for each perturbed pathway containing only the genes present in the microarray. Then, the connections between DEGs through shortest paths were found, and finally not DEGs were grouped in PIRSFs. The models generated were tested and improved using an integrate approach based on the combination between SEM and other type of evidences as explicated in the STRING database. Once a good model has been determined, a two-groups SEM analysis has been performed to unveil significant differences between groups. Studying each link present in the final perturbed model, we hypothesized which connections could be altered. For example in FTLD-U experiment, we found the connection between SHANK2 and PSD-95, and in MS we hypothesized that the genes IL-1b, ARF6 and CRKL could be involved in a same pathological mechanism. These results confirmed and could be able to elucidate the mechanisms that lead to the pathogenesis and progression of the observed diseases.
The framework illustrated, being a composition of different methods, could be easily adapted to new solutions. The idea is to have a general and modular framework where different other methods could be taken in consideration in each step of the pipeline.
First, alternative ways to select DEGs and perturbed pathways could be considered. In [59] a unified framework was proposed to jointly find significant perturbed pathways and DEGs by sparse Linear Discriminant Analysis (sLDA). Other methods for identifying DEGs and differential connections, based on Graphical Gaussian Modeling (GGM), are the following: a first one generates networks directly from very high dimensional data, determining the pattern of zeros in the inverse covariance matrix [60]; a second one defines Bayesian networks (directed acyclic graph: DAGs) on a structure of dependence derived from external resources [61]; the last one derives DAGs from external resources converting them into undirected cycle-free graphs [62]. An additional approach for conducting a differential analysis of networks directly from data by measuring gene association/interaction with connectivity scores, based on Partial Least Squares (PLS), was suggested by [63].
Second, we used PCA to create new observed composite variables that represent PIRSFs. Another plan could be to take advantage of the potentiality of SEM in the creation of latent variables as proposed by [64] in transcriptional regulation of protein-DNA interactions. A valid alternative to the modification indices of SEM could be the PC-algorithm [65], that allow to infer causal information from data. The idea in this case is to fix an initial model, provided by the perturbed pathways model, and then use the PC-algorithm to add new links supervised by STRING knowledge. Lastly, since in SEM multigroup analysis numerous hypothesis are tested, multiple testing control procedures using the method of [66] can be desirable.
Conclusions
The pipeline proposed introduces in the analysis of gene expression data the main principles that govern biological networks as well described by [14]. Otherwise from reverse-engineering gene regulatory networks, that build networks directly from data, our initial models are obtained by a biological curated pathway database (KEGG) and then modified on the basis of the knowledge provided by another database (STRING). The principal evidence is that the manifestation of a particular phenotype depends on the interactions existing among many causal agents. This was obtained taking in consideration not only DEGs but also how genes interact. The pipeline has been validated on two expression datasets. In both the cases, we tested the models, improved them and individuated the gene expression levels and the connections that were perturbed and that could justify the different phenotypes observed. The results were satisfactory and strongly coherent with experimental findings available in literature, considering that most of the genes in the model are known to characterize the phenomena analyzed and that links perturbed were previously connected to the progression of the diseases.
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
DP defined the procedure to generate models. MG supervised the work, in particular the statistical part, and together with DP, developed the procedure to test the models. DP and MG wrote jointly the paper. All the authors read and approved the final manuscript.
Supplementary Material
R codes for a demonstration of the procedure for the Multiple Sclerosis (MS) example.
Contributor Information
Daniele Pepe, Email: danielepepe84@gmail.com.
Mario Grassi, Email: mario.grassi@unipv.it.
Acknowledgements
This work was funded by Laboratory of Multivariate Statistics, Department of Brain and Behavioural Sciences, Medical and Genomic Statistics Unit, University of Pavia, Pavia- Italy. We want to thank, the PhD student Fernando Palluzzi and the PhD Valeria Carossa for their advice and helping.
References
- Barabási AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Hartemink AJ. Reverse engineering gene regulatory networks. Nat biotech. 2005;23:554–555. doi: 10.1038/nbt0505-554. [DOI] [PubMed] [Google Scholar]
- Shipley B. Cause and Correlation in Biology. A User’s Guide to Path Analysis, Structural Equations, and Causal Inference. New York: Cambridge University Press; 2000. [Google Scholar]
- Rosa GJ, Valente BD, de los Campo G, Wu XL, Gianola D, Silva MA. Inferring causal phenotype networks using structural equation models. Gen Select Evol. 2011;43:6. doi: 10.1186/1297-9686-43-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Namkung J, Lee S, Park T. Application of structural equation models to genome-wide association analysis. Genomics & Informatics. 2010;8:150–158. doi: 10.5808/GI.2010.8.3.150. [DOI] [Google Scholar]
- Mi X, Eskridge KM, George V, Wang D. Structural equation modeling of gene–environment interactions in coronary heart disease. Ann Hum Genet. 2011;75:255–265. doi: 10.1111/j.1469-1809.2010.00634.x. [DOI] [PubMed] [Google Scholar]
- Rao DC, Province MA. The future of path analysis, segregation analysis, and combined models for genetic dissection of complex traits. Hum Hered. 1999;50(1):34–42. doi: 10.1159/000022889. [DOI] [PubMed] [Google Scholar]
- Province MA, Rice TK, Borecki IB, Gu C, Kraja A, Rao DC. Multivariate and multilocus variance components method, based on structural relationships to assess quantitative trait linkage via SEGPATH. Genet Epidemiol. 2003;24(2):128–138. doi: 10.1002/gepi.10208. [DOI] [PubMed] [Google Scholar]
- Li R, Tsaih SW, Shockley K, Stylianou IM, Wergedal J, Paigen B, Churchill GA. Structural model analysis of multiple quantitative traits. PLoS Genet. 2006;2(7):e114. doi: 10.1371/journal.pgen.0020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Bentler PM. Covariance structure models for gene expression microarray data. Struct Equ Model. 2003;10:566–582. doi: 10.1207/S15328007SEM1004_5. [DOI] [Google Scholar]
- Xiong M, Li J, Fang X. Identification of genetic networks. Genetics. 2004;166(2):1037–1052. doi: 10.1534/genetics.166.2.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Sharpe K, Zhang T, Chen H, Zhu W, Li E, Taghavi S, Van Der Lelie D. Proceeding of IEEE First International Conference on Computational Advances in Bio and Medical Sciences (ICCABS 2011): 3–5 February 2011; Orlando. Washington, DC, USA: IEEE Computer Society; 2011. Comparative genetic pathway analysis using structural equation modeling; pp. 190–195. [Google Scholar]
- Khatri P, Sirota M, Butte A. Ten years of pathway analysis: current approaches and outstanding challenges. Plos Comp Bio. 2012;8:15–22. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolskaya AN, Arighi CN, Huang H, Barker WC, Wu CH. PIRSF family classification system for protein functional and evolutionary analysis. Evol Bioinform Online. 2006;2:197–209. [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan S, Mital P, Kim J, Kim C, Kusanovic JP, Romero R. A novel signaling pathway impact analysis for microarray experiments. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Chu G, Balasubramanian N, Jun L. samr: SAM: Significance Analysis of Microarrays. R reference manual: August 29, 2013. [ http://cran.r-project.org/web/packages/samr/samr.pdf]
- Tarca AL, Kathri P, Draghici S. SPIA: Signaling Pathway Impact Analysis (SPIA) using combined evidence of pathway over-representation and unusual signaling perturbations. R reference manual: May 27, 2014. [ http://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf]
- Wright SS. Correlation and causation. J Agric Res. 1921;20:557–585. [Google Scholar]
- Bollen KA. Structural Equations with Latent Variables. New York: Wiley; 1989. [Google Scholar]
- Pearl J. Graphs, causality, and structural equation models. Socio Meth Res. 1998;27:226–284. doi: 10.1177/0049124198027002004. [DOI] [Google Scholar]
- Csardi G, Nepusz T. igraph: network analysis and visualization. R reference manual: April 22, 2014. [ http://cran.r-project.org/web/packages/igraph/igraph.pdf]
- Rigdon E. A necessary and sufficient identification rule for structural models estimated in practice. Multivar Behav Res. 1995;30:359–383. doi: 10.1207/s15327906mbr3003_4. [DOI] [PubMed] [Google Scholar]
- Brito C, Pearl J. A new identification condition for recursive models with correlated errors. Struct Equ Model. 2002;9:459–474. doi: 10.1207/S15328007SEM0904_1. [DOI] [Google Scholar]
- Grace JB, Bollen KA. Representing general theoretical concepts in structural equation models: the role of composite variables. Environ Ecol Stat. 2008;15:191–213. doi: 10.1007/s10651-007-0047-7. [DOI] [Google Scholar]
- Sales G, Calura E, Romualdi C. Graphite: GRAPH Interaction from pathway Topological Environment. R reference manual: May 27, 2014, [ http://www.bioconductor.org/packages/release/bioc/manuals/graphite/man/graphite.pdf]
- R Development Core Team: The R Stats package. In A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. http://cran.r-project.org/ [Google Scholar]
- Bollen KA, Stine RA. Bootstrapping goodness-of-fit measures in structural equation models. Sociol Methods Res. 1992;21:205–229. doi: 10.1177/0049124192021002004. [DOI] [Google Scholar]
- Browne MW, Cudeck R. Alternative ways of assessing model fit. Sage Focus Editions. 1993;154:136. [Google Scholar]
- Hu LT, Bentler PM. Cutoff criteria for fit indexes in covariance structure analysis: conventional criteria versus new alternatives. Struct Equ Modeling. 1999;6:1–55. doi: 10.1080/10705519909540118. [DOI] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Jensen LJ. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(D1):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosseel Y. lavaan: An R Package for Structural Equation Modeling. J Stat Softw. 2012;48(2):1–36. http://www.jstatsoft.org/v48/i02/ [Google Scholar]
- Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Lee VMY. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008;17:1349–1362. doi: 10.1093/hmg/ddn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130(Suppl 4):1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int. 2004;45:583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Ferrer I. Neurons and their dendrites in frontotemporal dementia. Dement Geriatr Cogn Disord. 1999;10(Suppl 1):55–60. doi: 10.1159/000051214. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Tunon T, Serrano MT, Casas R, Alcantara S, Zujar MJ, Rivera RM. D-28 k and parvalbumin immunoreactivity in the frontal cortex in patients with frontal lobe dementia of non-Alzheimer type associated with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1993;56:257–261. doi: 10.1136/jnnp.56.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriaens ME, Jaillard M, Waagmeester A, Coort SL, Pico AR, Evelo CT. The public road to high-quality curated biological pathways. Drug Discov Today. 2008;13:856–862. doi: 10.1016/j.drudis.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Lippa CF. Review: disruption of the postsynaptic density in Alzheimer’s disease and other neurodegenerative dementias. Am J Alzheimers Dis Other Demen. 2010;25:547–555. doi: 10.1177/1533317510382893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen EY, Wexler EM, Versano R, Coppola G, Gao F, Winden KD, Geschwind DH. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron. 2011;71:1030–1042. doi: 10.1016/j.neuron.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Gong Y, Lippa CF, Zhu J, Lin Q, Rosso AL. Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer’s disease. Brain Res. 2009;1292:191–198. doi: 10.1016/j.brainres.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn-Hardy K, Fung HC, Lin X, Hernandez D, Simon-Sanchez J, Wood NW, Giunti P, Rafferty I, Hardy J, Storey E, Gardner RJ, Forrest SM, Fisher EM, Russell JT, Cai H, Singleton AB. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007;3:e108. doi: 10.1371/journal.pgen.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GP, Bennett JL, Gilden DH, Burgoon MP. The B cell response in multiple sclerosis. Neurol Res. 2006;28:236–244. doi: 10.1179/016164106X98099. [DOI] [PubMed] [Google Scholar]
- von Büdingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, Cree BA, Gourraud PA, Schwartzburg A, Huerta G, Telman D, Sundar PD, Casey T, Cox DR, Hauser SL. B cell exchange across the blood–brain barrier in multiple sclerosis. J Clin Investig. 2012;122:4533. doi: 10.1172/JCI63842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, Diamond B. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J Exp Med. 2006;203:2157–2164. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg B, Jelčić I, Baerenwaldt A, Oertel WH, Sommer N, Nimmerjahn F, Lünemann JD. Impaired inhibitory Fcγ receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci. 2009;106:4788–4792. doi: 10.1073/pnas.0807319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, London NR, Gibson CC, Davis CT, Tong Z, Sorensen LK, Shi DS, Guo J, Smith MC, Grossmann AH, Thomas KR, Li DY. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature. 2012;492:252–255. doi: 10.1038/nature11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F, Nisticò R, Mandolesi G, Piccinin S, Mango D, Kusayanagi H, Berretta N, Bergami A, Gentile A, Musella A, Nicoletti CG, Nicoletti F, Buttari F, Mercuri NB, Martino G, Furlan R, Centonze D. Interleukin-1β promotes long-term potentiation in patients with multiple sclerosis. NeuroMolecular Med. 2013. Epub ahead of print. [DOI] [PubMed]
- Iglesias AH, Camelo S, Hwang D, Villanueva R, Stephanopoulos G, Dangond F. Microarray detection of E2F pathway activation and other targets in multiple sclerosis peripheral blood mononuclear cells. J Neuroimmunol. 2004;150:163–177. doi: 10.1016/j.jneuroim.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Gilli F. Role of differential expression of interferon receptor isoforms on the response of multiple sclerosis patients to therapy with interferon beta. J Interf Cytokine Res. 2010;30:733–741. doi: 10.1089/jir.2010.0098. [DOI] [PubMed] [Google Scholar]
- Lamorte L, Rodrigues S, Sangwan V, Turner CE, Park M. Crk associates with a multimolecular Paxillin/GIT2/β-PIX complex and promotes Rac-dependent relocalization of Paxillin to focal contacts. Mol Biol Cell. 2003;14:2818–2831. doi: 10.1091/mbc.E02-08-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P. Impaired PtdIns(4,5)P(2) synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431:415–422. doi: 10.1038/nature02896. [DOI] [PubMed] [Google Scholar]
- Terao C, Ohmura K, Kawaguchi Y, Nishimoto T, Kawasaki A, Takehara K, Mimori T. PLD4 as a novel susceptibility gene for systemic sclerosis in a Japanese population. Arthritis Rheum. 2013;65:472–480. doi: 10.1002/art.37777. [DOI] [PubMed] [Google Scholar]
- Wu MC, Zhang L, Wang Z, Christiani DC, Lin X. Sparse linear discriminant analysis for simultaneous testing for the significance of a gene set/pathway and gene selection. Bioinformatics. 2009;25:1145–1151. doi: 10.1093/bioinformatics/btp019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiiveri HT. Multivariate analysis of microarray data: differential expression and differential connection. BMC Bioinforma. 2011;12:42. doi: 10.1186/1471-2105-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D, Wang L, Sørensen P. Network-enabled gene expression analysis. BMC Bioinforma. 2012;13:167. doi: 10.1186/1471-2105-13-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini P, Sales G, Massa MS, Chiogna M, Romualdi C. Along signal paths: an empirical gene set approach exploiting pathway topology. Nucl Acids Res. 2013;41:e19. doi: 10.1093/nar/gks866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Datta S, Datta S. A statistical framework for differential network analysis from microarray data. BMC Bioinforma. 2010;11:95. doi: 10.1186/1471-2105-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aburatani S. Application of structure equation modeling for inferring a serial transcriptional regulation in yeast. Gene Regul Syst Bio. 2011;5:75–88. doi: 10.4137/GRSB.S7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch M, Mächler M, Colombo D, Maathuis MH, Bühlmann P. Causal inference using graphical models with the R package pcalg. J Stat Softw. 2011;47:1–26. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JR Stat Soc Ser B(Methodological) 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
R codes for a demonstration of the procedure for the Multiple Sclerosis (MS) example.