

Abstract
The Corynebacterium glutamicum ATCC 31831 araBDA operon consists of three l-arabinose catabolic genes, upstream of which the galM, araR, and araE genes are located in opposite orientation. araR encodes a LacI-type transcriptional regulator that negatively regulates the l-arabinose-inducible expression of araBDA and araE (encoding an l-arabinose transporter), through a mechanism that has yet to be identified. Here we show that the AraR protein binds in vitro to three sites: one upstream of araBDA and two upstream of araE. We verify that a 16-bp consensus palindromic sequence is essential for binding of AraR, using a series of mutations introduced upstream of araB in electrophoretic mobility shift assays. Moreover, the DNA-binding activity of AraR is reduced by l-arabinose. We employ quantitative reverse transcription-PCR (qRT-PCR) analyses using various mutant strains deficient in l-arabinose utilization genes to demonstrate that the prominent upregulation of araBDA and araE within 5 min of l-arabinose supplementation is dependent on the uptake but independent of the catabolism of l-arabinose. Similar expression patterns, together with the upregulation by araR disruption without l-arabinose, are evident with the apparent galM-araR operon, although attendant changes in expression levels are much smaller than those realized with the expression of araBDA and araE. The AraR-binding site upstream of araB overlaps the −10 region of the divergent galM promoter. These observations indicate that AraR acts as a transcriptional repressor of araBDA, araE, and galM-araR and that l-arabinose acts as an intracellular negative effector of the AraR-dependent regulation.
INTRODUCTION
Corynebacterium glutamicum is a Gram-positive Actinobacteria species with a high G+C content in its genomic DNA. It is currently used in large-scale industrial production of amino acids such as l-glutamate and l-lysine (1, 2). Moreover, investigation into its application in the production of fuels and commodity chemicals has started to bear fruit (3–5). This is predicated on its potential to utilize cellulosic biomass, a renewable energy source that should play an important role in the global renewable energy of the future (6, 7). Cellulosic biomass contains significant amounts of pentose sugars, such as d-xylose and l-arabinose, that are often difficult for conventional bioprocesses to exploit. Recent advances in metabolic engineering of C. glutamicum have helped to open up new possibilities for efficient utilization of substrates containing mixtures of d-glucose, d-xylose, and l-arabinose (8–11). An understanding of the transcriptional regulation of sugar metabolism genes in C. glutamicum is an attractive avenue toward optimization of utilization of such sugar mixtures.
l-Arabinose is utilized by a Gram-negative bacterium, Escherichia coli, and a low-G+C Gram-positive bacterium, Bacillus subtilis, via a common pathway. l-Arabinose taken up by cells is sequentially converted to l-ribulose, l-ribulose 5-phosphate, and d-xylulose 5-phosphate by the actions of l-arabinose isomerase (encoded by araA), l-ribulokinase (encoded by araB), and l-ribulose 5-phosphate 4-epimerase (encoded by araD), respectively. d-Xylulose 5-phosphate is further catabolized through the pentose phosphate pathway. Despite the conservation of this pathway among various bacteria, two distinct regulatory systems of the l-arabinose utilization genes have been well established so far. In E. coli, AraC acts as an activator and as a repressor that regulates transcription of the genes required for the catabolism (araBAD) and uptake (araE and araFGH) of l-arabinose (12). AraC is composed of an N-terminal arabinose-binding and dimerization domain and a C-terminal DNA-binding domain. The expression of the araBAD operon and the AraC-encoding gene (araC), which are divergently transcribed, is repressed by binding of AraC to the promoter region forming a DNA loop in the absence of l-arabinose (13–15). In the presence of l-arabinose, three promoters, ParaBAD, ParaE, and ParaFGH, are activated, and ParaC is derepressed (16). In B. subtilis, AraR, which is structurally different from E. coli AraC, controls the transcription of several genes involved in the uptake and degradation of arabinose-containing polysaccharides, in addition to the genes corresponding to the E. coli AraC regulon (17–19). AraR comprises an N-terminal DNA-binding domain homologous to the GntR family proteins and a C-terminal effector-binding domain that shows similarity to members of the LacI/GalR family (20, 21). B. subtilis AraR binds to five different promoter regions with distinct affinities (ParaABDLMNPQ-abfA, ParaE, PabnA, Pxsa, and ParaR) (18, 19, 22). Moreover, an l-arabinose transporter encoded by araE under the control of AraR is also responsible for the uptake of d-xylose and d-galactose (23). Therefore, AraR is thought to play a central role in the regulation of utilization of the multiple sugars in B. subtilis. It is noted that the l-arabinose catabolic and uptake genes are also subject to carbon catabolite repression, controlled by the cyclic AMP (cAMP)-dependent transcriptional regulator CRP in E. coli (24) and the LacI family transcriptional regulator CcpA in B. subtilis (25). These distinct regulatory systems are responsible for inhibition of l-arabinose utilization in the presence of d-glucose.
Most strains of C. glutamicum tested cannot grow using l-arabinose as the sole carbon source owing to the lack of the pentose sugar utilization pathway (26). However, C. glutamicum ATCC 31831 was recently shown to efficiently utilize l-arabinose due to the presence of araBDA and araE in its genome (26). The expression of these genes associated with l-arabinose utilization is induced in the presence of l-arabinose and is also upregulated by inactivation of araR, encoding a LacI family transcriptional regulator, in the absence of l-arabinose (26). Although the AraR protein is suggested to be involved in the regulation of the l-arabinose-inducible genes in this strain, whether or not AraR directly regulates the relevant genes remains to be verified. It should be noted that AraR of C. glutamicum ATCC 31831 and AraR of B. subtilis belong to distinct families, although the latter protein reveals a mosaic structure with a C-terminal effector-binding domain that has some similarity to the former, LacI family protein. Furthermore, C. glutamicum ATCC 31831 simultaneously utilizes l-arabinose and d-glucose (26), apparently in contrast to the glucose repression of l-arabinose utilization genes in E. coli and B. subtilis described earlier. Thus, elucidation of the regulatory mechanism of the l-arabinose utilization genes in C. glutamicum ATCC 31831 should extend our understanding of the regulatory systems of sugar metabolism diversified among bacterial species.
In this study, we show that C. glutamicum ATCC 31831 AraR binds in vitro to 16-bp consensus palindromic sequences found separately at one site upstream of araBDA and at two sites upstream of araE. The AraR-binding site upstream of araB overlaps the promoter of the apparent galM-araR operon, divergently transcribed from araBDA. The binding activity of AraR is reduced by l-arabinose. In addition, we show effects of araR, araB, araD, araA, or araE deletion on l-arabinose-inducible gene expression in vivo that suggest that AraR acts as a transcriptional repressor of araBDA, araE, and galM-araR. The AraR-mediated expression of these multiple transcriptional units is coordinately derepressed in the presence of l-arabinose in response to uptake of the sugar, but independent of its catabolism.
MATERIALS AND METHODS
Bacterial strains.
C. glutamicum ATCC 31831 was used as a wild-type strain in this study. The araB, araD, and araR single-deletion mutant strains were described previously (26). E. coli strains JM109 (TaKaRa), JM110 (27), and BL21(DE3) (28) were used for cloning and/or expression of genes of interest.
Culture conditions.
For genetic manipulations, E. coli strains were grown at 37°C in Luria-Bertani (LB) medium (27). C. glutamicum strains were grown at 33°C in nutrient-rich A medium [2 g/liter yeast extract, 7 g/liter Casamino Acids, 2 g/liter (NH2)2CO, 7 g/liter (NH4)2SO4, 0.5 g/liter KH2PO4, 0.5 g/liter K2HPO4, 0.5 g/liter MgSO4·7H2O, 6 mg/liter FeSO4·7H2O, 4.2 mg/liter MnSO4·H2O, 0.2 mg/liter biotin, 0.2 mg/liter thiamine] (29) with 4% glucose. Where appropriate, the culture medium was supplemented with 50 μg ml−1 of kanamycin.
For analytical purposes, a C. glutamicum starter culture was grown aerobically in 10 ml of nutrient-rich A medium in a 100-ml test tube overnight. The cells were inoculated into fresh medium at a dilution of 100-fold or higher. The cells were cultured in 100 ml of nutrient-rich A medium at 33°C in a 500-ml flask. To assess the response to sugars, the medium was supplemented with l-arabinose and/or d-glucose at the stated concentrations.
DNA technique.
Chromosomal DNA was isolated from C. glutamicum cells by using a GenomicPrep cell and tissue DNA isolation kit (GE Healthcare, Buckinghamshire, England) according to the manufacturer's instructions, but modified by using 10 mg ml−1 lysozyme at 37°C for 1 h. Plasmid DNA was isolated using a QIAprep Spin miniprep kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions, but modified for extraction from C. glutamicum cells by using 10 mg ml−1 lysozyme at 37°C for 1 h.
PCR was performed using a model 480 DNA thermal cycler (PerkinElmer, MA). After the reaction mixture was heated at 96°C for 2 min, the PCR proceeded with 30 cycles of 15 s at 96°C, 30 s at 55°C, and 2 min at 68°C by using DNA polymerase KOD Plus (Toyobo, Osaka, Japan).
C. glutamicum cells were transformed by electroporation as described previously (30). E. coli cells were transformed by the CaCl2 procedure (27).
DNA sequencing was performed by the dideoxy chain termination method (31), using an ABI Prism 3100xl genetic analyzer (Applied Biosystems, Foster City, CA) and a BigDye Terminator, version 3.1, cycle sequencing kit (Applied Biosystems). DNA sequence data were analyzed with the Genetyx program (Genetyx Corporation, Tokyo, Japan). Sequences were aligned by use of the ATGC program (Genetyx Corporation).
Mutant construction.
The upstream and downstream regions of the target gene for deletion were amplified using sets of primers summarized in Table 1. The resultant amplicons were fused and cloned into pCRD206 (32), a suicide vector for markerless gene disruption. The resultant plasmids, pCRF500 and pCRF501, were used for in-frame deletion of araA and araE, respectively. C. glutamicum was subsequently transformed with the respective plasmid DNA, and screening for deletion mutants was performed as described previously (32). Deletion of the target genes was confirmed by PCR.
TABLE 1.
Primers used in this study
| Primer purpose and name | Sequence (5′–3′) | Overhanging restriction sitea |
|---|---|---|
| Plasmid construction for gene deletion | ||
| araA-ML-U-F | GCTCTAGAGTGCTGACGATGATGGCGGA | XbaI |
| araA-ML-U-R | CTCTACTAGTTCCATGTCGATCGAAGACCA | SpeI |
| araA-ML-D-F | GCTCTAGAAGTGCTATGGCAGTGCAACT | XbaI |
| araA-ML-D-R | CTCTACTAGTACTCACTGCGAACAAGGTCA | SpeI |
| araE-ML-U-F | GCTCTAGATCGCTCGGACACCGTCACAT | XbaI |
| araE-ML-U-R | GCGGATCCACCGAAGAAGTAGATGTACG | BamHI |
| araE-ML-D-F | CGGGATCCATCCCGCTCATCGTCGAGAA | BamHI |
| araE-ML-D-R | GCGATATCTGAGAGATGCCGTTGGTGCT | EcoRV |
| Plasmid construction for gene expression | ||
| AraR-Ex-F | GGAATTCCATATGAGCTCCACCCAG | NdeI |
| AraR-Ex-R | GGAATTCTCAGAGCTGTGTGAAATC | EcoRI |
| RT-PCR analysis | ||
| 16S-F | CAGGTCTCTGGGCAGTAACTGA | |
| 16S-R | CGTTTACGGCATGGACTACCA | |
| araA-F | GCGCCAATGACAACGTCAT | |
| araA-R | TCATTAGCCTGGGTGTGAAGGT | |
| araB-F | GGATCGAGCTCTTCAGTGAGGTT | |
| araB-R | CACCGGAGACGAAGTTGTAGGT | |
| araD-F | GCGATGGTGGTGTGCAACA | |
| araD-R | GGTCGAATGAGTGTGCACGAT | |
| araE-F | CATGTGGCCGATCATCCA | |
| araE-R | CGGCGATGACCATGAACA | |
| araR-F | GCACGCTGTCAAGCATTGA | |
| araR-R | GGTCTCTTCGGCATTTTTCATC | |
| galM-F | GGTCTTCGCCACCTTCTCTCT | |
| galM-R | GTGGTGTCGCCGACATTGT | |
| 5′-RACE | ||
| araB-RA-RTb | TGAGAACTAGGTCG | |
| araB-RA-F1 | ACGGAGTTGAAAGTATGGAG | |
| araB-RA-R1 | GTTCGGTTCCTTGATGAC | |
| araB-RA-F2 | AATTCGGATCGACCCGCATT | |
| araB-RA-R2 | TTTGAGAGCGGAGCTTCGTA | |
| araE-RA-RTb | GATCATCGTCTGGT | |
| araE-RA-F1 | CGACCGTACATCTACTTCTT | |
| araE-RA-R1 | GTTGAACAGTCTCTGTCATC | |
| araE-RA-F2 | TCCTCTTCTCTGCGCTGGTA | |
| araE-RA-R2 | GCTGTCTGAGCGGATGATGT | |
| galM-RA-RTb | TGCCATCGGTGTAGA | |
| galM-RA-F1 | TCCACGGTGGTGTTCTCGTT | |
| galM-RA-R1 | TTGATCGTGACCTGTTTTCC | |
| galM-RA-F2 | CCGACGGCATCTACACCGAT | |
| galM-RA-R2 | CTTCTGGGGCGATGTGTCAT | |
| Electrophoretic mobility shift assay | ||
| PA-F | TGGTGCTGCCATCGGTGTAG | |
| PA-R | AGCTTCGTATTGGTGTTATCGCT | |
| PB-F | CCTCGAGTGAGTAGCTCCAG | |
| PB-R | CTTCTGGGGCGATGTGTCAT | |
| PC-F | TCTGGGTTCTCGGGTGATAA | |
| PC-R | GCTGTCTGAGCGGATGATGT | |
| PD-F | ACCGTCTCCTCCCGATCAGA | |
| PD-R | TGGGTGGAGCTCATGGTGAT | |
| P2-F | TGCGATCCGGTCACT | |
| P3-F | AATGCGGGTCGATCCGAA | |
| P4-F | ACGGTCCACGACACT | |
| P5-F | GTTCGGTTCCTTGATGAC | |
| P6-R | GTGTGATGCTTGCTTGAC | |
| P7-R | GATGGCCGTTAAGAAC | |
| P8-R | CTCAAATGTGATCCGAAGC | |
| P9-R | ACGGAGTTGAAAGTATGGAG | |
| DNase I footprinting analysis | ||
| pUC-IR-Fw | TTGTAAAACGACGGCCAGTG | |
| pUC-IR-Rvc | GGAAACAGCTATGACCATGA | |
| araB-DF-F | CTTCTGGGGCGATGTGTCAT | |
| araB-DF-R | AGCTTCGTATTGGTGTTATCGCT | |
| araE-DF-F | CGAGCGACAAACACGACA | |
| araE-DF-R | TTGTCCTGGTTCTTCGAC |
The restriction site overhangs used in the cloning procedure are underlined in the sequences.
5′-Phosphorylated primer.
5′-IRD700-labeled primer.
Quantitative reverse transcription-PCR (qRT-PCR) analysis and rapid amplification of cDNA ends (RACE).
Total RNA was prepared from C. glutamicum cells by using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. Single-stranded cDNA was synthesized from 0.2 μg of total RNA by using PrimeScript reverse transcriptase (TaKaRa, Osaka, Japan) with a random hexamer mixture as a primer in 20 μl of reaction mixture, and then 2 μl of the cDNA mixture was added as a template to 18 μl of reaction mixture containing each primer (0.3 μM) and Power SYBR green PCR master mix (Applied Biosystems). After the reaction mixture was heated at 95°C for 10 min, PCRs proceeded via 40 cycles of 15 s at 95°C and 40 s at 60°C. The amount of amplified DNA was monitored by fluorescence at the end of each cycle by using an Applied Biosystems 7500 Fast real-time PCR system. The primers used are listed in Table 1. The relative abundances of the target mRNAs were quantified based on the cycle threshold value. To standardize the results, the relative abundance of 16S rRNA was used as the internal standard.
The 5′ end of mRNA was determined by RACE, using the primers summarized in Table 1. Using a 5′-Full RACE core set (TaKaRa, Osaka, Japan), single-stranded cDNA synthesized from total RNA by using the 5′-phosphorylated primer was self-ligated with T4 RNA ligase. The first PCR proceeded using inverted primers, and then the second PCR proceeded using nested inverted primers. The amplified DNA was inserted into pUC118 HincII/BAP by using a Mighty Cloning Reagent set (TaKaRa, Osaka, Japan). More than 10 clones from E. coli transformed with the resulting plasmid were sequenced.
Purification of the AraR protein expressed in E. coli.
A DNA fragment containing the araR gene was amplified by PCR using primers AraR-Ex-F and AraR-Ex-R (Table 1). The amplified DNA was digested with NdeI and EcoRI and was inserted into the corresponding site of the pET-28a expression vector (Merck KGaA, Darmstadt, Germany). The resulting plasmid, pCRF502, contains the araR gene fused to a His tag sequence at the N terminus. E. coli BL21(DE3) cells transformed with pCRF502 were grown at 37°C in 100 ml of LB medium supplemented with kanamycin (50 μg ml−1). The recombinant gene was expressed in exponentially growing cells (optical density at 610 nm [OD610] = 0.6) by adding 1 mM isopropyl-β-d-thiogalactopyranoside. After 1 h of incubation, the cells were harvested by centrifugation. The His-tagged AraR protein was extracted and purified by affinity column chromatography, using a Ni-nitrilotriacetic acid (Ni-NTA) Fast Start kit (Qiagen). The AraR protein was loaded onto a gel filtration column (PD-10 column; GE Healthcare UK Ltd., Little Chalfont, United Kingdom) equilibrated with buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol and eluted with the same buffer. The resultant AraR protein was used for an electrophoretic mobility shift assay (EMSA).
EMSA.
The purified His-tagged AraR protein at the indicated concentrations was incubated with a DNA probe in 20 μl of binding buffer containing 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 2.5 mM MgCl2, 0.45 mM EDTA, 0.5 mM dithiothreitol (DTT), 0.05% (vol/vol) Nonidet P-40, and 10% (vol/vol) glycerol for 25 min at 25°C. The binding reaction mixture was subjected to electrophoresis on a 6% polyacrylamide gel containing 5% (vol/vol) glycerol in 0.5× Tris-borate-EDTA (TBE) electrophoresis buffer, and the DNA probe was detected with SYBR green. DNA probes were prepared by PCRs using plasmids containing the promoter regions of interest as templates. Primers used are summarized in Table 1.
DNase I footprinting analysis.
A labeled DNA fragment was prepared by PCR, using pUC-IR-Fw and a 5′-IRD700-labeled primer, pUC-IR-Rv. pUC118 (TaKaRa) containing the region between positions +90 and −34 with respect to the transcription start site of araB or the region between positions +85 and −104 with respect to the transcription start site of araE was used as the template. The purified AraR protein at the stated concentrations was incubated with a DNA probe in 20 μl of binding buffer containing 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 2.5 mM MgCl2, 0.45 mM EDTA, 0.5 mM dithiothreitol, 0.05% Nonidet P-40, and 10% glycerol for 25 min at room temperature. Four microliters of binding buffer containing 1 to 2 mU of DNase I (TaKaRa), 5 mM MgCl2, and 10 mM CaCl2 was then added and incubated for 1 min, followed by the addition of 2 μl of 325 mM EDTA and subsequent heating at 80°C for 10 min. The samples were mixed with IR2 stop solution (Li-Cor, Lincoln, NE), heated at 95°C for 3 min, and separated in a 5.5% KB Plus gel matrix (Li-Cor), using a Li-Cor 4300 DNA analyzer. DNA sequencing reaction mixtures using the same IRD700-labeled primer and a DYEnamic direct cycle sequencing kit with 7-deaza-dGTP (GE Healthcare UK Ltd.) were subjected to the same gel analysis.
RESULTS
l-Arabinose-inducible expression of the l-arabinose utilization cluster genes.
On the chromosome of C. glutamicum ATCC 31831, six genes, i.e., araA, araB, araD, araE, galM, and araR, are located in a cluster (Fig. 1) (26). araA, araB, and araD, each of which is essential for l-arabinose utilization in this strain (26), encode arabinose isomerase, ribulokinase, and ribulose-5-phosphate 4-epimerase, respectively. An H+ symporter encoded by araE is involved in l-arabinose uptake (8). galM and araR encode a putative aldose 1-epimerase and a LacI-type transcriptional regulator, respectively. The araB gene is oriented in the same direction as its two downstream genes, araD and araA. The araB-araD and araD-araA intergenic regions are 13 and 35 bp long, respectively. Upstream of the araB gene, galM is divergently oriented. araR and araE are located downstream of galM, in the same direction. The galM-araR and araR-araE intergenic regions are 26 and 234 bp long, respectively.
FIG 1.
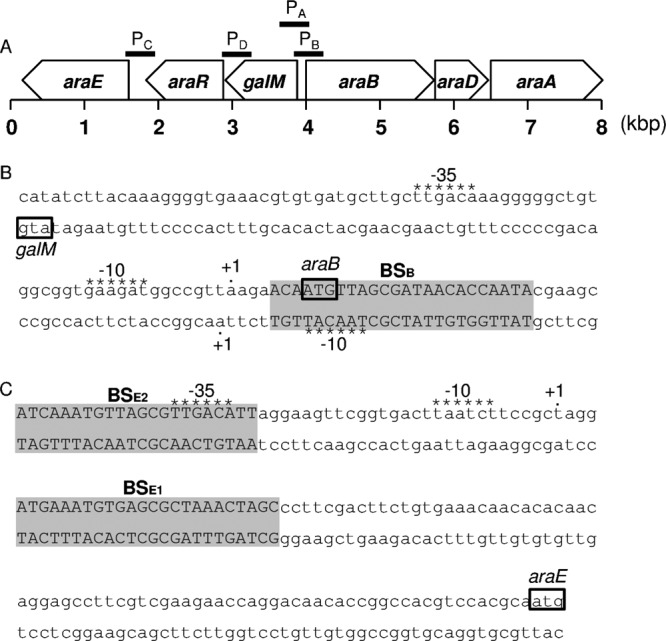
Cluster of l-arabinose utilization genes on the chromosome of C. glutamicum ATCC 31831. (A) The locations and directions of transcription of the six genes (araA, araB, araD, araE, araR, and galM), based on a previous study (26), are shown, and the fragments used as DNA probes (PA, PB, PC, and PD) in electrophoretic mobility shift assays are indicated by solid lines. The nucleotide sequences of the araB-galM intergenic region (B) and the araE upstream region (C) are shown. The translation start codons are boxed, and the putative −10 and −35 regions, based on the transcription start sites (+1) determined, are indicated by asterisks above the sequences. The regions protected by AraR in DNase I footprinting analysis are indicated in capital letters against a gray background.
We previously reported that the expression levels of araBDA and araE in this l-arabinose utilization gene cluster are dramatically increased in exponentially growing cells in minimal medium supplemented with l-arabinose compared with that in cells grown on d-glucose (26). Here we examined changes in the expression of these genes within 15 min of addition of l-arabinose by using qRT-PCR. C. glutamicum ATCC 31831 wild-type cells were cultured in nutrient-rich A medium for 4 h, and exponentially growing cells were then supplemented with l-arabinose or d-glucose to a final concentration of 2% (wt/vol). The levels of araB, araD, and araA mRNAs increased >20-fold within 5 min and then decreased to some extent in the subsequent 10 min (Fig. 2A to C). The expression levels of these mRNAs after 15 min of incubation with l-arabinose were nearly 10-fold higher than their levels before l-arabinose addition. Thus, it is likely that araB, araD, and araA are transcribed as a tricistronic mRNA. A very similar expression pattern in response to l-arabinose was observed for araE (Fig. 2D). The expression levels of galM and araR mRNAs increased about 4-fold within 5 min and then decreased in the subsequent 10 min (Fig. 2E and F). The levels of these mRNAs 15 min after l-arabinose addition were 2-fold higher than their levels before the addition of l-arabinose. These results indicate that galM and araR are upregulated in response to l-arabinose, similar to the other genes in this cluster, but to a much smaller extent. Therefore, it is likely that araE is transcribed under the control of its own promoter, independent of the probable upstream galM-araR operon. Addition of d-glucose in place of l-arabinose resulted in a decrease in the expression levels of these five genes during 15 min of cultivation (Fig. 2A to F), and the effects of glucose on l-arabinose-inducible gene expression were examined further as described below.
FIG 2.
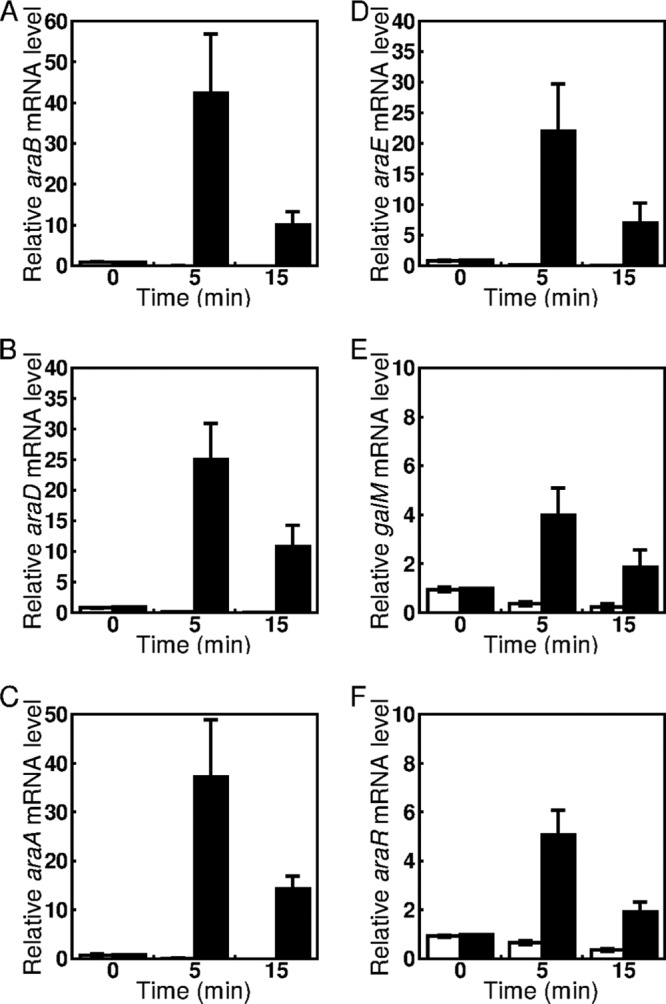
Changes in expression levels of araB (A), araD (B), araA (C), araE (D), galM (E), and araR (F) in response to l-arabinose. The C. glutamicum ATCC 31831 wild-type strain was grown in nutrient-rich A medium for 4 h, and exponentially growing cells were then supplemented with d-glucose (white bars) or l-arabinose (black bars) to a final concentration of 2% (wt/vol). The mRNA levels in cells incubated with sugars for 0, 5, and 15 min were determined by qRT-PCR and are presented relative to the value obtained for the cells before addition of l-arabinose. Mean values and standard deviations for at least three independent cultures are shown.
Exponentially growing cells cultured in nutrient-rich A medium were supplemented with l-arabinose and/or d-glucose at 2% (wt/vol) (each) and then incubated for 60 min. Addition of d-glucose resulted in at least a 2-fold decrease in the levels of araA and araE mRNAs within 30 min, but after 60 min of incubation, these mRNAs reverted to their levels before the addition of d-glucose (Fig. 3). The prominent upregulation of araA and araE observed within 30 min of l-arabinose supplementation was strongly suppressed by supplementation of d-glucose simultaneously. However, the levels of these mRNAs were increased within 60 min of incubation in the presence of both l-arabinose and d-glucose, to the same levels as those in the presence of only l-arabinose.
FIG 3.
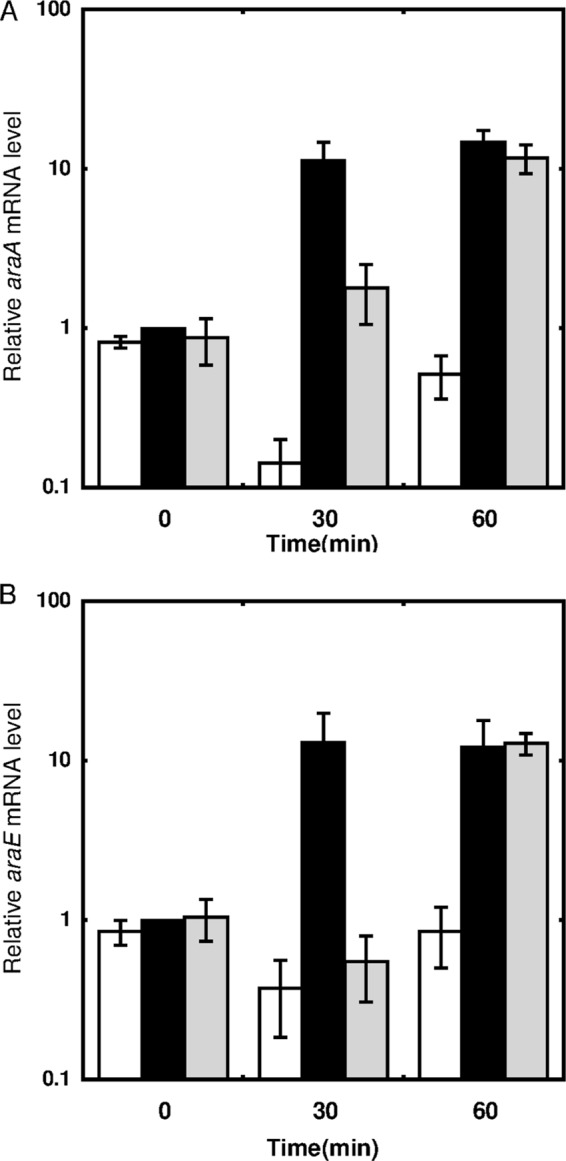
Effects of d-glucose on l-arabinose-dependent upregulation of araA (A) and araE (B). The C. glutamicum ATCC 31831 wild-type strain was grown in nutrient-rich A medium for 4 h, and exponentially growing cells were then supplemented with d-glucose (white bars), l-arabinose (black bars), or l-arabinose plus d-glucose (gray bars), to a final concentration of 2% (wt/vol) for each sugar. The mRNA levels in cells incubated with sugars for 0, 30, and 60 min were determined by qRT-PCR and are presented relative to the values obtained for the cells before addition of l-arabinose. Mean values and standard deviations for at least three independent cultures are shown.
Next, to confirm the involvement of AraR in l-arabinose-inducible gene expression, we compared the levels of araB, araE, and galM mRNAs in an araR deletion mutant strain (ΔaraR) with those in the wild-type strain after 5 min of incubation in the presence or absence of l-arabinose. In the absence of l-arabinose, the expression level of araB in the ΔaraR strain was 50-fold higher than that in the wild-type strain (Fig. 4A). The level of araB mRNA in the ΔaraR strain was even higher than the l-arabinose-induced level in the wild-type strain but decreased significantly in the presence of l-arabinose. The same results were observed for araE (Fig. 4B). Deletion of araR also resulted in an increase in the level of galM mRNA in the absence of l-arabinose, and the AraR-independent downregulation in response to l-arabinose was observed (Fig. 4C), but these changes in galM expression were much smaller than those in the case of araB and araE.
FIG 4.
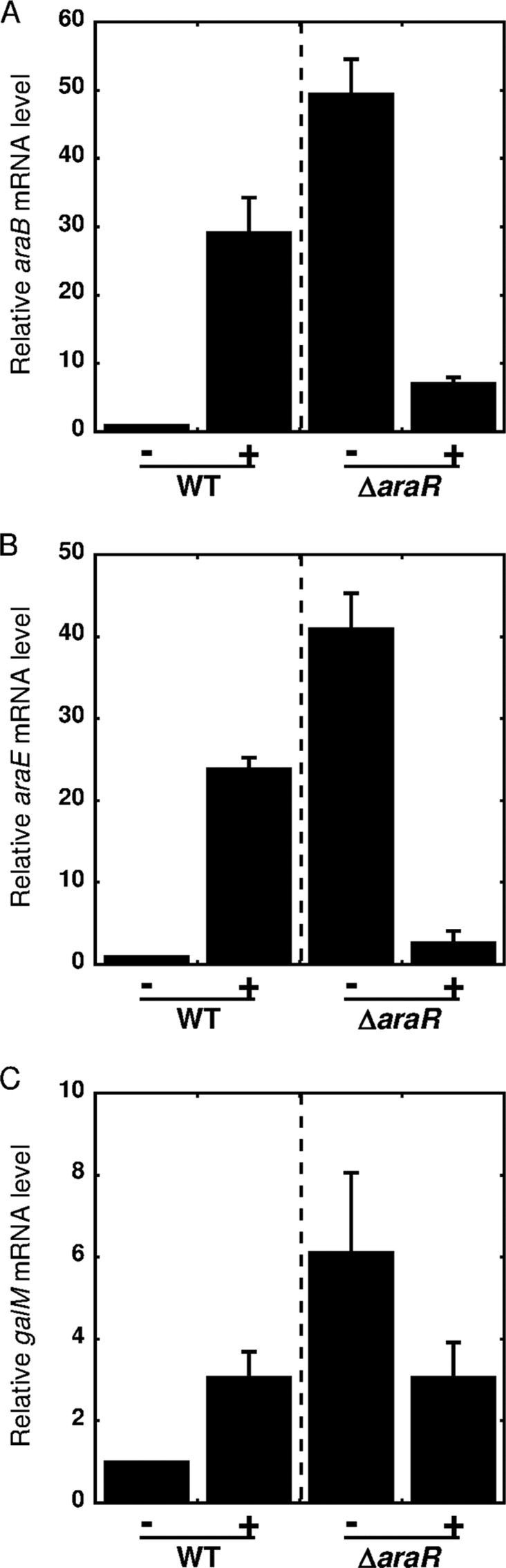
Effects of araR disruption on expression of araB (A), araE (B), and galM (C). The C. glutamicum ATCC 31831 wild-type strain (WT) and the araR deletion mutant strain (ΔaraR) were grown in nutrient-rich A medium for 4 h, and exponentially growing cells were then supplemented with l-arabinose to a final concentration of 2% (wt/vol). The mRNA levels in cells incubated with l-arabinose for 0 min (−) or 5 min (+) were determined by qRT-PCR and are presented relative to the values obtained for the wild-type cells before addition of l-arabinose. Mean values and standard deviations for at least three independent cultures are shown.
The AraR protein binds to the upstream regions of araB, galM, and araE.
To confirm the ability of the AraR protein to bind to the upstream regions of the l-arabinose-inducible genes, EMSA was performed using the C. glutamicum ATCC 31831 AraR protein, which was expressed in E. coli and purified. DNA fragments of 381 bp (PA; between positions −353 and +28 with respect to the araB translation start site), 400 bp (PB; between positions −380 and +20 with respect to the galM translation start site), 394 bp (PC; between positions −364 and +30 with respect to the araE translation start site), and 399 bp (PD; between positions −388 and +11 with respect to the araR translation start site) were used as DNA probes (Fig. 1A). The AraR protein reduced the electrophoretic mobility of the araB upstream DNA fragment; the amount of the AraR-DNA complex increased as the concentration of the AraR protein increased (Fig. 5A). The EMSAs also revealed that the AraR protein bound to the galM and araE upstream DNA fragments (Fig. 5B and C). In contrast, no complex of AraR with the araR upstream DNA fragment was detected (Fig. 5D). These results indicated that AraR binds to the upstream regions of araB, galM, and araE in a sequence-specific manner.
FIG 5.
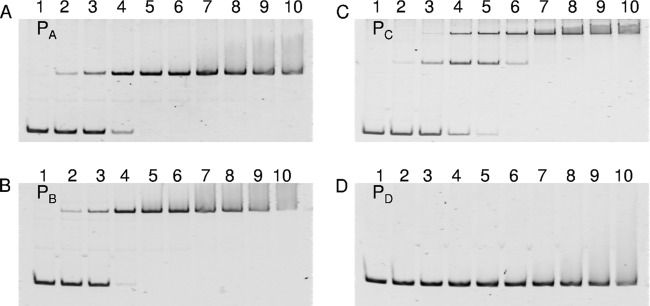
In vitro binding of AraR to the upstream regions of the l-arabinose utilization genes. EMSA was carried out with the AraR protein. The DNA probes used were the upstream regions of araB (PA) (A), galM (PB) (B), araE (PC) (C), and araR (PD) (D) shown in Fig. 1A. Each 20-μl binding reaction mixture, containing the DNA probe at 1 nM and the AraR protein at various concentrations (lanes 1, 0 nM; lanes 2, 10 nM; lanes 3, 25 nM; lanes 4, 50 nM; lanes 5, 75 nM; lanes 6, 100 nM; lanes 7, 125 nM; lanes 8, 150 nM; lanes 9, 175 nM; and lanes 10, 200 nM), was subjected to electrophoresis on a 6% polyacrylamide gel.
There was a 124-bp overlap between the upstream regions of araB and galM used for the DNA probes, each of which formed a complex with AraR as detected as a single shifted band (Fig. 5A and B). We performed EMSAs using a deletion series of the galM upstream region as DNA probes (Fig. 6A). The AraR protein was incubated with the respective DNA fragments and subjected to gel electrophoresis. The results of these EMSAs are shown in Fig. 6B; positive or no binding of AraR is indicated by + or −, respectively, in Fig. 6A. The EMSAs revealed that the AraR protein bound to all these 5′-deletion fragments (P2, P3, P4, and P5 in Fig. 6). A shifted band corresponding to a complex with AraR was observed for two of the 3′-deletion fragments (P6 and P7 in Fig. 6), but not for the other two (P8 and P9 in Fig. 6). These results indicate that the AraR-binding site upstream of galM is located within the 50-bp region between positions −59 and −109 with respect to the translation start site of galM. This AraR-binding site is also included in the araB upstream DNA fragment (PA) used for the EMSAs described above.
FIG 6.

EMSA with the AraR protein and various 5′- and 3′-deletion fragments of the galM upstream region. (A) The DNA fragments (P1 to P9; P1 is the same as PB in Fig. 1A and 5), with their EMSA results indicated (+, positive for binding of AraR; and −, no binding of AraR), corresponded to the regions located at the positions indicated with respect to the translation start site of galM. (B) Each 20-μl binding reaction mixture, containing 40 ng of the DNA probe with (+) or without (−) the AraR protein at 100 nM, was subjected to electrophoresis on a 6% polyacrylamide gel.
AraR-binding activity is reduced by l-arabinose.
The effects of l-arabinose on the DNA-binding activity of the AraR protein were examined by EMSA (Fig. 7). At a concentration of 5 mM, l-arabinose clearly reduced the formation of the complex of AraR with the araB upstream DNA fragment. In contrast, d-xylose did not affect the DNA-binding activity of AraR even at 15 mM. The binding of AraR to the araB upstream DNA fragment was inhibited as a function of the concentration of l-arabinose, from 1 mM to 15 mM. These results indicate that l-arabinose acts as a negative effector of the binding activity of AraR.
FIG 7.
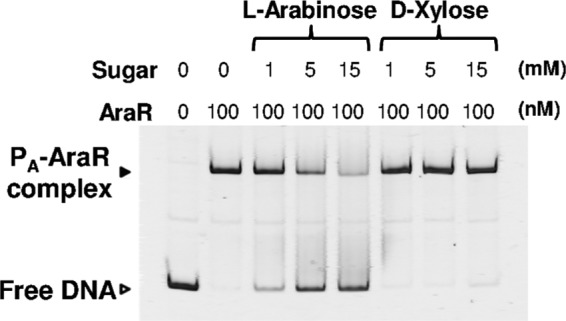
Effects of l-arabinose and d-xylose on the DNA-binding activity of AraR. EMSA was carried out with the AraR protein and the araB upstream region. Each 20-μl binding reaction mixture, containing the DNA probe PA (Fig. 5) at 1 nM with or without the AraR protein at 100 nM, was subjected to electrophoresis on a 6% polyacrylamide gel. The reaction mixture was supplemented with l-arabinose or d-xylose at various concentrations (1, 5, and 15 mM). The free DNA and the DNA-protein complex are indicated by white and black arrowheads, respectively.
Identification of AraR-binding sites in the region upstream of araB and araE.
The transcription start sites of the araB, araE, and galM genes determined by 5′-RACE were located 7, 104, and 69 bp upstream of the respective translation start sites (Fig. 1B and C). The consensus sequence of the −10 region of C. glutamicum SigA-dependent promoters (33) is found in their 5′-upstream regions. The consensus sequence of the −35 region is found in the araB and araE promoters but not in the galM promoter.
Next, we performed DNase I footprinting analysis to determine the AraR-binding site within the intergenic region between araB and galM (Fig. 8A). The DNA region protected against DNase I digestion by the binding of AraR was identified between positions +4 and +28 with respect to the transcription start site of araB. This protected region also corresponds to the region between positions −5 and −29 with respect to the transcription start site of galM, which is located divergently from that of araB (Fig. 1B and 8A). Therefore, the single AraR-binding site overlaps not only the translation start site of araB but also the −10 region of the divergent galM promoter. This AraR-binding site is included in the overlap between the araB and galM upstream DNA fragments used in the EMSAs described earlier (Fig. 5A and B).
FIG 8.

DNase I footprinting analysis with the AraR protein and the araB-galM intergenic region (A) or the araE upstream region (B). Each 20-μl binding reaction mixture, containing 80 ng (A) or 120 ng (B) of the DNA probe and the AraR protein at various concentrations (in panel A, lane 1, no protein; lane 2, 500 nM; and lane 3, 1,000 nM; and in panel B, lane 1, no protein; lane 2, 600 nM; and lane 3, 1,300 nM), was subjected to DNase I treatment followed by electrophoresis on a 5.5% sequencing gel. The sequence of each of these DNA regions was determined with the same labeled primer and plasmid as those for the footprinting probe and is shown to the left. The protected region and the transcription start site are indicated by a solid line and a bent arrow, respectively.
DNase I footprinting analysis using the araE promoter region revealed that the AraR protein protects two regions: one between positions +5 and +28 and one between positions −28 and −46 with respect to the transcription start site of araE (Fig. 1C and 8B). This is consistent with the results of EMSA using the araE promoter region, which showed two shifted bands corresponding to complexes with AraR (Fig. 5C). These results indicate that one of the two AraR-binding sites corresponds to the 5′-untranslated region and the other overlaps the −35 region of the araE promoter (Fig. 1C).
We found the consensus sequence ATGTtAGCGnTaAcat based on alignment of the three AraR-binding sites: one in the araB upstream region and two in the araE upstream region (Fig. 9A). This 16-bp consensus sequence has an imperfect inverted repeat of the 8-bp element. The palindromic sequence has some similarity to the operator sequences of the LacI/GalR family transcriptional regulators (34, 35). Effects of mutations in the putative recognition sequence in the araB upstream region on the binding of AraR were examined by EMSAs (Fig. 9B to H). Three consecutive base pairs at various positions within the 16-bp consensus sequence were mutated, and the resulting araB upstream fragments carrying the mutations (mut1 to mut6 in Fig. 9B) were used as DNA probes in EMSAs. The EMSAs revealed that the AraR protein bound to the mut1 and mut6 fragments (Fig. 9C and H) with the same affinity as that for the wild-type araB upstream fragment, as shown earlier (Fig. 5A). In contrast, lower binding affinities were observed for the mut2, mut3, and mut5 fragments (Fig. 9D, E, and G), and no binding of AraR to the mut4 fragment was detected at all (Fig. 9F). These results indicate that the CG nucleotides located in the center of the palindrome are essential for the binding of AraR and that the nucleotides located in the 5′ half of the palindrome are more important for AraR binding than those of the 3′ half, consistent with the conservation between the target sequences identified here.
FIG 9.

The AraR-binding motif. (A) The three AraR-binding sites determined by DNase I footprinting analyses, located in the araB (BSB) and araE (BSE1 and BSE2) upstream regions, are aligned with the consensus sequence, in which the nucleotides in capital and lowercase letters are identical in all and two of the three sites, respectively, and “n” stands for any nucleotide. Various mutations (mut1 to mut6) were introduced into the AraR-binding site within the araB upstream region (B) and used for EMSAs (C to H). An inverted repeat sequence is indicated by arrows under (A) or above (B) the sequence. Each 20-μl binding reaction mixture, containing the DNA probe at 1 nM and the AraR protein at various concentrations (lanes 1, no protein; lanes 2, 10 nM; lanes 3, 25 nM; lanes 4, 50 nM; lanes 5, 75 nM; lanes 6, 100 nM; and lanes 7, 125 nM), was subjected to electrophoresis on a 6% polyacrylamide gel. The free DNA and the DNA-protein complex are indicated by white and black arrowheads, respectively.
l-Arabinose-dependent induction of AraR-mediated gene expression is independent of catabolism of l-arabinose.
The role of l-arabinose catabolism in the l-arabinose-dependent induction of gene expression was examined by qRT-PCR, using the araA, araB, and araD deletion mutants (ΔaraA, ΔaraB, and ΔaraD strains, respectively). Previously, we reported that these deletion mutants cannot grow on l-arabinose as the sole carbon source (26). For qRT-PCR analysis, these strains were grown in nutrient-rich A medium, and then exponentially growing cells were supplemented with 2% (wt/vol) l-arabinose, as described earlier. In the absence of l-arabinose, the level of araB mRNA in the ΔaraA and ΔaraD strains was the same as that in the wild-type strain. Within 5 min of l-arabinose supplementation, araB mRNA was markedly upregulated in the ΔaraA and ΔaraD strains, to an extent comparable to that in the case of the wild-type strain (Fig. 10A). However, in the subsequent 10 min, the expression level of araB remained high in both of the deletion mutants, in contrast to the decreased level in the wild-type strain described earlier. Similar results were observed for araA expression in either the ΔaraB (Fig. 10A) or ΔaraD (data not shown) strain, and the same was observed for araD expression in either the ΔaraA or ΔaraB strain (data not shown). Furthermore, the expression of araE and araR was also upregulated within 5 min of l-arabinose supplementation in each of the ΔaraA, ΔaraB, and ΔaraD strains, as in the wild-type strain (Fig. 10B and C). After induction, the expression levels of these genes in the mutant strains hardly decreased in the subsequent 10 min, in contrast to the case in the wild-type strain. These results indicate that catabolism of l-arabinose taken up by cells is not required for the l-arabinose-dependent induction of these genes under the control of AraR but is required for the subsequent downregulation observed for the wild-type strain.
FIG 10.
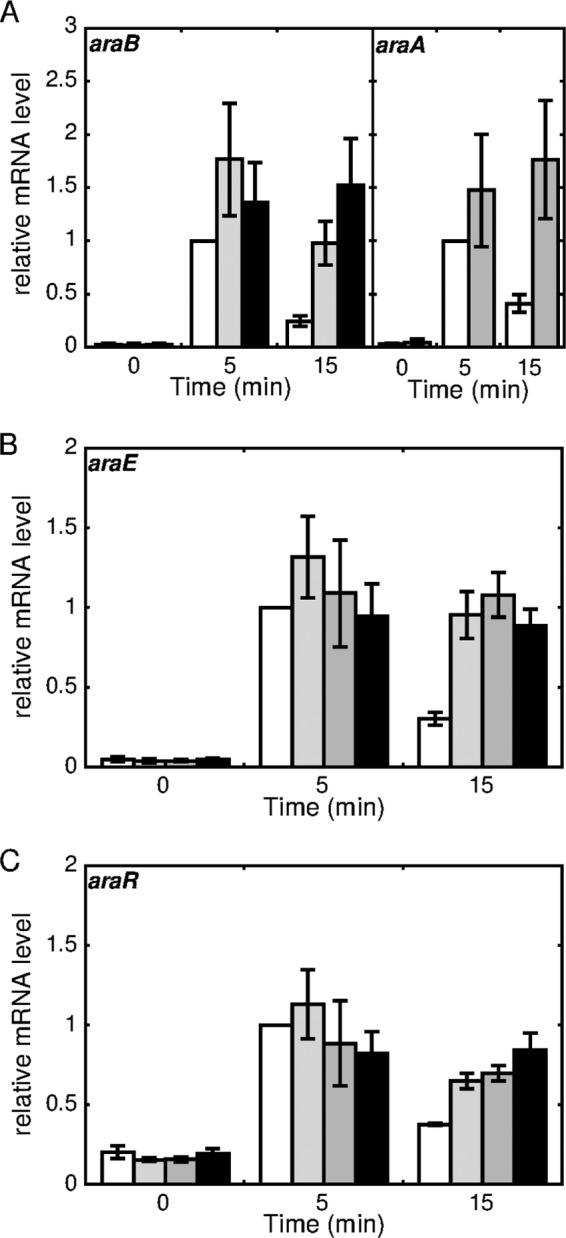
Effects of disruption of l-arabinose catabolic genes on l-arabinose-inducible gene expression. The C. glutamicum ATCC 31831 wild-type (white bars), araA (gray bars), araB (dark gray bars), and araD (black bars) deletion mutant strains were grown in nutrient-rich A medium for 4 h, and exponentially growing cells were then supplemented with l-arabinose at a final concentration of 2% (wt/vol). The levels of araB and araA (A), araE (B), and araR (C) in cells incubated with l-arabinose for 0, 5, and 15 min were determined by qRT-PCR. The mRNA levels are presented relative to the values obtained for wild-type cells incubated with l-arabinose for 5 min. Mean values and standard deviations for at least three independent cultures are shown.
Upregulation of l-arabinose utilization genes is dependent on uptake of l-arabinose.
Previously, we reported that deletion of araE, encoding an l-arabinose transporter, causes a low growth rate and reduces the final cell density in the presence of a low concentration of l-arabinose (26). Effects of different concentrations of l-arabinose on araB, araE, and araR expression were examined using the araE deletion mutant strain (ΔaraE) and the wild-type strain. Exponentially growing cells cultured in nutrient-rich A medium were supplemented with l-arabinose to final concentrations of 0.5 mM, 5 mM, 50 mM, and 200 mM and subsequently incubated for 5 min (Fig. 11). qRT-PCR analysis revealed that before the supplementation of l-arabinose, there was no apparent difference in the expression levels of each of the araB, araE, and araR genes between the ΔaraE strain and the wild-type strain. The expression of araE was detected using a pair of qRT-PCR primers corresponding to a region upstream of the deleted region in the ΔaraE strain. With the wild-type strain, lower concentrations of l-arabinose tended to be more effective for induction of the araB and araE genes. The expression of these genes was also markedly induced by l-arabinose at concentrations of 5 mM, 50 mM, and 200 mM in the ΔaraE strain, to an extent comparable to that in the case of the wild-type strain. However, addition of 0.5 mM l-arabinose was much less effective for induction of the expression of araB and araE in the ΔaraE strain than in the wild-type strain. Similar effects of araE inactivation on l-arabinose-dependent induction were observed for araR, although its dose response to l-arabinose in the wild-type strain was minimal, in contrast to the case of araB and araE as described above (Fig. 11). These results indicate that the uptake of l-arabinose is required for the induction of these genes under the control of AraR.
FIG 11.
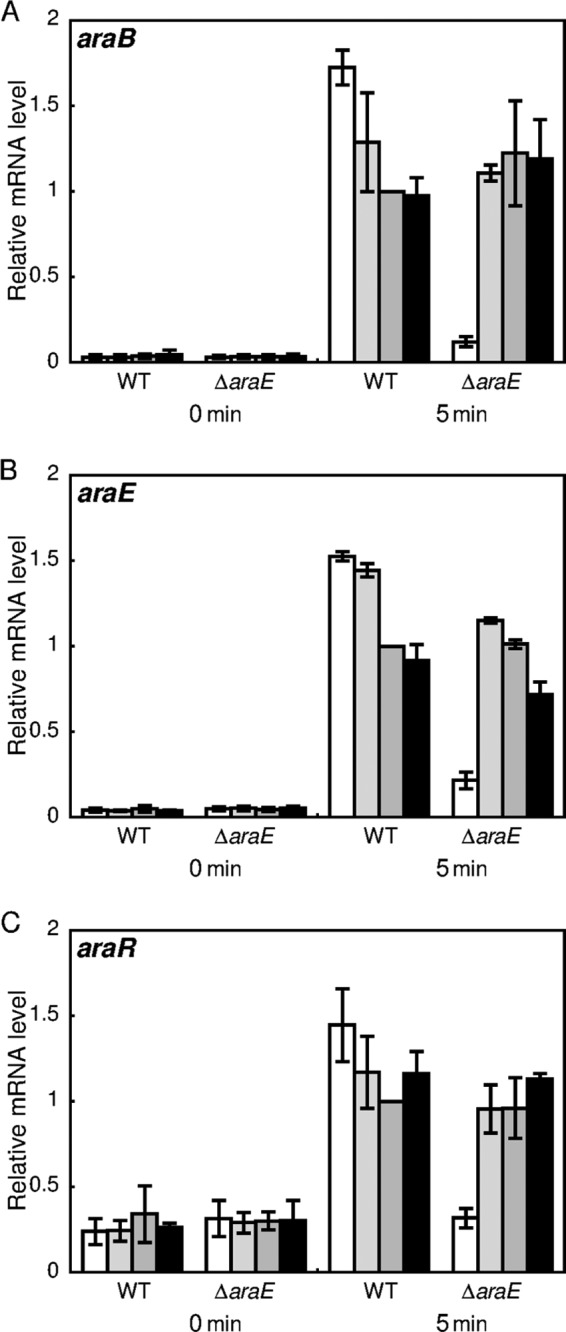
Effects of araE disruption on l-arabinose-dependent upregulation of araB (A), araE (B), and araR (C). The C. glutamicum ATCC 31831 wild-type (WT) and araE deletion mutant (ΔaraE) strains were grown in nutrient-rich A medium for 4 h, and exponentially growing cells were then supplemented with l-arabinose to final concentrations of 0.5 mM (white bars), 5 mM (light gray bars), 50 mM (dark gray bars), and 200 mM (black bars). The mRNA levels in the cells incubated with l-arabinose for 0 and 5 min were determined by qRT-PCR and are presented relative to the values obtained for wild-type cells incubated with 50 mM l-arabinose. Mean values and standard deviations for at least three independent cultures are shown.
DISCUSSION
In this study, we showed that the C. glutamicum ATCC 31831 AraR protein, belonging to the LacI family of transcriptional regulators, binds to the araB and araE upstream regions and that the in vitro DNA-binding activity of AraR is reduced by l-arabinose. We previously reported that the expression of araBDA and araE is upregulated in the presence of l-arabinose compared to that in the presence of d-glucose and by inactivation of araR in the absence of l-arabinose (26). These results indicate that AraR acts as a transcriptional repressor of the l-arabinose catabolic and uptake genes (araBDA and araE) and that AraR-mediated expression is derepressed in the presence of l-arabinose. This is consistent with the rapid induction of these genes after supplementation with l-arabinose, as shown in this study. It should be noted that l-arabinose induced these genes under the control of AraR in mutant strains deficient in l-arabinose catabolism, as well as in the wild-type strain (Fig. 10). Furthermore, in the strain deficient in araE, encoding an l-arabinose transporter, the induction of these genes by a low concentration of l-arabinose was markedly suppressed (Fig. 11), suggesting that the uptake of l-arabinose is required for the derepression of AraR-mediated gene expression. It is noted that the inactivation of araE results in slow growth with a low concentration of l-arabinose as the sole carbon source (26). However, this mutant strain grows as well as the wild-type strain in the presence of a high concentration of l-arabinose (26), which is consistent with the high induction levels of the l-arabinose utilization genes shown in this study. Taken together, our data establish that l-arabinose acts as an efficient inducer of AraR-mediated gene expression in C. glutamicum ATCC 31831 in vivo.
As in C. glutamicum ATCC 31831, l-arabinose plays a key role in the regulation of l-arabinose utilization genes as a primary inducer in E. coli and B. subtilis (36–39). However, their l-arabinose-responsive transcriptional regulators belong to distinct families. The AraC protein in E. coli functions as an activator in the presence of l-arabinose but as a repressor in the absence of l-arabinose (13, 14, 16). In B. subtilis, the oligomerization and effector-binding domains, located in the C terminus of the l-arabinose-responsive transcriptional regulator AraR, have sequence similarity to the LacI/GalR family proteins, but the DNA-binding domain, located in the N terminus, has sequence similarity to members of the GntR family (21, 40). As shown in this study, the LacI-type transcriptional regulator AraR in C. glutamicum ATCC 31831 recognizes a 16-bp palindromic sequence which has common features of the recognition sequences described so far for the other members of this family (35). It has been reported that the LacI/GalR family members coordinate available nutrients with expression of catabolic genes, but some regulate processes as diverse as nucleotide biosynthesis and toxin expression (41–43). As established for this family of transcriptional regulators, it is likely that binding of l-arabinose to the C-terminal domain in the C. glutamicum ATCC 31831 AraR protein results in a conformational change in the N-terminal DNA-binding domain. Thereby, the DNA-binding activity is inhibited in the presence of l-arabinose. This is the first report of the l-arabinose-dependent changes in DNA-binding activity of the LacI/GalR family regulators. The gene cluster associated with l-arabinose utilization has never been found in other strains of C. glutamicum so far. However, Mycobacterium smegmatis and Streptomyces venezuelae have homologs of the l-arabinose catabolic genes of C. glutamicum ATCC 31831, and LacI-type transcriptional regulators encoded near these genes on their chromosomes have 44% and 41% amino acid sequence identity, respectively, to C. glutamicum ATCC 31831 AraR. In M. smegmatis, which is phylogenetically related to C. glutamicum, the l-arabinose catabolic genes are upregulated by l-arabinose (44, 45), but the involvement of the AraR homolog in this regulation remains to be investigated. Many other strains of actinobacteria identified to date lack the l-arabinose catabolic genes, but the AraR-dependent regulatory system of l-arabinose utilization reported here might be conserved among other strains that have yet to be unidentified.
On the chromosome of C. glutamicum ATCC 31831, the araR gene is located immediately downstream of galM, encoding a putative aldose 1-epimerase. We showed here that araR and galM are similarly upregulated by l-arabinose. These results suggest that araR is cotranscribed with galM under the control of the l-arabinose-inducible galM promoter. Since the AraR-binding site is located within the intergenic region between araB and galM (Fig. 1), AraR may regulate the expression of the divergently transcribed araBDA and galM-araR genes through the same mechanism. However, l-arabinose supplementation and/or inactivation of araR showed much smaller effects on the expression of galM and araR than the effects on the expression of araBDA (Fig. 4). The AraR-binding site in the galM promoter overlaps its −10 region (Fig. 1) but is located downstream of the transcription start site of araB. The difference in the location of the AraR-binding site may explain the different degree of transcriptional repression. Furthermore, our previous study showed that the expression of araR, but not that of galM, is upregulated in cells grown in minimal medium with l-arabinose as the sole carbon source compared to that in cells grown on d-glucose (26). Thus, it is possible that the araR gene is under the control of its own promoter, although no binding of AraR to the araR upstream region was observed (Fig. 5). Further studies are needed to elucidate the role of AraR in galM promoter activity. The other regulatory mechanism independent of AraR should also be considered.
We found the 16-bp consensus palindromic sequence ATGTtAGCGnTaAcat based on the alignment of the three binding sites of C. glutamicum ATCC 31831 AraR identified here. This motif is present between positions +8 and +23 bp with respect to the araB transcription start site and in two positions, one between positions +10 and +25 bp and the other between positions −29 and −44 bp, with respect to the araE transcription start site (Fig. 1). Tremendous efforts have been devoted to characterizing the LacI/GalR transcriptional regulators so far, and the results indicate that a few bases in the center of the palindromic structure are strictly conserved among the members of this family (34, 35). However, the peripheral nucleotides in the palindrome are less conserved, which allows these regulators to specifically recognize their own target sequences. The consensus sequence of the multiple C. glutamicum ATCC 31831 AraR-binding sites has CG in the center of the palindrome, which is conserved among the LacI/GalR family members. As expected, the introduction of mutations in the center of the palindromic sequence resulted in no formation of a complex with AraR (Fig. 9F). The AraR-binding site is located downstream of the transcription start sites of araB and araE, suggesting that binding of AraR prevents the elongation of transcription by RNA polymerase. It is noted that a loop structure formed by the cooperative binding of a transcriptional repressor to separate sites in its target promoter is one of the most efficient mechanisms to repress transcription, as described for the LacI family (e.g., LacI and GalR in E. coli) (46, 47). This was also reported for the l-arabinose utilization genes under the control of different types of l-arabinose-responsive transcriptional regulators, i.e., AraC in E. coli (12) and AraR in B. subtilis (19). It is interesting that the second distal binding site of C. glutamicum ATCC 31831 AraR overlaps the −35 region of the l-arabinose transporter gene araE promoter, with a 37-bp space from its proximal binding site (Fig. 1C). This distal site may act cooperatively with the proximal one, although binding of AraR to the former site may prevent RNA polymerase from the interaction with the promoter, irrespective of the latter site. In this context, it should be noted that no similar distal AraR-binding site was found in the upstream region of the l-arabinose catabolic genes araBDA.
We found that in the araR deletion mutant strain, the expression levels of araB and araE markedly decreased within 5 min of the addition of l-arabinose, although in the absence of l-arabinose these mRNAs in this mutant were significantly elevated compared with those in the wild-type strain (Fig. 4). This AraR-independent regulatory mechanism may be involved in the decline after the rapid induction of the araBDA operon and araE within 15 min of the addition of l-arabinose to the wild-type cells (Fig. 2), although fluctuation of the intracellular level of l-arabinose may also affect the AraR-dependent expression of these genes in the beginning of l-arabinose catabolism. In E. coli, the induction of the l-arabinose uptake and catabolic genes was followed by repression during 1 h of incubation with l-arabinose (16). It has been suggested that the cAMP-dependent transcriptional regulator CRP is responsible for the l-arabinose-dependent repression. The expression of the l-arabinose-inducible genes in E. coli is severely inhibited in the presence of d-glucose, through a carbon catabolite repression mechanism mediated by CRP (48–50). We found that the l-arabinose catabolic and uptake genes in C. glutamicum ATCC 31831 were transiently downregulated upon the addition of d-glucose (Fig. 3). Moreover, the addition of l-arabinose plus d-glucose resulted in a delay of the upregulation of l-arabinose-inducible gene expression (Fig. 3). However, the steady-state expression level upregulated in the presence of both l-arabinose and d-glucose was comparable to that in the presence of only l-arabinose. This is consistent with our previous study showing the simultaneous utilization of l-arabinose and d-glucose, at the same rates (26). It should be noted that the global carbon catabolite repression system, mediated by CRP and CcpA in E. coli and B. subtilis, respectively, has never been found in C. glutamicum. Moreover, C. glutamicum has the ability to consume various carbon sources simultaneously (51–54). At present, a search for another transcriptional regulator involved in the probable carbon source-responsive multilayered control of the l-arabinose utilization genes is under way in our laboratory. An understanding of this regulatory system of l-arabinose utilization genes in C. glutamicum ATCC 31831 should provide critical insights into the unique control of genes involved in sugar metabolism in this industrially useful microorganism and a basis for the development of efficient bioprocesses.
ACKNOWLEDGMENTS
We thank Crispinus A. Omumasaba (RITE) for critically reading the manuscript.
This work was financially supported in part by the New Energy and Industrial Technology Development Organization (NEDO), Japan.
Footnotes
Published ahead of print 4 April 2014
REFERENCES
- 1.Yukawa H, Inui M, Vertès AA. 2007. Genomes and genome-level engineering of amino acid-producing bacteria, p 349–401 In Wendisch VF. (ed), Amino acid biosynthesis—pathways, regulation and metabolic engineering, vol 5 Springer, Berlin, Germany [Google Scholar]
- 2.Sahm H, Eggeling L, Eikmanns B, Krämer R. 1995. Metabolic design in amino acid producing bacterium Corynebacterium glutamicum. FEMS Microbiol. Rev. 16:243–252. 10.1111/j.1574-6976.1995.tb00171.x [DOI] [Google Scholar]
- 3.Inui M, Kawaguchi H, Murakami S, Vertès AA, Yukawa H. 2004. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J. Mol. Microbiol. Biotechnol. 8:243–254. 10.1159/000086705 [DOI] [PubMed] [Google Scholar]
- 4.Jojima T, Inui M, Yukawa H. 2013. Biorefinery applications of Corynebacterium glutamicum, p 149–172 In Yukawa H, Inui M. (ed), Corynebacterium glutamicum, vol 23 Springer, Berlin, Germany [Google Scholar]
- 5.Okino S, Inui M, Yukawa H. 2005. Production of organic acids by Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 68:475–480. 10.1007/s00253-005-1900-y [DOI] [PubMed] [Google Scholar]
- 6.Aristidou A, Penttila M. 2000. Metabolic engineering applications to renewable resource utilization. Curr. Opin. Biotechnol. 11:187–198. 10.1016/S0958-1669(00)00085-9 [DOI] [PubMed] [Google Scholar]
- 7.Elander RT, Dale BE, Holtzapple M, Ladisch MR, Lee YY, Mitchinson C, Saddler JN, Wyman CE. 2009. Summary of findings from the Biomass Refining Consortium for Applied Fundamentals and Innovation (CAFI): corn stover pretreatment. Cellulose 16:649–659. 10.1007/s10570-009-9308-y [DOI] [Google Scholar]
- 8.Sasaki M, Jojima T, Kawaguchi H, Inui M, Yukawa H. 2009. Engineering of pentose transport in Corynebacterium glutamicum to improve simultaneous utilization of mixed sugars. Appl. Microbiol. Biotechnol. 85:105–115. 10.1007/s00253-009-2065-x [DOI] [PubMed] [Google Scholar]
- 9.Kawaguchi H, Vertès AA, Okino S, Inui M, Yukawa H. 2006. Engineering of a xylose metabolic pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 72:3418–3428. 10.1128/AEM.72.5.3418-3428.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasaki M, Jojima T, Inui M, Yukawa H. 2008. Simultaneous utilization of d-cellobiose, d-glucose, and d-xylose by recombinant Corynebacterium glutamicum under oxygen-deprived conditions. Appl. Microbiol. Biotechnol. 81:691–699. 10.1007/s00253-008-1703-z [DOI] [PubMed] [Google Scholar]
- 11.Kawaguchi H, Sasaki M, Vertès AA, Inui M, Yukawa H. 2008. Engineering of an l-arabinose metabolic pathway in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 77:1053–1062. 10.1007/s00253-007-1244-x [DOI] [PubMed] [Google Scholar]
- 12.Schleif R. 2010. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol. Rev. 34:779–796. 10.1111/j.1574-6976.2010.00226.x [DOI] [PubMed] [Google Scholar]
- 13.Lee NL, Gielow WO, Wallace RG. 1981. Mechanism of araC autoregulation and the domains of two overlapping promoters, PC and PBAD, in the l-arabinose regulatory region of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 78:752–756. 10.1073/pnas.78.2.752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn S, Schleif R. 1983. In vivo regulation of the Escherichia coli araC promoter. J. Bacteriol. 155:593–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seedorff J, Schleif R. 2011. Active role of the interdomain linker of AraC. J. Bacteriol. 193:5737–5746. 10.1128/JB.05339-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson CM, Schleif RF. 1995. In vivo induction kinetics of the arabinose promoters in Escherichia coli. J. Bacteriol. 177:3438–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inácio JM, Sá-Nogueira I. 2008. Characterization of abn2 (yxiA), encoding a Bacillus subtilis GH43 arabinanase, Abn2, and its role in arabino-polysaccharide degradation. J. Bacteriol. 190:4272–4280. 10.1128/JB.00162-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raposo MP, Inácio JM, Mota LJ, Sá-Nogueira I. 2004. Transcriptional regulation of genes encoding arabinan-degrading enzymes in Bacillus subtilis. J. Bacteriol. 186:1287–1296. 10.1128/JB.186.5.1287-1296.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mota LJ, Tavares P, Sá-Nogueira I. 1999. Mode of action of AraR, the key regulator of l-arabinose metabolism in Bacillus subtilis. Mol. Microbiol. 33:476–489. 10.1046/j.1365-2958.1999.01484.x [DOI] [PubMed] [Google Scholar]
- 20.Franco IS, Mota LJ, Soares CM, Sá-Nogueira I. 2006. Functional domains of the Bacillus subtilis transcription factor AraR and identification of amino acids important for nucleoprotein complex assembly and effector binding. J. Bacteriol. 188:3024–3036. 10.1128/JB.188.8.3024-3036.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franco IS, Mota LJ, Soares CM, Sá-Nogueira I. 2007. Probing key DNA contacts in AraR-mediated transcriptional repression of the Bacillus subtilis arabinose regulon. Nucleic Acids Res. 35:4755–4766. 10.1093/nar/gkm509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mota LJ, Sarmento LM, Sá-Nogueira I. 2001. Control of the arabinose regulon in Bacillus subtilis by AraR in vivo: crucial roles of operators, cooperativity, and DNA looping. J. Bacteriol. 183:4190–4201. 10.1128/JB.183.14.4190-4201.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krispin O, Allmansberger R. 1998. The Bacillus subtilis araE protein displays a broad substrate specificity for several different sugars. J. Bacteriol. 180:3250–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hahn S, Dunn T, Schleif R. 1984. Upstream repression and CRP stimulation of the Escherichia coli l-arabinose operon. J. Mol. Biol. 180:61–72. 10.1016/0022-2836(84)90430-3 [DOI] [PubMed] [Google Scholar]
- 25.Inácio JM, Costa C, Sá-Nogueira I. 2003. Distinct molecular mechanisms involved in carbon catabolite repression of the arabinose regulon in Bacillus subtilis. Microbiology 149:2345–2355. 10.1099/mic.0.26326-0 [DOI] [PubMed] [Google Scholar]
- 26.Kawaguchi H, Sasaki M, Vertès AA, Inui M, Yukawa H. 2009. Identification and functional analysis of the gene cluster for l-arabinose utilization in Corynebacterium glutamicum. Appl. Environ. Microbiol. 75:3419–3429. 10.1128/AEM.02912-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113. 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- 29.Inui M, Murakami S, Okino S, Kawaguchi H, Vertès AA, Yukawa H. 2004. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J. Mol. Microbiol. Biotechnol. 7:182–196. 10.1159/000079827 [DOI] [PubMed] [Google Scholar]
- 30.Vertès AA, Inui M, Kobayashi M, Kurusu Y, Yukawa H. 1993. Presence of mrr- and mcr-like restriction systems in coryneform bacteria. Res. Microbiol. 144:181–185. 10.1016/0923-2508(93)90043-2 [DOI] [PubMed] [Google Scholar]
- 31.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74:5463–5467. 10.1073/pnas.74.12.5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okibe N, Suzuki N, Inui M, Yukawa H. 2011. Efficient markerless gene replacement in Corynebacterium glutamicum using a new temperature-sensitive plasmid. J. Microbiol. Methods 85:155–163. 10.1016/j.mimet.2011.02.012 [DOI] [PubMed] [Google Scholar]
- 33.Pátek M, Nešvera J. 2011. Sigma factors and promoters in Corynebacterium glutamicum. J. Biotechnol. 154:101–113. 10.1016/j.jbiotec.2011.01.017 [DOI] [PubMed] [Google Scholar]
- 34.Camas FM, Alm EJ, Poyatos JF. 2010. Local gene regulation details a recognition code within the LacI transcriptional factor family. PLoS Comput. Biol. 6:e1000989. 10.1371/journal.pcbi.1000989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weickert MJ, Adhya S. 1992. A family of bacterial regulators homologous to Gal and Lac repressors. J. Biol. Chem. 267:15869–15874 [PubMed] [Google Scholar]
- 36.Sá-Nogueira I, Ramos SS. 1997. Cloning, functional analysis, and transcriptional regulation of the Bacillus subtilis araE gene involved in l-arabinose utilization. J. Bacteriol. 179:7705–7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sá-Nogueira I, Nogueira TV, Soares S, Lencastre H. 1997. The Bacillus subtilis l-arabinose (ara) operon: nucleotide sequence, genetic organization and expression. Microbiology 143:957–969. 10.1099/00221287-143-3-957 [DOI] [PubMed] [Google Scholar]
- 38.Doyle ME, Brown C, Helling RWHRB 1972. Induction of the ara operon of Escherichia coli B-r. J. Bacteriol. 110:56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schleif R, Hess W, Finkelstein S, Ellis D. 1973. Induction kinetics of the l-arabinose operon of Escherichia coli. J. Bacteriol. 115:9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Procházková K, Čermáková K, Pachl P, Sieglová I, Fábry M, Otwinowski Z, Řezáčová P. 2012. Structure of the effector-binding domain of the arabinose repressor AraR from Bacillus subtilis. Acta Crystallogr. D Biol. Crystallogr. 68:176–185. 10.1107/S090744491105414X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swint-Kruse L, Matthews KS. 2009. Allostery in the LacI/GalR family: variations on a theme. Curr. Opin. Microbiol. 12:129–137. 10.1016/j.mib.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colmer JA, Hamood AN. 1998. Characterization of ptxS, a Pseudomonas aeruginosa gene which interferes with the effect of the exotoxin A positive regulatory gene, ptxR. Mol. Genet. Genomics 258:250–259. 10.1007/s004380050729 [DOI] [PubMed] [Google Scholar]
- 43.Meng LM, Nygaard P. 1990. Identification of hypoxanthine and guanine as the co-repressors for the purine regulon genes of Escherichia coli. Mol. Microbiol. 4:2187–2192. 10.1111/j.1365-2958.1990.tb00580.x [DOI] [PubMed] [Google Scholar]
- 44.Takata G, Poonperm W, Rao D, Souda A, Nishizaki T, Morimoto K, Izumori K. 2007. Cloning, expression, and transcription analysis of l-arabinose isomerase gene from Mycobacterium smegmatis SMDU. Biosci. Biotechnol. Biochem. 71:2876–2885. 10.1271/bbb.70177 [DOI] [PubMed] [Google Scholar]
- 45.Izumori K, Elbein KYAD. 1976. Pentose metabolism in Mycobacterium smegmatis: specificity of induction of pentose isomerases. J. Bacteriol. 128:587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oehler S, Eismann ER, Krámer H, Müller-Hill B. 1990. The three operators of the lac operon cooperate in repression. EMBO J. 9:973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Semsey S, Geanacopoulos M, Lewis DEA, Adhya S. 2002. Operator-bound GalR dimers close DNA loops by direct interaction: tetramerization and inducer binding. EMBO J. 21:4349–4356. 10.1093/emboj/cdf431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Görke B, Stülke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6:613–624. 10.1038/nrmicro1932 [DOI] [PubMed] [Google Scholar]
- 49.Hahn S, Hendrickson W, Schleif R. 1986. Transcription of Escherichia coli ara in vitro. The cyclic AMP receptor protein requirement for PBAD induction that depends on the presence and orientation of the araO2 site. J. Mol. Biol. 188:355–367 [DOI] [PubMed] [Google Scholar]
- 50.Lobell RB, Schleif RF. 1991. AraC-DNA looping: orientation and distance-dependent loop breaking by the cyclic AMP receptor protein. J. Mol. Biol. 218:45–54. 10.1016/0022-2836(91)90872-4 [DOI] [PubMed] [Google Scholar]
- 51.Wendisch VF, de Graaf AA, Sahm H, Eikmanns BJ. 2000. Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J. Bacteriol. 182:3088–3096. 10.1128/JB.182.11.3088-3096.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dominguez H, Cocaign-Bonsquet M, Lindley ND. 1997. Simultaneous consumption of glucose and fructose from sugar mixtures during batch growth of Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 47:600–603. 10.1007/s002530050980 [DOI] [Google Scholar]
- 53.Stansen C, Uy D, Delaunay S, Eggeling L, Goergen J-L, Wendisch VF. 2005. Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl. Environ. Microbiol. 71:5920–5928. 10.1128/AEM.71.10.5920-5928.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teramoto H, Inui M, Yukawa H. 2009. Regulation of expression of genes involved in quinate and shikimate utilization in Corynebacterium glutamicum. Appl. Environ. Microbiol. 75:3461–3468. 10.1128/AEM.00163-09 [DOI] [PMC free article] [PubMed] [Google Scholar]