

Abstract
Little is known about cell division in Clostridium difficile, a strict anaerobe that causes serious diarrheal diseases in people whose normal intestinal microbiome has been perturbed by treatment with broad-spectrum antibiotics. Here we identify and characterize a gene cluster encoding three cell division proteins found only in C. difficile and a small number of closely related bacteria. These proteins were named MldA, MldB, and MldC, for midcell localizing division proteins. MldA is predicted to be a membrane protein with coiled-coil domains and a peptidoglycan-binding SPOR domain. MldB and MldC are predicted to be cytoplasmic proteins; MldB has two predicted coiled-coil domains, but MldC lacks obvious conserved domains or sequence motifs. Mutants of mldA or mldB had morphological defects, including loss of rod shape (a curved cell phenotype) and inefficient separation of daughter cells (a chaining phenotype). Fusions of cyan fluorescent protein (CFP) to MldA, MldB, and MldC revealed that all three proteins localize sharply to the division site. This application of CFP was possible because we discovered that O2-dependent fluorescent proteins produced anaerobically can acquire fluorescence after cells are fixed with cross-linkers to preserve native patterns of protein localization. Mutants lacking the Mld proteins are severely attenuated for pathogenesis in a hamster model of C. difficile infection. Because all three Mld proteins are essentially unique to C. difficile, they might be exploited as targets for antibiotics that combat C. difficile without disrupting the intestinal microbiome.
INTRODUCTION
Clostridium difficile is a strictly anaerobic, Gram-positive, spore-forming bacterium that has become the leading cause of hospital-acquired diarrhea in developed countries. The annual impact of C. difficile infections in the United States has been estimated at 14,000 deaths and over $1 billion in excess medical costs (1). Both the severity and the frequency of C. difficile infections are increasing (2), and a recent report on the impact of antibiotic resistance classified the organism as an “urgent threat,” the highest threat level (1).
C. difficile infections typically occur in people who have been treated with antibiotics that disrupt the flora of the gastrointestinal tract (3, 4). Although C. difficile is resistant to many antibiotics, the infection usually resolves upon treatment with metronidazole or oral vancomycin (5). Unfortunately, disease recurs in ∼20% of patients, and the prognosis for this cohort is poor (4, 6, 7). The high rate of recurrence has been attributed to germination of C. difficile spores after antibiotic therapy is ended but before restoration of the normal flora (2, 8). For this reason, there is interest in developing antibiotics that target C. difficile selectively and in treatments such as fecal transplants which work by restoring a healthy microbiome (4, 7, 9, 10).
Here we describe a cluster of three genes found in C. difficile that is important for morphogenesis, cell division, and pathogenesis. We named the genes in this cluster mldA, mldB, and mldC, since they encode midcell localizing division proteins. Because these genes are restricted to C. difficile and a few of its closest relatives, drugs that inhibit the Mld proteins might target C. difficile without disrupting the intestinal microbiome. We also describe a method for using green fluorescent protein (GFP) and cyan fluorescent protein (CFP) to study protein localization in strict anaerobes. Because the various color variants of GFP all require O2 for chromophore development (11), their use has been largely restricted to aerobic bacteria or to anaerobes that tolerate transient (e.g., 20-min) exposure to air (12). The extension of O2-dependent fluorescent proteins to strict anaerobes should facilitate studies of protein localization, gene expression, and high-throughput screens for antibiotics in this very important class of bacteria.
MATERIALS AND METHODS
Strains, media, and growth conditions.
Bacterial strains used in this study are listed in Table 1. All C. difficile strains are derived from the erythromycin-sensitive JIR8094 isolate, which is in turn derived from the 630 sequenced strain (13, 14). C. difficile was routinely grown in tryptone yeast extract (TY) or brain heart infusion (BHI) media, supplemented as needed with thiamphenicol at 10 μg/ml, erythromycin at 5 μg/ml, kanamycin at 50 μg/ml, or cefoxitin at 16 μg/ml. TY medium consisted of 0.4% tryptone, 0.5% yeast extract, 0.1% l-cysteine, and (where indicated) 0.5% NaCl. BHI medium consisted of 3.7% brain heart infusion medium (Gibco) supplemented with 0.5% yeast extract, 0.4% glucose, and 0.1% l-cysteine. For solid media, agar was added at a 2% final concentration. C. difficile spores were germinated on cycloserine cefoxitin fructose agar (CCFA) plates containing 1.5% agar, 4% protease peptone, 0.5% sodium phosphate dibasic, 0.1% monopotassium phosphate, 0.2% NaCl, 0.003% neutral red, 0.006% magnesium sulfate, 0.6% fructose, 0.1% l-cysteine, 16 μg/ml cefoxitin, 125 μg/ml cycloserine, and 0.1% taurocholate (15). C. difficile strains were maintained at 37°C in an anaerobic chamber (Coy Laboratory products) in an atmosphere of 10% H2, 5% CO2, and 85% N2.
TABLE 1.
Strains used in this study
| Strain | Genotype and description | Reference or source |
|---|---|---|
| E. coli | ||
| EC448 | MC4100 Δ(λattL-lom)::bla lacIq P204::ftsZ-gfp | 62 |
| Omnimax-2 T1R | F′ [proAB+ lacIq lacZΔM15 Tn10(TetR) Δ(ccdAB)] mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 Δ(lacZYA-argF)U169 endA1 recA1 supE44 thi-1 gyrA96 relA1 tonA panD | Invitrogen |
| HB101/pRK24 | F− mcrB mrr hsdS20(rB− mB−) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20 | 20 |
| RAN334 | Omnimax/pRAN334 (Ptet::CFPopt cat) | This study |
| RAN357 | Omnimax/pRAN357 (Ptet::CFPopt-MCS cat) | This study |
| RAN410 | Omnimax/pRAN410 (Ptet::CFPopt-mldA cat) | This study |
| RAN460 | Omnimax/pRAN460 (Ptet::CFPopt-mldB cat) | This study |
| RAN461 | Omnimax/pRAN461 (Ptet::CFPopt-mldC cat) | This study |
| C. difficile | ||
| JIR8094 | Spontaneous erythromycin-sensitive derivative of C. difficile strain 630 | 13 |
| RAN125 | JIR8094 mldA248::ItrB::ermB | This study |
| RAN258 | JIR8094 mldB153::ItrB::ermB | This study |
| RAN427 | JIR8094/pRAN414 (Ptet::mldABC cat) | This study |
| RAN428 | JIR8094/pRAN415 (Ptet::mldAB cat) | This study |
| RAN429 | JIR8094/pRAN416 (Ptet::mldA cat) | This study |
| RAN430 | JIR8094/pRAN417 (Ptet::mldBC cat) | This study |
| RAN431 | JIR8094/pRAN418 (Ptet::mldB cat) | This study |
| RAN432 | JIR8094/pRAN419 (Ptet::mldC cat) | This study |
| RAN433 | JIR8094/pRPF185 (Ptet::gusA cat) | This study |
| RAN446 | RAN125 (mldA::ItrB::ermB)/pRAN414 (Ptet::mldABC cat) | This study |
| RAN447 | RAN125 (mldA::ItrB::ermB)/pRAN415 (Ptet::mldAB cat) | This study |
| RAN448 | RAN125 (mldA::ItrB::ermB)/pRAN416 (Ptet::mldA cat) | This study |
| RAN449 | RAN125 (mldA::ItrB::ermB)/pRAN417 (Ptet::mldBC cat) | This study |
| RAN450 | RAN125 (mldA::ItrB::ermB)/pRAN418 (Ptet::mldB cat) | This study |
| RAN451 | RAN125 (mldA::ItrB::ermB)/pRAN419 (Ptet::mldC cat) | This study |
| RAN452 | RAN125 (mldA::ItrB::ermB)/pRPF185 (Ptet::gusA cat) | This study |
| RAN453 | RAN258 (mldB::ItrB::ermB)/pRAN414 (Ptet::mldABC cat) | This study |
| RAN454 | RAN258 (mldB::ItrB::ermB)/pRAN415 (Ptet::mldAB cat) | This study |
| RAN455 | RAN258 (mldB::ItrB::ermB)/pRAN416 (Ptet::mldA cat) | This study |
| RAN456 | RAN258 (mldB::ItrB::ermB)/pRAN417 (Ptet::mldBC cat) | This study |
| RAN457 | RAN258 (mldB::ItrB::ermB)/pRAN418 (Ptet::mldB cat) | This study |
| RAN458 | RAN258 (mldB::ItrB::ermB)/pRAN419 (Ptet::mldC cat) | This study |
| RAN459 | RAN258 (mldB::ItrB::ermB)/pRPF185 (Ptet::gusA cat) | This study |
| RAN346 | JIR8094/pRAN334 (Ptet::CFPopt cat) | This study |
| RAN424 | JIR8094/pRAN410 (Ptet::CFPopt-mldA cat) | This study |
| RAN462 | JIR8094/pRAN460 (Ptet::CFPopt-mldB cat) | This study |
| RAN463 | JIR8094/pRAN461 (Ptet::CFPopt-mldC cat) | This study |
| RAN465 | RAN125 (mldA::ItrB::ermB)/pRAN410 (Ptet::CFPopt-mldA cat) | This study |
| RAN466 | RAN125 (mldA::ItrB::ermB)/pRAN460 (Ptet::CFPopt-mldB cat) | This study |
| RAN467 | RAN125 (mldA::ItrB::ermB)/pRAN461 (Ptet::CFPopt-mldC cat) | This study |
| RAN469 | RAN258 (mldB::ItrB::ermB)/pRAN410 (Ptet::CFPopt-mldA cat) | This study |
| RAN470 | RAN258 (mldB::ItrB::ermB)/pRAN460 (Ptet::CFPopt-mldB cat) | This study |
| RAN471 | RAN258 (mldB::ItrB::ermB)/pRAN461 (Ptet::CFPopt-mldC cat) | This study |
Escherichia coli strains were grown in LB medium at 37°C with ampicillin at 200 μg/ml or chloramphenicol at 20 μg/ml as needed. LB medium contained 10% tryptone, 5% yeast extract, 1% NaCl, and (for plates) 1.5% agar.
Plasmid and strain construction.
The oligonucleotide primers used in this work are listed in Table S1 in the supplemental material and were synthesized by Integrated DNA Technologies (Coralville, IA). All plasmids were verified by DNA sequencing and are listed in Table 2.
TABLE 2.
Plasmids used in this study
| Plasmid | Relevant feature(s) | Reference or source |
|---|---|---|
| pRPF185 | E. coli-C. difficile shuttle vector with tetracycline-inducible promoter; Ptet::gusA cat CD6ori RP4oriT-traJ pMB1ori | 21 |
| pCE240 | E. coli-C. difficile shuttle vector for creating C. difficile mutants using TargeTron mutagenesis; ltrB::ermB::RAM ltrA cat pIP404ori pMB1ori RP4oriT | 18 |
| pMC123 | E. coli-C. difficile shuttle vector; cat bla CD6ori RP4oriT pMB1ori | 17 |
| pTHE1037 | E. coli-C. difficile shuttle vector; cat CD6ori RP4oriT pMB1ori | This study |
| pBL100 | E. coli-C. difficile shuttle vector for creating C. difficile mutants using TargeTron mutagenesis; ltrB::ermB::RAM ltrA cat bla CD6ori RP4oriT pMBlori | 19 |
| pSW4-CFPopt | Template for PCR of CFPopt | 22 |
| pDSW1215 | pCD240/mldA248 | This study |
| pRAN101 | pTHE1037/mldA248 ltrB::ermB::RAM ltrA | This study |
| pRAN243 | pBL100/mldB153 | This study |
| pRAN414 | pRPF185 Ptet::mldABC | This study |
| pRAN415 | pRPF185 Ptet::mldAB | This study |
| pRAN416 | pRPF185 Ptet::mldA | This study |
| pRAN417 | pRPF185 Ptet::mldBC | This study |
| pRAN418 | pRPF185 Ptet::mldB | This study |
| pRAN419 | pRPF185 Ptet::mldC | This study |
| pRAN334 | pRPF185 Ptet::CFPopt | This study |
| pRAN357 | pRPF185 Ptet::CFPopt-MCS | This study |
| pRAN410 | pRPF185 Ptet::CFPopt-mldA | This study |
| pRAN460 | pRPF185 Ptet::CFPopt-mldB | This study |
| pRAN461 | pRPF185 Ptet::CFPopt-mldC | This study |
C. difficile null mutants of mldA and mldB were constructed using modified TargeTron procedures (Sigma-Aldrich) to insert a group II intron conferring erythromycin resistance (16). Primers for retargeting the group II intron were designed using algorithms generously provided by Rob Britton (Michigan State University) or ClosTron (16). First, an E. coli-C. difficile shuttle vector designated pTHE1037 was constructed by digesting pMC123 (17) with BspHI and self-ligating, which results in loss of bla. To retarget the intron to insert after nucleotide 248 of mldA, the intron template was amplified by PCR as outlined in the TargeTron user manual (Sigma-Aldrich) using the EBS CDE914 universal primer in combination with primers P1447, P1448, and P1449. The resulting PCR product was digested with HindIII and BsrG1 and then ligated into pCE240 (18) that had been digested with the same enzymes, resulting in pDSW1215. pDSW1215 was then digested with SphI and SfoI to obtain a TargeTron fragment, which was cloned into pTHE1037 that had been digested with EcoRI, made blunt-ended using a Klenow fragment, and then digested with SphI. The resulting plasmid was designated pRAN101. A plasmid that targets the group II intron to nucleotide 153 of mldB was constructed similarly except as follows: the primer set was CDE914, RP114, RP115, and RP116, and the PCR product was cloned onto HindIII-BsrG1-digested pBL100 (19), resulting in pRAN243.
Plasmids pRAN101 (mldA248) and pRAN243 (mldB153) were transformed into the E. coli HB101/pRK24 conjugation donor strain (20). pRAN101 and pRAN243 were then transferred to C. difficile JIR8094 via conjugation and selection for erythromycin resistance as described previously (16, 18). The insertion of introns into mldA and mldB was confirmed by PCR using primer pair RP1 and RP2 (mldA248) or primer pair RP28 and RP29 (mldB153). Finally, loss of the TargeTron plasmid was confirmed by thiamphenicol sensitivity.
For complementation studies, we constructed a set of plasmids that carry various combinations of mldABC. The plasmids are all derivatives of pRPF185, which has a tetracycline-inducible promoter (21). Genes were amplified using the following primer sets: for mldABC, RP164 and RP165; for mldAB, RP164 and RP187; for mldA, RP164 and RP186; for mldBC, RP184 and RP165; for mldB, RP184 and RP187; and for mldC, RP185 and RP165. The resulting PCR products were digested with SacI and BamHI and then ligated into pRPF185 digested with the same enzymes. The resulting plasmids were designated pRAN414 (mldABC), pRAN415 (mldAB), pRAN416 (mldA), pRAN417 (mldBC), pRAN418 (mldB), and pRAN419 (mldC). They were introduced into C. difficile strains by conjugation from strain HB101/pRK24, selecting for thiamphenicol resistance.
To study the localization of Mld proteins, we constructed gene fusions to a low-GC codon-optimized variant of cyan fluorescent protein, CFPopt (22). A basic CFPopt expression vector was constructed by using PCR to amplify cfpopt from template pSW4-CFPopt (22) with primers RP160 and RP161. The resulting PCR product was digested with SacI and BamHI and ligated into pRPF185 that had been cut with the same enzymes. The resulting plasmid, pRAN334, expresses CFPopt followed by a stop codon under the control of a tetracycline-inducible promoter. A plasmid designated pRAN357, in which the coding sequence of CFPopt is followed by a multicloning site for making fusions of CFPopt to the N terminus of target proteins, was constructed similarly using primers RP161 and RP171. CFPopt-Mld fusions were constructed by PCR amplifying the mld genes using the following primer pairs: for mldA, RP178 and RP179; for mldB, RP196 and RP197; and for mldC, RP198 and RP199. The resulting PCR products were digested with SphI and AscI and cloned onto pRAN357 digested with the same enzymes, resulting in CFPopt-MldA (pRAN410), CFPopt-MldB (pRAN460), and CFPopt-MldC (pRAN461). These plasmids were introduced into C. difficile by conjugation from HB101/pRK24, selecting for thiamphenicol resistance.
Intergenic RT-PCR and qRT-PCR.
RNA was harvested from logarithmically growing cells using an RNeasy system (Qiagen). The Access RT-PCR system (Promega) was used for reverse transcriptase PCR (RT-PCR). Real-time quantitative RT-PCR (qRT-PCR) experiments were performed as previously described (18) using Sybr green master mix (Applied Biosystems) and the following gene-specific primer pairs: for mldA, RP34 and RP35; for mldB, RP36 and RP37; and for mldC, RP38 and RP39. Data were normalized to mRNA levels of the C. difficile rpoB housekeeping gene (primer pair TEQ009 and TEQ010).
Morphology of mldA::erm and mldB::erm mutants.
Cultures grown overnight in TY medium were diluted 1:100 into TYN medium (TY medium containing 0.5% NaCl). When the optical density at 600 nm (OD600) of the culture reached ∼0.6, 500 μl was transferred to a microcentrifuge tube containing 5 μl of a 500 μg/ml stock solution of FM4-64FX (Life Technologies)–dimethyl sulfoxide (DMSO). (This derivative of FM4-64 is fixable, but we found that fixation caused a very splotchy staining appearance, so we omitted the fixation step.) Samples were then removed from the anaerobic chamber. Cells were collected by centrifugation, 450 μl of supernatant was removed, and the pellet was suspended in the residual volume of 50 μl. For microscopy, a 2-μl sample was transferred to a thin agarose pad (23) and imaged using a filter for Texas Red (Chroma no. U-N41004). Complementation experiments were performed similarly, except the overnight cultures contained 10 μg/ml of thiamphenicol (to select for the plasmid) and the day cultures contained both thiamphenicol and tetracycline (to induce plasmid-borne genes). The best complementation was achieved using tetracycline at 200 ng/ml.
Transmission electron microscopy.
Wild-type and mldA::erm strains grown overnight in TY medium were diluted 1:100 into 10 ml of TYN medium. When cultures reached an OD600 of ∼0.7, cells were fixed in the media using 2.5% glutaraldehyde for 30 min. Cells were centrifuged at 5,000 rpm, washed with phosphate-buffered saline (PBS), and resuspended in 50 μl of PBS. Warm 2% agarose was added at a 1:1 ratio. Once removed from the anaerobic chamber, this mixture was allowed to solidify at room temperature and subsequently at 4°C for 30 min each time. Samples were prepared as described previously (24). Samples were examined by the use of a JEM-1230 transmission electron microscope (Japan Electron Optics Laboratory Co., Peabody, MA) at the University of Iowa Central Microscopy Research Facility.
Localization of CFP-Mld fusion proteins in C. difficile.
C. difficile strains containing CFP fusion plasmids were grown overnight in BHI medium containing thiamphenicol at 10 μg/ml. The next morning, cultures were diluted 1:100 in the same medium supplemented with tetracycline at 500 ng/ml. When cultures reached an OD600 of ∼0.6, a 500-μl sample was transferred to a microcentrifuge tube containing a mixture of phosphate-buffered saline (PBS), paraformaldehyde, and glutaraldehyde as described previously (25). Samples were then removed from the anaerobic chamber and held at room temperature for 15 min followed by 30 min on ice. Fixed cells were washed 3 times with PBS, resuspended in 100 μl of PBS, and left in the dark at room temperature for >14 h to allow chromophore formation. The cells were immobilized on thin agarose pads for microscopy (23). Our microscope, camera, and software have been described previously (26). The filter set for CFPopt was ECFP/DEAC (Chroma no. 31044v2).
Localization of FtsZ-GFP in E. coli.
Strain EC448 was grown overnight in TY medium in a Coy anaerobic chamber. The next morning, the cells were diluted 1:500 into TY medium containing 2.5 μM IPTG (isopropyl-β-d-thiogalactopyranoside) (to induce the chromosomal ftsZ-gfp fusion) and grown to an OD600 of ∼0.6. Cells were then fixed as described for localization of the CFP-Mld fusions in C. difficile. Samples were photographed using a GFP filter set (Chroma no. 41017) at 0, 5, 15, and 20 h after removal from the anaerobic chamber. To verify that GFP fluorescence resulted from maturation of protein produced anaerobically, an aliquot of the fixed cells was spread onto an LB plate and incubated overnight at 37°C. Only a few colonies appeared, corresponding to 1 CFU per 107 cells in the fixed sample.
Immunoblot analysis.
For immunoblot analysis of CFP fusions, C. difficile strains carrying various CFPopt plasmids were grown to an OD600 of 0.6 to 0.8 in 5 ml of BHI medium supplemented with thiamphenicol and tetracycline. Cultures were then removed from the anaerobic chamber for further processing. Cells were collected by centrifugation, and the resulting pellet was resuspended in ∼0.5 ml of Laemmli sample buffer to achieve a sample concentration of 6.0 OD600 units per ml. Cells were lysed using sets of 10 brief pulses (∼0.5 s each) of sonication (Branson Sonifier 150; power output, 8.5). Lysates were loaded onto an Any kD polyacrylamide gel (Bio-Rad), and proteins were separated by SDS-PAGE. Proteins were then transferred to nitrocellulose. Blots were blocked with 5% nonfat dry milk–phosphate-buffered saline containing 0.1% Tween 20 (PBS-T). CFPopt was detected with polyclonal anti-GFP antibody diluted 1:10,000 in blocking buffer followed by goat anti-rabbit IgG (H+L) IRDye 800CW obtained from Li-Cor. Finally, blots were imaged on an Odyssey CLx infrared imaging system (Li-Cor).
For immunoblot analysis of TcdA toxin production, strains were grown for 5 days in 10 ml of TY medium or TY medium containing 1% glucose. Glucose was included to repress toxin production and served as a specificity control since we did not have an isogenic tcdA null mutant (27). After removal of cultures from the anaerobic chamber, cells were pelleted by centrifugation and the resulting supernatant was concentrated using Centricon 100 columns (Microcon; Millipore, Bedford, MA) to a final volume of 50 to 100 μl. The concentrated supernatant was mixed with 6× Laemmli sample buffer, and proteins were separated by SDS-PAGE (Mini-Protean TGX Precast gels from Bio-Rad; 7.5% polyacrylamide). Subsequent steps were as described above. The primary antibody was anti-Clostridium difficile toxin A antibody (PCG4) (ab19953; Abcam, Cambridge, MA) diluted 1:1,000 in PBS and incubated with the blot overnight. The secondary antibody was goat anti-mouse IRDye 800CW (Li-Cor) diluted 1:10,000 in PBS and incubated for 1 h.
Spore preparation.
C. difficile spores for virulence assays were prepared as previously described (28). Briefly, cells were grown on TY plates for 5 days. Plates were then removed from the anaerobic chamber and allowed to sit in air for at least 2 h. Cells and spores were suspended in PBS and washed, and any remaining viable vegetative cells were killed by heat treatment at 65°C under aerobic conditions for 10 min. Spore preparations were stored at 4°C until use. The concentration of viable spores in the preparations was quantified by germination on TY medium with 0.1% taurocholate. The yield was typically 10 to 25 million spores per plate.
To compare the overall efficiencies of sporulation and germination of the wild-type and mldA::erm mutant strains, spores were prepared as described above using either TY or TYN plates. Dilutions of these spore preparations were then plated on either TY or TYN plates, each containing 0.1% taurocholate, and colony formation was assessed after incubation of the plates anaerobically for 24 h at 37°C.
Syrian hamster model of C. difficile infection.
For pathogenesis assays, C. difficile spores were prepared as previously described (28). The number of viable spores was determined by plating on TY medium containing 0.1% taurocholate and 1% cysteine (15). Syrian gold hamsters (Harlan Sprague-Dawley, Inc., Indianapolis, IN) (80 to 125 g) were treated with a single oral dose of clindamycin (30 mg/kg of body weight) 5 days prior to infection with 10,000 spores of the C. difficile wild-type, mldA::erm mutant, or mldB::erm mutant strain (28, 29). Hamsters were monitored twice daily for morbidity signs: wet tail, diarrhea, ruffled fur, and lethargy. Moribund hamsters were euthanized. The data were graphed as Kaplan-Meier survival analyses (30) and compared for statistical significance using the log-rank test and GraphPad Prism 6 software (GraphPad Software, San Diego, CA). To ascertain whether hamsters were colonized with C. difficile, fecal samples were obtained from each hamster beginning 1 day prior to clindamycin treatment and continuing until day 4 postinoculation. Fecal samples were homogenized in sterile PBS and serial dilutions plated on CCFA containing 0.1% taurocholate. The animal experiments performed in this study were approved by the University of Iowa Institutional Animal Care and Use Committee.
RESULTS
Bioinformatic identification of a potential cell division gene cluster nearly unique to C. difficile.
In many bacteria, proteins containing a SPOR domain are prominent components of the septal ring that mediates cell division (31–34). SPOR domains (Pfam 05036) are about 75 amino acids (aa) long and bind to the peptidoglycan sacculus (35–37). The sacculus, also called the cell wall, surrounds bacterial cells, giving them their characteristic shape and protecting them from lysis due to turgor pressure. The genome of C. difficile strain 630 (ribotype 12) encodes a single putative SPOR domain protein, which is designated CD2717 and has no assigned function (14, 36, 38). cd2717 sits immediately upstream of two additional genes of unknown function, annotated cd2716 and cd2715a. As we demonstrate below, all three genes encode midcell localizing division proteins, so we refer to them here as MldA, MldB, and MldC (for CD2717, CD2716, and CD2715a, respectively).
MldA is predicted to be an 847-aa protein with a single transmembrane helix, three coiled-coil domains that might mediate protein-protein interactions, and a SPOR domain near the C terminus (Fig. 1C). The assignment of the putative SPOR domain in MldA is supported by an E value of 1.009e-8 and a manual alignment of the domain to the Pfam HMM SPOR domain logo (36) (see Fig. S1A in the supplemental material). MldA also has a strikingly high content of charged amino acids—of the 847 residues in MldA, 341 are D, E, K, or R, corresponding to 40% of the sequence (see Fig. S1B). MldB and MldC are predicted to encode cytosolic proteins of 663 and 106 residues, respectively (Fig. 1C). MldB has two predicted coiled-coil regions (39), but bioinformatic analyses failed to identify any conserved domains or sequence motifs in MldC.
FIG 1.
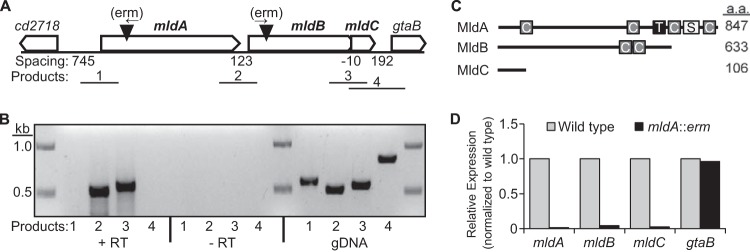
The mldABC gene cluster. (A) Arrangement of mldABC and neighboring genes. Filled triangles depict the location of erm insertions used to inactivate mldA and mldB. Arrows above the triangles show the direction of erm transcription. Numbered lines depict the approximate locations of products from intergenic RT-PCR. (B) mldABC probably comprise a transcription unit as determined by intergenic RT-PCR. +RT, reverse transcriptase step included; −RT, reverse transcriptase step omitted; gDNA, genomic DNA used as the template. (C) Domain organization of the Mld proteins. For clarity, the predicted domains are not drawn to scale. C, coiled-coil; T, transmembrane helix; S, SPOR domain; a.a., amino acid. There are no predicted domains in MldC. (D) Polarity of the mldA::erm mutant as assayed by qRT-PCR.
BLAST searches revealed that MldA, MldB, and MldC are found in all sequenced clinical isolates of C. difficile and in the closely related species C. hiranonis, Peptostreptococcus anaerobius, and Peptostreptococcus stomatis (see Table S2 in the supplemental material). Moreover, the order and spacing of the respective genes are conserved across these organisms, suggesting that they comprise an operon and encode proteins that function together in the same pathway. Outside the fact that SPOR domains are widely conserved, homologs of the Mld proteins could not be identified in any other sequenced bacteria, even in pathogenic clostridia such as C. perfringens, C. botulinum, and C. tetani. The restricted phylogenetic distribution of the Mld proteins is interesting in light of a recent proposal to move C. difficile into the family Peptostreptococcaceae (40). It also raises the possibility that inhibitors of the Mld proteins might target C. difficile without disrupting the intestinal microbiome.
The mldABC genes are cotranscribed.
To determine whether the mld genes are cotranscribed in C. difficile, RNA was extracted from cells grown to mid-logarithmic phase and RT-PCR was performed using primer pairs spanning the intergenic regions (Fig. 1A). Amplification products were obtained between mldA and mldB and between mldB and mldC but not upstream of mldA or downstream of mldC (Fig. 1B). These products were not the result of DNA contamination since they were not obtained in the absence of reverse transcriptase (−RT) (Fig. 1B). As a positive control for each primer pair, we demonstrated that PCR products could be obtained with genomic DNA (gDNA) as a template (Fig. 1B). We conclude that mldA, mldB, and mldC probably constitute a transcription unit. Additionally, the absence of an RT-PCR amplification product using the upstream-most primer pair (primer pair 1 in Fig. 1A) implies that the promoter is within about 350 bp of the mldA start codon. This inference stems from the location of the 5′ primer, which spanned −362 to −338 with respect to the GTG start codon of mldA and was too far upstream to capture the 5′ end of the transcript.
mld mutations impair cell division, cell elongation, and cell shape.
To determine whether mldA is important for cell division, we used TargeTron mutagenesis (16) to insert a 1.7-kb group II intron (ltrB) carrying an erythromycin cassette (erm) after nucleotide 248 of the mldA gene (Fig. 1A). Because the phenotypic changes in morphology resulting from gene mutants of E. coli and Bacillus subtilis often depend on the concentration and nature of salts in the growth media (see, e.g., references 41, 42, 43, and 44), we examined our mutant in TY medium and TYN medium (TY medium containing 0.5% NaCl). In TY medium, the mldA::erm mutant and the wild type grew at similar rates, although examination of cells under the microscope revealed subtle abnormalities in the mutant (Fig. 2A and B). But when grown in TYN medium, the mutant exhibited a modest reduction in growth rate and striking morphological defects (Fig. 2A and C and Table 3). These defects were accentuated by prolonging growth in TYN medium, so to quantify the phenotypic changes, we standardized growth by diluting overnight cultures grown in TY medium 1:100 into TYN medium and incubating for about 7 h until the OD600 reached 0.6.
FIG 2.

Phenotypes of mldA::erm and mldB::erm mutants. (A) Growth curves in TY and TYN. Each data point represents the mean ± standard deviation (s.d.) of results from triplicate cultures, but in most cases the error bars are not visible because they are smaller than the symbols. The data are representative of the results of at least three experiments. (B) Fluorescence micrographs of TY-grown cells stained with FM4-64. The caret indicates an off-center division septum in the wild type. (C) Fluorescence micrographs of TYN-grown cells stained with FM4-64. The white arrow indicates a highly curved segment of an mldA::erm mutant cell. Similar deformations were observed in 3% of the cells. (D) Transmission electron micrographs of septal regions from the wild type and an mldA::erm mutant grown in TYN. Black arrows point to regions where the peptidoglycan appears to be peeling away from the cell in the mutant. Similar defects were observed in 6 of 22 micrographs of septa from the mldA::erm mutant but in none of 18 micrographs of septa from the wild type.
TABLE 3.
Chaining phenotypes of mld mutants
| C. difficile strain | Total no. of cells or filamentsa | Overall lengthb (μm) | Cell lengthc (μm) | % of cells with indicated no. of septa per cell or filament |
|||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4–5 | ≥6 | ||||
| Wild type | 970 | 8.5 ± 2.8 | 5.1 | 31 | 61 | 6 | 1 | 0 | 0 |
| mldA::erm | 908 | 11.8 ± 4.5 | 4.3 | 8 | 38 | 22 | 21 | 9 | 3 |
| mldB::erm | 915 | 9.6 ± 3.6 | 4.3 | 13 | 54 | 19 | 9 | 4 | 1 |
At least 200 cells (or filaments of cells) of each strain were scored in 4 separate experiments, and data were pooled.
Data represent means ± standard deviations of the results determined with the pooled data set (end-to-end length of each cell [or each filament of the cell] regardless of the number of septa present).
Data represent the distance between the cell poles for an isolated cell or the distance between septa for a filament of a cell. Each cell or filament was measured, and the result was divided by the number of septa plus 1.
The wild-type strain grew as straight rods with an average length of ∼9 μm. Staining with the membrane dye FM4-64 revealed that over 90% of the cells had either zero or one division septum, indicating that daughter cells had separated shortly after cytokinesis. In contrast, almost all of the cells of the mldA::erm mutant had an irregular or curved contour, and ∼3% of the cells exhibited severe bends (white arrow in Fig. 2). The mldA::erm mutant also exhibited inefficient separation of daughter cells (a “chaining” phenotype), so that the mutant often appeared as a filament consisting of 2 to 8 cells (Fig. 2C and Table 3). On average, the cells of the mldA::erm mutant were 17% shorter than those of the wild type. Prolonged growth in TYN exacerbated these defects and resulted in some lysis. Transmission electron microscopy suggested that lysis was associated with separation of the peptidoglycan layer from the cell near the division septa (Fig. 2D). Despite the modest lysis seen in TYN broth, the plating efficiency on TYN was the same as that seen with the wild type, although the colonies were smaller. Collectively, these phenotypes indicate that MldA is required for proper elongation and efficient separation of daughter cells at the end of cytokinesis.
We inactivated mldB by inserting a group II intron immediately after nucleotide 153 of the gene (Fig. 1A). The mldB::erm mutant exhibited morphological defects similar to those of the mldA::erm mutant, although they were less severe (Fig. 2 and Table 3). We were unable to construct an mldC mutant despite four attempts that targeted different insertion sites. We doubt that mldC is essential, because mutants with polar insertions in mldA and mldB are viable (polarity is documented below). More likely, the potential TargeTron insertion sites in this short (∼300-bp) gene are not very efficient.
During the course of these studies, we noticed that C. difficile does not regulate division site placement very carefully. This can be seen in Fig. 2B, where one of the wild-type cells has an eccentric division septum, and in Fig. 2C, where the distribution of septa in chains of the mld mutants is sometimes uneven. Division site placement has been studied most intensively in the rod-shaped bacteria E. coli and B. subtilis, which, in contrast to C. difficile, divide with high fidelity at the midpoint of the cell (45, 46). We do not know the genetic basis or physiological significance of relaxed division site selection in C. difficile, but it is not an artifact of the JIR8094 strain background because we also observed irregular constriction site placement in strain R20291.
Complementation of insertions in mldA or mldB requires the entire mldABC cluster.
Quantitative RT-PCR revealed that the erm insertion in mldA is polar on mldB and mldC but not on the next downstream gene, gtaB (Fig. 1D). This supports the operon model proposed above on the basis of intergenic RT-PCR (Fig. 1B) but also raised the possibility that the morphology defects seen in the mldA::erm mutant may result from polarity on mldB and/or mldC. To address this, we first constructed a set of plasmids that contained various combinations of mldA, mldB, and mldC together with 295 bp of DNA from upstream of mldA, which we presumed would include the promoter. However, none of these plasmids complemented the mldA::erm mutant. Specifically, when all three genes were present on the plasmid, the cells became filamentous, suggesting that overexpression of mldABC was interfering with cell division. Subsets of genes did not cause filamentation, but they did not rescue any defects either.
To get better control over gene expression, we cloned the various combinations of mldA, mldB, and mldC into a plasmid with a tetracycline-inducible promoter (21). The shape and chaining defects of the mldA::erm and mldB::erm mutant backgrounds were largely corrected when all three mld genes were expressed (Fig. 3; see also Table S3 in the supplemental material). Weak complementation was observed with the mldAB construct, but plasmids expressing mldA, mldB, or mldC alone did not affect the phenotypes. Notably, complementation was very sensitive to the amount of tetracycline used as an inducer. We observed complementation only with 200 ng/ml tetracycline; 100 ng/ml did not correct the morphological defects, while 400 ng/ml caused cells to become filamentous in some cases (see below).
FIG 3.

Complementation of mldA::erm and mldB::erm. (A) Fluorescence micrographs of cells stained with FM4-64. Tetracycline was used at 200 ng/ml to induce expression of plasmid-borne genes. (B) Quantitative summary of the extent of chaining. (C) Overview of complementation results obtained with different combinations of mld genes. Restoration of nearly wild-type morphology is indicated by +++, while the absence of complementation is indicated by −. Quantitative data are presented in Table S3 in the supplemental material. (D) The experiment was performed as described for panel A, but tetracycline was used at 500 ng/ml.
We were surprised that expressing mldB and mldC together failed to complement the mldB::erm mutant. To explore this further, we performed qRT-PCR on the mldB::erm mutant, which revealed that the level of mldA mRNA was 3-fold lower than that seen with the wild type (see Fig. S2 in the supplemental material). Decreased expression of mldA in the mldB::erm mutant may be due to altered transcript stability. In sum, proper expression of mldA, mldB, and mldC was required for complementation, suggesting that all three genes contribute to cell shape and division.
Overproduction of MldAB impairs cell division.
During the course of the complementation experiments, we observed a striking filamentation phenotype when plasmids carrying either mldABC or mldAB were induced with high levels of tetracycline (Fig. 3D). For example, when an overnight culture grown in TY medium was diluted 1:100 into TYN medium with 500 ng/ml tetracycline, some filaments were over 100 μm long when the culture reached an OD600 = 0.5 and with prolonged growth could reach a length of 1 mm. Staining with FM4-64 revealed very few septa in these filaments, indicating that division was impaired prior to constriction. This defect is fundamentally different from the late stage (chaining) defect observed in mld mutants (Fig. 2). We do not know why overproduction of MldABC impairs division, but potential mechanisms include sequestration of an essential division protein and impaired access to the peptidoglycan. A similar filamentation phenotype was reported in E. coli when a SPOR domain protein named DamX was overproduced (34, 47). Induction of individual mld genes with high levels of tetracycline did not result in elongation.
The Mld proteins localize to the midcell.
We next wanted to ascertain whether the Mld proteins are components of the septal ring structure that mediates cytokinesis in bacteria. This issue would be addressed in most bacteria by constructing fusions to GFP, but C. difficile is a strict anaerobe and GFP requires O2 for chromophore maturation. A potential solution to this problem was foreshadowed in an early paper on GFP by Tsien and colleagues, who demonstrated that GFP produced from a plasmid in E. coli growing anaerobically became fluorescent over a period of several hours once air was admitted to the system (48). However, we considered it unlikely that simply expressing a GFP fusion in C. difficile and then exposing the bacteria to air would reveal septal localization, because the bacterial division apparatus is highly dynamic. For example, the FtsZ ring turns over with a half-life measured in seconds (49), suggesting that septal ring proteins would delocalize if a strict anaerobe were exposed to air for several hours to allow GFP to mature. These considerations prompted us to ask whether GFP could still acquire fluorescence if cells grown anaerobically were fixed with cross-linking agents. Indeed, a pilot experiment using E. coli engineered to produce FtsZ-GFP from a single-copy chromosomal ftsZ-gfp allele revealed that cells grown and fixed anaerobically were nonfluorescent at first but that the FtsZ ring became visible after exposure to air overnight (Fig. 4A).
FIG 4.

Septal localization of GFP and CFPopt fusions to division proteins. (A) Phase and fluorescence micrographs of E. coli cells producing an FtsZ-GFP fusion. Cells were grown and fixed anaerobically and then exposed to air for 5 or 20 h, as indicated. (B) Fluorescence micrographs of wild-type C. difficile cells producing the indicated CFPopt-Mld fusion proteins. Cells were grown and fixed anaerobically and then exposed to air overnight prior to imaging. Bright bands near the midcell demonstrate localization of the tagged Mld protein to a division site. These photographs used an exposure time of 4 s (CFP fusions) or 2 s (CFP alone).
To extend this approach to C. difficile, whose genome has a G+C content of only 29%, we obtained a derivative of cyan fluorescent protein called “CFPopt” that was codon optimized for expression in bacteria with AT-rich genomes (14, 22). We chose CFP because the intrinsic fluorescence of C. difficile interferes with visualization of yellow and green fluorescent proteins. A plasmid that produces CFP-MldA under the control of a tetracycline-inducible promoter was introduced into wild-type C. difficile by conjugation. Cells were grown to an OD600 of ∼0.5, fixed with paraformaldehyde/glutaraldehyde, removed from the anaerobic chamber, transferred to phosphate-buffered saline (PBS), and left at room temperature in the dark overnight to allow chromophore maturation. When examined the next morning, ∼34% of the cells in the population contained a single fluorescent band, indicating that CFP-MldA localized to the septal ring (Fig. 4B and Table 4). No other prominent localization patterns were apparent. We observed similar frequencies of midcell localization for both CFP-MldB and CFP-MldC (Fig. 4B and Table 4). If CFPopt was not fused to anything, fluorescence was distributed uniformly throughout the cytoplasm (Fig. 4B). If the fixation step was omitted, cells lysed when incubated aerobically overnight in PBS.
TABLE 4.
Dependency of localization on the presence of other Mld proteins
| C. difficile host strain | % of total |
|||
|---|---|---|---|---|
| CFPopt-MldA | CFPopt-MldB | CFPopt-MldC | CFPopt | |
| Wild type | 34 | 38 | 38 | 0a |
| mldA::erm | 45 | 0a | 0a | NDb |
| mldB::erm | 64 | 4 | 0a | ND |
Over 120 cells were scored, so the detection limit is ∼1%.
ND, not determined.
To determine whether MldA, MldB, and MldC exhibited any interdependencies for septal localization, we introduced the CFP-Mld fusion constructs into the mldA::erm and mldB::erm mutants. Western blotting verified that all fusions were produced at similar levels in the wild-type and mutant backgrounds (see Fig. S3 in the supplemental material). CFP-MldA localized to the midcell slightly better in the mutants than in the wild type (Table 4), indicating that MldA does not require either MldB or MldC for septal localization. In contrast, CFP-MldB failed to localize to the midcell in the mldA::erm mutant and localized only infrequently (∼4%) in the mldB::erm mutant (Table 4), which, as noted above, probably has reduced expression of mldA. Similarly, CFP-MldC failed to localize in either mutant (Table 4). Thus, localization of MldB and MldC to the midcell clearly requires the presence of MldA and may require the presence of all three Mld proteins. The increased frequency of CFP-MldA localization in the mutant backgrounds likely reflects the fact that these cells have more septa than wild-type cells (Fig. 2B), but it is not obvious why septal localization was observed more often in the mldB mutant than in the mldA mutant. In any case, the most important observation is that MldA localizes well independently of MldB or MldC, while the latter two proteins exhibit strong localization dependencies.
mld mutants are attenuated for pathogenesis in a hamster model of C. difficile infection.
We used the Syrian hamster model of C. difficile disease to assess the effect of mld mutations on pathogenesis (28). Hamsters were given a single oral dose of clindamycin to disrupt the intestinal microbiome. Five days later, each hamster received an oral dose of 10,000 C. difficile spores (as assessed by colony formation), and the animals were monitored for 30 days for signs of illness. Fecal pellets were collected to determine the presence of C. difficile spores, an indicator of colonization. The results are summarized in Fig. 5. Of the 18 hamsters inoculated with wild-type spores, all were colonized. Seventeen animals developed diarrhea, and 15 ultimately required euthanasia. Of the 20 hamsters that received mldA::erm spores, 19 were colonized, 4 developed diarrhea, and 3 were euthanized. Ten hamsters received mldB::erm spores. Of these, 9 were colonized, 3 developed diarrhea, and 1 was euthanized.
FIG 5.
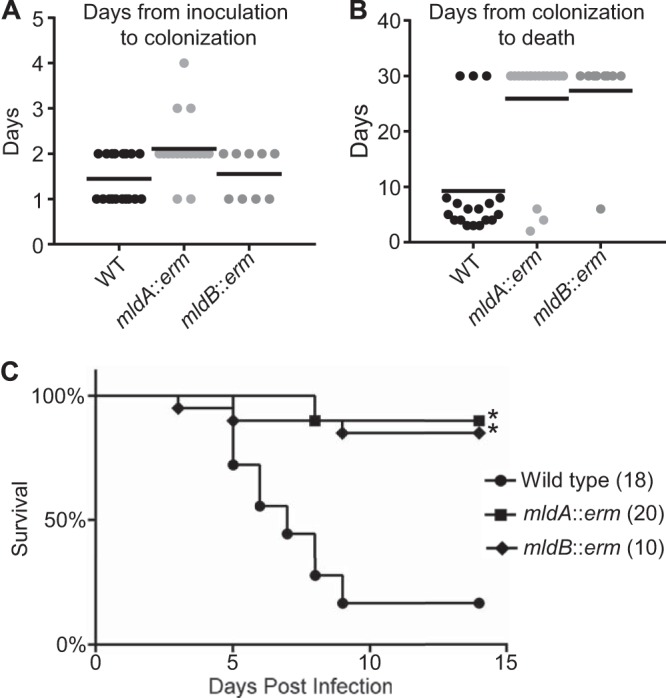
Mutants of mldA and mldB are attenuated in hamsters. Syrian hamsters inoculated orally with 10,000 viable spores were monitored for colonization for up to 4 days and morbidity for up to 30 days. (A) Time from inoculation to detection of C. difficile spores in fecal pellets. Each point represents one hamster, and the bar represents the mean. Colonization rates were 18/18 for the wild type (WT), 19/20 for the mldA::erm strain, and 9/10 for the mldB::erm strain. Data from the two hamsters that were not colonized were excluded from calculations of means. (B) Time from colonization to death. The surviving hamsters were sacrificed on day 30, and their data were included in calculations of means. (C) Kaplan-Meier survival plot indicating time from inoculation to death. Numbers in parentheses refer to the number of hamsters in each treatment group. Data for the wild-type and mldA::erm strains were pooled from two experiments; the mldB::erm mutant was assayed once. *, P ≤ 0.0003 for comparisons of data from the respective mutants and the wild type.
We did not attempt to complement the virulence defect because of issues related to plasmid instability and the requirement for a precise concentration of tetracycline to induce proper gene expression. However, the fact that we obtained similar results with two completely independent mutants (mldA::erm and mldB::erm) argues strongly that attenuation was due to loss of mld function rather than to an unknown secondary mutation elsewhere in the genome.
To begin exploring potential explanations for the pathogenesis defect, we assayed sporulation, germination, and production of toxin A. No defects were found (see Fig. S4 and S5 in the supplemental material). Note, however, that these results come with an important caveat—the phenotypic changes in mld mutants are sensitive to the growth conditions, and it is likely that the media we used in the laboratory (TY and TYN) do not faithfully reflect conditions in the intestinal tract of a hamster.
DISCUSSION
Multiple observations argue that the mldABC gene cluster contributes to proper cell division and elongation in C. difficile. First, MldA is predicted to contain a small, peptidoglycan-binding domain known as a SPOR domain. Proteins that contain a SPOR domain have been implicated in cell division in a variety of bacteria (31–34). The only exceptions of which we are aware concern proteins involved in other aspects of morphogenesis, namely, sporulation or swarmer cell differentiation (37, 50). Second, mldA::erm and mldB::erm null mutants have a late-stage division defect known as a chaining phenotype. Third, overexpression of mldABC inhibits an early stage of cell division. Fourth, the Mld proteins localize to the division site, implying they are part of the apparatus that mediates cell division. Fifth, cells in chains of the mldA::erm and mldB::erm mutants are short, indicating that they do not elongate very efficiently. Finally, these cells usually have a curved or irregular contour, indicating that they have lost control of lateral wall growth.
A chaining phenotype can result from either slow constriction or delayed separation of daughter cells. Because the Mld proteins contribute to both division and elongation, we cannot be certain of where they act in the division process. Nevertheless, our findings point to a separation defect because if chaining were a consequence of slow inward growth of the septum, the cells that make up the chains would probably be longer than normal; instead, they are shorter. Moreover, the Mld proteins localized in ∼35% of the cells, whereas staining with FM4-64 revealed septa in ∼65% of the cells, suggesting the Mld proteins are needed for a late stage of cell division (such as daughter cell separation) rather than an early stage (such as initiation of constriction).
An important issue for the future is to determine the mechanism by which MldABC facilitates cell division and proper extension of the lateral wall. In principle, defects in almost any process associated with biogenesis of the cell envelope could impair growth and division. Perturbations in teichoic acid metabolism are known to cause shape and division defects in B. subtilis, Listeria monocytogenes, and Staphyloccus aureus (reviewed in reference 51). Alternatively, C. difficile has a proteinaceous S-layer that is anchored to the peptidoglycan wall and could plausibly affect elongation and division (52, 53). But several observations suggest that the most likely role for MldABC is related to biogenesis of the peptidoglycan cell wall. MldA is predicted to have a peptidoglycan-binding SPOR domain, and transmission electron microscopy revealed disintegration of the cell wall at sites of septation in an mldA::erm mutant. Moreover, the shape and chaining defects are reminiscent of those of previously reported mutants with defects in peptidoglycan hydrolases (or proteins that govern peptidoglycan hydrolases) in other bacterial species (54–60). Whatever the biochemical activity of the Mld proteins turns out to be, we suspect all three proteins function as a complex, as reflected in the localization dependencies, the requirement for all three genes to complement the mldA::erm and mldB::erm mutants, and the presence of predicted coiled-coil regions in both MldA and MldB.
Our data indicate that mldA::erm and mldB::erm mutants are severely attenuated in the Syrian hamster model of C. difficile infection. We can envision several ways in which the Mld proteins of C. difficile might contribute to virulence. Because the Mld proteins affect cell division and rod morphology, they might make a general contribution to growth and fitness. Nevertheless, the mldA::erm mutant and, even more, the mldB::erm mutant colonized hamsters about as well as the parental wild-type C. difficile strain (Fig. 5A). It is possible that cell wall abnormalities alter the inflammatory response, attachment, toxin secretion, sporulation, or germination. One challenge in addressing these ideas is that the phenotypic changes caused by mld mutations are sensitive to growth conditions (e.g., TY medium versus TYN medium), making it difficult to extrapolate to the hamster model of infection, where the extent of the morphological defects is not known. Thus, although the mldA::erm mutant appeared normal in assays of sporulation, germination, and production of toxin A (see Fig. S4 and S5 in the supplemental material), whether these processes are impaired during growth in a hamster remains to be determined.
Early studies of GFP chromophore formation demonstrated that the protein is not fluorescent when expressed anaerobically in E. coli but acquires fluorescence after O2 is admitted to the system (48). This was recently confirmed for an O2-dependent red fluorescent protein (tdTOMATO) produced in C. difficile (61). We have extended this finding by showing that GFP and CFP acquire fluorescence even after fixation with a mixture of paraformaldehyde and glutaraldehyde. These fixatives work by cross-linking primary amines, especially lysine residues, which are absent from the chromophore of GFP family proteins (11). The advantage of fixation is that it preserves the proper localization of proteins after the cells are exposed to air and die. This opens the door for using GFP and related proteins to study protein localization at relatively high resolution in a variety of strict anaerobes. Although in the present study we applied fluorescent proteins only to the problem of determining the subcellular localization of proteins, our approach can probably be adapted to a wide array of studies where anaerobiosis is pertinent, such as exploring the architecture of biofilms or as a reporter in high-throughput screens for new antibiotics.
Supplementary Material
ACKNOWLEDGMENTS
We thank Theresa Ho for pTHE1037, Rob Britton for the group II intron-targeting algorithm, Neil Fairweather for pRPF185, Inka Sastalla and Stephen H. Leppla for pSW4-CFPopt, Linc Sonenshein for pMC123 and pBL100, and Ryan Arends, Linda McCarter, and members of the Ellermeier and Weiss laboratories for discussions. During the early phases of this study, D.S.W. was a Guest Professor at the M.P.I. for Terrestrial Microbiology in Marburg, Germany, and thanks R. K. Thauer for his hospitality and insights into anaerobic microbiology.
K.B.W. was supported by NIH T32 AI07511. This work was supported by National Institutes of Health grants GM-083975 to D.S.W. and AI-087834 to C.D.E.
Footnotes
Published ahead of print 11 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00038-14.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA [Google Scholar]
- 2.Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N. Engl. J. Med. 359:1932–1940. 10.1056/NEJMra0707500 [DOI] [PubMed] [Google Scholar]
- 3.Owens RC, Jr, Donskey CJ, Gaynes RP, Loo VG, Muto CA. 2008. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin. Infect. Dis. 46(Suppl 1):S19–S31. 10.1086/521859 [DOI] [PubMed] [Google Scholar]
- 4.van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ. 2013. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 368:407–415. 10.1056/NEJMoa1205037 [DOI] [PubMed] [Google Scholar]
- 5.Nelson RL, Kelsey P, Leeman H, Meardon N, Patel H, Paul K, Rees R, Taylor B, Wood E, Malakun R. 2011. Antibiotic treatment for Clostridium difficile-associated diarrhea in adults. Cochrane Database Syst. Rev. 2011:CD004610. 10.1002/14651858.CD004610.pub4 [DOI] [PubMed] [Google Scholar]
- 6.Fekety R, McFarland LV, Surawicz CM, Greenberg RN, Elmer GW, Mulligan ME. 1997. Recurrent Clostridium difficile diarrhea: characteristics of and risk factors for patients enrolled in a prospective, randomized, double-blinded trial. Clin. Infect. Dis. 24:324–333. 10.1093/clinids/24.3.324 [DOI] [PubMed] [Google Scholar]
- 7.O'Horo JC, Jindai K, Kunzer B, Safdar N. 10 July 2013. Treatment of recurrent Clostridium difficile infection: a systematic review. Infection 10.1007/s15010-013-0496-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carroll KC, Bartlett JG. 2011. Biology of Clostridium difficile: implications for epidemiology and diagnosis. Annu. Rev. Microbiol. 65:501–521. 10.1146/annurev-micro-090110-102824 [DOI] [PubMed] [Google Scholar]
- 9.Chilton CH, Crowther GS, Freeman J, Todhunter SL, Nicholson S, Longshaw CM, Wilcox MH. 3 September 2013. Successful treatment of simulated Clostridium difficile infection in a human gut model by fidaxomicin first line and after vancomycin or metronidazole failure. J. Antimicrob. Chemother. 10.1093/jac/dkt347 [DOI] [PubMed] [Google Scholar]
- 10.Goldstein EJ, Citron DM, Tyrrell KL, Merriam CV. 2013. Comparative in vitro activities of SMT19969, a new antimicrobial agent, against Clostridium difficile and 350 Gram-positive and Gram-negative aerobic and anaerobic intestinal flora isolates. Antimicrob. Agents Chemother. 57:4872–4876. 10.1128/AAC.01136-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsien RY. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. 10.1146/annurev.biochem.67.1.509 [DOI] [PubMed] [Google Scholar]
- 12.Hartman AH, Liu H, Melville SB. 2011. Construction and characterization of a lactose-inducible promoter system for controlled gene expression in Clostridium perfringens. Appl. Environ. Microbiol. 77:471–478. 10.1128/AEM.01536-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Connor JR, Lyras D, Farrow KA, Adams V, Powell DR, Hinds J, Cheung JK, Rood JI. 2006. Construction and analysis of chromosomal Clostridium difficile mutants. Mol. Microbiol. 61:1335–1351. 10.1111/j.1365-2958.2006.05315.x [DOI] [PubMed] [Google Scholar]
- 14.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38:779–786. 10.1038/ng1830 [DOI] [PubMed] [Google Scholar]
- 15.Wilson KH, Kennedy MJ, Fekety FR. 1982. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J. Clin. Microbiol. 15:443–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 80:49–55. 10.1016/j.mimet.2009.10.018 [DOI] [PubMed] [Google Scholar]
- 17.McBride SM, Sonenshein AL. 2011. Identification of a genetic locus responsible for antimicrobial peptide resistance in Clostridium difficile. Infect. Immun. 79:167–176. 10.1128/IAI.00731-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho TD, Ellermeier CD. 2011. PrsW is required for colonization, resistance to antimicrobial peptides, and expression of extracytoplasmic function sigma factors in Clostridium difficile. Infect. Immun. 79:3229–3238. 10.1128/IAI.00019-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouillaut L, Self WT, Sonenshein AL. 2013. Proline-dependent regulation of Clostridium difficile Stickland metabolism. J. Bacteriol. 195:844–854. 10.1128/JB.01492-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trieu-Cuot P, Arthur M, Courvalin P. 1987. Origin, evolution and dissemination of antibiotic resistance genes. Microbiol. Sci. 4:263–266 [PubMed] [Google Scholar]
- 21.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J. Biol. Chem. 286:27483–27493. 10.1074/jbc.M111.263889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sastalla I, Chim K, Cheung GY, Pomerantsev AP, Leppla SH. 2009. Codon-optimized fluorescent proteins designed for expression in low-GC gram-positive bacteria. Appl. Environ. Microbiol. 75:2099–2110. 10.1128/AEM.02066-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarry M, Arends SJ, Roversi P, Piette E, Sargent F, Berks BC, Weiss DS, Lea SM. 2009. The Escherichia coli cell division protein and model Tat substrate SufI (FtsP) localizes to the septal ring and has a multicopper oxidase-like structure. J. Mol. Biol. 386:504–519. 10.1016/j.jmb.2008.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsiao CH, Ueno N, Shao JQ, Schroeder KR, Moore KC, Donelson JE, Wilson ME. 2011. The effects of macrophage source on the mechanism of phagocytosis and intracellular survival of Leishmania. Microbes Infect. 13:1033–1044. 10.1016/j.micinf.2011.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pogliano K, Harry E, Losick R. 1995. Visualization of the subcellular location of sporulation proteins in Bacillus subtilis using immunofluorescence microscopy. Mol. Microbiol. 18:459–470. 10.1111/j.1365-2958.1995.mmi_18030459.x [DOI] [PubMed] [Google Scholar]
- 26.Mercer KL, Weiss DS. 2002. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J. Bacteriol. 184:904–912. 10.1128/jb.184.4.904-912.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dupuy B, Sonenshein AL. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol. Microbiol. 27:107–120. 10.1046/j.1365-2958.1998.00663.x [DOI] [PubMed] [Google Scholar]
- 28.Sambol SP, Tang JK, Merrigan MM, Johnson S, Gerding DN. 2001. Infection of hamsters with epidemiologically important strains of Clostridium difficile. J. Infect. Dis. 183:1760–1766. 10.1086/320736 [DOI] [PubMed] [Google Scholar]
- 29.Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. 10.1038/nature07822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaplan EL, Meier P. 1958. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 53:457–481. 10.1080/01621459.1958.10501452 [DOI] [Google Scholar]
- 31.Dai K, Xu Y, Lutkenhaus J. 1993. Cloning and characterization of ftsN, an essential cell division gene in Escherichia coli isolated as a multicopy suppressor of ftsA12(Ts). J. Bacteriol. 175:3790–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Möll A, Thanbichler M. 2009. FtsN-like proteins are conserved components of the cell division machinery in proteobacteria. Mol. Microbiol. 72:1037–1053. 10.1111/j.1365-2958.2009.06706.x [DOI] [PubMed] [Google Scholar]
- 33.Gerding MA, Liu B, Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. 2009. Self-enhanced accumulation of FtsN at division sites and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. J. Bacteriol. 191:7383–7401. 10.1128/JB.00811-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arends SJ, Williams K, Scott RJ, Rolong S, Popham DL, Weiss DS. 2010. Discovery and characterization of three new Escherichia coli septal ring proteins that contain a SPOR domain: DamX, DedD, and RlpA. J. Bacteriol. 192:242–255. 10.1128/JB.01244-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ursinus A, van den Ent F, Brechtel S, de Pedro M, Höltje JV, Löwe J, Vollmer W. 2004. Murein (peptidoglycan) binding property of the essential cell division protein FtsN from Escherichia coli. J. Bacteriol. 186:6728–6737. 10.1128/JB.186.20.6728-6737.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301. 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishima M, Shida T, Yabuki K, Kato K, Sekiguchi J, Kojima C. 2005. Solution structure of the peptidoglycan binding domain of Bacillus subtilis cell wall lytic enzyme CwlC: characterization of the sporulation-related repeats by NMR. Biochemistry 44:10153–10163. 10.1021/bi050624n [DOI] [PubMed] [Google Scholar]
- 38.Monot M, Boursaux-Eude C, Thibonnier M, Vallenet D, Moszer I, Medigue C, Martin-Verstraete I, Dupuy B. 2011. Reannotation of the genome sequence of Clostridium difficile strain 630. J. Med. Microbiol. 60:1193–1199. 10.1099/jmm.0.030452-0 [DOI] [PubMed] [Google Scholar]
- 39.Lupas A. 1996. Prediction and analysis of coiled-coil structures. Methods Enzymol. 266:513–525. 10.1016/S0076-6879(96)66032-7 [DOI] [PubMed] [Google Scholar]
- 40.Yutin N, Galperin MY. 2013. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ. Microbiol. 15:2631–2641. 10.1111/1462-2920.12173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ricard M, Hirota Y. 1973. Process of cellular division in Escherichia coli: physiological study on thermosensitive mutants defective in cell division. J. Bacteriol. 116:314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray T, Popham DL, Setlow P. 1998. Bacillus subtilis cells lacking penicillin-binding protein 1 require increased levels of divalent cations for growth. J. Bacteriol. 180:4555–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Formstone A, Errington J. 2005. A magnesium-dependent mreB null mutant: implications for the role of mreB in Bacillus subtilis. Mol. Microbiol. 55:1646–1657. 10.1111/j.1365-2958.2005.04506.x [DOI] [PubMed] [Google Scholar]
- 44.Reddy M. 2007. Role of FtsEX in cell division of Escherichia coli: viability of ftsEX mutants is dependent on functional SufI or high osmotic strength. J. Bacteriol. 189:98–108. 10.1128/JB.01347-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu XC, Margolin W. 1999. FtsZ ring clusters in min and partition mutants: role of both the Min system and the nucleoid in regulating FtsZ ring localization. Mol. Microbiol. 32:315–326. 10.1046/j.1365-2958.1999.01351.x [DOI] [PubMed] [Google Scholar]
- 46.Migocki MD, Freeman MK, Wake RG, Harry EJ. 2002. The Min system is not required for precise placement of the midcell Z ring in Bacillus subtilis. EMBO Rep. 3:1163–1167. 10.1093/embo-reports/kvf233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lyngstadaas A, Lobner-Olesen A, Boye E. 1995. Characterization of three genes in the dam-containing operon of Escherichia coli. Mol. Gen. Genet. 247:546–554. 10.1007/BF00290345 [DOI] [PubMed] [Google Scholar]
- 48.Heim R, Prasher DC, Tsien RY. 1994. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 91:12501–12504. 10.1073/pnas.91.26.12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson DE, Gueiros-Filho FJ, Erickson HP. 2004. Assembly dynamics of FtsZ rings in Bacillus subtilis and Escherichia coli and effects of FtsZ-regulating proteins. J. Bacteriol. 186:5775–5781. 10.1128/JB.186.17.5775-5781.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gode-Potratz CJ, Kustusch RJ, Breheny PJ, Weiss DS, McCarter LL. 2011. Surface sensing in Vibrio parahaemolyticus triggers a programme of gene expression that promotes colonization and virulence. Mol. Microbiol. 79:240–263. 10.1111/j.1365-2958.2010.07445.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown S, Santa Maria JP, Jr, Walker S. 2013. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 67:313–336. 10.1146/annurev-micro-092412-155620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calabi E, Ward S, Wren B, Paxton T, Panico M, Morris H, Dell A, Dougan G, Fairweather N. 2001. Molecular characterization of the surface layer proteins from Clostridium difficile. Mol. Microbiol. 40:1187–1199. 10.1046/j.1365-2958.2001.02461.x [DOI] [PubMed] [Google Scholar]
- 53.Fagan RP, Fairweather NF. 2014. Biogenesis and functions of bacterial S-layers. Nat. Rev. Microbiol. 12:211–222. 10.1038/nrmicro3213 [DOI] [PubMed] [Google Scholar]
- 54.Meisner J, Montero Llopis P, Sham LT, Garner E, Bernhardt TG, Rudner DZ. 2013. FtsEX is required for CwlO peptidoglycan hydrolase activity during cell wall elongation in Bacillus subtilis. Mol. Microbiol. 89:1069–1083. 10.1111/mmi.12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Domínguez-Cuevas P, Porcelli I, Daniel RA, Errington J. 2013. Differentiated roles for MreB-actin isologues and autolytic enzymes in Bacillus subtilis morphogenesis. Mol. Microbiol. 89:1084–1098. 10.1111/mmi.12335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sham LT, Barendt SM, Kopecky KE, Winkler ME. 2011. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc. Natl. Acad. Sci. U. S. A. 108:E1061–E1069. 10.1073/pnas.1108323108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang DC, Peters NT, Parzych KR, Uehara T, Markovski M, Bernhardt TG. 2011. An ATP-binding cassette transporter-like complex governs cell-wall hydrolysis at the bacterial cytokinetic ring. Proc. Natl. Acad. Sci. U. S. A. 108:E1052–E1060. 10.1073/pnas.1107780108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Höltje JV. 2001. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 41:167–178. 10.1046/j.1365-2958.2001.02499.x [DOI] [PubMed] [Google Scholar]
- 59.Uehara T, Parzych KR, Dinh T, Bernhardt TG. 2010. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 29:1412–1422. 10.1038/emboj.2010.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cloud KA, Dillard JP. 2004. Mutation of a single lytic transglycosylase causes aberrant septation and inhibits cell separation of Neisseria gonorrhoeae. J. Bacteriol. 186:7811–7814. 10.1128/JB.186.22.7811-7814.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barra-Carrasco J, Olguin-Araneda V, Plaza-Garrido A, Miranda-Cardenas C, Cofre-Araneda G, Pizarro-Guajardo M, Sarker MR, Paredes-Sabja D. 2013. The Clostridium difficile exosporium cysteine (CdeC)-rich protein is required for exosporium morphogenesis and coat assembly. J. Bacteriol. 195:3863–3875. 10.1128/JB.00369-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181:508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.