

Abstract
Anodic aluminum oxide (AAO) filters have high porosity and can be manufactured with a pore size that is small enough to quantitatively capture viruses. These properties make the filters potentially useful for harvesting total microbial communities from water samples for molecular analyses, but their performance for nucleic acid extraction has not been systematically or quantitatively evaluated. In this study, we characterized the flux of water through commercially produced nanoporous (0.02 μm) AAO filters (Anotop; Whatman) and used isolates (a virus, a bacterium, and a protist) and natural seawater samples to test variables that we expected would influence the efficiency with which nucleic acids are recovered from the filters. Extraction chemistry had a significant effect on DNA yield, and back flushing the filters during extraction was found to improve yields of high-molecular-weight DNA. Using the back-flush protocol, the mass of DNA recovered from microorganisms collected on AAO filters was ≥100% of that extracted from pellets of cells and viruses and 94% ± 9% of that obtained by direct extraction of a liquid bacterial culture. The latter is a minimum estimate of the relative recovery of microbial DNA, since liquid cultures include dissolved nucleic acids that are retained inefficiently by the filter. In conclusion, we demonstrate that nucleic acids can be extracted from microorganisms on AAO filters with an efficiency similar to that achievable by direct extraction of microbes in suspension or in pellets. These filters are therefore a convenient means by which to harvest total microbial communities from multiple aqueous samples in parallel for subsequent molecular analyses.
INTRODUCTION
Direct filtration is one of the most common methods for harvesting microorganisms from natural water samples for molecular analyses (1–3), because it is technically simple and efficiently concentrates microorganisms onto a small surface area for subsequent nucleic acid extraction. Most direct filtration protocols focus on the capture of cells (bacteria, archaea, and eukaryotes) using filters with pore sizes of ≥0.2 μm (4). Since most viruses in aquatic environments are <0.2 μm in diameter (5), they are not quantitatively represented in the nucleic acids recovered by these protocols. Collection of viruses for molecular analyses is more commonly achieved using tangential flow ultrafiltration (6). This method can also be used to harvest viruses and cells simultaneously (7), but the method is time-intensive and requires relatively expensive equipment, making it unsuitable for high-throughput sampling. Other methods for harvesting cells or viruses from water include adsorption/elution or flocculation (8), but many of these methods have highly variable recovery efficiencies depending on the microorganism, and they have been typically used for water quality monitoring rather than comprehensive community analysis. An iron flocculation procedure was recently shown to concentrate viruses in natural communities with high efficiency (9), but it specifically targeted only the viral fraction.
A simple, direct filtration procedure that effectively captures all of the microorganisms, including viruses, from a water sample would be useful for many applications (e.g., studies of microbial ecology, monitoring of recreational and drinking water quality, or testing for pathogens in the process streams of food, biotechnology, and biopharmaceutical industries). Some of the membrane materials typically used for molecular analysis of microbial communities (e.g., polyvinyldifluoride and polycarbonate) can be manufactured with pore sizes small enough to retain most or all viruses but have such a low flux that they are less practical for routine collections. The best option, at present, for simultaneous capture of viruses and cellular microorganisms appears to be nanoporous anodic aluminum oxide (AAO) filters. These filters have high porosity with uniform pore size (10) and are commercially manufactured with pores as small as 0.02 μm, which is close to the lower limit for known viruses (11). They are also available in disposable housings with luer-lock fittings, making them convenient for field collections by in-line filtration. Many samples can be processed in parallel by this method using multichannel peristaltic pumps.
Two of us reported previously on the use of AAO filters for molecular analyses of aquatic viruses (12, 13). Extraction of the nucleic acids was accomplished using a commercial kit (MasterPure complete DNA and RNA purification kit; Epicentre) that is based on a salting-out extraction protocol (14). The salting-out procedure employs a buffer containing an ionic detergent (SDS) to solubilize tissues, cells, or viruses. Following the lysis step (usually at 37 to 65°C with proteinase K), SDS-protein complexes are precipitated by the addition of salt and pelleted by centrifugation. Nucleic acids in the supernatant are then precipitated with alcohol. Many variations on the salting-out procedure have been published (15–19).
We chose the salting-out extraction procedure, rather than one of the popular guanidinium-based extraction methods, because guanidinium salts have been reported to cause irreversible binding of DNA to AAO (20, 21). We later described a modification of our filter-extraction protocol (22) in which extraction buffer is back flushed through the membrane rather than being pushed through in the same direction as the sample. This modification was adopted out of concern that the exceptionally small pore size of the filters may trap high-molecular-weight nucleic acids. Although there were reasonable theoretical bases for our choices about the extraction buffer composition and the direction in which it is applied to the filter, there has been no empirical evidence that these choices actually affect the yield of nucleic acids. In fact, there are no data at all on the efficiency with which nucleic acids can be extracted from microorganisms collected on AAO filters.
Given the utility of these membranes for harvesting microorganisms and viruses (23–25, 47), we felt that an assessment of their performance was worthwhile. In this study, we tested the flow characteristics and loading capacity of commercially produced AAO filters with a 0.02-μm pore size, and we systematically tested a number of factors that we predicted would influence the recovery of nucleic acids from microorganisms collected on the filters. This included a comparison of DNA yields when extraction buffer was pushed through the filter in the same direction as filtration (forward flush) versus being pulled through in the opposite direction (back flush) and a comparison of recoveries using SDS- versus guanidinium-based extraction buffers. We also compared the efficiencies with which DNA and RNA were recovered from microorganisms when they were collected on filters versus being suspended in liquid or pelleted, and we looked at the effects of lysozyme treatment on recovery of nucleic acids from samples containing cells or only viruses. We then evaluated the linearity of DNA yield for various volumes of sample filtered and evaluated the tendency of the filters to trap dissolved DNA. From these experiments, we provide an assessment of the performance of AAO filters for nucleic acid extractions and a recommended protocol for conducting such extractions.
MATERIALS AND METHODS
Filters.
We used 25-mm-diameter filter membranes sealed by thermal welding in polypropylene housings with integral luer-lock fittings. Depending on the experiment, the filter material was either AAO (0.02- or 0.2-μm pore size) (Anotop 25; Whatman) or polyethersulfone (PES) (0.2 μm) (Puradisc 25; Whatman). For one experiment comparing flow rates, an AAO membrane disc (0.02-μm pore size, 25-mm diameter) with a plastic support ring (Anodisc; Whatman) was mounted in a reusable polypropylene filter housing (Advantec MFS). The details of the flow rate and extraction experiments are presented in the following sections and are summarized in Table 1.
TABLE 1.
Summary of samples extracted, variables tested, and controls used
| Filter test | Variable(s)a | IDb | Sample typec | Vol processed, ml (type of processing)d | Control(s)e |
|---|---|---|---|---|---|
| Flow rate | Temp (7–35°C), pressure (25–214 kPa) | Seawater, AW | 96–97 (F) | ||
| Seawater, ALOHA | 1,000–1,100 (F) | ||||
| Pure water | Variable | ||||
| Extraction efficiency | Flush direction | Seawater (<0.22 μm), KB | 200 (F) | ||
| Seawater, KB | 200 (F) | ||||
| A | Phage | 2 (F) | Liquid | ||
| B | Bacterial culture | 3 (F) | Liquid | ||
| C | Protist culture | 1 (F) | Pelleted* | ||
| % recovery | D, E | Seawater, KB | 11.7 (F) | Liquid, pelleted** | |
| Vol loaded | F, G | Seawater, KB | 10, 50, 250, 500, 1,000 (F) | Liquid, pelleted** | |
| H | Protist culture | 1, 2, 4 (F) | Liquid | ||
| Buffer chemistry (SDS vs GuHCl), filter material (AAO, PES) | I, J | Bacterial culture | 3 (F) | Liquid | |
| Proteinase K, lysozyme | Seawater, AW | 2 (C), 50 (F) | Pelleted* | ||
| Buffer chemistry (SDS vs LB3) | Seawater, KB | 500 (F) | |||
| Buffer chemistry (SDS vs LB1) | K | Seawater, AW | 50 (F) | Pelleted* | |
| DNA trapping | DNA size | DNA, 5-kb ladder | 0.5 (F) | Unfiltered ladder |
For buffer chemistry, SDS refers to extractions using the MasterPure kit, GuHCl refers to extractions using the DNeasy kit, and LB1 and LB3 refer to nonproprietary lysis buffers. The SDS-versus-LB1 extraction was conducted with and without lysozyme.
Experiments for which extraction efficiency was calculated are assigned an identification (ID) letter (A to K) for cross-referencing to Table 3.
For environmental samples, locations were Ala Wai Canal (AW), Station ALOHA (ALOHA), and Kāne‘ohe Bay (KB).
Processing consisted of filtration (F) or centrifugation (C).
Controls for extraction efficiency tests consisted of direct extractions of cells or viruses, including the liquid in which they were suspended (liquid) or after centrifuging to sediment primarily cells (*) or cells and viruses (**) as indicated (pelleted).
Filter flow rate and capacity.
The relationship between pressure and flow rate for 0.02-μm Anotop 25 AAO filters was tested by pumping purified water (NanoPure; Barnstead) through the filters at various temperatures (7, 22, and 35°C) at gauge pressures ranging from 25 to 214 kPa using a peristaltic pump (MasterFlex digital drive and L/S 14 PharMed tubing; Cole-Parmer). To evaluate filter-to-filter variability, the flow rate at 22°C was tested for three separate filters (Whatman catalog no. 6809-4002, lot no. D123932). Pressure was monitored with a flowthrough pressure transducer (GE Healthcare Life Sciences) connected immediately upstream of the filter. To account for pressure pulsing from the peristaltic pump, mean pressure was calculated from the highest and lowest pressure readings observed at a given pump speed. Flow rates were determined by measuring the volume of filtrate collected in a timed interval (1 to 3 min) in a 25-ml graduated cylinder (accuracy of ±0.1 ml).
The practical capacity of the filters was tested by pumping whole seawater through AAO filters (0.02-μm Anotop 25) in duplicate from each of two locations differing in their productivity. The first sample was collected at 25-m depth in the oligotrophic open ocean (Station ALOHA, located 100 km north of O‘ahu, HI; 22°45′00″N, 158°00′00″W) and the second from the upper decimeter of a eutrophic estuarine drainage canal (Ala Wai Canal, located in Honolulu, HI; 21°16′31.3″N, 157°49′03.8″W). Pressures and flow rates were recorded during the course of the filtration. To characterize the differences in microbial concentrations in these two habitats, we quantified prokaryotes and viruses by epifluorescence microscopy (26) and chlorophyll a by fluorescence after extraction in acetone (Ala Wai Canal) or by in situ fluorescence using a sensor calibrated to extracted chlorophyll (Station ALOHA) as described in the Hawaii Ocean Time Series Field & Laboratory Protocols (http://hahana.soest.hawaii.edu/hot/protocols/chap12.html).
Preparation of samples for filter extraction tests. (i) Coastal seawater.
Seawater samples were collected in polycarbonate carboys from a pier in Kāne‘ohe Bay on the windward side of O‘ahu, HI (21°25′46.80″N, 157°47′31.51″W) on multiple occasions between February 2010 and January 2013. For some experiments, whole seawater (10 ml to 1 liter) was directly filtered through Anotop 25 (0.02-μm) filters. Replicate samples were filtered in parallel using a multichannel peristaltic pump. In other cases, the seawater (200 ml) was first filtered through 0.22-μm PES membrane filter capsules (Sterivex; Millipore) prior to filtration through Anotop filters. Samples were filtered with a peristaltic pump as described above at approximately 14 ± 2 ml min−1. Residual sample was evacuated from all filters using air pressure from a hand-operated syringe, and then the filters were wrapped in Parafilm M (Pechiney), placed in Whirl-Paks (Nasco), and stored at −80°C until they were extracted.
(ii) Cultivated bacteriophage.
Vibrio phage VvAW1, a podovirus with a double-stranded DNA genome (38 kb) and a capsid diameter of 43 to 45 nm (27), was purified from plate lysates in a CsCl equilibrium buoyant density gradient (28), buffer exchanged into 3 ml SM (0.4 M NaCl, 0.02 M MgSO4, 0.05 M Tris, pH 7.5), and then filtered onto Anotop 25 (0.02-μm) filters in triplicate for each method and stored as noted above.
(iii) Bacterial culture.
A strain of Vibrio vulnificus (V93D1V) isolated from coastal waters of Oahu (29) was grown at 37°C with shaking for approximately 3 h or until the culture reached stationary phase as determined by the optical density (OD) reading. For experiments with washed cells, 12 ml of culture was centrifuged at 4,000 × g for 10 min. The pelleted cells were washed twice with 1× phosphate-buffered saline (PBS) (13.7 mM NaCl, 0.27 mM KCl, 1 mM phosphate buffer; 0.02-μm filtered), then resuspended in 1× PBS. For experiments with unwashed cells, 500 μl of liquid culture was diluted with 3 ml of 0.02-μm-filtered SM buffer. Suspended cells were filtered onto AAO (0.02-μm or 0.2-μm Anotop 25) or PES (0.2-μm Puradisc 25) syringe-tip filters with a sterile syringe in triplicate for each method and stored as noted above.
(iv) Cultivated protist.
A unialgal, nonaxenic culture of a dinoflagellate (Gymnodinium strain AL-DI06, isolated from Station ALOHA) was grown to a cell density of ∼1.2 × 105 cells ml−1 in f/2 medium at 25°C under a 12-h/12-h light/dark cycle. Subsamples of the culture (1, 2, or 4 ml, depending on the experiment) were filtered onto Anotop 25 (0.02-μm) filters with a sterile syringe in triplicate for each method and stored as noted above.
Back flush versus forward flush.
AAO filters through which whole seawater, 0.22-μm-filtered seawater, purified virus, bacterial culture, or dinoflagellate culture had been filtered were extracted using an SDS-based extraction buffer and the reagents in a commercially available kit (MasterPure; Epicentre). The back-flush protocol was conducted as described previously (22), in which case the buffer was introduced through the outlet side of the filter and recovered via the inlet, opposite to the direction in which the sample was loaded. For the forward-flush protocol, the extraction buffer was introduced through the inlet side of the filter and recovered via the outlet.
For each filter, 1 ml of tissue and cell lysis solution (MasterPure) containing 100 μg ml−1 proteinase K were loaded into a 3-ml-capacity sterile syringe (the injection syringe) and gently pushed through the filter via the appropriate opening until liquid just reached the other opening. The other opening was then sealed with a second 3-ml syringe (the aspiration syringe) and liquid pulled through to further saturate the filter membrane. The sealing of the filter on both ends with syringes allowed for thermal expansion without leakage of the buffer during the high-temperature incubation. The assembly was incubated at 65°C for 15 min while attached to a rotisserie in a hybridization oven rotating at full speed (ca. 16 rpm). The lysis buffer was then recovered by drawing it into the aspiration syringe. The extract was transferred to a 1.7-ml microcentrifuge tube and placed on ice. Salt-induced precipitation of protein-detergent complexes followed by an alcohol precipitation was then carried out following the manufacturer's instructions. All AAO filters from this study were extracted using this protocol, unless otherwise stated.
Trapping of dissolved DNA on AAO filters.
To determine whether dissolved DNA is trapped on the AAO membrane filters, we filtered 1 μg of a 5-kb DNA ladder (CHEF DNA size standard; Bio-Rad) resuspended in 500 μl of 1× Tris-EDTA (TE) through Anotop 25 (0.02-μm) filters in triplicate using a hand-operated syringe. We measured the quantity of DNA in the initial flowthrough and in three 500-μl washes of 1× TE. We then extracted the filters using the back-flush protocol and measured the quantity of DNA in the extract to estimate how much of the material retained on the membrane would be released. To determine whether there is a difference in the size distribution of the DNA trapped on the filter and on the material that passes through, we analyzed the initial flowthrough, the first wash, and the extracted material via pulsed-field gel electrophoresis (PFGE). Because the yields of DNA in the extract were low, we first concentrated the extracts by centrifugal ultrafiltration. To account for any losses resulting from the extract concentration step, we diluted triplicate aliquots of the 5-kb ladder in extraction buffer and then concentrated them in the same manner as the extract. We loaded approximately equal total masses of DNA (49 to 51 ng) of the 5-kb control ladder (in duplicate) and the concentrated control ladder, filtrates, washes, and extracts (all in triplicate) onto a 1% agarose gel with Blue Juice as the loading buffer (final concentration, 1×; Life Technologies). The ladders were resolved by PFGE in lithium borate buffer (1× LB; Faster Better Media, LLC) at 14°C, 6 V cm−1, and a 120° included angle for 18 h with a 1- to 6-s switch time. The gel was poststained with 1× SYBR gold (Life Technologies) and imaged with the Molecular Gel Logic 200 imaging system (Kodak). We measured the relative intensities of the bands using Gaussian curve fitting with Molecular Imaging software v.4.0.3 (Kodak) and then scaled the intensities within a sample according to the average recovery determined by fluorometry. We then calculated the percent recovery for each band in a sample by dividing its scaled intensity by that of the corresponding band in the appropriate control ladder.
Effects of filter loading on nucleic acid recovery.
Various volumes of whole seawater (10, 50, 250, 500, and 1,000 ml) from Kāne‘ohe Bay or a dinoflagellate culture (1, 2, or 4 ml) were filtered onto Anotop 25 (0.02-μm) filters in triplicate and extracted by the back-flush method as described above. For whole seawater, extraction efficiency was determined relative to two different controls using parallel subsamples of the same seawater, i.e., (i) direct extraction (1.8 ml) and (ii) extraction of a cell and virus pellet formed via ultracentrifugation (11.7 ml), under the conditions specified in “Estimating extraction efficiencies” below. Controls for the dinoflagellate culture were either directly extracted from liquid culture or a cell pellet produced by centrifugation at 16,000 × g for 10 min. All nucleic acid extractions were performed using the MasterPure kit, following the manufacturer's instructions.
Effects of buffer chemistry and filter material on nucleic acid recovery. (i) Direct extraction controls.
Equal portions of washed cells of V. vulnificus (V93D1V) were transferred into 1.7-ml microcentrifuge tubes, and half were extracted using the MasterPure kit, following the manufacturer's instructions for cell samples. The MasterPure kit is based on the protocol by Miller et al. (14) and uses an SDS-containing lysis buffer (30). The others were extracted with the DNeasy blood and tissue kit (Qiagen), following the manufacturer's instructions for cultured cells under the “Purification of total DNA from animal blood or cells” protocol. The DNeasy kit uses a guanidinium-based lysis buffer (guanidinium chloride [GuHCl]).
(ii) Filter extractions.
Aliquots from the same batch of washed cells were filtered onto AAO filters (0.2 μm and 0.02 μm) (Anotop 25; Whatman) or PES filters (0.2 μm) (Puradisc 25) with a sterile syringe. Half of the filters were extracted in triplicate with the MasterPure kit buffers using both the back- and forward-flush methods described above. The rest of the filters were extracted in triplicate using the back- and forward-flush methods described above but with buffers from the DNeasy kit, following the manufacturer's reaction conditions for cultured cells. For each filter, 1 ml of 0.02-μm-filtered lysis buffer (500 μl buffer AL and 500 μl 1× PBS) containing 40 μl proteinase K (DNeasy kit) was loaded into a 3-ml-capacity sterile syringe (the injection syringe) and gently pushed through the filter in the same manner as described above for either the back- or forward-flush filter extraction method. The other opening of the filter was sealed with a second syringe (the aspiration syringe), and the assembly was incubated at 56°C for 10 min while attached to a rotisserie in a hybridization oven rotating at full speed (ca. 16 rpm). The lysis buffer was recovered with the aspiration syringe and transferred into a 1.7-ml microcentrifuge tube, and 500 μl 100% ethanol was added to the sample. The mixture was vortexed and loaded onto a DNeasy minispin column and centrifuged at 10,000 × g for 1 min. Buffer washes were then performed according to the manufacturer's protocol, and nucleic acids were recovered by eluting twice with 200 μl buffer AE.
Effects of proteinase K digestion time on nucleic acid yields from cells.
Replicate subsamples of water were collected from Ala Wai Canal in Honolulu, HI, on multiple occasions in 2013 and 2014. For some experiments, either whole or filtered (<0.22 μm) seawater was filtered through Anotop 25 (0.02-μm) filters as described above. Other samples of 2 ml were centrifuged at 20,000 × g for 20 min to pellet cells. The supernatant was discarded, samples were frozen at −80°C and then thawed, and total nucleic acids were extracted from the pellets and filters in triplicate using the MasterPure kit but varying the proteinase K digestion time (15 or 30 min or 1, 3, 6, 12, 18, or 24 h).
Effects of lysozyme on nucleic acid yields from cells.
To test the combined effects of lysozyme and proteinase K digestions on nucleic acid recoveries, the proteinase K time series described in the previous section was conducted with and without a 30-min lysozyme treatment. Replicate subsamples of water collected from Ala Wai Canal were filtered or centrifuged as described above to collect plankton or pellet cells. Total nucleic acids were extracted from the filters or pellets in triplicate by one of four treatments using combinations of two variables: (i) with and without an initial lysozyme treatment (250 U sample−1) (Ready-Lyse lysozyme solution; Epicentre) and (ii) with a short (15-min) or long (1- or 12-h) proteinase K digestion. For the filter extraction with lysozyme treatment, the back-flush method described above was followed with a slight modification. After thawing the filters, 500 μl of TES (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 100 mM NaCl) with 1 μl Ready-Lyse lysozyme (250 U μl−1 in TES buffer) was carefully loaded onto the filter with a 3-ml syringe. The aspiration syringe was connected to the outlet to act as a seal for the buffer, and the filter was incubated at room temperature for 30 min. An aliquot of 500 μl 2× tissue and cell lysis solution (MasterPure) with 100 μg proteinase K was then added to the filter using the same injection syringe, and the filter-syringe assembly was incubated at 65°C for the indicated duration of proteinase K digestion. The rest of the protocol is the same as described above for extracting from filters or pelleted cells.
Comparison of a proprietary and a published salting-out extraction procedure.
Replicate subsamples of seawater were filtered through Anotop 25 (0.02-μm) filters or centrifuged to collect cells as described above. One set of filters and cell pellets were extracted using the back-flush method with the MasterPure kit as described above, with or without lysozyme treatment. Another set was extracted in the same manner (same volumes, temperatures, and centrifugation conditions) but using defined buffers derived from the literature, with or without lysozyme treatment. The first lysis buffer (LB3) contained a relatively high SDS concentration (50 mM Tris-HCl, 5 mM EDTA, 3% SDS, pH 8). This was intended to mimic the concentration of SDS in the MasterPure lysis buffer (estimated by the volume of the SDS pellet after salting out) and is the same buffer formulation as that used by Yu and Mohn (15). The second lysis buffer (LB1) contained a lower SDS concentration (50 mM Tris-HCl, 5 mM EDTA, 1% SDS, pH 8). The nucleic acid yield with LB3 was determined with no lysozyme treatment. The yield using LB1 was determined with or without an initial lysozyme treatment (Ready-Lyse lysozyme solution; Epicentre). In all cases, the extraction buffers were supplemented with proteinase K (100 μg ml−1 final concentration) just prior to use in the extractions. Salt-induced precipitation of protein-detergent complexes was accomplished by addition of ammonium acetate (2 M final concentration) and centrifugation at 16,000 × g for 10 min. The supernatant was transferred to a fresh 1.7-ml microcentrifuge tube, and linear acrylamide (20 μg ml−1 final concentration; Ambion) was added to the sample just prior to the addition of one volume of isopropanol. Nucleic acids were pelleted by centrifugation at 16,000 × g for 30 min. The nucleic acid pellet was then washed twice with 500 μl of 70% ethanol, dried completely, and resuspended in 20 μl TE.
Estimating extraction efficiencies.
To test for losses of DNA attributable specifically to the filter matrix (e.g., trapping and adsorption), we determined the yield of nucleic acids extracted from microorganisms collected on filters relative to the yields in the absence of a filter (liquid suspensions or pellets). Extraction efficiencies were also determined for all other filter extraction tests in which a control extraction was performed, as indicated in Table 1.
(i) Whole seawater.
Subsamples (11.7 ml) of seawater from Kāne‘ohe Bay were transferred to polyallomer tubes and centrifuged at 35,000 × g for 105 min in a swinging-bucket rotor (SW 41) in an ultracentrifuge (Beckman Optima XL-80K) to pellet cells and viruses. These conditions are sufficient to completely pellet particles with a sedimentation coefficient of ≥100S. While those samples were in the centrifuge, additional triplicate subsamples (11.7 ml), were filtered onto Anotop 25 (0.02-μm) filters and extracted using the back-flush protocol as described above. For the centrifuged samples, the supernatant was removed and saved. The pellet was immediately extracted using the MasterPure kit, as described in the manufacturer's instructions for cell samples. Nucleic acids remaining in the supernatant (presumed to be dissolved) were assayed as previously described (31). In brief, the supernatant was diluted with 2 ml of 0.5 M EDTA and then concentrated in a centrifugal ultrafiltration device (30-kDa cutoff) (Amicon 15; Millipore) by centrifuging at 2,500 × g for 25 min. The concentrates were then washed by adding 1 ml of 1× TE (10 mM Tris-HCl [pH 8], 1 mM EDTA) to the retentate cup and centrifuging again at 2,500 × g for 20 min. Each sample was then further concentrated by transferring it to a smaller centrifugal ultrafiltration unit (30-kDa cutoff) (Amicon Ultra-0.5; Millipore) and centrifuging at 10,000 × g for 10 min. The concentrate was washed by adding 500 μl of 1× TE, and centrifuging again at 10,000 × g for 30 min. The final concentrate was recovered by inverting the filter cartridge into a fresh tube and centrifuging at 1,000 × g for 3 min.
(ii) Bacterial cultures.
Cells in a culture of V. vulnificus (V93D1V) were washed and resuspended in 1× PBS, filtered onto Anotop 25 (0.02-μm) filters in triplicate, and then extracted as described above. Equal volumes of the same washed, resuspended cells were transferred into 1.7-ml microcentrifuge tubes for direct nucleic acid extraction using the MasterPure kit, following the manufacturer's instructions for suspended cell samples.
Quantification of nucleic acids.
Fluorometric assays were used to estimate the yields of nucleic acids from all of the experiments. DNA was quantified using a Quant-iT DNA assay kit (Life Technologies) and a Perkin-Elmer 2030 multilabel reader. To quantify RNA, samples were first treated with Turbo DNase (Life Technologies) to eliminate cross-reaction from DNA (32). RNA was then quantified using a Quant-iT RNA assay kit (Life Technologies) and a TD-700 fluorometer.
RESULTS
Filter flow rate and capacity.
The rate of filtration of pure water through an Anotop 25 (0.02-μm) filter increased linearly with pressure (Fig. 1A) and temperature (Fig. 1A, inset) over the ranges we tested (25 to 214 kPa and 7 to 35°C). The maximum flow rate in the tested range of conditions was 25 ml min−1 (202 kPa, 35°C). Under conditions similar to those that we use for routine collection of field samples (150 kPa, 22°C), the flow rate was approximately 13 ± 1.4 ml min−1 (mean ± standard deviation [SD], n = 3). From the area of the filter, we calculated a flux at standard ambient temperature and pressure (25°C, 100 kPa) of 1,180 ± 130 liters m−2 h−1 (mean ± SD). The normalized flux for an Anodisc mounted in a reusable housing was about 2.4 times greater than that of the Anotop (data not shown).
FIG 1.

Flow rates of pure water (A) or environmental water samples (B) through 0.02-μm Anotop 25 filters. The flow rate of pure water is shown as a function of pressure at various temperatures (A) and as a function of temperature at a fixed pressure of 100 kPa (inset). Data for 22°C are from triplicate filters to show variability among filters. The flow rates of environmental samples from the open ocean and an urban estuary (B) are normalized to a pressure of 150 kPa and plotted as a function of the volume of sample filtered. Also shown are the concentrations of viruses (Vir) and prokaryotes (Pro) in units of 109 liter−1 and of chlorophyll a (Chl) in units of μg liter−1 in the two samples (inset).
Filtration of water from two environments differing in plankton concentrations resulted in increasing pressures over time at the fixed initial flow rate of approximately 14 ml min−1. For the estuarine sample, pressure quickly exceeded the recommended maximum for the tubing (270 kPa), so the pump speed was reduced and filtration continued (total filtration time of <20 min for 100 ml). The pump speed was held constant for the open-ocean sample, and pressure steadily increased from 140 to 270 kPa over the 75 min filtration time, during which approximately 1 liter of sample was filtered. By normalizing all flow rates for both samples to a constant pressure of 150 kPa, we found that the flow rate declined linearly as a function of the volume filtered, and the decline was much steeper for estuarine water (Fig. 1B), which had higher concentrations of plankton than open-ocean water (Fig. 1B, inset). The normalized flow rate dropped by an order of magnitude after filtering approximately 0.1 liter of the estuarine sample but was reduced by only half after filtering 1 liter of open-ocean water.
DNA yield by forward versus back flush.
The back-flush extraction method for Anotop 25 (0.02-μm) filters yielded significantly more DNA than the forward-flush method when the sample consisted of cultured cells of a bacterium (t test, P = 0.034) or a dinoflagellate (t test, P = 0.017) (Fig. 2). There was no significant difference in DNA recovered from Anotop 25 (0.02-μm) filters by the forward- and back-flush methods when the sample was whole seawater (t test, P = 0.473), 0.22-μm-filtered seawater (t test, P = 0.214), or a phage isolate (t test, P = 0.920) (Fig. 2). Although there was no evidence of any improvement from back flushing when a sample was dominated by viral DNA (0.22-μm-filtered seawater and the phage isolate), the back flush resulted in a higher mean recovery in every trial where cells were present in the sample. The improvement was sometimes small and not statistically significant relative to the variability in yields among triplicates within a given experiment. To improve the statistical power, we normalized the yields of the extractions of all cell-containing samples by expressing them as a percentage of the push-through average within each experiment. When the cell extraction experiments (cell cultures and whole seawater) were then considered in aggregate, the higher yields from back flushing were found to be significant (t test, P = 0.003), with the difference between forward and back flush increasing in the order phage < bacterium < protist (Fig. 3).
FIG 2.

Total DNA recovered from cultures and whole or filtered (<0.22 μm) seawater using the forward- and back-flush methods of extraction. Values shown are means and standard deviations from triplicate assays conducted once for the phage and seawater and two times (effectively six replicates) for the bacteria and protist. An asterisk denotes the situations where the recoveries for the two methods were significantly different.
FIG 3.
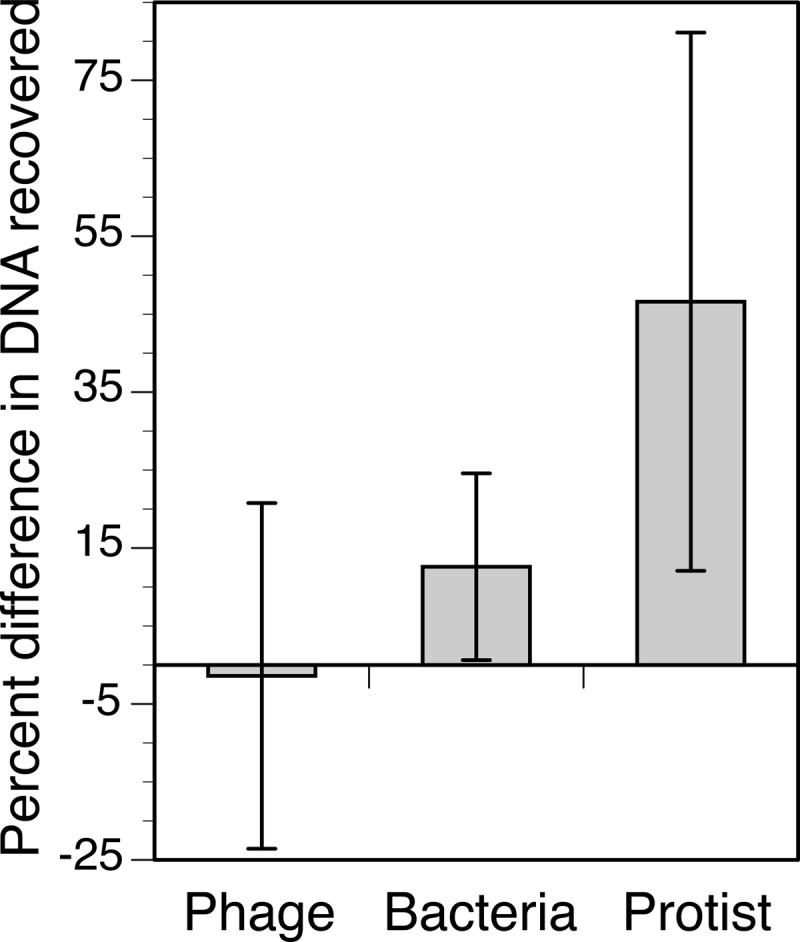
Improvement in DNA recovery for phage, bacteria, and protist cultures when using the back-flush relative to the forward-flush method. Shown are the average yields from the back-flush method expressed as a percentage of the forward-flush average for each experiment. Error bars are the standard deviations from triplicate assays conducted once for the phage and two times (effectively six replicates) for the bacteria and protist.
Trapping of DNA.
The 5-kb DNA ladder had similar masses of DNA in discrete bands ranging in size from 4.9 to ca. 98 kb, which together accounted for 78% of the total mass. Some additional higher-molecular-weight DNA (>80 kb; 16% of the total) migrated into the gel but was not well resolved, and some DNA (6% of the total) remained trapped in the well (Fig. 4A). Based on bulk estimates of DNA mass by fluorometry, approximately 77% ± 13% (mean ± SD) of the DNA passed through the filter (64% in the initial filtrate and the remainder in two washes), and 7% ± 2% (mean ± SD) was recovered from the filter by the back-flush extraction protocol (Table 2). The filtrates, washes, and extract together accounted for 84% ± 12% (mean ± propagated error) of the total loaded. Analysis of the DNA size distribution in the ladders, filtrates, washes, and extracts showed that for the smaller bands (5 to 54 kb) a similar large percentage of the DNA was recovered in the filtrate (≥80%) (Fig. 4), and there was no significant relationship between size and recovery (Spearman rank correlation, r = −0.021; P > 0.5). An additional small percentage (≤11%) of the material in those bands was recovered in the extract. For larger DNA that entered the gel (from 49 to >80 kb) there was a negative correlation between size and the percentage of DNA in the filtrate (Spearman rank correlation, r = −1.00; P < 0.001). The missing material from these bands was not accounted for in the extract (Fig. 4B). For the form of DNA that remained trapped in the well under the specified separation conditions, 45% was accounted for in the filtrate plus wash and 46% in the extract.
FIG 4.

Changes in the size distribution of the DNA in a 5-kb ladder as a result of filtration through and extraction from a 0.02-μm Anotop filter, shown as pulsed-field gel images (A) and as calculated percent recoveries for each band size (B). (A) Representative gel images of unconcentrated and concentrated control ladders, the material passing through the 0.02-μm filter, and the material extracted from the filter. An approximately equal mass of material was loaded in each lane on the same gel and separated using conditions indicated in the text. (B) Recoveries for each size band were calculated as a percentage of the intensity in the corresponding control ladder after scaling intensity to the total DNA recovered as determined by fluorometry. W refers to material trapped in the well.
TABLE 2.
Recovery of DNA in a 5-kb ladder recovered after filtration through AAO (0.02-μm Anotop 25) filters
| Sample | % recoverya |
|---|---|
| Filtrate | 64 (12) |
| Wash 1 | 10 (2) |
| Wash 2 | 2 (0.5) |
| Wash 3 | 0.6 (0.3) |
| Extract | 7 (2) |
| Total | 84 (12) |
Percentages of DNA in the filtrate and washes represent material passing through the filters, and that in the extract represents material recovered from the filters using a back-flush protocol. Values are means (standard deviations) from triplicate assays.
Effect of sample volume on extraction yield.
There was a positive, linear relationship between the volume of whole-seawater sample added to an Anotop 25 (0.02-μm) filter and the amount of DNA extracted from it (Fig. 5A). The percent recoveries were variable, with an average of 101% ± 24%, and there did not appear to be a consistent trend in extraction efficiency with volume (Fig. 5B). The sample-to-sample variability and the high variance in the smaller-volume samples may be a result of heterogeneity in the natural sample. However, even with volumes of up to 1 liter, recoveries were high relative to the reference estimate of the DNA concentration in the plankton (extraction from pelleted cells and viruses).
FIG 5.

Recovery of DNA from AAO filters as a function of volume of sample filtered, expressed as either the total (A) or the estimated percentage (B) of DNA recovered from seawater or the total (C) or percentage (D) of DNA recovered from a dinoflagellate culture. Percent recoveries for the seawater samples were calculated relative to an estimate of the concentration of planktonic DNA in the seawater obtained by direct extraction of cells and viruses that were pelleted by ultracentrifugation from a subsample of the same water. To calculate percent recoveries from the dinoflagellate culture, the concentration of DNA in the culture was estimated by direct liquid extraction of a culture subsample. Values shown are means and standard deviations from triplicate assays.
To minimize the problems of heterogeneity seen in the natural sample, the experiment was repeated using a culture. We again observed a positive, linear relationship between the volume of culture loaded on the AAO filters and the amount of DNA extracted (Fig. 5C). Although there appeared to be a trend of decreasing efficiency with increasing culture volume, this trend was not significant because of the high variance (Fig. 5D). The calculated minimum efficiencies for the sample ranged from 72 to 89% relative to the reference estimate of DNA in the culture obtained by direct liquid extraction (cells plus dissolved DNA).
Guanidinium- versus SDS-based buffers.
When DNA was extracted directly from washed bacterial cells suspended in liquid, the DNA yield was approximately 55% lower for the DNeasy kit than for the MasterPure kit. We assumed that the higher value obtained by the latter kit most closely represented the true amount of nucleic acids in the culture, and that number was used as the control value relative to which efficiencies for all of the filter extractions were calculated, regardless of the chemistry used.
The amount of DNA recovered from the AAO filters (0.2- or 0.02-μm Anotop 25) using the MasterPure kit was significantly higher than when using the DNeasy kit, whether the forward- or back-flush method was used (t test, P = 0.008 for forward flush on 0.02-μm Anotop 25 and P ≤ 0.005 for all other tests) (Fig. 6). The difference found between the two kits when extracting from PES filters was significant as well (t test, P ≤ 0.005). When comparing the forward- versus back flush-methods in the AAO filter extractions for the DNeasy kit, the amount of DNA recovered from the back-flush method was significantly higher (t test, P = 0.005 for 0.02 μm and P = 0.038 for 0.2 μm). However, this was not true for the PES filters (0.2 μm) using the DNeasy kit (t test, P = 0.198).
FIG 6.

Total mass of DNA recovered by direct extraction of a liquid bacterial culture or extraction of an equivalent volume of culture collected on three types of filters (0.02-μm or 0.2-μm AAO and 0.2-μm PES). These assays were conducted with the forward-flush (FF) and back-flush (BF) methods using either the MasterPure complete DNA and RNA purification kit (Epicentre) or the DNeasy blood and tissue kit (Qiagen). Values shown are means and standard deviations from triplicate assays.
Effects of proteinase K digestion time and lysozyme on nucleic acid yields from cells.
Yields of nucleic acids from pelleted cells after 15 min of digestion were not improved by increasing proteinase K digestions up to 6 h (analysis of variance [ANOVA] with post hoc Tukey test, P > 0.05) without addition of lysozyme treatment. The yields after 12 h of digestion were significantly higher than those at 15 min, by 23 to 25% (ANOVA with post hoc Tukey test, P < 0.05). In another experiment with the addition of lysozyme treatment, yields of nucleic acids were not significantly improved with proteinase K digestion times from 15 min up to 18 h, but the effect was significant after 24 h (Tukey test, P < 0.05).
In a second set of tests comparing interacting effects of lysozyme and proteinase K digestion, the effect of increasing the proteinase K digestion time from 15 min to 12 h was not significant for RNA or DNA, except when an initial lysozyme digestion was included. An initial lysozyme digestion did not improve the yields of DNA or RNA when the proteinase K digestion was 15 min, but it improved yields of DNA and RNA by 40 and 50% when the subsequent proteinase K incubation time was 12 h (ANOVA with post hoc Tukey test, P = 0.024 for DNA and P = 0.006 for RNA).
The effects of lysozyme (30 min) and proteinase K (15 min or 1 h) were also tested for seawater plankton (total or <0.22 μm) collected on AAO filters. The addition of lysozyme was significant only for the whole seawater in the 15-min proteinase K incubation, increasing yields of DNA by 23% (ANOVA with post hoc Tukey test, P = 0.031).
Because of the variable results among individual experiments, the data from multiple experiments were normalized to yields at 15 min and considered in aggregate. For the larger pooled data set, the lysozyme treatment significantly improved yields for the whole-seawater samples (t test, n = 20, P = 0.008) but not the filtered (0.22 μm) seawater samples (t test, n = 6, P = 0.985). When total seawater plankton was extracted in the absence of lysozyme, there was no increase in DNA yield with increasing proteinase K digestion time (ANOVA, n = 42, P > 0.25). However, when an initial lysozyme treatment was included, there was a significant positive relationship (ANOVA, n = 44, P ≤ 0.0025) between DNA yield and proteinase K digestion time, with an increase in yield of 1% ± 0.6% (mean ± 95% CI) per hour of incubation over the range tested (15 min to 24 h).
MasterPure versus modified salting-out extraction.
In a direct comparison of the DNA yield from AAO filters using the proprietary MasterPure buffers versus the yield using nonproprietary buffers derived from the literature, we found the yields with the MasterPure buffers to be consistently higher. The yield from the MasterPure buffers was 2.3 times higher (t test, P = 0.032) than for the first salting-out method with a higher SDS concentration (3%, LB3). However, when using the second method with a lower SDS concentration (1%, LB1), we found that the yield with the MasterPure buffers was not significantly different with or without lysozyme (ANOVA, P = 0.462). When extracting from pelleted cells, the MasterPure extraction method with lysozyme treatment resulted in significantly higher yields than the LB1 modified salting-out method without lysozyme treatment (ANOVA with post hoc Tukey test, P = 0.021). The addition of lysozyme resulted in significantly higher yields (by 27%) for the LB1 modified salting-out method on the pelleted cells (ANOVA with post hoc Tukey test, P = 0.018). When the data for the pelleted cell extraction and filter extraction procedures both with and without lysozyme were pooled, the MasterPure method and the second, nonproprietary method with 1% SDS were not significantly different (ANOVA, P = 0.231).
DNA and RNA extraction efficiencies.
In an experiment designed to constrain relative extraction efficiency, the masses of DNA and RNA recovered from total seawater plankton collected on Anotop 25 (0.02-μm) filters were 112% ± 5% and 134% ± 13%, respectively (t test, P = 0.014 for DNA and P = 0.006 for RNA), of the yields recovered from plankton pelleted from an equal volume of seawater by ultracentrifugation. To test whether the higher yields from the filters might have been attributable to dissolved DNA that was trapped by the filters but which failed to pellet, we directly measured the nucleic acids in the supernatant after ultracentrifugation. When we summed the quantity of nucleic acids detected in the supernatant of the controls and the amount recovered from the extracted pellet, the total mass of nucleic acids was indistinguishable from that recovered on the filter (t test, P = 0.088).
In a second experiment using a cultured bacterium, the estimated recoveries of nucleic acids from the Anotop 25 (0.02-μm) filters were 84% ± 8% for DNA and 73% ± 5% for RNA relative to an equal volume of cell culture extracted directly. In this experiment the cells were unwashed so the sample would contain nucleic acids from the cells plus any nucleic acids dissolved in the medium.
In addition to these two tests designed solely to test extraction efficiency, we also estimated extraction efficiencies in all other extraction tests where a control extraction was conducted (Table 3). For all tests in which a cell (or cell-plus-virus) pellet was used as a control, the average extraction efficiency was 102% ± 19% for DNA. When the bacterial culture extraction data were combined, the average percent recovery of DNA was 94% ± 9% (Table 3, B, I, and J). The average percent recoveries of DNA and RNA for all experiments using liquid culture extracts as a control were 93% ± 24% and 82% ± 12%, respectively.
TABLE 3.
Recovery of nucleic acids from microorganisms collected on AAO (0.02-μm Anotop 25) filters
| Controla | Sampleb | ID | Extraction methodc | Variable | % recoveryd |
|
|---|---|---|---|---|---|---|
| DNA | RNA | |||||
| Liquid | Phage | A | MP | Flush direction | 73 (10) | |
| Bacteria | B | MP | Flush direction | 96 (7) | ||
| I | MP | Chemistry, 1 | 84 (8) | 73 (5) | ||
| J | MP | Chemistry, 2 | 102 (4) | 91 (4) | ||
| Protist | H | MP | Vol, 1 ml | 89 (12) | ||
| H | MP | Vol, 2 ml | 82 (14) | |||
| H | MP | Vol, 4 ml | 72 (11) | |||
| Seawater, KB | D | MP | % recovery | 117 (23) | ||
| G | MP | Vol, 10 ml | 130 (52) | |||
| G | MP | Volume 50 ml | 68 (53) | |||
| G | MP | Vol, 250 ml | 93 (30) | |||
| G | MP | Vol, 500 ml | 113 (36) | |||
| G | MP | Vol, 1 liter | 95 (33) | |||
| All liquid | 93 (24) | 82 (12) | ||||
| Pelleted* | Protist | C | MP | Flush direction | 144 (12) | |
| Seawater, AW | K | MP, 60 | Chemistry + Lys | 91 (11) | ||
| K | MP, 60 + Lys | Chemistry + Lys | 92 (14) | |||
| K | LB1, 60 | Chemistry + Lys | 110 (38) | |||
| K | LB1, 60 + Lys | Chemistry + Lys | 65 (19) | |||
| Pelleted** | Seawater, KB | E | MP | % recovery | 112 (7) | 134 (15) |
| F | MP | Vol,10 ml | 132 (33) | |||
| F | MP | Vol, 50 ml | 69 (49) | |||
| F | MP | Vol, 250 ml | 95 (6) | |||
| F | MP | Vol, 500 ml | 115 (7) | |||
| F | MP | Vol, 1 liter | 97 (15) | |||
| All pelleted | 102 (19) | 134 (15) | ||||
| Pelleted** + supernatant | Seawater, KB | E | MP | % recovery | 106 (7) | |
Extraction efficiency controls consisted of direct extractions of microorganisms, including the liquid in which they were suspended (liquid) or after centrifuging to pellet primarily cells (*) or cells and viruses (**) as indicated (pelleted).
For environmental samples, locations were Ala Wai Canal (AW) and Kāne‘ohe Bay (KB).
The extraction methods refer to the standard MasterPure protocol with 15 min of proteinase K incubation and no lysozyme treatment (MP), unless otherwise indicated by 60 (60 min of proteinase K incubation), + Lys (lysozyme treatment), or LB1 (nonproprietary extraction method).
Values are means (standard deviations) from triplicate assays, expressed as a percentage of control values.
DISCUSSION
In this study, we (i) characterized the flow characteristics and capacities of commercially produced AAO filters, (ii) empirically verified the hypothesis that both physical trapping and adsorption can be important sources of loss when extracting nucleic acids from microorganisms harvested on these filters, and (iii) demonstrated that a previously reported back-flushing protocol (22) can effectively avoid those losses. The results of our experiments to test the effects of extraction chemistry, flow direction, and enzyme treatment on yields of nucleic acids provide insights that may be useful for researchers developing extraction protocols for other types of samples or filters. We first discuss our results and their implications and then comment on our recommended protocols for extractions of nucleic acids from AAO filters using proprietary or nonproprietary buffers (Fig. 7).
FIG 7.

Recommended extraction protocol for obtaining nucleic acids from AAO filters.
Filter flow characteristics.
Our estimate of the flux of 0.02-μm Anotop 25 filter units is only about 30% of the reported inherent flux for the Anopore membranes in the filter units (manufacturer's technical literature). We presume that the lower flux is a result of the manner in which the membranes are mounted and supported in the housing. We observed higher flux rates for an Anodisc membrane mounted in a reusable housing (ca. 70% of the reported Anopore flux), which implies that there is something specific about the manufacture of the Anotop units that leads to occlusion of a large fraction of the pores. Nevertheless, we find that the flux is sufficient for our applications and that the small disposable units are especially convenient for field sampling and subsequent extraction. Another type of filter (modified hydrophilic polyvinylidene difluoride [PVDF]) with the same nominal 20-nm pore size is offered in a very similar format (Optiscale-25 with Viresolve NFP membrane; Millipore). We considered the use of these as an alternative to the Anotop 25, but they have a normalized flux that is four times lower (unpublished observation) and cost several times more per filter, so we did not evaluate them further.
Our tests of the Anotop 25 filter capacity illustrate how different types of samples will influence the amount of water that can be filtered and how long one can expect filtration to take. Extrapolation of the lines in Fig. 1B suggests that the practical capacity of the 0.02-μm Anotop 25 filter was about 100 ml for the highly productive, turbid waters of an urban estuary but around 2 liters for lower-productivity open ocean waters. This is consistent with the observation that 1 to 2 liters of surface water at Station ALOHA can be filtered through a 0.02-μm Anotop 25, with the amount varying seasonally (C. Schvarcz, personal communication). In one experiment for this study, we were able to filter a liter of water from Kāne‘ohe Bay through a 0.02-μm Anotop 25, but on other occasions we have managed to filter only 200 to 550 ml of water from the same location in the bay (24). This is consistent with the variable conditions in the bay, which range from nutrient poor with low biomass during periods of low rainfall to nutrient enriched with large plankton blooms following terrestrial runoff from rain events (33).
Physical trapping.
The relationship we observed between DNA size and filter capture is consistent with other studies (34), but we were still surprised that there was no discernible loss of DNA with a forward-flush extraction of the bacteriophage or a natural viral assemblage. Even though viruses have comparatively small genomes, the persistence length of double-stranded DNA (ca. 35 nm [35]) is greater than the pore size, and we anticipated significant losses for the viral DNA. The relative high efficiency with which DNAs of all tested microorganisms and a DNA ladder passed through the filters is likely a result of DNA elongation driven by the converging flow fields as the molecules approach the pores (36).
Our experiments with the DNA ladder suggest that there was size selection occurring during filtration of DNA in the range typical of viruses, but only for DNA that was >50 kb. Since the virus isolate we used for testing was ca. 38 kb (27) and the majority of viral genomes in seawater are ≤50 kb (37), this would explain why we found a small, but statistically insignificant, difference in overall yield of DNA from viruses in the forward- versus back-flushing tests. We presume that the material in the 5-kb ladder that did not enter the gel was of unusually high molecular weight (38). We do not know why this material was recovered with higher efficiency than material of intermediate size in the 60- to ca. 100-kb size range, but based on the observed patterns of recovery, one plausible scenario is that smaller DNA molecules readily pass the filter and end up primarily in the filtrate, intermediate sizes become trapped within the pores of the filter and remain associated with the membrane, and a greater proportion of very large molecules are trapped on the upper surface of the filter, such that they can be more efficiently released into the extract by back flushing. Regardless of the mechanism, our data suggest that there is size selection that occurs when DNA is forced through the AAO filter membrane.
In addition to the effect of size, differences in DNA topology (circular versus linear or degree of supercoiling) can have a large influence on filter passage (39, 40). A likely consequence is that viruses having circular or very large genomes could be underrepresented in an extract if the nucleic acids are forced to pass through a small-pore-size filter. Forward flushing will also generally reduce the yield of DNA from cells. Therefore, for most routine extractions we recommend back flushing to minimize the chance of representational bias. This is particularly important for quantitative analyses of community composition. One must bear in mind, however, that the trapping and release of at least some fraction of dissolved DNA over a broad size range mean that not all of the nucleic acids recovered from a filter will necessarily derive from intact viruses or cells. This is an issue that has been recognized even with large-pore-size filters (41–43).
Adsorption.
The reversible adsorption of nucleic acids to silica in the presence of chaotropic salts (e.g., iodide or guanidinium ions) is a well-known phenomenon (44, 45) and serves as the basis for some of the most popular DNA and RNA purification kits. However, the binding of nucleic acids to AAO in the presence of guanidinium appears to be irreversible (21). This phenomenon might be exploited for solid-phase amplifications (20), but for our application it is a possible source of loss.
The changes in yields that we observed in response to varying the (i) extraction buffer chemistry, (ii) direction of buffer flow, (iii) filter material, or (iv) filter pore size were are all consistent with nucleic acids binding to the AAO in the presence of guanidinium. In contrast to the relatively small losses of DNA with forward flushing in the SDS-based buffer (apparently a function of physical trapping), the loss with forward flushing in the guanidinium-based buffer was dramatic, but only for AAO filters. Presumably forward flushing was a particular problem with the guanidinium buffer, because it increases the contact of the DNA with the filter, dramatically increasing adsorption. The loss was less pronounced, but still significant, for forward flushing with the 0.2-μm Anotop 25 filter, which we attribute to decreases in both physical trapping and lower adsorption resulting from the larger pore size. The absence of significant loss when forward flushing through PES membranes, regardless of buffer, suggests that the adsorptive losses are specific to AAO membranes as hypothesized.
Buffer chemistry.
Although back flushing effectively eliminated the severe adsorption loss when extracting DNA from AAO filters with the guanidinium-based DNeasy kit, we found that the yields were consistently lower than with the alternative SDS-based MasterPure kit by about 50%. In another preliminary experiment we found that the yields between the two kits we used were similar for direct extraction of cultured cells (data not shown), so the difference between the kits appears to vary. The lower yields obtained with the DNeasy kit are not likely a result of exceeding the binding capacity of the silica, since the reported capacity (30 μg) is 10 times the yield we achieved. Although we do not have an explanation for them, our observations are consistent with those of others who have also reported relatively poor yields using the Qiagen spin column kit compared to a liquid SDS-proteinase K extraction protocol (41).
We used two commercial extraction kits for our experiments out of convenience and because of their popularity. However, we recognize that a reliance on proprietary kits is not always desirable, and their availability and cost can be an issue for many labs. We therefore decided to compare the MasterPure kit with nonproprietary buffers derived from the literature. We tested a buffer with 3% SDS initially, because this appeared to be most similar to concentration of SDS in the MasterPure buffer (roughly estimated from the volume of the SDS pellet after salting out). That produced poor results, so we reduced the SDS concentration to 1%. This appears to have the advantage of reducing the inactivation of proteinase K itself, while still sufficiently denaturing other proteins to achieve near-maximal protein degradation rates (46). A coprecipitant was added to the nonproprietary protocol to enhance the yield of nucleic acids during the final alcohol precipitation step. A coprecipitant was not added to the MasterPure protocol, because the buffers already include an unspecified reagent to enhance recoveries of small amounts of nucleic acids (30). The yields using the MasterPure kit buffers were consistently higher than those achieved with the nonproprietary buffers, but the difference between the second formulation with 1% SDS (LB1) and the MasterPure buffer was relatively small and not statistically significant. The failure to reject the null hypothesis (no difference in yields between the two methods), may be a result of the relatively large sample-to-sample variance among the triplicate samples, but for investigators averse to using proprietary buffers or for whom access to the MasterPure kit is difficult, our protocol with a nonproprietary 1% SDS lysis buffer seems a reasonable alternative. We prefer using the commercial kit, however, because of convenience, consistency in preparation in dedicated clean facilities, and a cost per extraction that is still quite reasonable compared to that with spin column kits.
Extraction efficiency.
A previous comprehensive study of DNA extraction from microorganisms collected on 0.2-μm PES membranes (41) identified a number of key variables affecting extraction yields. Two of these were enzymatic digestion steps (lysozyme and proteinase K). Although our tests are not directly comparable to those of Boström et al. (41) because of differences in experimental design, the results of the two studies are consistent and suggest that using an initial lysozyme digestion followed by a long incubation with proteinase K and SDS (up to 24 h) can significantly increase yields of DNA from cells. We further show that these conditions also increased yields of RNA and found that increasing the proteinase K digestion time in the absence of lysozyme was of marginal value. We found some degradation of RNA with these increased proteinase K digestion times (data not shown) and therefore recommend increasing the proteinase K incubation to only about 1 h (or at most ≤6 h). As expected, the lysozyme treatment did not result in a significant increase of yields in the absence of cells, so this step could be eliminated if one wishes to extract nucleic acids only from viruses. For quantitative analyses of community composition, on the other hand, we would recommend including a lysozyme digestion, since we found no evidence that it reduces total yields and it may facilitate the lysis of some populations that would be otherwise underrepresented.
Because our primary goal in this study was to test specific hypotheses about the interaction between nucleic acids and the 0.02-μm AAO filters, we most often employed the simpler protocol with no initial lysozyme digestion and a 15-min proteinase K digestion, the standard conditions recommended by the manufacturer of the MasterPure kit. The yields under these conditions are likely not as high as they could be, but they were sufficient for the comparative extraction tests to determine the extent to which the filter matrix influenced yields. However, even these simple comparisons of yields between microorganisms on filters versus those suspended in liquid or in a pellet are complicated by the presence of dissolved DNA in the samples, a variable fraction of which is physically trapped by the filters.
Despite our inability to definitively distinguish contributions of truly dissolved nucleic acids on the filters or in microorganism pellets, we can constrain the relative extraction efficiencies. In cases where the direct extraction of suspended microorganisms included all of the dissolved DNA in a sample, our filter extractions were underestimates (93% ± 24%), and in cases where dissolved DNA was depleted prior to direct extraction, our filter extraction efficiencies were overestimates (102% ± 19%). Given these ranges for the under- and overestimates, the relative filter extraction efficiency appears to be very high, with yields close to 100% of those from direct extraction. This suggests that our recommended protocol effectively avoids interference from the filter matrix.
Conclusions.
We have shown that Anotop 25 filters with a 0.02-μm AAO membrane can be used to capture microorganisms from tens to thousands of milliliters of water, depending on the load of microorganisms in the sample, and at flow rates on the order of 10 to 20 ml min−1 or more depending on the water temperature and desired pressure. When extracting nucleic acids from cells and viruses trapped on the filters, yields can be significantly affected by the extraction buffer, filter material, direction of flow, and nucleic acid size. The most serious losses occurred when a guanidinium-containing extraction buffer was combined with AAO filters and the extract was forced through the filter. Among the conditions tested, the highest relative yields were achieved using the buffers of the MasterPure total DNA and RNA extraction kit and back flushing the filter (yields using nonproprietary buffers were similar). Using this preferred protocol, which employs an SDS-based extraction buffer, we showed that nucleic acids could be extracted from microorganisms collected on AAO filters at the same efficiency as from microorganisms in pellets or in liquid suspension. To ensure maximum yields from bacteria, one should consider including a lysozyme digestion step and increasing the proteinase K digestion time. Because yields of nucleic acids were linear over a broad range of mass and volume filtered, collection of microorganisms from liquid samples onto AAO filters is a simple and effective way to increase the mass of nucleic acids harvested without sacrificing efficiency.
ACKNOWLEDGMENTS
We thank O. Nigro for kindly providing cultures of the bacterium Vibrio vulnificus (V93D1V) and its phage (VvAW1) and C. Schvarcz for providing cultures of the Gymnodinium dinoflagellate strain (AL-DI06).
This work was funded by NSF grants to G.F.S. and A.I.C. (OCE 08-26650) and to G.F.S. (ANT 09-44851) and by the Center for Microbial Oceanography: Research and Education (EF 04-24599).
Footnotes
Published ahead of print 18 April 2014
REFERENCES
- 1.Fuhrman J, Comeau D, Hagström A, Chan A. 1988. Extraction from natural planktonic microorganisms of DNA suitable for molecular biological studies. Appl. Environ. Microbiol. 54:1426–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pichard SL, Frischer ME, Paul JH. 1993. Ribulose-bisphosphate carboxylase gene expression in subtropical marine phytoplankton populations. Mar. Ecol. Prog. Ser. 101:55. 10.3354/meps101055 [DOI] [Google Scholar]
- 3.Somerville CC, Knight IT, Straube WL, Colwell RR. 1989. Simple, rapid method for direct isolation of nucleic acids from aquatic environments. Appl. Environ. Microbiol. 55:548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valentin K, John U, Medlin LK. 2005. Nucleic acid isolation from environmental aqueous samples. Methods Enzymol. 395:15–37. 10.1016/S0076-6879(05)95002-7 [DOI] [PubMed] [Google Scholar]
- 5.Brum JR, Schenck RO, Sullivan MB. 2013. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 7:1738–1751. 10.1038/ismej.2013.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wommack KE, Sime-Ngando T, Winget DM, Jamindar S, Helton RR. 2010. Filtration-based methods for the collection of viral concentrates from large water samples, p 110–117 In Suttle CA, Wilhelm SW, Weinbauer MG. (ed), Manual of aquatic viral ecology. American Society of Limnology and Oceanography, Waco, TX [Google Scholar]
- 7.Suzuki MT, Preston CM, Béjà O, de la Torre JR, Steward GF, DeLong EF. 2004. Phylogenetic screening of ribosomal RNA gene-containing clones in bacterial artificial chromosome (BAC) libraries from different depths in Monterey Bay. Microb. Ecol. 48:473–488. 10.1007/s00248-004-0213-5 [DOI] [PubMed] [Google Scholar]
- 8.Percival SL, Chalmers RM, Embrey M, Hunter PR, Sellwood J, Wyn-Jones P. 2004. Microbiology of waterborne diseases. Elsevier, London, United Kingdom [Google Scholar]
- 9.John SG, Mendez CB, Deng L, Poulos B, Kauffman AKM, Kern S, Brum J, Polz MF, Boyle EA, Sullivan MB. 2011. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 3:195–202. 10.1111/j.1758-2229.2010.00208.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jani AMM, Losic D, Voelcker NH. 2013. Nanoporous anodic aluminium oxide: advances in surface engineering and emerging applications. Prog. Mater. Sci. 58:636–704. 10.1016/j.pmatsci.2013.01.002 [DOI] [Google Scholar]
- 11.Steward GF, Culley AI, Wood-Charlson EM. 2013. Marine viruses, p 127–144 In Levin SA. (ed), Encyclopedia of biodiversity, 2nd ed, vol 5 Elsevier, Waltham, MA [Google Scholar]
- 12.Culley AI, Steward GF. 2007. New genera of RNA viruses in subtropical seawater, inferred from polymerase gene sequences. Appl. Environ. Microbiol. 73:5937–5944. 10.1128/AEM.01065-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culley AI, Asuncion BF, Steward GF. 2009. Detection of inteins among diverse DNA polymerase genes of uncultivated members of the Phycodnaviridae. ISME J. 3:409–418. 10.1038/ismej.2008.120 [DOI] [PubMed] [Google Scholar]
- 14.Miller SA, Dykes DD, Polesky HF. 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:1215. 10.1093/nar/16.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Z, Mohn WW. 1999. Killing two birds with one stone: simultaneous extraction of DNA and RNA from activated sludge biomass. Can. J. Microbiol. 45:269–272. 10.1139/w98-211 [DOI] [Google Scholar]
- 16.Ferrara GB, Murgia B, Parodi AM, Valisano L, Cerrano C, Palmisano G, Bavestrello G, Sara M. 2006. The assessment of DNA from marine organisms via a modified salting-out protocol. Cell. Mol. Biol. Lett. 11:155–160. 10.2478/s11658-006-0013-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. 1989. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 17:8390. 10.1093/nar/17.20.8390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laitinen J, Samarut J, Hölttä E. 1994. A nontoxic and versatile protein salting-out method for isolation of DNA. Biotechniques 17:316–322 [PubMed] [Google Scholar]
- 19.Aljanabi SM, Martinez I. 1997. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 25:4692–4693. 10.1093/nar/25.22.4692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dames S, Bromley LK, Herrmann M, Elgort M, Erali M, Smith R, Voelkerding KV. 2006. A single-tube nucleic acid extraction, amplification, and detection method using aluminum oxide. J. Mol. Diagn. 8:16–21. 10.2353/jmoldx.2006.040398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerdes JC, Marmaro JM, Roehl CA. September 2001. Nucleic acid archiving. US patent 6,291,166. [Google Scholar]
- 22.Steward GF, Culley AI. 2010. Extraction and purification of nucleic acids from viruses, p 154–165 In Wilhelm SW, Weinbauer MG, Suttle CA. (ed), Manual of aquatic viral ecology. American Society of Limnology and Oceanography, Waco, TX [Google Scholar]
- 23.Culley AI, Asuncion BA, Steward GF. 2009. Detection of inteins among diverse DNA polymerase genes of uncultivated members of the Phycodnaviridae. ISME J. 3:409–418. 10.1038/ismej.2008.120 [DOI] [PubMed] [Google Scholar]
- 24.Culley AI, Steward GF. 2007. New genera of RNA viruses in subtropical seawater inferred from polymerase gene sequences. Appl. Environ. Microbiol. 73:5937–5944. 10.1128/AEM.01065-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Needham DM, Chow C-ET, Cram JA, Sachdeva R, Parada A, Fuhrman JA. 2013. Short-term observations of marine bacterial and viral communities: patterns, connections and resilience. ISME J. 7:1274–1285. 10.1038/ismej.2013.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel A, Noble RT, Steele JA, Schwalbach MS, Hewson I, Fuhrman JA. 2007. Virus and prokaryote enumeration from planktonic aquatic environments by epifluorescence microscopy with SYBR green I. Nat. Protoc. 2:269–276. 10.1038/nprot.2007.6 [DOI] [PubMed] [Google Scholar]
- 27.Nigro OD, Culley AI, Steward GF. 2012. Complete genome sequence of bacteriophage VvAW1, which infects Vibrio vulnificus. Stand. Genomic Sci. 6:415–426. 10.4056/sigs.2846206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawrence JE, Steward GF. 2010. Purification of viruses by centrifugation, p 166–181 In Wilhelm SW, Weinbauer MG, Suttle CA. (ed), Manual of aquatic viral ecology. American Society of Limnology and Oceanography, Waco, TX [Google Scholar]
- 29.Nigro OD. 2012. Environmental controls on Vibrio vulnificus and other pathogenic vibrios in tropical and subtropical coastal waters. Dissertation. University of Hawai‘i at Mānoa, Manoa, HI [Google Scholar]
- 30.Watson J, Schanke J, Grunenwald H, Meis R, Hoffman L, Lewandowska-Skarbek M, Moan E. 1998. A new method for DNA and RNA purification. J. Lig. Assay 21:394–403 [Google Scholar]
- 31.Brum JR, Steward GF, Karl DM. 2004. A novel method for the measurement of dissolved deoxyribonucleic acid in seawater. Limnol. Oceanogr. Methods 2:248–255. 10.4319/lom.2004.2.248 [DOI] [Google Scholar]
- 32.Steward GF, Culley AI, Mueller JA, Wood-Charlson EM, Belcaid M, Poisson G. 2013. Are we missing half of the viruses in the ocean? ISME J. 7:672–679. 10.1038/ismej.2012.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Carlo EH, Hoover DJ, Young CW, Hoover RS, Mackenzie FT. 2007. Impact of storm runoff from tropical watersheds on coastal water quality and productivity. Appl. Geochem. 22:1777–1797. 10.1016/j.apgeochem.2007.03.034 [DOI] [Google Scholar]
- 34.Rudinger G, Blazek ER. 2002. Fluid mechanics of DNA double-strand filter elution. Biophys. J. 82:19–28. 10.1016/S0006-3495(02)75370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brinkers S, Dietrich HRC, de Groote FH, Young IT, Rieger B. 2009. The persistence length of double stranded DNA determined using dark field tethered particle motion. J. Chem. Phys. 130:215105. 10.1063/1.3142699 [DOI] [PubMed] [Google Scholar]
- 36.Latulippe DR, Zydney AL. 2011. Separation of plasmid DNA isoforms by highly converging flow through small membrane pores. J. Colloid Interface Sci. 357:548–553. 10.1016/j.jcis.2011.02.029 [DOI] [PubMed] [Google Scholar]
- 37.Steward GF, Montiel JL, Azam F. 2000. Genome size distributions indicate variability and similarities among marine viral assemblages from diverse environments. Limnol. Oceanogr. 45:1697–1706. 10.4319/lo.2000.45.8.1697 [DOI] [Google Scholar]
- 38.Gurrieri S, Smith SB, Bustamante C. 1999. Trapping of megabase-sized DNA molecules during agarose gel electrophoresis. Proc. Natl. Acad. Sci. U. S. A. 96:453–458. 10.1073/pnas.96.2.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higuchi A, Kato K, Hara M, Sato T, Ishikawa G, Nakano H, Satoh S, Manabe S-I. 1996. Rejection of single stranded and double stranded DNA by porous hollow fiber membranes. J. Membr. Sci. 116:191–197. 10.1016/0376-7388(96)00051-8 [DOI] [Google Scholar]
- 40.Watson MP, Winters MA, Sagar SL, Konz JO. 2006. Sterilizing filtration of plasmid DNA: effects of plasmid concentration, molecular weight, and conformation. Biotechnol. Prog. 22:465–470. 10.1021/bp050280s [DOI] [PubMed] [Google Scholar]
- 41.Boström KH, Simu K, Hagström Å, Riemann L. 2004. Optimization of DNA extraction for quantitative marine bacterioplankton community analysis. Limnol. Oceanogr. Methods 2:365–373. 10.4319/lom.2004.2.365 [DOI] [Google Scholar]
- 42.Liang Z, Keeley A. 2013. Filtration recovery of extracellular DNA from environmental water samples. Environ. Sci. Technol. 47:9324–9331. 10.1021/es401342b [DOI] [PubMed] [Google Scholar]
- 43.Sørensen N, Daugbjerg N, Richardson K. 2013. Choice of pore size can introduce artefacts when filtering picoeukaryotes for molecular biodiversity studies. Microb. Ecol. 65:964–968. 10.1007/s00248-012-0174-z [DOI] [PubMed] [Google Scholar]
- 44.Vogelstein B, Gillespie D. 1979. Preparative and analytical purification of DNA from agarose. Proc. Natl. Acad. Sci. U. S. A. 76:615–619. 10.1073/pnas.76.2.615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boom R, Sol CJA, Salimans MMM, Jansen CL, Wertheim-van Dillen PME, Van der Noorda J. 1990. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28:495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hilz H, Wiegers U, Adamietz P. 1975. Stimulation of proteinase K action by denaturing agents: application to the isolation of nucleic acids and the degradation of proteins. Eur. J. Biochem. 56:103–108. 10.1111/j.1432-1033.1975.tb02211.x [DOI] [PubMed] [Google Scholar]
- 47.Tucker KP, Parsons R, Symonds EM, Breitbart M. 2011. Diversity and distribution of single-stranded DNA phages in the North Atlantic Ocean. ISME J. 5:822–830. 10.1038/ismej.2010.188 [DOI] [PMC free article] [PubMed] [Google Scholar]