

Key Points
Lman1 tissue-specific knockout mice reveal that endothelial cells, not hepatocytes, are the primary source of FVIII biosynthesis.
F8 gene expression is heterogeneous among endothelial cell populations in different tissues.
Abstract
The primary cellular source of factor VIII (FVIII) biosynthesis is controversial, with contradictory evidence supporting an endothelial or hepatocyte origin. LMAN1 is a cargo receptor in the early secretory pathway that is responsible for the efficient secretion of factor V (FV) and FVIII to the plasma. Lman1 mutations result in combined deficiency of FV and FVIII, with levels of both factors reduced to ∼10% to 15% of normal in human patients. We generated Lman1 conditional knockout mice to characterize the FVIII secretion profiles of endothelial cells and hepatocytes. We demonstrate that endothelial cells are the primary biosynthetic source of murine FVIII and that hepatocytes make no significant contribution to the plasma FVIII pool. Utilizing RiboTag mice and polyribosome immunoprecipitation, we performed endothelial cell–specific messenger RNA isolation and quantitative polymerase chain reaction analyses to confirm that endothelial cells highly express F8 and to explore the heterogeneity of F8 expression in different vascular beds. We demonstrate that endothelial cells from multiple, but not all, tissues contribute to the plasma FVIII pool in the mouse.
Introduction
Plasma coagulation factor VIII (FVIII) is tightly associated with von Willebrand factor (VWF). Though it is well established that VWF is synthesized exclusively in endothelial cells and megakaryocytes, the cellular source of FVIII has been debated for decades. The delayed rise of FVIII in von Willebrand disease patients infused with VWF demonstrates that VWF and FVIII do not require synthesis in the same cell for complex formation.1 Dozens of conflicting reports have implicated numerous cell and tissue types as the source of FVIII, though the majority of evidence points to either an endothelial and/or hepatocyte origin of FVIII synthesis.2-4
Multiple curative organ transplantation studies in hemophilia A patients and experimental dog models have shown that the liver contributes significantly to FVIII biosynthesis,5-8 though the failure of FVIII levels in hemophilia A liver transplant recipients to respond to desmopressin suggested FVIII localization to a different compartment than VWF.9 As early as 1971, Webster et al demonstrated extrahepatic FVIII synthesis by showing that FVIII synthesis was maintained after replacing the livers of normal dog recipients with hemophilic livers.10 Similarly, extrahepatic FVIII production was shown in humans after transplantation of a hemophilia A donor liver into a nonhemophilic recipient.11 Spleen and lung transplantation has also been shown to restore FVIII plasma levels in several animal models of hemophilia,12-15 and FVIII production also has been documented by human purified microvascular endothelial cells from lung in vitro.16
Immunoradiometric assays localized FVIII antigen to guinea pig liver, spleen, lung, kidney, and isolated hepatocytes2,17 and to human lymph nodes, lung, liver, and spleen,18 whereas microscopy studies suggested synthesis in human hepatocytes3 or human liver sinusoidal endothelial cells (LSECs).19,20 F8 messenger RNA (mRNA) transcripts have been detected in purified murine LSECs and hepatocytes at comparable levels, but not in Kupffer cells,4 and by others in human spleen, lymph nodes, kidney, and isolated hepatocytes, but not in white blood cells or cultured umbilical vein endothelial cells.2 F8 mRNA transcripts have also been shown to be synthesized in hemophilia A mouse livers by hepatocytes and LSECs that were derived from transplanted wild-type bone marrow progenitor cells.21
We took advantage of a genetically modified mouse model that is deficient for lectin, mannose-binding protein 1 (LMAM1), also known as endoplasmic reticulum/Golgi intermediate compartment (ERGIC)-53). LMAN1 is part of a ubiquitously expressed cargo receptor that cycles between the ERGIC in the early secretory pathway. LMAN1 is required for the efficient secretion of FVIII as well as coagulation FV to the plasma. Mutations in LMAN1 cause an autosomal-recessive bleeding disorder known as combined deficiency of coagulation FV and FVIII (F5F8D). Although FV and FVIII are synthesized at markedly different levels (human plasma FV ∼7.0 μg/mL and FVIII ∼200 ng/mL) and potentially in different tissues, loss of the LMAN1 cargo receptor in F5F8D leads to parallel reduction of both of these factors to ∼10% to 15% of normal levels.22,23
We previously reported a genetically engineered mouse carrying a gene-trap interruption of the Lman1 gene.24 Mice homozygous for this gene-trap allele (Lman1gt1/gt1) exhibit reduced FV and FVIII levels that are ∼50% of normal, in contrast to the ∼10% to 15% levels in human F5F8D patients. As expected, this modest reduction in FV and FVIII results in a much milder phenotype than reported for hemophilia A mice carrying a F8 gene–targeted allele leading to complete absence of FVIII.25 We now describe a second, independent conditional Lman1 allele and analysis of mice with LMAN1 deficiency induced specifically in the hepatocyte or endothelial cell compartments. Deletion of Lman1 only in hepatocytes results in reduced FV but normal plasma FVIII, whereas endothelial Lman1 deletion reduces FVIII, but not FV, plasma levels. In addition, analysis of purified, endothelial-specific polysomes by quantitative polymerase chain reaction (qPCR) confirms enrichment of F8 mRNA expression in endothelial cells (relative to total tissue lysates), with considerable heterogeneity in the level of F8 mRNA expression across different vascular beds.
Methods
Lman1 conditional knockout mice
Embryonic stem cell clone B06 (C57BL/6 genetic background) carrying a conditional Lman1 gene-trap allele [Lman1cgt, also known as Lman1tm1a(KOMP)Wtsi] with a gene-trap insertion into Lman1 intron 1 and loxP sites flanking exons 2 and 3 was obtained from the Knockout Mouse Project (KOMP) (Figure 1A). Correct targeting of the conditional gene-trap construct was confirmed by long-range PCR at both the 5′ and 3′ ends of the targeted allele with Phusion Hot Start high-fidelity DNA polymerase (Finnzymes) (supplemental Figure 1A, available at the Blood Web site) using the PCR primers listed in supplemental Table 1. The embryonic stem cell clone was expanded and then injected into C57BL/6J blastocysts at the University of Michigan Transgenic Mouse Core. Germline transmission was achieved by mating male chimeric founders with C57BL/6J albino female mice [B6(Cg)-Tyrc-2J/J; stock no. 000058, The Jackson Laboratory]. The resulting germline transmitted Lman1cgt allele was maintained on a C57BL/6J genetic background, as were all subsequent distinct alleles derived from Lman1cgt. Primer trio LMAN1 C/D/E was used to distinguish between the wild-type Lman1+ allele and the targeted Lman1cgt allele (Figure 1B). Primer set LMAN1 A/B also distinguishes between the Lman1+ and Lman1cgt alleles (Figure 1C). Genotyping for all animals reported in this study was performed with mouse tail-clip DNA using Go-Taq Green Master Mix (Promega) and the relevant primers in supplemental Table 1, with the PCR products resolved by 2% agarose gel electrophoresis. The expected PCR amplicon sizes for all Lman1 alleles generated with primer sets C/D/E and A/B/C are listed in Table 1.
Figure 1.
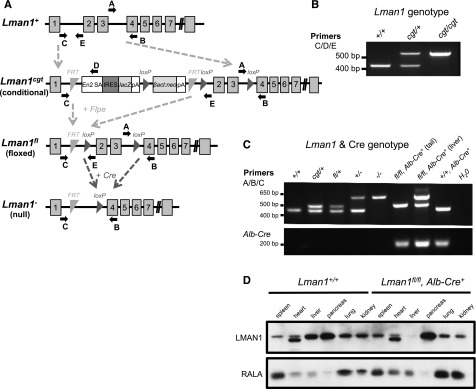
Lman1 mutant alleles. (A) The Lman1 conditional gene-trap allele (Lman1cgt) contains a gene-trap insertion in intron 1 flanked by 2 FRT sites. Mice carrying this allele were crossed to β-actin FLP transgenic mice. Mice heterozygous for the resulting Lman1 floxed allele (Lman1fl) were crossed to Alb-Cre+ or Tek-Cre+ transgenic mice to excise exons 2 and 3, generating the Lman1 null allele (Lman1−) in selected tissues. Gray blocks represent exons. A, B, C, D, and E represent genotyping primers. (Adapted from the Knockout Mouse Project. General conditional gene targeting scheme: https://www.komp.org/alleles.php#conditional-promoter; Lman1 targeting: https://www.komp.org/geneinfo.php?geneid=66654); cgt, conditional gene trap; fl, floxed; En2 SA, splice acceptor of mouse En2 exon 2; IRES, encephalomyocarditis virus (EMCV) internal ribosomal entry site; lacZ, Escherichia coli β-galactosidase gene; pA, SV40 polyadenylation signal; βact:neo, human β-actin promoter-driven neomycin cassette. (B) A 3-primer PCR assay (primers C, D, and E) distinguishes the Lman1+ allele (434 bp) from Lman1cgt (565 bp). (C) PCR genotyping assay used for tail-snip genomic DNA from a Lman1fl/fl, Alb-Cre+ mouse, with the addition of a liver biopsy genomic DNA sample from the Lman1fl/fl, Alb-Cre+ mouse to demonstrate excision of Lman1 exons 2 and 3 in the liver. A 3-primer PCR assay (primers A, B, and C) distinguishes the Lman1+ (444 bp), Lman1cgt (508 bp), Lman1fl (508 bp), and Lman1− (635 bp) alleles. Alb-Cre primers were used to determine the Cre genotype of offspring from matings of Lman1fl/+, Alb-Cre+ mice with Lman1fl/fl mice. Comparison of the signal in DNA from liver tissue of an Lman1fl/fl, Alb-Cre+ mouse (lane 7) to tail DNA from the same animal (lane 6) indicates a high degree of specific excision in the liver. A similar PCR genotyping strategy of tail-snip genomic DNA was used to identify Lman1fl/fl, Tek-Cre+ mice, with primer trio A/B/C demonstrating the Lman1 genotype and with Tek-Cre–specific primers rather than Alb-Cre primers (not shown). (D) Western blot of spleen, heart, liver, pancreas, lung, and kidney tissues from a wild-type C57BL/6J mouse and a Lman1fl/fl, Alb-Cre+ (hepatocyte-knockout) mouse, demonstrating nearly complete and tissue-specific Lman1 knockout in the liver of Lman1fl/fl, Alb-Cre+ mice. The lower-molecular-weight band observed in the heart samples represents a nonspecific band due to cross-reactivity with the LMAN1 antibody, because this same band is observed in the heart tissue of ubiquitous Lman1 knockout mice (not shown).
Table 1.
Expected PCR amplicon size (bp) for Lman1 alleles
| Primer set | Lman1+ | Lman1cgt | Lman1fl | Lman1− |
|---|---|---|---|---|
| C/D/E | 434 | 565 | 591 | No product |
| A/B/C | 444 | 508 | 508 | 635 |
Transgenic mice (C57BL/6J background) carrying Flpe (flippase) recombinase driven by an actin promoter were obtained from The Jackson Laboratory [B6;SJL-Tg(ACTFLPe)9205Dym/J; stock no. 003800]. Intercrosses of Lman1cgt/+ mice and Flpe transgenic mice generated the Lman1fl allele [also known as Lman1tm1c(KOMP)Wsti] (Figure 1A). Transgenic Tek-Cre+ (Tie2-Cre) recombinase mice [B6.Cg-Tg(Tek-cre)12Flv/J; stock no. 004128] and Alb-Cre+ recombinase mice [B6.Cg-Tg(Alb-cre)21Mgn/J; stock no. 003574] were obtained from The Jackson Laboratory. Primer trio LMAN1 A/B/C was used to distinguish between the Lman1+ allele and the Lman1fl and Lman1− alleles derived from Lman1cgt (Figure 1C) for the identification of conditional knockout Lman1 mice, and Alb-Cre or Tek-Cre primers (supplemental Table 1) were used to determine the Cre genotype. The Alb-Cre forward/reverse primers generate a 207-bp PCR amplicon. The Tek-Cre multiplex primer trio generates an internal control 200-bp PCR product from the endogenous Tek gene and a 579-bp PCR product from the Tek-Cre transgene. Tek-Cre–driven recombinase activity results in the deletion of loxP-flanked target sequences in endothelial cells (of all tissues) and hematopoietic cells, but also to a lesser extent in the germline. Only male Lman1fl/+, Tek-Cre+ mice were crossed with female Lman1fl/fl mice to generate Lman1fl/fl, Tek-Cre+ offspring, because the Tek-Cre recombinase activity occurs at a lower frequency in the male germline than in the female germline.26
All animals were housed according to the University of Michigan Unit of Laboratory Animal Medicine guidelines, and the University Committee on the Use and Care of Animals approved all animal studies under protocol number 08571.
Western blot antibodies
Western blot antibodies were a monoclonal antibody against human β-actin (Santa Cruz Biotechnology), a monoclonal antibody against the RalA GTPase (Sigma-Aldrich), a goat anti-mouse immunoglobulin G horseradish peroxidase antibody, a polyclonal antibody against LMAN1 (Sigma-Aldrich), and a polyclonal rabbit antibody against the heavy chain of murine FV.27 FV antigen levels in plasma and platelets were assessed by western blot analysis using the FV antibody.
Measurement of FV and FVIII levels
Blood was collected into 4% sodium citrate solution (9:1) (Sigma-Aldrich) via inferior vena cava puncture essentially as previously described,28 under pentobarbital anesthesia (Nembutal sodium solution) with a 1-mL syringe. Platelet-poor plasma was isolated as previously described,29 snap frozen, and stored at −80°C. Platelet-rich plasma and platelet pellets were isolated and processed as follows: 2 mL of room-temperature buffered saline glucose citrate (129 mM NaCl, 13.6 mM Na3 citrate, 11.1 mM glucose, 1.6 mM KH2PO4, 8.6 mM NaH2PO4 [pH 7.3]) were placed in a 5-mL polypropylene tube, to which was added 1 to 1.5 mL of whole blood. Buffered saline glucose citrate was added to a final volume of 4 mL and then gently mixed by inversion. The tubes were centrifuged at 180g for 10 minutes at room temperature. The supernatant (semi–platelet-rich plasma) was removed and subjected to a platelet count on an Advia 120 Automated Hematology Analyzer (Siemens), and the number of platelets was normalized between samples before centrifuging in fresh tubes at 700g for 10 minutes. Platelet pellets were lysed with Triton-X 100 on ice (final concentration 0.2%) for 30 minutes and frozen as previously described.24 Plasma and platelet samples from previously described F5−/− mice carrying either a platelet- or hepatocyte-specific F5 transgene,30 respectively, were used as experimental controls.
Plasma FV and FVIII activity levels were determined in prothrombin time– and partial thromboplastin time–based assays, respectively. Individual mouse plasma samples were mixed with human plasma deficient for the respective factor (George King) and analyzed on a KC-4 Coagulation Analyzer (Sigma-Aldrich). A pooled C57BL/6J plasma stock was diluted into human FV- or FVIII-deficient plasma (George King) to generate a reference standard curve.
RiboTag mice
RiboTag mice31 (B6.129-Rpl22tm1.1Psam/J) were a kind gift from Dr Paul Amieux (University of Washington) and were also obtained from The Jackson Laboratory (stock no. 011029). Genotyping primers for the RiboTag mice are listed in supplemental Table 1. RiboTag mice (Rpl22HA/HA) in this study are homozygous for a modified Rpl22 gene (ribosomal protein L22) that contains both a wild-type exon 4 flanked by loxP sites as well as an alternate hemagglutinin (HA)-tagged exon 4 sequence.
RNA isolation
Mice were anesthetized followed by intracardiac perfusion with 12 mL of 100 μg/mL cycloheximide (Sigma-Aldrich) in PBS, after which tissues were isolated and snap frozen in liquid nitrogen. Immunoprecipitation of endothelial cell–specific polysomes (and bound mRNA) from whole liver, kidney, brain, and heart lysates was performed essentially as previously described, with minor modifications.31 Frozen tissues were pulverized, and the resulting powders were transferred to tubes containing polysome buffer (50 mM Tris [pH 7.5], 100 mM KCl, 12 mM MgCl2, 1% Nonidet P-40, 1 mM dithiothreitol, 200 U/mL Promega RNasin, 1 mg/mL heparin, 100 μg/mL cycloheximide, Sigma protease inhibitor mixture). Cleared lysate was incubated with a purified mouse monoclonal antibody against the HA tag (HA.11 clone 16B12 [Covance], 3 μg per sample) for 1 hour with rotation at 4°C. Protein G magnetic beads (New England Biolabs) equilibrated in polysome buffer were then added for an additional 30-minute rotated incubation at 4°C. The beads were washed in high-salt buffer (50 mM Tris [pH 7.5], 300 mM KCl, 12 mM MgCl2, 1% Nonidet P-40, 1 mM dithiothreitol, 100 μg/mL cycloheximide), and ribosome-associated mRNA was subsequently eluted from the beads by adding Qiagen buffer RLT supplemented with 2-mercapto-ethanol (1% vol/vol). Total RNA was isolated from the whole-tissue lysates (input) and immunoprecipitated polyribosome samples with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol, and DNase treatment was performed on column (Qiagen). RNA samples were quantified on a spectrophotometer (Nanodrop), and the integrity of the RNA was assessed by gel electrophoresis. Total RNA from whole blood of C57BL/6J mice was extracted by using TRIzol LS and purified on RNeasy spin columns (Qiagen).
cDNA conversion and qPCR analysis
Total mRNA (100 ng) from input lysates and HA-tagged polysome samples was converted to complementary DNA (cDNA) with the Superscript First-Strand system (Invitrogen) and oligoDT primers. Gene-specific primers (supplemental Table 1) were designed with Primer Express Software (Applied Biosystems) and qPCR was executed with SYBR-Green RT-PCR Master Mix (Applied Biosystems) on a 7900HT Fast Real-Time PCR machine (Applied Biosystems). Data were analyzed using the comparative threshold cycle method with Rpl37 (ribosomal protein L37), Rpl38 (ribosomal protein L38), and Eif3e (eukaryotic translation initiation factor 3, subunit E) as internal controls,32 and the ΔCt value (Ct value of sequence of interest − Ct value of reference sequence) of the bead elution was related to the ΔCt value of its respective total tissue lysate to determine the fold-change. One microgram of mRNA from whole blood was used for subsequent cDNA conversion with oligoDT primers.
Statistical analysis
A χ2 test was used to evaluate for statistical deviation from expected Mendelian ratios for the genotypes of offspring from matings between Lman1fl/fl mice and Lman1fl/+, Cre+ mice. Analysis of the FV and FVIII activity data was performed using the Student t test comparing pooled levels from Lman1fl/fl and Lman1fl/+ (control) mice relative to levels from the other experimental genotypes (Lman1fl/fl, Cre+ and Lman1fl/+, Cre+). To evaluate the qPCR data with respect to enrichment or depletion in the endothelial cell fraction relative to the whole-tissue lysate, a paired t test was performed. A Student t test was used to analyze the qPCR data for whole-blood relative to whole-tissue lysates. A P value < .05 was considered to be statistically significant for all analyses.
Results
FVIII is synthesized in murine endothelial cells
Long-range PCR confirmed the correct genomic targeting of the Lman1cgt KOMP targeting construct (supplemental Figure 1B). Lman1cgt/+ mice were crossed to Flpe recombinase transgenic mice to excise the gene-trap cassette, generating the Lman1fl allele in which exons 2 and 3 are flanked by loxP sites but no other gene-trap sequence is retained (Figure 1A). The Lman1fl allele may be converted to the null Lman1− allele in a conditional manner by tissue-specific Cre-mediated excision of exons 2 and 3. Heterozygous Lman1fl/+ mice were crossed with male transgenic Tek-Cre+ (Tie2-Cre) mice that carry the Cre recombinase gene driven by the Tek promoter to generate Lman1fl/+, Tek-Cre+ mice with a single Tek-Cre allele. To delete Lman1 in endothelial cells of all tissues, Lman1fl/+, Tek-Cre+ mice were subsequently crossed to Lman1fl/fl mice to generate Lman1fl/fl, Tek-Cre+ endothelium-specific knockout mice. The same general mating scheme was used to generate hepatocyte-specific Lman1 knockout mice by utilizing an Alb-Cre recombinase. Lman1fl/fl, Tek-Cre+ mice (endothelial-knockout) and Lman1fl/fl, Alb-Cre+ (hepatocyte-knockout) mice are viable and were observed in the expected Mendelian ratios (Table 2). As demonstrated by PCR analysis of genomic DNA isolated from an Lman1fl/fl, Alb-Cre+ mouse tail snip and liver tissue, the conversion of the Lman1fl allele to the Lman1− null allele by genetic excision of Lman1 exons 2 and 3 was very efficient and specific to the liver (Figure 1C). Similarly, western blot analysis of LMAN1 levels in multiple tissues harvested from wild-type C57BL/6J mice and Lman1fl/fl, Alb-Cre+ mice confirmed efficient loss of LMAN1 expression in the liver of Lman1fl/fl, Alb-Cre+ mice (Figure 1D). The equivalent levels of LMAN1 in other tissues of Lman1fl/fl mice compared with wild-type mice confirmed normal LMAN1 expression from the Lman1fl allele in the absence of Cre recombinase (Figure 1D). FV and FVIII activity levels for both Lman1fl/+, Tek-Cre+ and Lman1fl/+, Alb-Cre+ mice (heterozygous conditional knockout mice) were indistinguishable from control animals (Figure 2A-B), consistent with the autosomal-recessive inheritance of F5F8D and with previous results in Lman1gt1/+ mice.24 Lman1fl/fl and Lman1fl/+ mice were pooled together as controls for subsequent experiments.
Table 2.
Generation of Lman1 conditional knockout mice
| Crosses | Genotype distribution at 3 wk | P value (χ2) | |||
|---|---|---|---|---|---|
| Lman1fl/+, Cre− | Lman1fl/+, Cre+ | Lman1fl/fl, Cre− | Lman1fl/fl, Cre+ | ||
| Expected % | 25% | 25% | 25% | 25% | |
| Lman1fl/fl × Lman1fl/+, Alb-Cre+ | 24% (17) | 25% (18) | 19% (14) | 32% (23) | .5 (NS) |
| Lman1fl/fl × Lman1fl/+, Tek-Cre+ | 19% (16) | 28% (23) | 26.5% (22) | 26.5% (22) | .7 (NS) |
The χ2 test (degrees of freedom = 3) was based upon an expected genotype ratio of 3:1, with Lman1fl/fl/Cre+ mice expected to represent 25% of the offspring from each mating and all other genotypes cumulatively accounting for 75% of offspring.
NS, not significant.
Figure 2.

FV and FVIII activity levels for Lman1 conditional knockout mice. Lman1fl/+ and Lman1fl/fl mice were pooled as controls. Data for Lman1gt1/gt1 mice24 (previously reported) are included for comparison. (A) FV activity measured by prothrombin time–based coagulation assay. Lman1fl/fl, Alb-Cre+ mice exhibit a statistically significant reduction in plasma FV activity, relative to control mice (P < 1.5 × 10−10), and are indistinguishable from Lman1gt1/gt1 mice (P > .5). In contrast, FV activity levels for Lman1fl/fl, Tek-Cre+ mice are not reduced. (B) FVIII activity measured by partial thromboplastin time–based coagulation assay. Lman1fl/fl, Tek-Cre+ mice exhibit a statistically significant reduction in plasma FVIII activity, relative to control mice (P < 1.7 × 10−8), and are indistinguishable from Lman1gt1/gt1 mice (P > .3). FVIII activity levels for Lman1fl/fl, Alb-Cre+ mice are not reduced. Each symbol represents an individual animal. Horizontal lines indicate mean and error bars indicate standard error of the mean for each genotype.
As expected from the known synthesis of plasma FV in hepatocytes,30 Lman1fl/fl, Alb-Cre+ mice demonstrate significant reduction of plasma FV activity relative to Lman1fl/fl and Lman1fl/+ control mice (48.3% vs 96.8%, P < 1.5 × 10−10) (Figure 2A), with consistent reductions in plasma FV antigen level also observed by western blotting (Figure 3A). Plasma FV activity in Lman1fl/fl, Alb-Cre+ mice was indistinguishable from Lman1gt1/gt1 mice (P > .5; Figure 2A), suggesting highly efficient excision in hepatocytes, as directed by the Alb-Cre transgene. In contrast, plasma FVIII levels in Lman1fl/fl, Alb-Cre+ mice were indistinguishable from controls (90.2% vs 97.5%, P > .12) (Figure 2B). These data demonstrate that hepatocyte synthesis does not contribute significantly to the plasma FVIII pool in the mouse. The Lman1fl/fl, Tek-Cre+ mice exhibit normal plasma FV activity relative to Lman1fl/fl and Lman1fl/+ control mice (99.8% vs 96.8%, P > .5) (Figure 2A). In contrast, FVIII activity levels in Lman1fl/fl, Tek-Cre+ mice were markedly reduced (52.8% vs 97.5%, P < 1.7 × 10−8) and were indistinguishable from the levels previously reported in Lman1gt1/gt1 null mice (52.8% vs 47.9%, P > .30) (Figure 2B). These results suggest that endothelial cells are the primary biosynthetic source of FVIII in vivo.
Figure 3.

Relative FV antigen in plasma and platelets of Lman1 conditional knockout mice. (A) Platelet-poor plasma (equal volume) and (B) isolated platelets (equal number) from conditional Lman1 knockout mice and littermate control mice were analyzed for relative FV antigen levels by western blot. The same plasma samples were run twice as duplicates. Plasma immunoglobulin G serves as a loading control for the plasma samples, and β-actin serves as a loading control for platelets. Three adult mice were analyzed per genotype. Plasma from F5−/− mice carrying a platelet-specific F5 transgene30 was included as a negative control for the plasma samples. Similarly, platelets isolated from a F5−/− mouse carrying a hepatocyte-specific F5 transgene30 were included as a negative control for the platelet samples.
The murine plasma and platelet FV pools are biosynthetically distinct and are derived from synthesis in hepatocytes and megakaryocytes, respectively.33 Consistent with these previous reports, platelet FV antigen, as assessed by western blotting, appears roughly equivalent to control mice in Lman1fl/fl, Alb-Cre+ mice, with a modest reduction in Lman1fl/fl, Tek-Cre+ mice, (Figure 3B), consistent with the known expression of Tek-Cre in megakaryocytes.34
F8 mRNA is localized to endothelial cells and not hepatocytes
The previously reported RiboTag mouse31 was used to isolate endothelial cell–specific mRNA for qPCR analysis from a number of different murine tissues. In the absence of Cre-recombinase, RiboTag mice ubiquitously express the wild-type RPL22 ribosomal protein in all cell types. However, in the presence of Cre-recombinase, RPL22 is fused to an HA tag, which can be used to selectively immunoprecipitate polyribosomes derived only from cells expressing Cre.31 Mice homozygous for the RiboTag allele and hemizygous for the Tek-Cre transgene were generated, which should result in expression of the RPL22-HA fusion protein only in endothelial (and hematopoietic) cells.
qPCR analyses of endothelial cell mRNA isolated from unfractionated liver lysates from RiboTag, Tek-Cre+ mice (n = 4) (Figure 4A; supplemental Table 2) demonstrated depletion (5- to 10-fold) of transcripts from several known hepatocyte-specific genes, including albumin (Alb), prothrombin (F2), coagulation factor V (F5), coagulation FVII (F7), and coagulation FX (F10). In contrast, a 15- to 22-fold enrichment was observed for gene transcripts that are known to be endothelial specific, including cadherin 5 (Cdh5), TEK tyrosine kinase (Tek), vascular cell adhesion molecule 1 (Vcam1), and VWF (Vwf).
Figure 4.

F8 mRNA is expressed in selective endothelial cell beds. Plots show fold-change (enrichment or depletion) of mRNA transcripts for a number of genes in the endothelial cell mRNA isolated from (A) liver, (B) kidney, (C) heart, and (D) brain tissues of 4 RiboTag, Tek-Cre+ mice relative to the whole-tissue lysate input (set to 1.0). Values <1.0 indicate depletion in the endothelial cell fraction, relative to input. In contrast, values >1.0 indicate enrichment. Each symbol represents an independent animal. Values for tissue-specific, non–endothelial cell genes are shown as open circles. Values for endothelial cell–specific genes are shown as light gray squares. Values for F8 mRNA fold-change are shown as black diamonds. Horizontal lines indicate mean and error bars indicate standard error of the mean for each genotype.
Similarly, endothelial cell mRNA from kidney (Figure 4B; supplemental Table 2), heart (Figure 4C; supplemental Table 2), and brain (Figure 4D; supplemental Table 2) prepared from the same 4 RiboTag, Tek-Cre+ mice showed 10- to 50-fold depletion of corresponding tissue-specific non–endothelial cell genes and 4- to 20-fold enrichment of endothelial cell–specific genes (Cdh5, Tek, Vcam1, and Vwf).
qPCR analysis of F8 mRNA levels revealed an unexpected variation in the level of F8 expression across these vascular beds. F8 mRNA transcripts were highly enriched (relative to total tissue lysate) in liver endothelial cells (16.5-fold, P < 2.2 × 10−4), comparable to that of known endothelial-specific genes. F8 transcripts were also enriched in kidney endothelial cells (7.4-fold, P < 1.6 × 10−3). However, no F8 mRNA enrichment was observed in the endothelial cells of brain (0.25-fold, P > .1) or heart (0.67-fold, P > .15).
Circulating peripheral blood cells do not express F8 mRNA
Because Tek-Cre is also expressed in hematopoietic cells,34 qPCR was performed on whole blood to exclude contaminating blood as the source of the enriched F8 mRNA in RiboTag, Tek-Cre+ tissues. F8 mRNA in the blood was detected at <2% of the levels in liver and kidney, with normalization to internal control genes Rpl37, Rpl38, and Eif3e (Tables 3 and 4). Thus, peripheral blood cell mRNA can be excluded as the source of the F8 mRNA signal in RiboTag, Tek-Cre+ mouse tissues. The enrichment of mRNA transcripts for Vwf and F5 in whole-blood mRNA is consistent with the known expression patterns of these genes in murine megakaryocytes/platelets.30,35,36
Table 3.
Gene expression average fold-change in the whole-blood mRNA relative to total liver lysate (input)
| Gene | Expression | Fold-change ± SEM | Range | P value |
|---|---|---|---|---|
| Cdh5 | Endothelial | 0.19 ± 0.02 | 0.17-0.22 | <3.5 × 10−2 |
| Tek | Endothelial | 0.04 ± 0.01 | 0.034-0.046 | <2.0 × 10−3 |
| Vcam1 | Endothelial | 0.001 ± 0.004 | 0.001-0.009 | <5.6 × 10−3 |
| Vwf | Endothelial/megakaryocyte | 113.40 ± 7.09 | 106.6-120.7 | <3.0 × 10−6 |
| F5 | Hepatic/megakaryocyte | 1.31 ± 0.06 | 1.25-1.36 | >.3 |
| F8 | ? | 0.01 ± 0.01 | 0.007-0.012 | <1.0 × 10−4 |
To evaluate the potential contribution of blood contamination to transcript levels of endothelial-specific genes, the tissue lysates of liver samples were set as a reference and the ΔCt values of individual blood samples (n = 4) were related to the mean ΔCt of the reference group. SEM, standard error of the mean.
Table 4.
Gene expression average fold-change in the whole-blood mRNA relative to total kidney lysate (input)
| Gene | Expression | Fold-change ± SEM | Range | P value |
|---|---|---|---|---|
| Cdh5 | Endothelial | 0.08 ± 0.01 | 0.074-0.095 | <7.4 × 10−5 |
| Tek | Endothelial | 0.002 ± 0.001 | 0.007-0.010 | <2.1 × 10−5 |
| Vcam1 | Endothelial | 0.003 ± 0.004 | 0.001-0.009 | <3.2 × 10−3 |
| Vwf | Endothelial/megakaryocyte | 248.92 ± 15.56 | 233.2-264.9 | <3.3 × 10−9 |
| F5 | Hepatic/megakaryocyte | 73.41 ± 3.15 | 70.3-76.6 | <1.5 × 10−5 |
| F8 | ? | 0.02 ± 0.01 | 0.011-0.029 | <1.5 × 10−4 |
To evaluate the potential contribution of blood contamination to transcript levels of endothelial-specific genes, the tissue lysates of kidney samples were set as a reference and the ΔCt values of individual blood samples (n = 4) were related to the mean ΔCt of the reference group.
Discussion
The in vivo biosynthetic origin of FVIII has been controversial for decades.2-8,10,11,13,14,16-21 We took advantage of the dependence of efficient FV and FVIII secretion on LMAN1 to examine the cellular biosynthetic source of each protein in vivo. Although both the human plasma and platelet FV pools are produced by hepatocytes,37 murine plasma FV is synthesized exclusively in hepatocytes and murine platelet FV in megakaryocytes.30,33 In the absence of LMAN1, FV levels are reduced in both plasma and platelets in humans and mice.22,24,38
Consistent with these previous data, we observed that mice with Lman1 specifically deleted in the hepatocytes exhibit low plasma FV and normal platelet FV, whereas Lman1 deletion in the hematopoietic compartment results in low platelet FV and normal plasma FV. Our finding of normal FVIII in mice with hepatocyte-specific knockout of Lman1 excludes the hepatocyte as a major source of FVIII production in vivo. Conversely, the observation that endothelial deletion of Lman1 results in reduced FVIII levels that are comparable to the levels in LMAN1 null mice (Figure 2B) indicates that high-level, tissue-specific Lman1 excision was achieved with the Tek-Cre recombinase and suggests that the endothelial cell is the primary source of FVIII biosynthesis in vivo.
The Tek-Cre used for specific targeting to the endothelium is known to be expressed both in the endothelial and hematopoietic compartments.39,40 The absence of F8 mRNA enrichment in peripheral blood of wild-type C57BL/6J mice excludes trapped peripheral blood cells as the source for the FVIII endothelial cell signal, as does the absence of F8 mRNA enrichment in the brain or heart (Figure 4). The lack of F8 mRNA in the blood is consistent with a recent clinical report of allogeneic bone marrow transplantation in a child with severe aplastic anemia and hemophilia A. The child’s FVIII levels and coagulation test results remained essentially unchanged from his pretransplantation values when measured 4 months after bone marrow transplantation,41 indicating that the bone marrow is not an important source of FVIII synthesis in vivo.
Examination of affinity-selected endothelial cell mRNA from actively translating ribosomes isolated from multiple tissues confirmed endothelial FVIII biosynthesis in vivo. However, the levels of F8 mRNA observed in the endothelial cells of liver, kidney, brain, and heart tissues revealed surprising variation across these vascular beds, with the highest levels of expression in liver endothelial cells, intermediate levels in kidney, and no detectable enrichment of F8 mRNA transcripts in brain or heart endothelial cells.
Taken together, these data demonstrate that endothelial cells from multiple (but not all) tissues and vascular beds contribute to the plasma FVIII pool in the mouse, with a large contribution from hepatic endothelial cells. These results account for the successful reversal of hemophilia A by liver transplantation and presumably also by lung and spleen transplantation.5-7,13,14 These results are also consistent with the maintenance of normal FVIII levels in patients with parenchymal liver disease in whom the endothelium may still be intact.42-44 The variable levels of endothelial cell mRNA enrichment across tissues likely reflects differences in mRNA expression profiles and the relative ratio of endothelial to parenchymal cells in different vascular beds.45-47 Though the functional significance of this heterogeneity in FVIII endothelial production is unknown, it could reflect variation in local hemostatic demand across different vascular beds.
Our data are also consistent with several recent publications reporting the relative contributions of hepatocytes and endothelial cells to the plasma FVIII pool. Transplantation of unfractionated liver cells (a mixture of hepatocytes, LSECs, Kupffer cells, and hepatic stellate cells) into the peritoneal cavity of hemophilia A mice corrected the disease, but transplantation of a hepatocyte-enriched fraction alone had no effect.48 Similarly, transplanted normal endothelial cells are able to repopulate the liver endothelium and correct the bleeding phenotype of hemophilia A mice.49 In addition, FVIII activity levels were found to be 10- to 100-fold higher in human LSECs than in hepatocytes separated by flow-cytometry cell sorting.50 Nonetheless, other studies have detected FVIII in human hepatocytes,5-8,51 and differences in the pathway for FVIII production between humans and mice cannot be excluded.
A growing body of evidence suggests extensive heterogeneity among endothelial cells in vivo.52 For example, Vwf expression varies widely across different endothelial beds, with different tissue microenvironmental factors regulating its transcription.53,54 Similarly, the expression of dozens to hundreds of genes may vary among different specialized vascular beds.55 Although we observed modest variation in the expression levels of Vwf and 3 other endothelial-specific genes in our studies of liver, kidney, heart, and brain endothelial cells, we observed large differences in F8 gene expression among these 4 tissues. F8 mRNA transcripts were most highly enriched in hepatic endothelial cells, with intermediate enrichment in kidney endothelial cells and little to no enrichment in the endothelial cells of heart or brain. Our results are consistent with recent transcriptional profiling of tissue-specific endothelial cell populations isolated by flow sorting, where F8 mRNA transcripts were detected in endothelial cells from liver and glomeruli, but not in the testis, spleen, muscle, brain, lung, heart, or bone marrow.45 This heterogeneity of endothelial cell gene expression has important implications not only for FVIII biosynthesis but also for regulation of other tissue-specific endothelial cell functions.56,57 The finding that F8 and Vwf are both highly expressed in endothelial cells likely confers a regulatory mechanism for FVIII biosynthesis, consistent with previous reports that the coexpression of F8 and Vwf in vitro result in increased stable accumulation of FVIII activity.58 Finally, the establishment of endothelial cells as the primary site of FVIII biosynthesis also has important implications for the treatment of hemophilia A, particularly for gene-targeting approaches directed specifically to endothelial cells.59-61
Acknowledgments
The authors thank the University of Michigan Transgenic Animal Model Core staff for their work in generating the Lman1 conditional mice, and Dr Robert Montgomery and Scot Fahs for sharing their complementary studies and manuscript prior to publication and for providing helpful comments and suggestions in preparation of this manuscript.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL039693 and P01-HL057346. D.G. is an investigator of the Howard Hughes Medical Institute. Support for the University of Michigan Transgenic Animal Model Core was provided by the University of Michigan Cancer Center (P30 CA046592).
Footnotes
There is an Inside Blood Commentary on this article in this issue.
Presented in abstract form at the 55th annual meeting of the American Society of Hematology, New Orleans, LA, December 8, 2013.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.A.E., A.C.A.C., and D.G. designed the research; L.A.E., A.C.A.C., and R.N.K. performed the experiments; L.A.E. and D.G. wrote the manuscript; and all authors reviewed and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David Ginsburg, Life Sciences Institute, University of Michigan, 210 Washtenaw Ave, Ann Arbor, MI 48109; e-mail: ginsburg@umich.edu.
References
- 1.Haberichter SL, Montgomery RR. The biology of von Willebrand factor and factor VIII-regulated release. Haematologica Reports. 2005;1(6):9–14. [Google Scholar]
- 2.Wion KL, Kelly D, Summerfield JA, Tuddenham EG, Lawn RM. Distribution of factor VIII mRNA and antigen in human liver and other tissues. Nature. 1985;317(6039):726–729. doi: 10.1038/317726a0. [DOI] [PubMed] [Google Scholar]
- 3.Zelechowska MG, van Mourik JA, Brodniewicz-Proba T. Ultrastructural localization of factor VIII procoagulant antigen in human liver hepatocytes. Nature. 1985;317(6039):729–730. doi: 10.1038/317729a0. [DOI] [PubMed] [Google Scholar]
- 4.Do H, Healey JF, Waller EK, Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J Biol Chem. 1999;274(28):19587–19592. doi: 10.1074/jbc.274.28.19587. [DOI] [PubMed] [Google Scholar]
- 5.Bontempo FA, Lewis JH, Gorenc TJ, et al. Liver transplantation in hemophilia A. Blood. 1987;69(6):1721–1724. [PMC free article] [PubMed] [Google Scholar]
- 6.Marchioro TL, Hougie C, Ragde H, Epstein RB, Thomas ED. Hemophilia: role of organ homografts. Science. 1969;163(3863):188–190. doi: 10.1126/science.163.3863.188. [DOI] [PubMed] [Google Scholar]
- 7.Scharrer I, Encke A, Hottenrott C. Clinical cure of haemophilia A by liver transplantation. Lancet. 1988;2(8614):800–801. doi: 10.1016/s0140-6736(88)92456-7. [DOI] [PubMed] [Google Scholar]
- 8.Gordon FH, Mistry PK, Sabin CA, Lee CA. Outcome of orthotopic liver transplantation in patients with haemophilia. Gut. 1998;42(5):744–749. doi: 10.1136/gut.42.5.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamont PA, Ragni MV. Lack of desmopressin (DDAVP) response in men with hemophilia A following liver transplantation. J Thromb Haemost. 2005;3(10):2259–2263. doi: 10.1111/j.1538-7836.2005.01553.x. [DOI] [PubMed] [Google Scholar]
- 10.Webster WP, Zukoski CF, Hutchin P, Reddick RL, Mandel SR, Penick GD. Plasma factor VIII synthesis and control as revealed by canine organ transplantation. Am J Physiol. 1971;220(5):1147–1154. doi: 10.1152/ajplegacy.1971.220.5.1147. [DOI] [PubMed] [Google Scholar]
- 11.Madeira CL, Layman ME, de Vera RE, Fontes PA, Ragni MV. Extrahepatic factor VIII production in transplant recipient of hemophilia donor liver. Blood. 2009;113(21):5364–5365. doi: 10.1182/blood-2009-02-206979. [DOI] [PubMed] [Google Scholar]
- 12.Norman JC, Covelli VH, Sise HS. Transplantation of the spleen: experimental cure of hemophilia. Surgery. 1968;64(1):1–14. [PubMed] [Google Scholar]
- 13.Webster WP, Penick GD, Peacock EE, Brinkhous KM. Allotransplantation of spleen in hemophilia. N C Med J. 1967;28:505. [Google Scholar]
- 14.Veltkamp JJ, Asfaou E, van de Torren K, van der Does JA, van Tilburg NH, Pauwels EK. Extrahepatic factor VIII synthesis. Lung transplants in hemophilic dogs. Transplantation. 1974;18(1):56–62. doi: 10.1097/00007890-197407000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Groth CG, Hathaway WE, Gustafsson A, et al. Correction of coagulation in the hemophilic dog by transplantation of lymphatic tissue. Surgery. 1974;75(5):725–733. [PMC free article] [PubMed] [Google Scholar]
- 16.Jacquemin M, Neyrinck A, Hermanns MI, et al. FVIII production by human lung microvascular endothelial cells. Blood. 2006;108(2):515–517. doi: 10.1182/blood-2005-11-4571. [DOI] [PubMed] [Google Scholar]
- 17.Kelly DA, Summerfield JA, Tuddenham EG. Localization of factor VIIIC: antigen in guinea-pig tissues and isolated liver cell fractions. Br J Haematol. 1984;56(4):535–543. doi: 10.1111/j.1365-2141.1984.tb02178.x. [DOI] [PubMed] [Google Scholar]
- 18.Exner T, Rickard KA, Kronenberg H. Measurement of factor VIII CAg by immunoradiometric assay in human tissue extracts. Thromb Res. 1983;32(4):427–436. doi: 10.1016/0049-3848(83)90094-4. [DOI] [PubMed] [Google Scholar]
- 19.Stel HV, van der Kwast TH, Veerman EC. Detection of factor VIII/coagulant antigen in human liver tissue. Nature. 1983;303(5917):530–532. doi: 10.1038/303530a0. [DOI] [PubMed] [Google Scholar]
- 20.Hollestelle MJ, Thinnes T, Crain K, et al. Tissue distribution of factor VIII gene expression in vivo—a closer look. Thromb Haemost. 2001;86(3):855–861. [PubMed] [Google Scholar]
- 21.Yadav N, Kanjirakkuzhiyil S, Ramakrishnan M, Das TK, Mukhopadhyay A. Factor VIII can be synthesized in hemophilia A mice liver by bone marrow progenitor cell-derived hepatocytes and sinusoidal endothelial cells. Stem Cells Dev. 2012;21(1):110–120. doi: 10.1089/scd.2010.0569. [DOI] [PubMed] [Google Scholar]
- 22.Nichols WC, Seligsohn U, Zivelin A, et al. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93(1):61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B, Cunningham MA, Nichols WC, et al. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nat Genet. 2003;34(2):220–225. doi: 10.1038/ng1153. [DOI] [PubMed] [Google Scholar]
- 24.Zhang B, Zheng C, Zhu M, et al. Mice deficient in LMAN1 exhibit FV and FVIII deficiencies and liver accumulation of α1-antitrypsin. Blood. 2011;118(12):3384–3391. doi: 10.1182/blood-2011-05-352815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10(1):119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 26.de Lange WJ, Halabi CM, Beyer AM, Sigmund CD. Germ line activation of the Tie2 and SMMHC promoters causes noncell-specific deletion of floxed alleles. Physiol Genomics. 2008;35(1):1–4. doi: 10.1152/physiolgenomics.90284.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang TL, Cui J, Rehumtulla A, et al. The structure and function of murine factor V and its inactivation by protein C. Blood. 1998;91(12):4593–4599. [PubMed] [Google Scholar]
- 28.Adeghe AJ, Cohen J. A better method for terminal bleeding of mice. Lab Anim. 1986;20(1):70–72. doi: 10.1258/002367786781062016. [DOI] [PubMed] [Google Scholar]
- 29.Tchaikovski SN, VAN Vlijmen BJ, Rosing J, Tans G. Development of a calibrated automated thrombography based thrombin generation test in mouse plasma. J Thromb Haemost. 2007;5(10):2079–2086. doi: 10.1111/j.1538-7836.2007.02719.x. [DOI] [PubMed] [Google Scholar]
- 30.Sun H, Yang TL, Yang A, Wang X, Ginsburg D. The murine platelet and plasma factor V pools are biosynthetically distinct and sufficient for minimal hemostasis. Blood. 2003;102(8):2856–2861. doi: 10.1182/blood-2003-04-1225. [DOI] [PubMed] [Google Scholar]
- 31.Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci USA. 2009;106(33):13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kouadjo KE, Nishida Y, Cadrin-Girard JF, Yoshioka M, St-Amand J. Housekeeping and tissue-specific genes in mouse tissues. BMC Genomics. 2007;8:127. doi: 10.1186/1471-2164-8-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang TL, Pipe SW, Yang A, Ginsburg D. Biosynthetic origin and functional significance of murine platelet factor V. Blood. 2003;102(8):2851–2855. doi: 10.1182/blood-2003-04-1224. [DOI] [PubMed] [Google Scholar]
- 34.Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis. 2010;48(9):563–567. doi: 10.1002/dvg.20654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomer A. Human marrow megakaryocyte differentiation: multiparameter correlative analysis identifies von Willebrand factor as a sensitive and distinctive marker for early (2N and 4N) megakaryocytes. Blood. 2004;104(9):2722–2727. doi: 10.1182/blood-2004-02-0769. [DOI] [PubMed] [Google Scholar]
- 36.Murphy GJ, Leavitt AD. A model for studying megakaryocyte development and biology. Proc Natl Acad Sci USA. 1999;96(6):3065–3070. doi: 10.1073/pnas.96.6.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Camire RM, Pollak ES, Kaushansky K, Tracy PB. Secretable human platelet-derived factor V originates from the plasma pool. Blood. 1998;92(9):3035–3041. [PubMed] [Google Scholar]
- 38.Karimi M, Cairo A, Safarpour MM, et al. Genotype and phenotype report on patients with combined deficiency of factor V and factor VIII in Iran [published online ahead of print January 2, 2014]. Blood Coagul Fibrinolysis. doi: 10.1097/MBC.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 39.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193(6):741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiasson VL, Talreja D, Young KJ, Chatterjee P, Banes-Berceli AK, Mitchell BM. FK506 binding protein 12 deficiency in endothelial and hematopoietic cells decreases regulatory T cells and causes hypertension. Hypertension. 2011;57(6):1167–1175. doi: 10.1161/HYPERTENSIONAHA.110.162917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostronoff M, Ostronoff F, Campos G, et al. Allogeneic bone marrow transplantation in a child with severe aplastic anemia and hemophilia A. Bone Marrow Transplant. 2006;37(6):627–628. doi: 10.1038/sj.bmt.1705292. [DOI] [PubMed] [Google Scholar]
- 42.Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost. 2004;91(2):267–275. doi: 10.1160/TH03-05-0310. [DOI] [PubMed] [Google Scholar]
- 43.Rapaport SI, Ames SB, Mikkelsen S, Goodman JR. Plasma clotting factors in chronic hepatocellular disease. N Engl J Med. 1960;263:278–282. doi: 10.1056/NEJM196008112630604. [DOI] [PubMed] [Google Scholar]
- 44.Langley PG, Hughes RD, Williams R. Increased factor VIII complex in fulminant hepatic failure. Thromb Haemost. 1985;54(3):693–696. [PubMed] [Google Scholar]
- 45.Nolan DJ, Ginsberg M, Israely E, et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. 2013;26(2):204–219. doi: 10.1016/j.devcel.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100(2):158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 47.Aitsebaomo J, Portbury AL, Schisler JC, Patterson C. Brothers and sisters: molecular insights into arterial-venous heterogeneity. Circ Res. 2008;103(9):929–939. doi: 10.1161/CIRCRESAHA.108.184937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumaran V, Benten D, Follenzi A, Joseph B, Sarkar R, Gupta S. Transplantation of endothelial cells corrects the phenotype in hemophilia A mice. J Thromb Haemost. 2005;3(9):2022–2031. doi: 10.1111/j.1538-7836.2005.01508.x. [DOI] [PubMed] [Google Scholar]
- 49.Follenzi A, Benten D, Novikoff P, Faulkner L, Raut S, Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118(3):935–945. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shahani T, Covens K, Lavend’homme R, et al. Human liver sinusoidal endothelial cells but not hepatocytes contain FVIII. J Thromb Haemost. 2014;12(1):36–42. doi: 10.1111/jth.12412. [DOI] [PubMed] [Google Scholar]
- 51.Pandey GS, Yanover C, Miller-Jenkins LM, et al. PATH (Personalized Alternative Therapies for Hemophilia) Study Investigators. Endogenous factor VIII synthesis from the intron 22-inverted F8 locus may modulate the immunogenicity of replacement therapy for hemophilia A. Nat Med. 2013;19(10):1318–1324. doi: 10.1038/nm.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med. 2012;2(1):a006429. doi: 10.1101/cshperspect.a006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamamoto K, de Waard V, Fearns C, Loskutoff DJ. Tissue distribution and regulation of murine von Willebrand factor gene expression in vivo. Blood. 1998;92(8):2791–2801. [PubMed] [Google Scholar]
- 54.Aird WC, Edelberg JM, Weiler-Guettler H, Simmons WW, Smith TW, Rosenberg RD. Vascular bed-specific expression of an endothelial cell gene is programmed by the tissue microenvironment. J Cell Biol. 1997;138(5):1117–1124. doi: 10.1083/jcb.138.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chi JT, Chang HY, Haraldsen G, et al. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci USA. 2003;100(19):10623–10628. doi: 10.1073/pnas.1434429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kane NM, Xiao Q, Baker AH, Luo Z, Xu Q, Emanueli C. Pluripotent stem cell differentiation into vascular cells: a novel technology with promises for vascular re(generation). Pharmacol Ther. 2011;129(1):29–49. doi: 10.1016/j.pharmthera.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Bai H, Wang ZZ. Directing human embryonic stem cells to generate vascular progenitor cells. Gene Ther. 2008;15(2):89–95. doi: 10.1038/sj.gt.3303005. [DOI] [PubMed] [Google Scholar]
- 58.Kaufman RJ, Wasley LC, Davies MV, Wise RJ, Israel DI, Dorner AJ. Effect of von Willebrand factor coexpression on the synthesis and secretion of factor VIII in Chinese hamster ovary cells. Mol Cell Biol. 1989;9(3):1233–1242. doi: 10.1128/mcb.9.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du LM, Nurden P, Nurden AT, et al. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chuah MK, Nair N, VandenDriessche T. Recent progress in gene therapy for hemophilia. Hum Gene Ther. 2012;23(6):557–565. doi: 10.1089/hum.2012.088. [DOI] [PubMed] [Google Scholar]
- 61.Vandendriessche T, Chuah MK. Targeting endothelial cells by gene therapy. Blood. 2013;122(12):1993–1994. doi: 10.1182/blood-2013-08-518266. [DOI] [PubMed] [Google Scholar]