

Abstract
The PI3K/Akt/mTOR pathway is overactivated and heat shock protein (HSP) 90 is overexpressed in common cancers. We hypothesized that targeting both pathways can kill intrahepatic cholangiocarcinoma (CCA) cells. HSP90 and PTEN protein expression was evaluated by immunohistochemical staining of samples from 78 patients with intrahepatic CCA. CCA cell lines and a thioacetamide (TAA)-induced CCA animal model were treated with NVP-AUY922 (an HSP90 inhibitor) and NVP-BEZ235 (a PI3K/mTOR inhibitor) alone or in combination.
Both HSP90 overexpression and loss of PTEN were poor prognostic factors in patients with intrahepatic CCA. The combination of the HSP90 inhibitor NVP-AUY922 and the PI3K/mTOR inhibitor NVP-BEZ235 was synergistic in inducing cell death in CCA cells. A combination of NVP-AUY922 and NVP-BEZ235 caused tumor regression in CCA rat animal model. This combination not only inhibited the PI3K/Akt/mTOR pathway but also induced ROS, which may exacerbate the vicious cycle of ER stress. Our data suggest simultaneous targeting of the PI3K/mTOR and HSP pathways for CCA treatment.
Keywords: NVP-AUY922, NVP-BEZ235, heat-shock protein 90, phosphatase and tensin homolog
INTRODUCTION
Intrahepatic cholangiocarcinoma (CCA) is a relatively rare, but increasing more common, hepatobiliary cancer [1–3]. CCA, the second most common liver cancer, is an aggressive cancer typically diagnosed at an advanced stage with poor prognosis [4].
The ABC-02 trial showed improved overall survival of CCA patients treated with gemcitabine plus cisplatin compared to those treated with gemcitabine alone (11.7 versus 8 months), and defined gemcitabine plus platinum as a standard treatment for advanced biliary tract cancer [5]. Several molecular-targeted therapies have been assessed in clinical trials, with median progression-free survivals of 1.8–7 months [6, 7]. However, there is no standard therapy for refractory CCA, and additional therapeutic drugs are needed urgently.
The molecular chaperone heat shock protein 90 (HSP90) plays an important role in post-translational maturation and activation of many oncogenic client proteins that increase the survival, growth, and invasive potential of cancer cells [8, 9]. HSP90 inhibitors induce ubiquitination and proteosomal degradation of numerous oncoproteins, which consequently inhibit cancer cell growth and survival [10]. We had previously demonstrated that the HSP90 inhibitor NVP-AUY922 exerted antitumor effects in CCA cells in pathways involving the phosphatidylinositol 3-kinase catalytic subunit α (PI3K)/v-Akt murine thymoma viral oncogene homolog 1 protein kinase (Akt)/mammalian target of rapamycin (mTOR) and the v-Ki-ras2 Kirsten rat sarcoma viral oncogene (KRAS)/mitogen-activated protein kinase (MAPK) pathways [11]. Although HSP90 inhibitors are considered a novel therapeutic strategy for several cancers pre-clinically [11–15], HSP90 inhibitor mono-therapy has shown only limited clinical success [16, 17] due to suboptimal inhibition of target client proteins [18]. Combination approaches may be required for effective clinical use of HSP90 inhibitors [19, 20].
The PI3K/Akt/mTOR signaling cascade is central to cell survival, apoptosis, metabolism, motility, and angiogenesis [21]. This pathway is up-regulated in CCA [22] and is a key pathway for CCA drug development [6, 7]. PIK3CA (phosphoinositide 3-kinase, catalytic, α-polypeptide) activating mutations are rarely found in CCA [22], suggesting that additional mechanisms are involvement. Recently, an imidazo [4, 5-c] quinoline derivative dual PI3K/mTOR inhibitor, NVP-BEZ235 was shown to inhibit CCA cell growth [23]. However, similar to HSP90 inhibitor mono-therapy, PI3K inhibitor mono-therapy has not produced significant clinical responses [21, 24].
NVP-BEZ235 targets PI3K and mTOR equally in cancer [25], and we postulated that the combination of a PI3K/mTOR inhibitor and an HSP90 inhibitor might cooperatively inhibit tumor cell proliferation and induce apoptosis. Furthermore, the HSP90 inhibitor also induces endoplasmic reticulum (ER) stress, which leads to mitochondrial damage and subsequent apoptosis [26]. This process may be fueled by oxidative stress when combined with an mTOR inhibitor [27]. Therefore, the combination of an HSP90 inhibitor and a PI3K/mTOR dual inhibitor may promote irreversible ER stress and induce cell death.
Here, we investigated the effects of targeting the PI3K/mTOR pathway (with NVP-BEZ235) and HSP90 (with NVP-AUY922) in CCA, both in vitro and in vivo.
RESULTS
HSP90 and PTEN expression in human CCA
HSP90 and PTEN were diffusely expressed in the cytoplasm in human MF-CCA (Fig 1A, 1B). Two representative examples showing HSP90 and PTEN immunohistochemical staining in human CCA were graded from 0 to 3+, with 0 and 1+ indicating low expression and 2+ and 3+ indicating high expression (Figure 1A to 1B).
Figure 1. HSP90 and PTEN expression was correlated with survival in 78 patients with resectable MF-CCA.
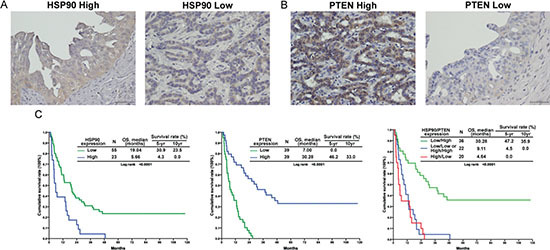
(A, B) Immunohistochemical staining of MF-CCA tumors with different intensity scores for HSP90 and PTEN expression; (C) The high-HSP90 group showed significantly worse overall survival (P < 0.001, left); The low-PTEN group showed significantly worse overall survival (P < 0.001, middle); The combined high-HSP90 and low-PTEN group showed the worst overall survival (P < 0.001, right).
Clinicopathological features and HSP90 and PTEN expression levels in patients with MF-CCA
Of the 78 specimens obtained from MF-CCA patients, 55 (70.5%) showed high HSP90 expression (2+ and 3+ positive), while 39 (50%) showed low PTEN expression. Clinicopathological features were similar between patients with low and high HSP90 and PTEN expression (Table 1 and 2).
Table 1. Clinicopathological features of 78 patients with high and low heat shock protein 90 (HSP90)-expressing mass-forming cholangiocarcinomas.
| Factor | HSP90 high (n = 55) | HSP90 low (n = 23) | P |
|---|---|---|---|
| Age (years) | 58.3 ± 12.2 | 63.2 ± 10.9 | 0.086 |
| Gender | 0.713 | ||
| Male | 24 (43.6) | 9 (39.1) | |
| Female | 31 (56.4) | 14 (60.9) | |
| Symptom | 0.097 | ||
| Positive | 44 (80.0) | 22 (95.7) | |
| Negative | 11 (20.0) | 1 (4.3) | |
| AST (IU/L) | 0.564 | ||
| ≤ 34 | 29 (52.7) | 10 (45.5) | |
| > 34 | 26 (47.3) | 12 (54.5) | |
| ALT (U/L) | 0.357 | ||
| ≤ 36 | 27 (52.9) | 13 (65.0) | |
| > 36 | 24 (47.1) | 7 (35.0) | |
| ALP (U/L) | 0.491 | ||
| ≤ 94 | 15 (28.3) | 8 (36.4) | |
| > 94 | 38 (71.7) | 14 (63.6) | |
| Bilirubin (total) (mg/dL) | 0.934 | ||
| ≤ 1.3 | 45 (81.8) | 19 (82.6) | |
| > 1.3 | 10 (18.2) | 4 (17.4) | |
| Albumin (g/dL) | 0.226 | ||
| ≤ 3.5 | 13 (26.5) | 9 (40.9) | |
| > 3.5 | 36 (73.5) | 13 (59.1) | |
| Serum CEA (ng/mL) | 0.984 | ||
| ≤ 5 | 17 (44.7) | 8 (44.4) | |
| > 5 | 21 (55.3) | 10 (55.6) | |
| Size (cm) | 0.843 | ||
| ≤ 5 | 23 (43.4) | 9 (40.9) | |
| > 5 | 30 (56.6) | 13 (59.1) | |
| Lymph node | 0.076 | ||
| Negative | 38 (69.1) | 11 (47.8) | |
| Positive | 17 (30.9) | 12 (52.2) | |
| Differentiated | 0.131 | ||
| Well | 1 (1.8) | 1 (4.3) | |
| Moderate | 29 (52.7) | 12 (52.2) | |
| Poorly | 25 (45.5) | 8 (34.8) | |
| Other | 0 | 2 (8.7) | |
| Post-op chemotherapy | 0.254 | ||
| No | 21 (38.2) | 12 (52.2) | |
| Yes | 34 (61.8) | 11 (47.8) | |
| Post-op radiotherapy | 0.266 | ||
| No | 46 (83.6) | 22 (95.7) | |
| Yes | 9 (16.4) | 1 (4.3) |
Table 2. Clinicopathological features of 78 patients with high and low phosphatase and tensin homolog (PTEN)-expressing mass-forming cholangiocarcinoma patients.
| Factor | PTEN high (n = 39) | PTEN low (n = 39) | P |
|---|---|---|---|
| Age (years) | 61.2 ± 10.8 | 58.3 ± 13.0 | 0.294 |
| Gender | 0.492 | ||
| Male | 15 (38.5) | 18 (46.2) | |
| Female | 24 (61.5) | 21 (53.8) | |
| Symptom | 0.209 | ||
| Positive | 35 (89.7) | 31 (79.5) | |
| Negative | 4 (10.3) | 8 (20.5) | |
| AST (IU/L) | 0.570 | ||
| ≤ 34 | 19 (48.7) | 21 (53.8) | |
| > 34 | 20 (51.3) | 18 (46.2) | |
| ALT (U/L) | 0.820 | ||
| ≤ 36 | 22 (56.4) | 21 (53.8) | |
| > 36 | 17 (43.6) | 18 (46.2) | |
| ALP (U/L) | 0.240 | ||
| ≤ 94 | 10 (25.6) | 14 (35.9) | |
| > 94 | 29 (74.4) | 25 (64.1) | |
| Bilirubin (total) (mg/dL) | 0.555 | ||
| ≤ 1.3 | 33 (84.6) | 31 (79.5) | |
| > 1.3 | 6 (15.4) | 8 (20.5) | |
| Albumin (g/dL) | 0.089 | ||
| ≤ 3.5 | 16 (41.0) | 9 (23.1) | |
| > 3.5 | 23 (59.0) | 30 (76.9) | |
| Serum CEA (ng/mL) | 0.063 | ||
| ≤ 5 | 14 (35.9) | 17 (58.6) | |
| > 5 | 25 (64.1) | 12 (41.4) | |
| Size (cm) | 0.109 | ||
| ≤ 5 | 13 (33.3) | 20 (51.3) | |
| > 5 | 26 (66.7) | 19 (48.7) | |
| Lymph node | 0.101 | ||
| Negative | 21 (53.8) | 28 (71.8) | |
| Positive | 18 (46.2) | 11 (28.2) | |
| Differentiated | 0.188 | ||
| Well | 2 (5.1) | 0 | |
| Moderate | 21 (53.8) | 20 (51.3) | |
| Poorly | 14 (35.9) | 19 (48.7) | |
| Other | 2 (5.1) | 0 | |
| Post-op chemotherapy | 0.819 | ||
| No | 16 (41.0) | 17 (43.6) | |
| Yes | 23 (59.0) | 22 (56.4) | |
| Post-op radiotherapy | 0.498 | ||
| No | 33 (84.6) | 35 (89.7) | |
| Yes | 6 (15.4) | 4 (10.3) |
Survival and prognostic analysis of MF-CCA patients who underwent a hepatectomy
Seventy-eight MF-CCA patients who had undergone hepatectomy were enrolled in the survival analysis study. The follow-up duration ranged from 1.4 to 94.1 (median, 13.6) months. Overall survival (OS) rates at 1, 3, and 5 years were 55.1%, 22.9%, and 14.9%, respectively (data not shown). Univariate log-rank analysis identified the following factors as negatively affecting OS: the presence of symptoms, high preoperative alkaline phosphatase and carcinoembryonic antigen levels, low albumin levels, tumor size >5 cm, positive surgical margin status, and high HSP90 and low PTEN expression (Table 3). Multivariate Cox proportional hazard analysis revealed that poor nutritional status, positive margin status, and high HSP90 and low PTEN expression independently predicted unfavorable OS in MF-CCA patients after hepatectomy (Figure 1C and Table 4).
Table 3. Univariate analysis of factors influencing the overall survival of 78 MF-CCA patients.
| Factor | Survival (months) | P | |||
|---|---|---|---|---|---|
| Median | 95% CI of median | 3-year (%) | 5-year (%) | ||
| Gender | 0.719 | ||||
| Male (n = 33) | 14.70 | 7.89–21.50 | 21.2 | 18.2 | |
| Female (n = 45) | 10.82 | 5.85–15.79 | 24.4 | 15.0 | |
| Symptoms | 0.006 | ||||
| Negative (n = 12) | 37.71 | 4.78–70.64 | 58.3 | 41.7 | |
| Positive (n = 66) | 10.46 | 5.84–15.07 | 16.7 | 11.9 | |
| AST (IU/L) | 0.197 | ||||
| ≤ 34 (n = 39) | 13.32 | 9.53–17.10 | 30.8 | 22.8 | |
| > 34 (n = 38) | 10.72 | 2.28–19.16 | 15.8 | 10.5 | |
| ALT (IU/L) | 0.625 | ||||
| ≤ 36 (n = 40) | 12.99 | 6.42–19.56 | 25.0 | 19.7 | |
| > 36 (n = 31) | 14.70 | 7.81–21.58 | 19.4 | 9.7 | |
| ALP (IU/L) | 0.009 | ||||
| ≤ 94 (n = 23) | 23.90 | 11.66–36.15 | 39.1 | 29.8 | |
| > 94 (n = 52) | 9.11 | 4.93–13.29 | 15.4 | 9.6 | |
| Bil (total) (mg/dL) | 0.581 | ||||
| ≤ 1.3 (n = 64) | 12.99 | 6.41–19.56 | 25.0 | 16.9 | |
| > 1.3 (n = 14) | 10.72 | 0.00–22.23 | 14.3 | 14.3 | |
| Albumin (g/dL) | 0.043 | ||||
| ≤ 3.5 (n = 22) | 4.70 | 3.12–6.29 | 18.2 | 13.6 | |
| > 3.5 (n = 49) | 19.04 | 13.31–24.76 | 24.5 | 15.9 | |
| Serum CEA (ng/dL) | 0.043 | ||||
| ≤ 5 (n = 25) | 18.51 | 2.09–34.93 | 40.0 | 27.0 | |
| > 5 (n = 31) | 10.29 | 4.05–16.53 | 6.5 | 6.5 | |
| Margin | < 0.0001 | ||||
| Negative (n = 54) | 19.43 | 14.97–23.89 | 33.3 | 23.8 | |
| Positive (n = 24) | 4.41 | 2.43–6.38 | 0.0 | 0.0 | |
| Size | 0.006 | ||||
| ≤ 5cm (n = 32) | 19.99 | 13.75–26.23 | 37.5 | 30.7 | |
| > 5cm (n = 43) | 9.11 | 1.97–16.25 | 14.0 | 7.0 | |
| Lymph node | 0.091 | ||||
| Negative (n = 49) | 19.43 | 13.70–25.16 | 28.6 | 18.0 | |
| Positive (n = 29) | 10.46 | 0.00–22.71 | 13.8 | 13.8 | |
| Histology | 0.207 | ||||
| Well (n = 2) | 2.73 | 0.0 | 0.0 | ||
| Moderate (n = 41) | 13.84 | 7.45–20.24 | 22.0 | 17.1 | |
| Poor (n = 33) | 12.99 | 5.25–20.72 | 27.3 | 17.3 | |
| Others* (n = 2) | 4.37 | 0.0 | 0.0 | ||
| HSP90 expression | <0.0001 | ||||
| Low (n = 55) | 19.04 | 11.66–26.41 | 30.9 | 23.5 | |
| High (n = 23) | 5.66 | 3.55–7.77 | 4.3 | 0.0 | |
| PTEN expression | < 0.0001 | ||||
| Low (n = 39) | 7.00 | 3.95–10.06 | 0.0 | 0.0 | |
| High (n = 39) | 30.28 | 12.30–48.26 | 46.2 | 33.0 | |
| HSP90/PTEN expression | < 0.0001 | ||||
| Low/high (n = 36) | 30.28 | 13.27–47.29 | 47.2 | 35.9 | |
| Low/low or high/high (n = 22) | 9.11 | 5.03–13.19 | 4.5 | 0.0 | |
| High/low (n = 20) | 4.64 | 1.90–7.37 | 0.0 | 0.0 |
HSP90: heat shock protein 90; PTEN: phosphatase and tensin homolog; AST: aspartate aminotransferase; ALT: alanine aminotransferase; ALP: alkaline phosphatase; CEA: carcinoembryonic antigen; CA 19–9: carbohydrate antigen; IU: international unit; op: operation
cystadenocarcinoma:1, musinous:1
Table 4. Cox's proportional hazards analysis.
| Factor | Hazard ratio (95% confidence interval) | P |
|---|---|---|
| Symptoms (positive/negative) | 1.469 (0.307–7.024) | 0.630 |
| Alkaline phosphatase (U/L) (> 94/≤ 94) | 1.742 (0.671–4.524) | 0.254 |
| Albumin (g/dL) (≤ 3.5/> 3.5) | 2.497 (1.001–6.297) | 0.049 |
| CEA (ng/dL) (> 5/≤ 5) | 1.306 (0.542–3.148) | 0.552 |
| Tumor size (cm) (> 5/≤ 5) | 1.065 (0.387–2.934) | 0.903 |
| Margin (positive/negative) | 2.543 (1.13–4.352) | 0.043 |
| Lymph node status (positive/negative) | 0.868 (0.371–2.034) | 0.745 |
| HSP90, PTEN (low, low or high, high/low, high) | 5.820 (2.138–15.844) | 0.001 |
| (high, low/low, high) | 5.471 (1.930–15.507) | 0.001 |
| Post-op radiotherapy (with/without) | 1.494 (0.454–4.922) | 0.509 |
HSP90: heat shock protein 90; PTEN: phosphatase and tensin homolog; NS: not significant
The combination of NVP-AUY922 and NVP-BEZ235 blocked proliferation and induced cytotoxicity of CCA cell lines
The effects of NVP-AUY922 or NVP-BEZ235 mono-therapy and the combination on the growth of CCA cell lines were determined. NVP-AUY922 and NVP-BEZ235 exhibited strong antiproliferative effects in both HuCCT1 and CGCCA cells. In HuCCT1 and CGCCA cells, the IC50 values of NVP-AUY922 were 15 and 85 nM, respectively, and those of NVP-BEZ235 were 97 and 99 nM, respectively (Figure 2A). NVP-AUY922 (at 7.5, 15, 30, 60, and 120 nmol/L) at NVP-BEZ235 (at 7.5, 15, 30, 60, and 120 nmol/L) both alone and in combination induced dose-dependent cytotoxicity in the CGCCCA cells (Figure 2B). The combination of NVP-AUY922 and NVP-BEZ235 induced synergistic cytotoxicity. Similar results were seen in the HuCCT1 cell line (Figure 2B). Although NVP-BEZ235 itself did not trigger cell death in either cell line, both PI-staining and PARP cleavage analysis (Figure 2C) revealed that it synergistically activated apoptotic activity with NVP-AUY922.
Figure 2. The combination of NVP-AUY922 and NVP-BEZ235 synergistically induced apoptosis in CCA cell lines.
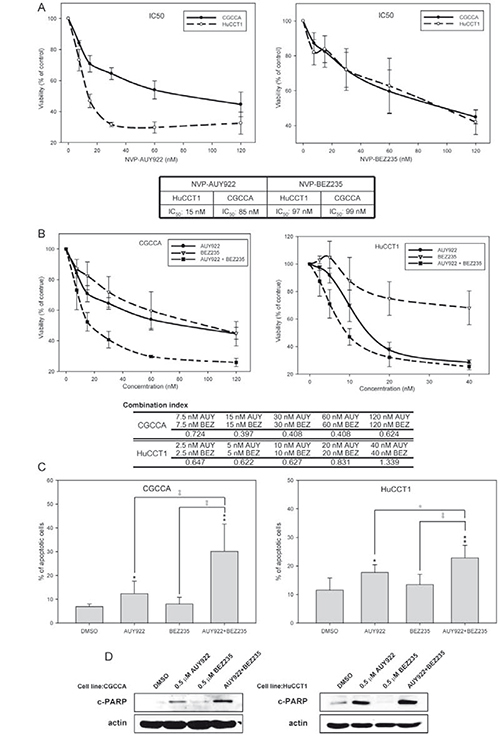
(A) CGCCA and HuCCT1 cells were incubated with various concentrations (0, 8, 16, 32, 64, or 128 nM) of either NVP-AUY922 or NVP-BEZ235 for 72 h; (B) CGCCA and HuCCT1 cells were incubated with either NVP-AUY922 or NVP-BEZ235 or both NVP-AUY922 and NVP-BEZ235 at various concentrations for 72 h. The combination index (CI) < 1, CI = 1, or CI > 1 indicate synergism, an additive effect, or antagonism, respectively; (C) CGCCA and HuCCT1 cells were treated with 0.5 uM of either NVP-AUY922 or NVP-BEZ235 or a combination of these 2 for 72 h. The number of apoptotic cells measured using the TACS Annexin V-FITC apoptosis detection kit is represented as a percentage of the total events. (D) The immunoblots are analyses of cleaved poly (ADP-ribose) polymerase (PARP). β-Actin was used as the loading control.
NVP-AUY922 and NVP-BEZ235 work together to block the PI3K/Akt/mTOR signaling pathway
To analyze the combinatorial effect of a PI3K/mTOR dual inhibitor and an HSP90 inhibitor, western blotting was performed to analyze the activation status of a series of proteins in the PI3K/Akt/mTOR pathway. NVP-BEZ235 efficiently inhibited both PI3K and mTOR activity within 2 h as indicated by the phosphorylation status of Akt, 4E-BP1, and S6K, respectively. However, this was abrogated in the 8–24-h time period (Figure 3B). On the other hand, NVP-AUY922 inhibited this pathway more slowly (Figure 3A). Interestingly, the kinetics of PI3K/Akt/mTOR signaling pathway inhibition by the combination treatment was similar to that of NVP-BEZ235 alone. Moreover, the combination markedly hindered abrogation of the effect of NVP-BEZ235 mono-therapy (Figure 3C). These results showed that NVP-BEZ235 and NVP-AUY922 work together to block the PI3K/Akt/mTOR signaling pathway.
Figure 3. NVP-AUY922 and NVP-BEZ235 functioned together to block the PI3K/Akt/mTOR signaling pathway.
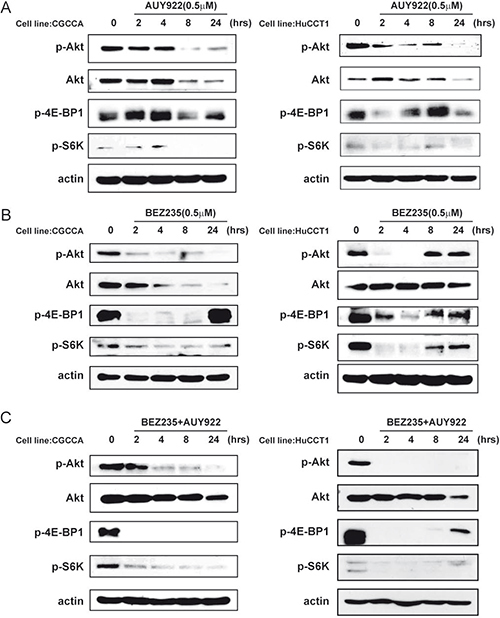
(A) Western blot analysis revealed the molecular signature of PI3K/mTOR inhibition induced by NVP-AUY922 in the CCA and HuCCT1 cell lines. Cell lysates from the CGCCA and HuCCT1 cell lines were treated with 0.5 uM NVP-AUY922 at various time points (0, 24, 48, 72 h); (B) Western blot analysis reveals the molecular signature induced by NVP-BEZ235 in CCA and HuCCT1 cell lines. Cell lysates from CGCCA and HuCCT1 cells were treated with 0.5 uM NVP-BEZ235 for various time points (0, 24, 48, and 72 h); (C) Western blot analysis revealed the molecular signature induced by NVP-BEZ235 and NVP-AUY922 treatment in CCA and HuCCT1 cell lines. Cell lysates from CGCCA and HuCCT1 cells were treated with 0.5 uM NVP-AUY922 and NVP-BEZ235 at various time points (0, 24, 48, and 72 h). β-Actin was used as the loading control.
NVP-AUY922 induced ER stress and disrupted mitochondrial homeostasis
We analyzed whether the HSP90 inhibitor NVP-AUY922 triggered ER stress in CCA cells. Indeed, NVP-AUY922 upregulated the expression of several chaperones such as Grp94, Grp78, IRE1α and phosphorylation of eIF2α (Figure 4A). These signaling signatures represent the unfolded protein response (UPR) [28] and indicate that CCA cells were struggling to survive under NVP-AUY922 treatment. However, CHOP and JNK activation indicated a shift to the ER stress-induced apoptosis response in CCA cells following NVP-AUY922 treatment [Figure 4A, 29–31]. These data are consistent with the observation that NVP-AUY922 alone triggered a collapse in mitochondrial membrane potential (Figure 4B) and induced CCA cell apoptosis (Figure 2C).
Figure 4. NVP-AUY922 induced ER stress and mitochondrial damage, which was fueled by oxidative stress when combined with NVP-BEZ235.
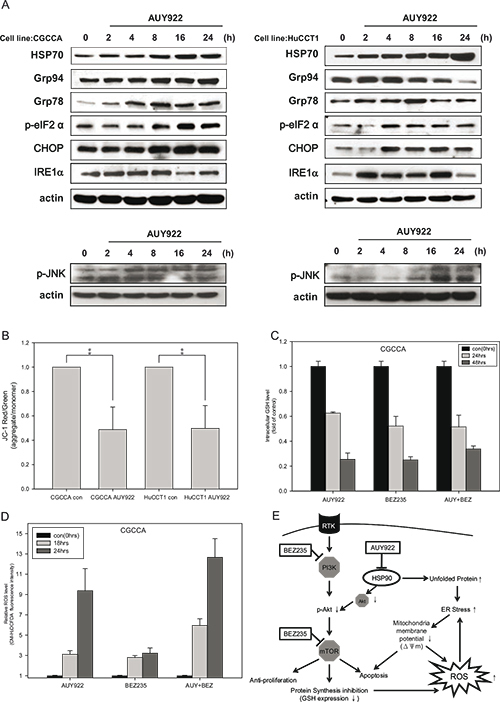
(A) CCA cells were incubated with 0.5 uM NVP-AUY922 for 0, 2, 4, 8, 16, and 24 h. Whole cell lysates were subjected to western blot analysis for HSP70, Grp94, Grp78, p-eIF2α, CHOP, IRE1α, and phosphor-JNK. β-Actin was used as the loading control; (B) CCA cells were incubated with 0.5 uM NVP-AUY922 for 48 h. The red and green color ratio of JC-1 reflects the change in the mitochondrial membrane potential (ΔΨm); (C) Relative levels of reduced glutathione (GSH) in CGCCA cell line treated with 0.5 uM NVP-AUY922 and NVP-BEZ235 alone or combined for 0, 24, and 48 h. (D) Reactive oxidative species (ROS) levels induced by 0.5 uM NVP-AUY922 and NVP-BEZ235 alone or combined for 0, 18, and 24 h in CGCCA cell line. (E) The model shows that NVP-AUY922 induces ER stress, which leads to mitochondrial damage, and ultimately to apoptosis. When combined with NVP-BEZ235 treatment, this process is fueled by oxidative stress. NVP-BEZ235 and NVP-AUY922 cooperate to induce apoptosis by vertically affecting the PI3K/Akt/mTOR signaling pathway at multiple nodes.
NVP-AUY922 and NVP-BEZ235 promoted excessive oxidative stress by inducing ROS and simultaneously suppressing the G6PD/glutathione antioxidant pathway
NVP-AUY922 induced UPR and ER stress, which led to mitochondrial damage and cell apoptosis. This process might be exacerbated by oxidative stress when combined with an mTOR inhibitor [27]. Oxidative stress is caused by an imbalance between ROS production and ROS clearance. We investigated whether NVP-BEZ235 might enhance the effects of NVP-AUY922 by suppressing endogenous antioxidant levels. Interestingly, Both NVP-BEZ235 and NVP-AUY922 alone or combined decreased the reduced form of glutathione (GSH), one of the most important endogenous antioxidants, in CCA cells (Figure 4C and supplemental Figure 1A). We also analyzed whether simultaneously inhibiting HSP90 and the PI3K/AKT pathway could induce strong ROS production. Our result demonstrated that the combination of NVP-AUY922 and NVP-BEZ235 triggered strong ROS production in CCA cells (Figure 4D and supplemental Figure 1B).
Evaluation of the antitumor effect of NVP-AUY922 and NVP-BEZ235 on CCA in vivo
After inducing CCA with TAA, tumors in the control and treated groups were evaluated by transverse, sagittal, and coronal PET-computed tomography (CT) images. As shown in Figure 5A, each group had at least one FDG-avid tumor in the liver after 20 weeks of TAA treatment, as demonstrated by animal the PET-CT coronal images. In the experimental groups, rats were given BEZ235 alone (Group 2), AUY922 alone (Group 3), BEZ235/AUY922 (Group 4), or gemcitabine/oxaliplatin (Group 5). The change in the tumor-to-liver (T/L) ratio, as indicated by the SUV for each group, is shown in Figure 5B. The T/L ratio of SUV in the control group (Group 1) elevated steadily until the last scan (a 14.5% to 21.7% increase, from the second to the fifth week after the experiments; Figure 5B). The T/L ratio of SUV in each treatment group tended to decrease until the last scan; the T/L ratio of SUV decreased 2 weeks after the experiment, ranging from 3% to 15%. Gemcitabine with oxaliplatin induced a significant decrease in the T/L ratio of SUV after 2 weeks compared with the control group (control, 14.7% versus group 5, −15.5%, p < 0.05; Figure 5B). However, after 5 weeks of treatment, NVP-AUY922 and NVP-BEZ235 induced a significant decrease in the T/L ratio of SUV compared to the control group (control, 21.7% versus group 4, −18.3%; p < 0.05; Figure 5B).
Figure 5. Detection of rat CCA by animal PET and changes in the tumor/liver SUV ratio.
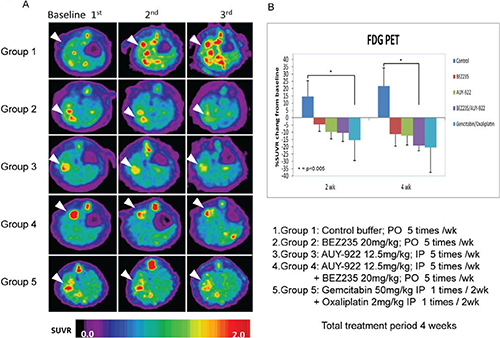
(A) Coronal views of fused CT and PET scans of control and experimental rats revealed the CCA-expressing areas of the liver in which the 18F-FDG uptake was higher than baseline at 2–5 wk after the experiment (i.e., wk 20, 22, and 25). (B) Change in the tumor-to-liver (T/L) ratio of SUV in the control and experiment groups at 2–5 wk after the experiment (i.e., wk 22 and 25).
DISCUSSION
In this study, we demonstrated that both high HSP90 expression and loss of PTEN expression were independent prognostic factors in CCA. This implies that HSP90 and the PTEN-related PI3K/Akt/mTOR pathway are potential therapeutic targets in CCA. We then investigated the effect of a combination of an HSP90 inhibitor (NVP-AUY922) and a PI3K/mTOR dual inhibitor (NVP-BEZ235) on CCA. We found that they exerted significant synergistic proapoptotic and antiproliferative effects in CCA cell lines in vitro. This combination worked in concert to inhibit the PI3K/Akt/mTOR pathway and induce ROS accumulation; these effects may exaggerate the vicious cycle of ER stress. We also demonstrated the synergistic effect of the NVP-AUY922 and NVP-BEZ235 in a TAA-induced CCA animal model.
HSP90 is a ubiquitously expressed chaperone that is involved in the post-translational folding and activation of numerous client proteins implicated in oncogenesis [32]. The prognostic value of HSP90 expression in several types of cancer has been discussed, but to our knowledge, there is the first report of the clinical significance of HSP90 expression in CCA prognosis. Lower HSP90 protein expression was associated with longer overall survival (Table 3 and Figure 1). Congruently, HSP90 expression was significantly associated with tumor aggressiveness and poor prognosis in gastric and breast cancer [33, 34], which indicates HSP90 is a good drug target in CCA [11, 35].
Moreover, the PI3K/Akt/mTOR pathway is upregulated in CCA cells [22] and is one of the most important targets for CAA drug development [6, 7]. Activating mutations in PIK3CA are only found in 9% of intrahepatic CCA cases [22, 36], suggesting that additional mechanisms may positively regulate this pathway. For instance, PTEN, a well-characterized human tumor suppressor gene, is an antagonist of the PI3K/Akt/mTOR pathway. Loss of PTEN and activation of SMAD4 or KRAS activation may induce CCA development in murine models [37, 38], however, clinical implications of decreased PTEN protein expression in intrahepatic CCA has not yet been investigated. In this study, univariate analyses indicated that loss of PTEN expression correlated with a worse survival in patients with intrahepatic CCA (Table 3 and Figure 1). This result also supports our published data; PI3K/Akt/mTOR pathway plays a critical role in CCA [11]. Moreover, patients with high HSP90 protein expression and PTEN loss had the worst survival according to multivariate analyses (Table 4), which suggested that combining a PI3K inhibitor with an HSP90 inhibitor may represent an effective treatment for CCA.
The primary function of HSP90 in cancer was thought to be stabilization of client oncoproteins, suggesting that this protein might be a good drug target. However, recent HSP90 inhibitor clinical trials have shown that use of HSP90-targeted drugs might not be an optimally effective therapeutic strategy [9]. Combining HSP90 inhibitors with other targeted therapies may block the compensatory signaling mechanisms and impart a clinical benefit [39]. One strategy is vertically targeting the same pathway [40]. For example, the survival of patients with melanoma and the BRAF V600 mutation improved when treated with a combined BRAF and MEK inhibitor [41]. In our previous study, we identified Akt as a client protein of HSP90 [11]. NVP-BEZ235 is a novel dual PI3K/mTOR inhibitor and, when combined with NVP-AUY922, may vertically target the pathway at multiple nodes (Figure 4E). Indeed, we observed that combining NVP-AUY922 and NVP-BEZ235 enhanced and prolonged the inhibition of the PI3K/Akt/mTOR pathway (Figure 3C).
A broad spectrum of insults, including nutrient deprivation, can trigger ER stress, leading to activation of adaptive pathways to alleviate the insults, restore ER homeostasis, and ensure survival. However, under extensive stress, this same system will trigger a pro-apoptotic pathway [29, 42]. Most cancer cells have chronically elevated baseline ER stress levels, as indicated by increased expression of GRP78 [43]. When treated with drugs that specifically trigger ER stress, the key pro-apoptotic ER stress protein, CHOP, can induce cancer cell apoptosis [44]. HSP90 is critical for several cellular functions, including protein folding and assembly. Thus, an HSP90 inhibitor may increase unfolded protein accumulation (UPR), increase ER stress, and cause apoptosis [26]. In our study, we found that NVP-AUY922 induced ER stress and activated the UPR, as demonstrated by the upregulation of GRP78, peIF2a, GRP94, IRE1α and CHOP, within 4 h (Figure 4A) in CCA cells. The ER stress induced apoptosis was via both the JNK and caspase 4 pathways (Figure 4A and supplemental Figure 2).
ER stress can lead to ROS production, which occurs subsequent to the accumulation of unfolded proteins in the ER. Mitochondrial ROS is also generated in after ER stress induced by Ca2+ release and depolarization of the inner mitochondrial membrane (ΔΨm) [26, 45]. Oxidative stress, along with unresolved ER stress, contributes to cell death. Furthermore, the HSP90 inhibitor IPI-504 and the mTOR inhibitor rapamycin exert synergic antitumor effects in Ras-driven tumors by promoting ER stress and mitochondrial damage, via IPI-504 mediated ROS increase and a rapamycin-dependent suppression of GSH [27]. We here observed that NVP-AUY922 induced ER stress and decreased the mitochondrial membrane potential (ΔΨm) (Figure 4A and 4B). Both NVP-AUY922 and NVP-BEZ235 inhibited GSH synthesis (Figure 4C and supplemental Figure 1A). Finally, both NVP-AUY922 and NVP-BEZ235 induced ROS production (Figure 4D and supplemental Figure 1B) and promoted cell death.
To investigate the effectiveness of NVP-AUY922, NVP-BEZ235, their combination and gemcitabine and oxaliplatin for CCA treatment, we used the previously established TAA-induced rat CCA model [46]. This model recapitulates the histological progression of human CCA [46], indicating that it is a good platform to investigate new CCA treatment regimens. PET was then used to measure the tumor response to treatment [47]. Because we could not detect lesions < 2 mm by PET and the invasive CCA border was indistinguishable from the normal liver background, the T/L ratio of the SUV was used to represent tumor growth [47]. The T/L ratio of the SUV was significantly lower in rats treated with NVP-AUY922 and NVP-BEZ235 than in control rats by 5 weeks after treatment (Figure 5B), suggesting that tumor growth was suppressed in rats treated with NVP-AUY922 and NVP-BEZ235.
In conclusion, both HSP90 protein overexpression and lower PTEN expression were poor prognostic factors, which implied that HSP90 inhibitors and PI3K/mTOR inhibitors are potential therapeutic agents in CCA. NVP-AUY922 and NVP-BEZ235 had potent synergic antitumor activity (in the low nanomolar range) against CCA cells. Marked tumor regression was demonstrated in the TAA-induced CCA rat animal model upon treatment with a combination of NVP-AUY922 and NVP-BEZ235. These preclinical findings provide a rationale to conduct clinical trials with NVP-AUY922 and NVP-BEZ235 in CCA patients.
METHODS
Clinicopathological features of 78 patients with mass-forming CCA (MF-CCA)
From the archives of the Chang Gung Memorial Hospital, 78 patients with MF-CCA who had undergone hepatectomy between 1989 and 2006 were selected based on the availability of sufficient quantities of tumor cells. Intrahepatic CCA was defined as a carcinoma that had arisen from the second-order or more distal branches of the intrahepatic ducts. CCAs were further classified into 3 types based on the macroscopic appearance of the tumors: MF-CCA, periductal-infiltrating CCA, and intraductal papillary CCA. Curative resection was defined as a negative resection margin observed during histopathological examination. Surgical mortality was defined as death within a month of surgery. Laboratory tests were conducted the day before the surgery. Serum carbohydrate antigen 19-9 (CA 19-9) and carcinoembryonic antigen (CEA) levels were measured by radioimmunoassay. Tumors were preoperatively evaluated by abdominal ultrasonography (US), endoscopic retrograde cholangiopancreatography, percutaneous transhepatic cholangiography, computed tomography (CT), magnetic resonance cholangiopancreatography (MRCP), or hepatic arteriography, as appropriate. Tumor stage was defined according to the pathological tumor node metastasis (pTNM) classification proposed by the American Joint Committee on Cancer (AJCC), 6th edition. Following either a positive section margin or local recurrence, adjuvant chemotherapy was systemically administered with a 5-flurouracil (5-FU)-based regimen. This clinical study was approved by the Institutional Review Board (IRB) of the Chang Gung Memorial Hospital (No. 99-2886B). All patients gave informed consent prior to inclusion in the immunostaining study.
Immunohistochemical staining of HSP90 and PTEN in 78 MF-CCA
Hematoxylin and eosin (H&E)-stained slides from each case were reviewed. Specimens from MF-CCA patients who had undergone hepatectomy were fixed in formalin and embedded in paraffin. A 4-μm section was stained for HSP90 and PTEN. Primary antibodies against HSP90 (#3098-100, 1:200 dilution; BioVision, Mountain View, CA) and PTEN (#ab31392, 1:100 dilution; Abcam, Cambridge, UK) were added to the slides, and the slides were incubated overnight at 4°C. The slides were then washed in TBST 3 times for 5 min each before visualization with the DAKO LSAB2 System, Peroxidase (no K0675; DAKO A/S, Glostrup, Denmark). Control slides were incubated with the secondary antibody only. After washing in TBST 3 times for 5 min each, the slides were mounted. The slides were analyzed blindly under microscopy, and the expression in the cholangiolar epithelium was classified as low or high according to the following: negative (< 1% cytoplasmic staining), 1+ (1%–20% cytoplasmic staining), 2+ (21%–50% cytoplasmic staining), or 3+ (> 50% membranous staining). Negative and 1+ immunostaining were arbitrarily classified as low expression, while all others were classified as high expression.
Follow-up study
The follow-up evaluation included a physical examination and blood chemistry tests at each visit. In addition, serum levels of CEA and CA 19-9 were measured, and the remnant liver was examined by US every 3 months. When a new lesion was detected by US or elevated levels of CEA/CA 19-9 were noted, abdominal CT or MRCP was performed for confirmation. Moreover, when patients complained of bone pain, bone scans were performed to detect metastasis. If any of the abovementioned procedures indicated recurrence, the patient was re-admitted for a more comprehensive assessment, including angiographic evaluation or magnetic resonance imaging (MRI). Treatment for recurrence included surgery, systemic chemotherapy, external beam radiotherapy, intraluminal radiotherapy, interventional radiological therapy, and conservative treatment.
Cell lines
Two intrahepatic CCA cell lines, HuCCT1 and CGCCA, were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan) and Chang Gung Memorial Hospital, respectively [46, 48]. HuCCT1 and CGCCA cells were routinely cultured in RPMI 1640 and Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY), respectively, supplemented with 10% heat-inactivated fetal bovine serum, 100 μg/mL streptomycin, and 100 μg/mL penicillin, in a humidified atmosphere containing 5% CO2 at 37°C.
Reagents
NVP-AUY922 and NVP-BEZ235 were provided by Novartis (Basel, Switzerland). Gemcitabine and oxaliplatin were purchased from TTY Biopharm (Taipei, Taiwan). For the in vitro experiments, 10 mM stock solutions of NVP-AUY922 and NVP-BEZ235 were prepared in 100% DMSO and stored at −20°C (NVP-AUY922) and 4°C (NVP-BEZ235). For administration, optimized NVP-AUY922 and NVP-BEZ235 salts with high solubility in aqueous solution were formulated in D5W. NVP-AUY922 was delivered by intraperitoneal (ip) injection in a volume of 6.25 mg/kg. NVP-BEZ235 was delivered orally (po) in a volume of 15 mg/kg. Gemcitabine (50 mg/kg) and oxaliplatin (2 mg/kg) were delivered by ip injection.
Cell viability assay
The viability of the cells was determined using the sulforhodamine B (SRB) (Sigma-Aldrich, St. Louis, USA) assay, it's based on measuring cellular protein content of live cells. The CGCCA and HuCCT1 cells were seeded with 2 × 103/well in 100 μl of culture medium into 96-well microplates, and allowed to adhere for 24 h. Next day, the cells were treated with various concentration of NVP-AUY922, NVP-BEZ235 or in combination for 72 h. After the incubation period, the medium were removed, and the cells were fixed with 10% (W/V) trichloroacetic acid (TCA) (Sigma-Aldrich, St. Louis, USA) for 30 min at 4°C. Then, cleared up TCA and washed the plates with water four times, allowed them to air-dry at room temperature. Next step, stained with SRB dissolved in 1.0% acetic acid for 30 min, after which, rinsed the plates repeatedly with 1.0% acetic acid to remove excess dye and air-dry. The protein-bound dye was dissolved in 10 mM Tris base solution, and determine optical density (OD) at 540 nm using a VersaMax microplate reader (Molecular Devices, Sunnyvale, USA).
Combination index (CI) determination
The CI values of NVP-AUY922 and NVP-BEZ235 were evaluated using a Biosoft CalcuSyn software program (Ferguson, MO) based on the method reported by Chou and Talalay [49]. Cells were treated with various concentrations of drugs either alone or in combination for 72 h. Data from cell viability assays were expressed as the fraction of antiproliferative activity (cell viability loss) induced by an individual drug, alone or in combination, and CI values were generated for the 2 drugs. CI < 1, CI = 1, and CI > 1 indicated synergism, an additive effect, or antagonism, respectively.
Measurement of reactive oxidative species (ROS)
ROS generation was quantified using an oxidant-sensitive fluorescent probe (CM-H2DCFDA; Invitrogen, Carlsbad, CA) as a substrate for measuring intracellular oxidant production [50]. Cells were seeded into 6-well plates (1 × 105 cells/well), incubated for 24 h, and then treated with NVP-AUY922 or NVP-BEZ235 for 18 or 24h. Next, these cells were incubated with CM-H2DCFDA (20 μM final concentration) for 30 min at 37°C, harvested, and washed twice with PBS; then, fluorescence was quantified by flow cytometry using a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ). ROS production was expressed as the mean fluorescence intensity. Experiments were performed in triplicate, and data are expressed as mean ± standard deviation.
Assay to detect apoptotic cell death
Apoptosis was measured using the TACS Annexin V-FITC apoptosis detection kit (Trevigen) according to the manufacturer's instructions. After a 24-h incubation, cells were treated with DMSO, NVP-AUY922, or NVP-BEZ235 for 48 h. Cells were collected and stained with Annexin V and propidium iodide (PI), and then analyzed using a FACSCalibur. The data were analyzed using CellQuest software (BD). Experiments were performed in triplicate, and data are expressed as mean ± standard deviation.
Measurement of mitochondrial membrane potential (ΔΨm)
Variation in ΔΨm was investigated using 5,5',6,6'-tetrachloro-1,1',3,3'- tetraethyl-benzimidazol-carbocyanine iodide (JC-1; BD Biosciences, San Diego, CA) according to the manufacturer's instructions [51]. Cells were seeded into 6-well plates at a density of 1 × 105 cells/well, incubated overnight, and then treated with DMSO or 0.5 uM NVP-AUY922 or NVP-BEZ235 or both. After treatment, the cells were collected and incubated with JC-1 for 15 min at 37°C. After washing twice with PBS, the cells were suspended in a total volume of 400 mL and analyzed using a FACSCalibur.
Measurement of glutathione content
Intracellular reduced glutathione (GSH) content was determined using the GSH-Glo™ Glutathione Assay kit (Promega, Madison, WI) [52]. The kit is a luminescence-based assay for the detection and quantification of GSH. Briefly, cells were seeded into 96-well white plates (SPL Life Sciences, Korea) at a concentration of 3,000 (HuCCT1) or 1,800 (CGCCA) cells/well in 100 μL of culture medium. At 24 h post-seeding, the cells were treated with 0.5 uM NVP-AUY922 or NVP-BEZ235 or both for 24 or 48 h, and then processed according to the manufacturer's instructions. The luminescence values were measured using the Infinite M1000 microplate reader (TECAN, Australia).
Western blotting
Whole cell lysates of CCA cell lines were obtained using Pierce RIPA buffer (Thermo Scientific, Rockford, IL). Protein samples were separated on 8%–12% gradient sodium dodecyl sulfate- polyacrylamide gels (SDS-PAGE) and transferred to Immobilon-P (Millipore, Bedford, MA) membranes. Antigen-antibody complexes were detected using the ECL blotting analysis system (Millipore). Primary antibodies against the following targets were used: AKT (9272; Cell Signaling, Danvers, MA), p-AKT (9271; Cell Signaling), cleaved poly (ADP-ribose) polymerase (c-PARP, 9541S; Cell Signaling), p-4E-BP1 (9459; Cell Signaling), p-p70S6K (9205; Cell Signaling), β-actin (Abcam ab6276; Abnova Corporation, Taipei), HSP70 (4872; Cell Signaling), Grp78 (BiP, 3183; Cell Signaling), Grp94 (2104; Cell Signaling), p-eIF2α (3597; Cell Signaling), Caspase 4 (4450; Cell Signaling), IRE1α (3294; Cell Signaling), phosphor-JNK (9255; Cell Signaling), and CCAAT/-enhancer-binding protein homologous protein (CHOP, GADD153, sc-575; Santa Cruz Biotechnology, Santa Cruz, CA).
Animal studies
All animal studies were approved by the experimental animal ethics committee of the Chang Gung Memorial Hospital, and were conducted in accordance with the US National Institute of Health (NIH) guidelines for the care and use of laboratory animals (Publication no. 85-23, revised 1996). Thirty adult male Sprague-Dawley (SD) rats (310 ± 14 g) were equally divided into 5 groups: the control (Group 1), BEZ235 (Group 2), AUY922 (Group 3), BEZ235/AUY922 (Group 4), and gemcitabine/oxaliplatin groups (Group 5). The rats were housed in an animal room under a 12-h light-dark cycle (light from 08:00 AM to 08:00 PM) at an ambient temperature of 22°C. Food and water were provided ad libitum. The rats were administered 300 mg thioacetamide (TAA)/L in drinking water daily for up to 25 weeks. Mice in the BEZ235, AUY922, and BEZ235/AUY922 groups were administered NVP-BEZ235 (15 mg/kg, po), NVP-AUY922 (6.25 mg/kg, ip), or a combination of these 2 agents, respectively, once daily 5 d per week over a 4-week period (specifically, the 21st week to the 24th week of the experimental period). The gemcitabine/oxaliplatin group received gemcitabine (50 mg/kg, ip) and oxaliplatin (2 mg/kg, ip) once every 2 weeks over a 4-week period. The control group rats received ip injections of buffer, according to the same schedule.
Positron emission tomography
To evaluate changes in glycolysis in live rats with liver tumors, we conducted 2-deoxy-2-[F-18] fluoro-d-glucose (FDG)-positron emission tomography (PET) studies at the molecular imaging center of Chang Gung Memorial Hospital. Thirty rats were treated with thioacetamide and subjected to serial PET scanning on weeks 20, 22, and 25 using the Inveon™ system (Siemens Medical Solutions USA Inc., Knoxville, TN). Equal numbers of animals were assigned to the control and treatment groups according to baseline PET findings. In other words, the control and treatment groups possessed similar PET-positive rates. The details regarding radioligand preparation, scanning protocols, and optimal scanning time determination have been described previously by our group [47]. Briefly, animals were fasted overnight prior to the scan. At 90 min post-18F-FDG injection (intravenous), 30-min static scans were obtained for all the animals. All imaging studies were performed using a temperature- (37°C) and anesthesia gas- (2% isoflurane in 100% oxygen) controlled imaging bed (Minerve System). PET images were reconstructed using the 2D ordered subset expectation-maximization method (4 iterations and 16 subsets) without attenuation and scatter corrections. All imaging data were processed using the PMOD image analysis workstation (PMOD Technologies Ltd., Zurich, Switzerland). The largest liver tumor was identified by careful investigation of 3 image sets for each rat. 18F-FDG uptake into the largest liver tumor and apparently normal liver tissue was quantified by calculating the standardized uptake value (SUV) according to the following formula:
These values were calculated according to the recommendations of the European Organization for Research and Treatment of Cancer [43]. The tumor regions of interest (ROIs) were determined using transverse images of the selected tumors and measuring the largest diameter. Normal liver ROIs were also determined using the same transverse images. The mean SUV (SUVmean) of the normal liver and tumor tissue was determined, and the tumor-to-liver radioactivity ratio was calculated for comparison.
Statistical analysis
All data are presented as mean ± standard deviation (SD). Differences between experimental animals and controls were calculated using the Mann-Whitney U test or the Kruskal-Wallis test. Overall survival was calculated using the Kaplan-Meier method. Eighteen clinicopathological variables were selected for difference analysis using the log-rank test (univariate). The Cox proportional hazards model was employed for multivariate regression analysis. SPSS statistical software for Windows was used for statistical analysis (SPSS version 13.0; Chicago, IL). P values ≤ 0.05 were considered statistically significant.
SUPPLEMENTARY FIGURES AND TABLES
Acknowledgments
Supported by the Taiwan Cancer Clinic Foundation and the Yen Tjing Ling Medical Foundation to Taipei Veterans General Hospital (V102B-036 to M. Chen); by the Chang Gung Memorial Hospital (CMRPG3B0362, CMRPG3B0532 and CMRPG2C0531 to C. Yeh and K. Chiang); by the grant from the National Science Council (NSC 102-2314-B-075-070 to M. Chen and NSC102-2627-B-010-001- to C.Y. Huang); by the grant from a grant Ministry of Education, Aim for the Top University Plan (103AC-T503 to C.Y. Huang)
REFERENCES
- 1.Ustundag Y, Bayraktar Y. Cholangiocarcinoma: a compact review of the literature. World J Gastroenterol. 2008;14:6458–6466. doi: 10.3748/wjg.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet. 2005;366:1303–1314. doi: 10.1016/S0140-6736(05)67530-7. [DOI] [PubMed] [Google Scholar]
- 3.Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33:1353–1357. doi: 10.1053/jhep.2001.25087. [DOI] [PubMed] [Google Scholar]
- 4.Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24:115–125. doi: 10.1055/s-2004-828889. [DOI] [PubMed] [Google Scholar]
- 5.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 6.Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol. 2010;28:3531–3540. doi: 10.1200/JCO.2009.27.4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu AX, Hezel AF. Development of molecularly targeted therapies in biliary tract cancers: reassessing the challenges and opportunities. Hepatology. 2011;53:695–704. doi: 10.1002/hep.24145. [DOI] [PubMed] [Google Scholar]
- 8.Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 9.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blagosklonny MV. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002;16:455–62. doi: 10.1038/sj.leu.2402415. [DOI] [PubMed] [Google Scholar]
- 11.Chen MH, Lin KJ, Yang WL, Kao YW, Chen TW, Chao SC, Chang PM, Liu CY, Tzeng CH, Chao Y, Chen MH, Yeh CN, Huang CY. Gene expression-based chemical genomics identifies heat-shock protein 90 inhibitors as potential therapeutic drugs in cholangiocarcinoma. Cancer. 2013;119:293–303. doi: 10.1002/cncr.27743. [DOI] [PubMed] [Google Scholar]
- 12.Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, Quadt C, Buckler A, Cozens R, Drysdale MJ, Garcia-Echeverria C, Chène P. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10:R33. doi: 10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaspar N, Sharp SY, Eccles SA, Gowan S, Popov S, Jones C, Pearson A, Vassal G, Workman P. Mechanistic evaluation of the novel HSP90 inhibitor NVP-AUY922 in adult and pediatric glioblastoma. Mol Cancer Ther. 2010;9:1219–1233. doi: 10.1158/1535-7163.MCT-09-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaiser M, Lamottke B, Mieth M, Jensen MR, Quadt C, Garcia-Echeverria C, Atadja P, Heider U, von Metzler I, Türkmen S, Sezer O. Synergistic action of the novel HSP90 inhibitor NVP-AUY922 with histone deacetylase inhibitors, melphalan, or doxorubicin in multiple myeloma. Eur J Haematol. 2010;84:337–344. doi: 10.1111/j.1600-0609.2009.01403.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee KH, Lee JH, Han SW, Im SA, Kim TY, Oh DY, Bang YJ. Antitumor activity of NVP-AUY922, a novel heat shock protein 90 inhibitor, in human gastric cancer cells is mediated through proteasomal degradation of client proteins. Cancer Sci. 2011;102:1388–1395. doi: 10.1111/j.1349-7006.2011.01944.x. [DOI] [PubMed] [Google Scholar]
- 16.Pacey S, Gore M, Chao D, Banerji U, Larkin J, Sarker S, Owen K, Asad Y, Raynaud F, Walton M, Judson I, Workman P, Eisen T. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs. 2012;30:341–349. doi: 10.1007/s10637-010-9493-4. [DOI] [PubMed] [Google Scholar]
- 17.Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N, Chapman PB. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14:8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demidenko ZN, Vivo C, Halicka HD, Li CJ, Bhalla K, Broude EV, Blagosklonny MV. Pharmacological induction of Hsp70 protects apoptosis-prone cells from doxorubicin: comparison with caspase-inhibitor- and cycle-arrest-mediated cytoprotection. Cell Death Differ. 2006;13:1434–41. doi: 10.1038/sj.cdd.4401812. [DOI] [PubMed] [Google Scholar]
- 20.Walsby E, Pearce L, Burnett AK, Fegan C, Pepper C. The Hsp90 inhibitor NVP-AUY922-AG inhibits NF-κB signaling, overcomes microenvironmental cytoprotection and is highly synergistic with fludarabine in primary CLL cells. Oncotarget. 2012 May;3(5):525–3. doi: 10.18632/oncotarget.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway—beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riener MO, Bawohl M, Clavien PA, Jochum W. Rare PIK3CA hotspot mutations in carcinomas of the biliary tract. Genes Chromosomes Cancer. 2008;47:363–367. doi: 10.1002/gcc.20540. [DOI] [PubMed] [Google Scholar]
- 23.Yothaisong S, Dokduang H, Techasen A, Namwat N, Yongvanit P, Bhudhisawasdi V, Puapairoj A, Riggins GJ, Loilome W. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumour Biol. 2013;34:3637–48. doi: 10.1007/s13277-013-0945-2. [DOI] [PubMed] [Google Scholar]
- 24.Venkannagari S, Fiskus W, Peth K, Atadja P, Hidalgo M, Maitra A, Bhalla KN. Superior efficacy of co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and pan-histone deacetylase inhibitor against human pancreatic cancer. Oncotarget. 2012 Nov;3(11):1416–27. doi: 10.18632/oncotarget.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 26.Taiyab A, Sreedhar AS, Rao Ch M. Hsp90 inhibitors, GA and 17AAG, lead to ER stress-induced apoptosis in rat histiocytoma. Biochem Pharmacol. 2009;78:142–152. doi: 10.1016/j.bcp.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 27.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, Maertens O, Jeong SM, Bronson RT, Lebleu V, Kalluri R, Normant E, Haigis MC, Manning BD, Wong KK, Macleod KF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–413. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 29.Schonthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Front Biosci (Schol Ed) 2012;4:412–431. doi: 10.2741/s276. [DOI] [PubMed] [Google Scholar]
- 30.Powers MV, Valenti M, Miranda S, Maloney A, Eccles SA, Thomas G, Clarke PA, Workman P. Mode of cell death induced by the HSP90 inhibitor 17-AAG (tanespimycin) is dependent on the expression of pro-apoptotic BAX. Oncotarget. 2013 Nov;4(11):1963–75. doi: 10.18632/oncotarget.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieto-Miguel T1, Fonteriz RI, Vay L, Gajate C, López-Hernández S, Mollinedo F. Endoplasmic reticulum stress in the proapoptotic action of edelfosine in solid tumor cells. Cancer Res. 2007;67:10368–78. doi: 10.1158/0008-5472.CAN-07-0278. [DOI] [PubMed] [Google Scholar]
- 32.Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel. 2006;9:483–495. [PubMed] [Google Scholar]
- 33.Wang J, Cui S, Zhang X, Wu Y, Tang H. High expression of heat shock protein 90 is associated with tumor aggressiveness and poor prognosis in patients with advanced gastric cancer. PLoS One. 2013;8:e62876. doi: 10.1371/journal.pone.0062876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conroy SE, Sasieni PD, Fentiman I, Latchman DS. Autoantibodies to the 90kDa heat shock protein and poor survival in breast cancer patients. Eur J Cancer. 1998;34:942–943. [PubMed] [Google Scholar]
- 35.Zhang J, Zheng Z, Zhao Y, Zhang T, Gu X, Yang W. The heat shock protein 90 inhibitor 17-AAG suppresses growth and induces apoptosis in human cholangiocarcinoma cells. Clin Exp Med. 2013;13:323–328. doi: 10.1007/s10238-012-0208-3. [DOI] [PubMed] [Google Scholar]
- 36.Thomas MB. Biological characteristics of cancers in the gallbladder and biliary tract and targeted therapy. Crit Rev Oncol Hematol. 2007;61:44–51. doi: 10.1016/j.critrevonc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Marsh V, Davies EJ, Williams GT, Clarke AR. PTEN loss and KRAS activation cooperate in murine biliary tract malignancies. J Pathol. 2013;230:165–173. doi: 10.1002/path.4189. [DOI] [PubMed] [Google Scholar]
- 38.Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S, Stiles B, Wang RH, Ohara N, Yoshino T, LeRoith D, Torbenson MS, Gores GJ, Wu H, Gao B, Deng CX. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116:1843–1852. doi: 10.1172/JCI27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demidenko ZN, An WG, Lee JT, Romanova LY, McCubrey JA, Blagosklonny MV. Kinase-addiction and bi-phasic sensitivity-resistance of Bcr-Abl- and Raf-1-expressing cells to imatinib and geldanamycin. Cancer Biol Ther. 2005;4:484–90. doi: 10.4161/cbt.4.4.1702. [DOI] [PubMed] [Google Scholar]
- 40.Yap TA, Omlin A, de Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013;31:1592–1605. doi: 10.1200/JCO.2011.37.6418. [DOI] [PubMed] [Google Scholar]
- 41.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA, 3rd, Falchook G, Algazi A, Lewis K, Long GV, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grant S. Enhancing proteotoxic stress as an anticancer strategy. Oncotarget. 2011 Apr;2(4):284–6. doi: 10.18632/oncotarget.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget. 2011 Mar;2(3):209–2. doi: 10.18632/oncotarget.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 46.Yeh CN, Maitra A, Lee KF, Jan YY, Chen MF. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: an animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis. 2004;25:631–636. doi: 10.1093/carcin/bgh037. [DOI] [PubMed] [Google Scholar]
- 47.Yeh CN, Lin KJ, Hsiao IT, Yen TC, Chen TW, Jan YY, Chung YH, Lin CF, Chen MF. Animal PET for thioacetamide-induced rat cholangiocarcinoma: a novel and reliable platform. Mol Imaging Biol. 2008;10:209–216. doi: 10.1007/s11307-008-0141-8. [DOI] [PubMed] [Google Scholar]
- 48.Yeh CN, Lin KJ, Chen TW, Wu RC, Tsao LC, Chen YT, Weng WH, Chen MF. Characterization of a novel rat cholangiocarcinoma cell culture model-CGCCA. World J Gastroenterol. 2011;17:2924–2932. doi: 10.3748/wjg.v17.i24.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 50.Robinson JP, Bruner LH, Bassoe CF, Hudson JL, Ward PA, Phan SH. Measurement of intracellular fluorescence of human monocytes relative to oxidative metabolism. J Leukoc Biol. 1988;43:304–310. doi: 10.1002/jlb.43.4.304. [DOI] [PubMed] [Google Scholar]
- 51.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD Jr, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Troyano A, Fernandez C, Sancho P, de Blas E, Aller P. Effect of glutathione depletion on antitumor drug toxicity (apoptosis and necrosis) in U-937 human promonocytic cells. The role of intracellular oxidation. J Biol Chem. 2001;276:47107–47115. doi: 10.1074/jbc.M104516200. [DOI] [PubMed] [Google Scholar]
- 53.Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA, Pruim J, Price P. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35:1773–1782. doi: 10.1016/s0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.