

Abstract
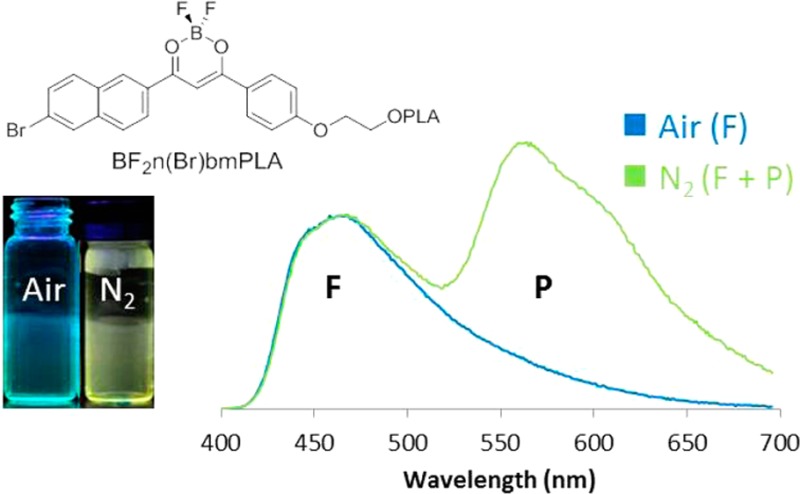
Luminescent materials are important for imaging and sensing. Aromatic difluoroboron β-diketonate complexes (BF2bdks) are classic fluorescent molecules that have been explored as photochemical reagents, two-photon dyes, and oxygen sensors. A series of BF2bdks with naphthyl and phenyl groups was synthesized, and photophysical properties were investigated in both methylene chloride and poly(lactic acid) (PLA). Polymer molecular weight and dye attachment site along with bromide heavy atom placement were varied to tune optical properties of dye–PLA materials. Systems without heavy atoms have long phosphorescence lifetimes, which is useful for lifetime-based oxygen sensing. Bromine substitution on the naphthyl ring resulted in intense, clearly distinguishable fluorescence and phosphorescence peaks important for ratiometric oxygen sensing and imaging.
Introduction
Luminescent materials are important for imaging and sensing. Both lifetime and intensity based methods have been exploited for oxygen sensing.1 A benefit of lifetime methods is that they are independent of dye concentration; however, they often require specialized and more costly instrumentation.2,3 Intensity-based methods are compatible with common fluorescence detection techniques, even standard digital cameras,4 but they often require calibration of local dye concentration versus standards. This criterion has been met by ratiometric methods that rely on multicomponent systems combining an oxygen-sensitive phosphor and an oxygen-insensitive standard in an inert, readily processable matrix.5−9 When fashioned as nanoparticles2,6−13 or films,4,14 these materials allow for oxygen imaging with enhanced spatial and temporal resolution.
Previously we reported a class of single-component dual-emissive oxygen sensing polymeric materials composed of boron difluoride β-diketonates (BF2bdk) linked to poly(lactic acid) (PLA), that combine sensor (phosphorescence, P), standard (fluorescence, F), and polymer matrix in one. Specifically, with BF2dbmPLA (dbm = dibenzoylmethane)10,11,15−19 and its halogenated, heavy atom congener, BF2dbm(I)PLA,14 we demonstrated that emission wavelengths, relative F/P intensities, and oxygen sensitivities are tunable with heavy atom substitution and polymer molecular weight11,15 and that these materials can be exploited for cellular,14,15 tissue, and in vivo imaging studies (e.g., tumor hypoxia14,18).
In a recent model study, blending BF2bdk dyes with PLA, it was noted that emission is comprised of π–π* and intramolecular charge transfer (ICT) processes. In symmetrical phenyl–phenyl (Ph–Ph) dyes like dbm, π–π* processes dominate, whereas in unsymmetrical Ph–Np or Ph–An systems (Np = naphthyl, An = anthracene), ICT processes dominate.20 Computational studies showed that electron density is localized on the larger arene donor (Np or An vs Ph) in the HOMO but is distributed across the molecule in the LUMO. Luminescence data support these claims, with additional ICT bands in absorption and emission spectra (i.e., for Ph–An systems) and longer radiative lifetimes. Building upon this observation that the larger arene donor dominates the ICT emission properties in unsymmetrical dye systems, a set of Ph–Np dyes with bromide heavy atom substitution were prepared and blended with PLA to explore the effects of heavy atom position on luminescence properties.21 It was demonstrated that lifetimes, intensities, and oxygen sensitivity can be systematically modulated by placing the heavy atom on the more electron-rich naphthyl versus phenyl ring, with naphthyl ring substitution showing the strongest heavy atom effect (i.e., more intense, shorter lived phosphorescence). More recently, this concept was generalized with Lewis acid–aryl ketone systems.22
Here we contribute new findings to the growing boron luminescent materials field1,23−35 by extending investigations to a series of naphthyl-phenyl dye–polymer conjugates. Covalent attachment of the dye to the polymer is preferred over blends for practical uses, given decreased dye leaching and dye/polymer phase separation at high loadings. Diketonate ligands (1–4), boron initiators (5–8), and associated polymers (9–12) were designed with the following fundamental and applied considerations in mind. First, extended conjugation afforded by the naphthyl ring in Np–Ph systems (vs Ph–Ph in dbm) can lead to more red-shifted dyes in new color regimes for multiplexing applications. Additionally, understanding ways that conjugation length correlates with emission wavelengths for BF2bdk–PLA materials is important for generating red-shifted materials for greater tissue penetration of light in biological contexts. Previous studies show that overall conjugation length is not the only determinative factor for emission wavelength, given ICT effects.20 Halide substitution also results in a slight bathochromic shift in emission.21

Second, substituents and their placement influence electron density at arene rings and thus material optical properties. Though modulation of triplet emission was achieved by heavy atom placement in Np–Ph dye/polymer blends, here we test whether this phenomenon extends to dye–polymer conjugates. In the present series, polymers are attached to dyes via electron-donating alkoxy linkages that can perturb the donor strength of the two arene rings. Thus, polymer and bromide substituent attachments at either phenyl or naphthyl rings are compared.
Third, for ratiometric oxygen sensing, strong phosphorescence intensity is desirable. Thus, bromide heavy atom substitution is compared to non-brominated systems, and phosphorescence is further modulated via molecular weight tuning, as previously reported.14 For example, low molecular weight polymers (i.e., higher dye loading) result in increased intersystem crossing from the singlet to the triplet state, increased relative phosphorescence intensity, and shorter lifetimes. Furthermore, for BF2dbm(I)PLA, nearly full range F/P intensity tuning was possible to generate materials for different kinds of sensing through molecular weight tuning alone. For instance, materials with strong phosphorescence versus fluorescence serve as turn on sensors, whereas systems with comparable, readily detectable F and P intensities are exploited for ratiometric tumor hypoxia imaging and other biomedical uses. However, the sensitivity of F/P to polymer molecular weight is not known for different dyes, and this is an important parameter for sensor materials design. This study explores that parameter for a series of Np–Ph dyes.
Fourth, discrete, clearly distinguishable fluorescence and phosphorescence peaks with measurable F/P ratios are important for ratiometric sensing. Here we probe ways that dye molecular structure correlates with singlet and triplet energies. Emission wavelengths and can be further modulated by molecular weight, with greater peak separation achievable with higher molecular weights (i.e., low dye loading). Importantly, systems with poor fluorescence and phosphorescence peak separation are nonetheless useful for gated emission and lifetime sensing modalities, and non-halogenated systems with longer lifetimes result in more sensitive materials for oxygen sensing. Materials with small singlet–triplet energy gaps are also of interest for improving efficiency in OLEDs.36,37
Experimental Section
Materials
1-[4-(2-Hydroxyethoxy)phenyl]-3-(2-naphthyl)propane-1,3-dione, nbmOH (1), and BF2nbmOH (5) were prepared as previously described.16 3,6-Dimethyl-1,4-dioxane-2,5-dione (d,l-lactide, Aldrich) was recrystallized twice from ethyl acetate and stored under nitrogen. Tin(II) 2-ethylhexanoate (Sn(oct)2, Spectrum), boron trifluoride diethyl etherate (Aldrich, purified, redistilled), and all other reagents and solvents were used as received without further purification. Solvents CH2Cl2 and THF were dried and purified by over 3 Å molecular sieves activated at 300 °C.38 All other chemicals were reagent grade from Sigma-Aldrich and were used without further purification.
Methods
1H NMR (300 MHz) spectra were recorded on a Unity Inova 300/51 instrument in CDCl3. 1H NMR were referenced to the signals for the residual protiochloroform at 7.26 ppm, protioDMSO at 2.50 ppm, and protioacetone at 2.09 ppm. Coupling constants are given in hertz. Polymer molecular weights were determined by gel permeation chromatography (GPC) (THF, 25 °C, 0.8 mL/min) using multiangle laser light scattering (MALS) (λ = 658 nm, 25 °C) and refractive index (RI) (λ = 658 nm, 25 °C) detection. Polymer Laboratories 5 μm mixed-C columns (guard column plus two columns) along with Wyatt Technology (Optilab T-rEX interferometric refractometer, miniDAWN TREOS multiangle static light scattering (MALS) detector, ASTRA 6.0 software) and Agilent Technologies instrumentation (series 1260 HPLC with diode array (DAD) detector, ChemStation) were used in GPC analysis. UV–vis spectra were recorded on a Hewlett-Packard 8452A diode-array spectrophotometer.
Steady-state fluorescence emission spectra were recorded on a Horiba Fluorolog-3 Model FL3-22 spectrofluorometer (double-grating excitation and double-grating emission monochromator). A 2 ms delay was used when recording the delayed emission spectra. Time-correlated single-photon counting (TCSPC) fluorescence lifetime measurements were performed with a NanoLED-370 (λex = 369 nm) excitation source and a DataStation Hub as the SPC controller. Phosphorescence lifetimes were measured with a 1 ms multichannel scalar (MCS) excited with a flash xenon lamp (λex = 369 nm; duration <1 ms). Lifetime data were analyzed with DataStation v2.4 software from Horiba Jobin Yvon. Fluorescence quantum yields (ΦF) of initiator and polymer samples in CH2Cl2 were calculated versus anthracene as a standard as previously described using the following values: ΦF(anthracene) = 0.27,39,40nD(EtOH) = 1.360, nD(CH2Cl2) = 1.424.41 Optically dilute CH2Cl2 solutions of the dyes were prepared in 1 cm path length quartz cuvettes with absorbances <0.1 au. Thin films were prepared on the inner wall of vials by dissolving polymers in CH2Cl2 (∼1 mg/mL) and then evaporating the solvent by slowly rotating the vial under a stream of nitrogen. The films were then dried in vacuo for ∼15 min before measurements were taken.
All compounds were modeled using the Gaussian 0942 suite of programs using density functional theory. B3LYP/6-31+G(d) was utilized for ground state geometry optimization with a Tomasi polarized continuum for dichloromethane solvent. The vibrational frequencies for the optimized geometries were all positive, assuring that the geometries are at least a local minimum. Single point energy calculations were used to generate the molecular orbital diagrams utilizing B3LYP/6-31G(d). Time-dependent density functional theory, TD-B3LYP/6-311+G(d), was employed for estimates of the absorption spectra, at the respective optimized geometries.43,44 The first three excited states were computed for each compound. Molecular orbitals were depicted by GaussView 5 software.45
β-Diketonate Ligand Synthesis
6-Hydroxy-2-acetonapthalene
The naphthyl precursor was prepared as previously described46 with the following exceptions. 6-Methoxy-2-acetonapthalene (1.5 g, 7.5 mmol) dissolved in 12 M HCl (500 mL) was heated at 90 °C for 2 h. The hot reaction mixture was filtered through a sintered glass frit (M) to remove a greenish-black precipitate. The filtrate was allowed to cool to room temperature, and then the flask was further cooled in an ice water bath. The resulting white solid was collected by filtration and was washed with copious quantities of distilled water until the pH of the wash fraction was neutral (pH ∼ 6.5). (Note: in certain preparations, when the sample was greenish or brownish in color, further purification by recrystallization from hexane/EtOAc was performed.) After drying in vacuo, a white powder was obtained: 1.27 g (91%). 1H NMR (300 MHz, CDCl3, ppm): δ 8.41 (s, 1H, 1-ArH), 8.00 (d, 1H, J = 8.7 Hz, 8-ArH), 7.89 (d, 1H, J = 9.0, 3-ArH), 7.72 (d, 1H, J = 9.0, 4-ArH), 7.20–7.16 (bm, 2H, 5, 7-ArH), 5.42 (s, 1H, OH), 2.71 (s, 3H, CH3).
6-(2-Hydroxy)ethoxy-2-acetonapthalene
6-Hydroxy-2-acetonapthalene (515 mg, 2.8 mmol) was dissolved in DMF (100 mL) in the presence of K2CO3 (1.22 g, 8.8 mmol) and KI (50 mg, 0.3 mmol) and was refluxed at 100 °C overnight (16 h). DMF was removed via H2O/CH2Cl2 extraction (1 L of H2O, 100 mL CH2Cl2). The organic layer was washed with H2O (2 × 20 mL) and brine (2 × 20 mL) and then was dried over Na2SO4, filtered, and concentrated via rotary evaporation. The crude product was purified by column chromatography (3:1 hexanes/EtOAc) and dried in vacuo to yield a white powder: 460 mg (72%). 1H NMR (300 MHz, CDCl3, ppm): δ 8.40 (s, 1H, 1-ArH), 8.02 (d, 1H, J = 9, 8-ArH), 7.88 (d, 1H, J = 9.0, 3-ArH), 7.76 (d, 1H, J = 9, 4-ArH), 7.23 (d, 1H, J = 9, 7-ArH), 7.18 (s, 1H, 5-ArH), 4.23 (t, J = 4.5, 2H, −ArOCH2CH2OH), 4.05 (t, J = 4.5, 2H, −ArOCH2CH2OH), 2.06 (s, 1H, −OH).
2-(6-(2-((Tetrahydro-2H-pyran-2-yl)oxy)ethoxy)naphthalen-2-ylethanone
The ligand precursor was prepared as previously described using 6-(2-hydroxy)ethoxy-2-acetonapthalene instead of 1-[4-(2-hydroxyethoxy)phenyl]ethanone. The crude product was purified by silica column chromatography (6:1 hexanes/EtOAc) to give a tan solid: 1.12 g (68%). 1H NMR (300 MHz, CDCl3, ppm): δ 8.40 (s, 1H, 1-ArH), 8.00 (d, 1H, J = 9, 8-ArH), 7.86 (d, 1H, J = 9.0, 3-ArH), 7.57 (d, 1H, J = 9.0, 4-ArH), 7.26 (d, 1H, J = 9.0, 7-ArH), 7.19 (s, 1H, 5-ArH), 4.74 (t, 1H, J = 3.5,–O–CH* (CH2−)O−), 4.31 (t, 2H, J = 4.5, −ArOCH2CH2OR), 3.92 (t, 2H, J = 4.2, −ArOCH2CH2OR), 3.55 (t, 2H, J = 6, OCH2–C3H6–CH (R)O−), 2.70 (s, 3H, −CH3), 1.80–1.65 (bm, 6H, OCH2–C3H6–CH (R)O−). HRMS (ESI, TOF) m/z calcd for C19H23O4 315.1596 [M]+; found 315.1596.
1-[4-(2-Hydroxyethoxy)phenyl]-3-(6-bromo-2-naphthyl)propane-1,3-dione (n(Br)bmOH) (2)
The BrNp-Ph ligand was prepared as described for 5. A cream colored powder was obtained: 520 mg (59%). 1H NMR (300 MHz, CDCl3): δ 17.03 (s, 1H, −OH), 8.49 (s, 1H, 1″-ArH) 8.10–7.90 (m, 4H, 2′,6′-ArH, 3″,5″-ArH,), 7.88–7.71 (m, 2H, 7″,8″-ArH), 7.63 (d, J = 9, 1H, 3″-ArH), 7.03 (d, J = 9, 2H, 3′, 5′-ArH), 6.93 (s, 1H, COCHCO), 4.19 (t, J = 4.2, −ArOCH2CH2OH), 4.03 (m, 2H, −ArOCH2CH2OH), 3.48 (s, 1H, CH2CH2OH). HRMS (ESI, TOF) m/z calcd for C21H18O4Br 413.0388 [M + H]+; found 413.0401.
1-[6-(2-Hydroxyethoxy)-2-naphthyl]-3-phenylpropane-1,3-dione (bnmOH) (3)
The Ph-Np ligand was prepared as described for 5. A white powder was obtained: 154 mg (48%). 1H NMR (300 MHz, CDCl3): δ 17.02 (s, 1H, −OH), 8.49 (s, 1H, 1″-ArH), 8.06–7.99 (bm, 3H, 8″-ArH, 2′,6′-ArH) 7.90 (d, J = 9, 1H, 3″-ArH) 7.81 (d, J = 9, 1H, 4″-ArH), 7.57–7.51 (bm, 3H, 3′,4′,5′-ArH), 7.25 (d, J = 9, 1H, 7″-ArH), 7.20 (s, 1H, 5″-ArH) 6.98 (s, 1H, −COCHCO−), 4.25 (t, J = 6, 2H, ArOCH2CH2OH), 4.06 (t, J = 6, 2H, ArOCH2CH2OH), 2.2 (s, 1H, ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd for C21H19O4 335.1283 [M]+; found 335.1289.
1-[6-(2-Hydroxyethoxy)-2-naphthyl]-3-(4-bromophenyl)propane-1,3-dione (b(Br)nmOH) (4)
The BrPh-Np ligand was prepared as described for 5. A tan powder was obtained: 110 mg (14%). 1H NMR (300 MHz, CDCl3): δ 16.96 (s, 1H, −OH), 8.48 (s, 1H, 1″-ArH), 8.00 (d, J = 8.7, 1H, 8″-ArH), 7.94–7.90 (m, 3H, 2′, 6′-ArH, 3″-ArH), 7.81 (d, J = 8.7, 1H, 4″-ArH), 7.64 (d, J = 8.7, 2H, 3′, 5′-ArH), 7.26 (d, J = 8.7,1H, 7″-ArH), 7.19 (s, 1H, 5″-ArH), 6.94 (s, 1H, COCHCO), 4.25 (t, J = 4.2, 2H, −ArOCH2CH2OH), 4.09–4.04 (m, 2H, −ArOCH2CH2OH), 2.05 (s, 1H, CH2CH2OH). HRMS (ESI, TOF) m/z calcd for C21H18O4Br 413.0388 [M + H]+; found 413.0376.
Boron Initiator Complexes
A representative preparation for difluoroboron initiators is provided for complex 6 below.47
BF2nbmOH (5)16
The Np-Ph complex was prepared as described in the literature. A yellow powder was obtained: 296 mg (84%). 1H NMR (300 MHz, CDCl3): δ 8.76 (s, 1H, 2″-ArH), 8.21 (d, 2H, 2′,6′-ArH), 7.98 (m, 5H, 4″,7″,9″,10″-ArH, COCHCO), 7.68 (m, 2H, 5″,6″-ArH), 7.08 (d, 2H, 3′,5′-ArH), 4.23 (t, J = 4.2, 2H, −ArOCH2CH2OH), 4.05 (t, J = 4.2, 2H, −ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd C21H16BO4F2Na 405.1099 [M + Na]+; found 405.1094.
BF2n(Br)bmOH (6)
The ligand nbmOH, 2 (250.0 mg, 0.605 mmol), was added to a flame-dried two-neck round-bottom flask under nitrogen and was dissolved in THF/CH2Cl2 (20/20 mL) to give a pale yellow solution. Boron trifluoride diethyl etherate (115 μL, 0.907 mmol) was added via syringe, and the solution turned bright yellow. The reaction was stirred at room temperature for 48 h. Solvents were removed via rotary evaporation, resulting in a yellow solid. The crude material was purified by recrystallization in 1:1 EtOAc/acetone to yield a yellow-orange powder: 143 mg (53%). The BrNp-Ph complex was prepared as described for 11. A yellow powder was obtained: 21 mg (37%). 1H NMR (300 MHz, DMSO): δ 9.07 (s, 1H, 1″-ArH), 8.43–8.38 (bm, 4H, 3″,8″-ArH, 2′,6′-ArH), 8.18–8.11 (bm, 2H, 5″,7″-ArH), 7.98 (s, 1H, COCHCO), 7.81 (d, J = 9, 1H, 4″-ArH), 7.23 (d, J = 9, 2H, 3′,5′-ArH), 4.98 (t, J = 5.4, 1H, −OH), 4.19 (t, J = 4.8, 2H, −ArOCH2CH2OH), 3.77 (t, J = 4.5, 2H, −ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd C21H16BO4F2BrNa 483.0191 [M + Na]+; found 483.0180.
BF2bnmOH (7)
The Ph-Np complex was prepared as described for 6. A yellow powder was obtained: 154 mg (48%). 1H NMR (300 MHz, DMSO): δ 9.07 (s, 1H, 1″-ArH), 8.39 (d, J = 9, 2H, 2′,6′-ArH), 8.31 (d, J = 9, 1H, 8″-ArH), 8.13 (d, J = 9, 1H, 3″-ArH), 8.03–7.98 (m, 2H, 4′-ArH, COCHCO), 7.81 (d, J = 9, 1H, 7″-ArH), 7.68 (t, J = 6, 2H, 3′,5′-ArH), 7.50 (s, 1H, 5″-ArH), 7.34 (d, J = 9, 1H, 4″-ArH), 5.00 (t, J = 6, 1H, OH), 4.18 (t, J = 6, 2H, −ArOCH2CH2OH), 3.80 (t, J = 6, 2H, −ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd C21H18BO4F2 383.1266 [M]+; found 383.1252.
BF2b(Br)nmOH (8)
The BrPh-Np complex was prepared as described for 6. A yellow powder was obtained: 41 mg (49%). 1H NMR (300 MHz, DMSO): δ 9.07 (s, 1H, 1″-ArH), 8.32- 8.28 (m, 3H, 2′,6′-ArH, 8″-ArH), 8.13 (d, J = 9.0, 1H, 3″-ArH), 8.03–7.99 (m, 2H, 7″-ArH, COCHCO), 7.90 (d, J = 9, 2H, 3′,5′-ArH), 7.50 (s, 1H, 5″-ArH), 7.32 (d, 1H, 4″-ArH), 5.0 (t, J = 6, 1H, −OH), 4.18 (t, J = 6, 2H, −ArOCH2CH2OH), 3.80 (t, J = 6, 2H, −ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd C21H16BO4F2BrNa 483.0191 [M + Na]+; found 483.0193.
Boron Polymer Synthesis
Preparative scale reactions were conducted as follows. The boron initiator and d,l-lactide were placed in a Kontes flask and sealed under N2. The bulb of the flask was entirely submerged in an oil bath at 130 °C. After the d,l-lactide melted, Sn(Oct)2 in hexanes was added, and the reaction was heated for the designated time (∼1–3 h). (See Table S1 for reagent loadings and reaction times for specific samples.) Crude polymer was purified by precipitation from CH2Cl2/cold MeOH (−20 °C). The polymer was collected by centrifugation, the filtrate was decanted, and the rubbery solid was redissolved in CH2Cl2 and reprecipitated in cold MeOH (−20 °C). The resulting solid was reprecipitated from CH2Cl2/hexanes, collected by centrifugation, the filtrate was decanted, and the residue was dried in vacuo to give the polymers as foams. The polymer molecular weight was determined by 1H NMR spectroscopy and GPC. Number-average molecular weights (Mn), polydispersity indices (PDIs), and yields for samples of different molecular weights are collected in Table S1. Representative 1H NMR spectral data are provided below for each dye initiator.
BF2nbmPLA16
Polymer 9a was obtained as a yellow crystalline solid: 81 mg (45%). Mn (GPC/RI) = 5400 Da, PDI = 1.05; Mn (NMR) = 3800 Da. 1H NMR (300 MHz, CDCl3): δ 8.76 (s, 1H, 1″-ArH), 8.20 (d, J = 9, 2H, 2′,6′-ArH), 8.10–7.90 (bm, 5H, 3″,4″,5″,8″-ArH, −COCHCO), 7.60–7.55 (bm, 2H, 6″,7″-ArH), 7.05 (d, J = 9, 2H, 3′,5′-ArH), 5.30–5.16 (bm, 48H, PLA −CH–CH3), 4.55 (m, 2H, −ArOCH2CH2OH), 4.35 (m, 2H, −ArOCH2CH2OH), 1.57 (bs, 146 H, PLA −CHCH3).
BF2n(Br)bmPLA (10)
Polymer 10a was obtained as a yellow crystalline solid: 151 mg (44%). Mn (GPC/RI) = 6800 Da, PDI = 1.03; Mn (NMR) = 6900 Da. 1H NMR (300 MHz, CDCl3): δ 8.72 (s, 1H, 1″-ArH), 8.20 (d, J = 9, 2H, 2′,6′-ArH), 8.10–8.08 (m, 2H, 3″,8″-ArH), 7.89–7.87 (m, 2H, 5″,7″-ArH), 7.69 (d, J = 9, 1H, 4″-ArH), 7.23 (s, 1H, −COCHCO−), 7.05 (d, J = 9, 2H, 3′,5′-ArH), 5.31–5.11 (bm, 89H, PLA −CHCH3), 4.55 (m, 2H, −ArOCH2CH2OH), 4.35 (m, 2H, −ArOCH2CH2OH), 1.57 (bs, 205H, PLA −CHCH3).
BF2bnmPLA (11)
Polymer 11a was obtained as a yellow-orange foam: 71 mg (64%). Mn (GPC/RI) = 5200 Da, PDI = 1.04; Mn (NMR) = 3300 Da. 1H NMR (300 MHz, CDCl3): δ 8.73 (s, 1H, 1″-ArH), 8.20 (d, J = 9, 2H 2′,6′-ArH), 8.10 (d, J = 9, 1H, 8″-ArH), 7.95 (d, J = 9, 1H, 3″-ArH), 7.85 (d, J = 9, 1H, 7″-ArH), 7.71 (t, J = 6, 1H, 4′-ArH), 7.58 (t, J = 6, 2H, 3′,5′-ArH), 7.31–7.25 (m, 2H, −COCHCO-, 4″-ArH), 7.19 (s, 1H, 5″-ArH), 5.30–5.16 (bm, 45 H, PLA −CHCH3), 4.55 (m, 2H, −ArOCH2CH2OH), 4.35 (m, 2H, −ArOCH2CH2OH), 1.57 (bs, 144 H, PLA −CHCH3).
BF2b(Br)nmPLA (12)
Polymer 12a was obtained as a bright yellow-green crystalline foam: 101 mg (40%). Mn (GPC/RI) = 5600 Da, PDI = 1.07; Mn (NMR) = 3100 Da. 1H NMR (300 MHz, CDCl3): δ 8.73 (s, 1H, 1″-ArH), 8.10–8.03 (bm, 4H, 8″-ArH, 2′,6′-ArH, −COCHCO−), 7.95 (d, J = 9, 1H, 3″-ArH), 7.85 (d, J = 9, 1H, 7″-ArH), 7.73 (d, J = 9, 2H, 3′,5′-ArH), 7.33 (d, J = 9, 1H, 4″-ArH), 7.19 (s, 1H, 5″-ArH), 5.21–5.11 (bm, 72 H, PLA −CHCH3), 4.58 (t, J = 4.8, 2H, −OCHH2CH2−), 4.35 (t, J = 4.8, 2H, −ArOCH2CH2OH), 1.57 (s, 241 H, PLA −CHCH3).
Kinetics Studies17
Kinetics studies were performed for initiator 6. BF2n(Br)bmOH (30 mg, 0.06 mmol) and d,l-lactide (0.467 g, 3.25 mmol) were placed in a Kontes flask sealed under N2. The bulb of the flask was entirely submerged in an oil bath at 130 °C. After the d,l-lactide melted, Sn(Oct)2 (0.24 mg, 0.60 μmol) in hexanes was added, and the flask was resealed under N2. Aliquots were drawn up into a pipet tip at the specified times (Table S2; Figure 1A–C) until ∼93% conversion. Percent monomer conversion was determined via 1H NMR spectroscopy by comparing the integration of monomer versus (monomer + polymer) peaks. Molecular masses of dye–polymer conjugates were measured by 1H NMR spectroscopy and GPC with DAD, MALS, and RI detectors.
Figure 1.

GPC trace (A) and kinetics plots (B, C) for lactide polymerization with initiator BF2n(Br)bmOH 6 (130 °C, 6:lactide:Sn(Oct)2 = 1:50:1/50). Aliquots were taken at 10, 30, 60, 80, 105, and 190 min as indicated.
Results and Discussion
Synthesis
A representative boron polymer synthesis via β-diketonate ligand 3 and boron complex initiator 7 is illustrated in Scheme 1 for BF2bnmPLA 11. Ligands are prepared via Claisen condensation of appropriate ketone and ester building blocks. Primary alcohols are installed on the ketones to serve as sites for d,l-lactide polymerization. Commercially available 4-hydroxyacetophenone, or 6-hydroxy-2-acetonapthone prepared by acid hydrolysis of 6-methoxy-2-acetonapthone, is functionalized with −OCH2CH2OH via Williamson ether synthesis and protected with 1,2-dihydropyran for Claisen condensation in the presence of NaH. Following deprotection and purification by recrystallization, the β-diketonate ligands (1–4) were boronated with BF3·OEt2. Reactions were stirred until ligands were completely consumed, according to TLC. In many instances, products precipitated from the reaction mixture over time. After purification by recrystallization powders 5–8 were obtained. The structure and purity of boron dye products were confirmed by MS and 1H NMR spectroscopy.
Scheme 1. Representative Synthesis of β-Diketonate Ligand, Boron Initiator, and Polymer Shown for bnmOH (3), BF2bnmOH (7), and BF2bnmPLA (11).

Hydroxyl-functionalized boron initiators 5–8 were used to produce dye–polymer conjugates 9–12 by solvent-free ring-opening polymerization of d,l-lactide using a tin octoate catalyst at 130 °C. Samples were purified by precipitation from CH2Cl2/hexane to remove the catalyst and multiple times with CH2Cl2/cold methanol until 1H NMR spectroscopy confirmed all monomer was removed. Reagent loadings, reaction times, and molecular weight data are provided in Table S1. Monomer loadings were increased to achieve higher molecular weights. To test the maximal molecular weight attainable with good molecular weight control, initiator 6 was loaded with 300 equiv of lactide monomer and monitored over time. After 24 h, a 21 kDa polymer was achieved (PDI 1.14; 84% conversion). Extended reaction time did not result in further chain extension (71 h, 20.3 kDa, PDI 1.17, 96% conversion); chain transfer or thermal depolymerization may occur.
GPC traces of purified polymer products revealed minor high molecular weight shoulders (Figure 1A). In previous studies, reactions were typically stopped at ∼70% conversion to avoid transesterification and broader PDIs at high monomer conversion,48−51 but here data suggest that other factors are at play. First, here too, kinetics studies (e.g., with 6, Figure 1B,C) reveal relatively linear Mn vs % conversion and pseudo-first-order kinetics plots to ∼70% conversion, but PDIs are higher than usual at the onset (∼1.33 for 23% conversion) and only drop after ∼25% conversion (PDI = 1.05). Second, the high molecular weight shoulders are consistently double the molecular weight of the main polymer peak. Furthermore, GPC analysis of aliquots shows evidence of this feature throughout this polymerization, not just at high conversions (Figure 1A). These findings point to initiator aggregation rather than diminished control as the polymerization progresses, though at very high conversions (e.g., ∼90%) slightly increased PDIs and nonlinear Mn vs conversion is observed, consistent with chain transfer and transesterification. In fact, the naphthyl dyes show lower solubility in common organic solvents compared to previous BF2dbmOH initiators. For all naphthyl complexes, residual solid initiator was evident in the lactide melt at the onset of polymerization reactions; however, the mixtures clarified and solids disappeared as the reactions progressed.
While this might point to slow initiation relative to propagation, in fact, no induction period is noted and no tailing is evident on the low molecular weight side of the main polymer peak. Attempts to facilitate the reaction with increased catalyst loading resulted in increased polymerization rate but showed no obvious effect on molecular weight control. Difluoroboron bdk dyes are reported to dimerize and form H-aggregates, which can influence the optical properties.52 Dimeric initiators could lead to polymer fractions with twice the targeted molecular weight. Certain naphthyl initiators show higher percentages of dimeric products than others in the eluting polymer fractions (8: 10–12% dimer vs 5–7: 5–7% dimer by mass). This correlates with noted differences in their solubility. Alternatively, polymer aggregation could occur during GPC analysis, polymer purification, or processing. That dimeric shoulders increase slightly in GPC traces for purified polymer samples versus analytical samples from kinetics runs lends some support for this hypothesis.
Optical Properties in Solution
Absorption spectra for boron initiators are compared in Figure 2. All complexes have high extinction coefficients (48 000–65 000 M–1 cm–1) typical for π–π* transitions in this family of dyes and high-energy features on dominant peaks. For initiators 5 and 6, the high-energy shoulders at 402 and 395 nm are more distinct. Previously this has been correlated with boron complex dimerization and ascribed to H-aggregates.52 This is consistent with decreased solubility for naphthyl complexes and GPC results in THF solution discussed above. Introduction of the bromide heavy atom onto the Np site of BF2nbmOH resulted in a slight bathochromic shift (3 nm) and increase in extinction coefficient, whereas bromination of the Ph site of BF2bnmOH showed a slightly greater red-shift (7 nm) but a decrease in molar absorptivity. Representative UV/vis spectral data for polymers (∼8 kDa samples) are also provided in Table 1. (See Table S3 for optical properties of polymers with other molecular weights.) Spectra are very similar to corresponding boron initiators with slightly decreased extinction coefficients, typical for these boron bdk PLA materials.14,18
Figure 2.
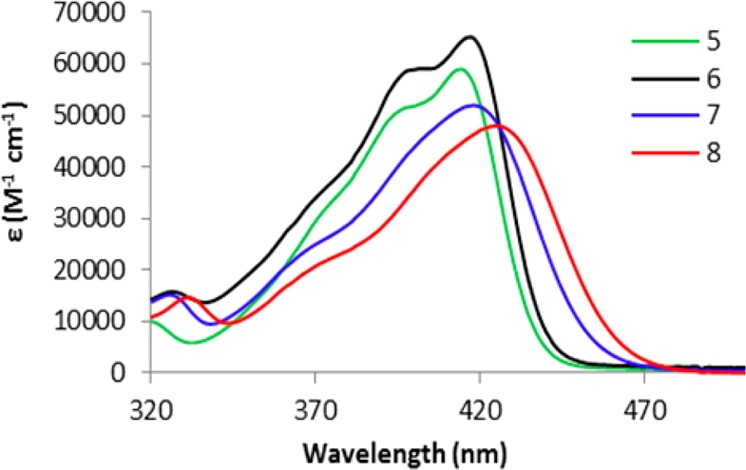
Absorption spectra for boron initiators in dilute CH2Cl2 solutions (Abs < 0.1).
Table 1. Absorption and Emission Data for Boron Initiators and Representative Polymer Samples in CH2Cl2.
| λabsa (nm) | εb (M–1 cm–1) | λemc (nm) | τFd (ns) | ΦFe | τradf | Stokes shift (cm–1) | ||
|---|---|---|---|---|---|---|---|---|
| BF2nbmOH | 5 | 414 | 59 000 | 452 | 1.55 | 0.40 | 3.88 | 2031 |
| BF2nbmPLA | 9b | 414 | 54 000 | 458 | 1.75 | 0.38 | 4.61 | 2321 |
| BF2n(Br)bmOH | 6 | 417 | 65 000 | 448 | 0.53 | 0.19 | 2.79 | 1659 |
| BF2n(Br)bmPLA | 10b | 416 | 50 000 | 448 | 0.52 | 0.18 | 2.89 | 1717 |
| BF2bnmOH | 7 | 418 | 52 000 | 505 | 3.41 | 0.64 | 5.33 | 4121 |
| BF2bnmPLA | 11b | 418 | 43 000 | 501 | 3.32 | 0.71 | 4.68 | 3963 |
| BF2b(Br)nmOH | 8 | 425 | 48 000 | 521 | 3.33 | 0.75 | 4.44 | 4336 |
| BF2b(Br)nmPLA | 12a | 425 | 25 000 | 514 | 3.27 | 0.72 | 4.54 | 4074 |
Absorption maxima.
Extinction coefficients calculated at the absorption maxima.
Fluorescence emission maxima.
Fluorescence lifetimes excited with a 369 nm light-emitting diode (LED) monitored at the emission maxima. All fluorescence lifetimes are fitted with single-exponential decay.
Relative quantum yields, with anthracene in EtOH as a standard.
Radiative lifetimes, where τrad = τF/ΦF.
Luminescence data for initiators and representative polymers in solution are given in Table 1. All samples exhibit intense emission under UV excitation (Figure 3). Initiators 5 and 6 with alkoxy donors on the phenyl rings show intense blue emission at 452 and 448 nm, respectively, while fluorescence for 7 and 8 with −OR donors on the naphthyl rings are red-shifted to 505 and 521 nm, respectively. Once PLA is grown from the initiators, the emission maxima slightly blue-shift (4–7 nm), which may be attributed to a solvatochromic effect or enhanced solubility and thus a diminished tendency to dimerize (Table 1).
Figure 3.

Boron dye initiators 5–8 in CH2Cl2 showing emission colors.
Typically, increased conjugation results in red-shifted emission.53 Naphthyl-phenyl systems here are red-shifted as predicted, compared to previously reported phenyl–phenyl (i.e., dbm) derivatives. For example, BF2dbmPLA15 showed an absorption maximum, λabs = 396 nm, and emission maximum, λem = 426 nm, which are both more blue-shifted than data for samples 9–12 red-shifted (Table 1; e.g. 9b: λabs = 414 nm, λem = 458 nm). Previous studies showed that a π-donating methoxy group red-shifts emission.20 Fluorescence wavelengths for samples 5–12 with the −OCH2CH2OR initiator (R = H) and polymer (R = PLA) tails are consistent with this trend. Alkoxy placement also plays a role in emission maxima. For example, 5 (Np-Ph) with alkoxy substitution on the phenyl ring emits at 452 nm and displays a small Stokes shift (2031 cm–1). In comparison, dye 7 (Ph-Np) with substitution on the naphthyl ring has further red-shifted emission (i.e., 505 nm) and shows a larger Stokes shift (4121 cm–1).
In addition to red-shifting absorbance, bromide heavy atom incorporation in dyes 6 and 8 also affects emission. It is well-known that halide heavy atoms result in decreased luminescence lifetimes and quantum yields via enhanced intersystem crossing.54 Results for brominated and parent (i.e., H) compounds are consistent with this trend. For example, the presence of bromide in BF2n(Br)bmOH 6 results in a decrease in fluorescence lifetime compared to BF2nbmOH 5 (i.e., 5: τ = 1.55 ns (no heavy atom) vs 6: τ = 0.53 ns (heavy atom)) and a decrease in fluorescence quantum yield (5: ΦF = 0.4 vs 6: ΦF = 0.19). A similar trend is noted in the corresponding polymers 9 and 10. In samples 6 and 10, heavy atom substitution is on the larger, more electron-rich naphthyl donor, but in 8 and 12, when the bromide is present on the smaller, less electron-rich phenyl donor, the heavy atom effect is much less pronounced.21 Lifetimes and quantum yields for 8 and 12 are very similar (Table 1). These results with initiators and polymer conjugates are consistent with prior BF2bdk model studies indicating moderate to strong ICT character for unsymmetrically substituted difluoroboron diketonates.20,55 A greater disparity in π-electron-donating ability of the two arene rings results in stronger ICT character dominated by the more electron-rich aryl group.
Computational Studies
Previously, it was shown that the electronic transitions of BF2bdks depend on their molecular symmetries.20 According to TD-SCF calculations, the reddest and strongest transitions are the HOMO to LUMO transitions. Stronger ICT is observed for complexes with unsymmetrical diarene ligands, where there is a disparity in electron donor ability from one arene ring (e.g., anthracene) compared to another (e.g., phenyl). On the other hand, when the bdk arene rings are comparable (i.e., Ph-Ph in dbm), a delocalized π–π* model is proposed. Similar trends are noted here for initiator complexes, 5–8, which were chosen as the subjects for computational modeling given their relative simplicity compared to the respective polymers. Molecular orbital diagrams for compounds 5, 7, and 8 suggest intramolecular charge transfer (ICT) (Figure 4). Electron density is localized on the stronger naphthyl donor in the HOMOs for 5, 7, and 8, whereas in the LUMOs it is distributed across the molecular structure. In contrast, for 6 the electron density is more distributed through the molecular structure on both HOMO and LUMO and the π to π* transition dominates (Figure 4). The electron-withdrawing bromide substituent may diminish electron density on the naphthyl ring, making donor capacity of Br–Np and Ph–OR rings comparable. Experimental absorption and emission data lend credence to this claim. Radiative lifetimes, τrad increase with increasing ICT character,56 and this is a trend that is observed for samples 5, 7, and 8 (3.88–5.33 ns) compared to 6 (2.89 ns), with 6 showing the shortest τrad. Additionally, both 7 and 8 have larger Stokes shifts compared to 5 and 6. This is consistent with a geometry change in the excited state that can be expected for compounds with stronger charge transfer character. As in naphthyl model studies,20 these systems show no distinct ITC features in absorption or emission spectra. Full data are provided in Tables S4 and S5.
Figure 4.

Molecular orbital diagrams for compounds 5–8 showing the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) in CH2Cl2.
Optical Properties in Films
Boron polymers were also studied in the solid state. Thin films were prepared in vials by slow evaporation of CH2Cl2 solutions, followed by drying in vacuo. Optical properties for polymers 9–12, including different molecular weights, are presented in Table 2. δFluorescence spectra and lifetime measurements were obtained under ambient conditions (e.g., air, ∼21% oxygen).
Table 2. Luminescence Data for Films Made from Boron Polymers of Different Molecular Weights.
| fluorescence |
RTP |
||||
|---|---|---|---|---|---|
| polymer | Mna (kDa) | λemb (nm) | τpw0c (ns) | λemd (nm) | τpw0e (ms) |
| 9a | 5.4 | 526 | 12.85 | 558 | 69.76 |
| 9b | 7.7 | 496 | 8.84 | 555 | 102.7 |
| 9c | 16.1 | 461 | 2.57 | 552 | 130.9 |
| 10a | 6.8 | 491 | 3.69 | 568 | 10.50 |
| 10b | 8.8 | 483 | 1.71 | 565 | 11.61 |
| 10c | 11.7 | 467 | 1.02 | 566 | 14.36 |
| 10d | 26.2 | 460 | 0.76 | 565 | 14.64 |
| 11a | 5.2 | 533 | 13.39 | 550 | 20.26 |
| 11b | 7.5 | 512 | 9.10 | 540 | 61.94 |
| 11c | 9.5 | 505 | 7.74 | 545 | 91.14 |
| 12a | 5.6 | 518 | 5.00 | 538 | 25.95 |
| 12b | 12.9 | 500 | 3.47 | 538 | 40.76 |
| 12c | 16.6 | 495 | 3.25 | 539 | 45.81 |
Number-average molecular weights detected by DAD, LS/RI detectors in THF solvent.
Steady-state fluorescence spectra emission maxima under air. Excitation source: 369 nm xenon lamp.
Fluorescence lifetimes excited with a 369 nm light-emitting diode (LED) monitored at the emission maxima. All fluorescence lifetimes are fitted with multiexponential decay. Detailed lifetime data are provided in Table S7.
Delayed emission spectra maxima under N2. Excitation source: xenon flash lamp.
Pre-exponential weighted RTP lifetimes. Excitation source: xenon flash lamp; RTP lifetime fit to triple-exponential decay.
For polymers 9–11 the emission wavelengths in PLA matrices are more red-shifted than in CH2Cl2 solution (for Mn ∼ 7–8 kDa, 9b: CH2Cl2: 458 nm, film: 526 nm; 10a: CH2Cl2: 448 nm, PLA: 491 nm; 11b: CH2Cl2: 501 nm, film: 533 nm). This trend is consistent with previous reports.14 Dye–polymer 12a (Mn = 5.6 kDa) is an anomaly; there is little difference between emission in solution (514 nm) versus film (518 nm), and the emission only blue-shifts further with increasing molecular weight. Previously and here for samples 9–11, emission wavelengths for polymers in solution and in films were only comparable for high polymer molecular weights (i.e., low dye loading), where dyes are diluted and monomeric dye structures are favored.20
Previous studies showed that BF2bdk luminescence properties are dependent upon the molecular weight of polylactide11 or dye loading in dye/polymer blends.19,20 Shorter dye–dye distances corresponding to low dye–PLA molecular weight resulted in lower energy green emission for BF2dbmPLA, whereas longer polymer chains (small dye loadings) with diminished dye–dye interactions correlate with blue-shifted emission.11,19 Polymers 9–12 also follow this trend, namely, emission wavelength blue-shifts with increasing polymer molecular weight. For example, polymers 9a–c display emission wavelengths of 526, 496, and 461 nm, respectively. Furthermore, the sensitivity to dye loading (i.e., Mn) varies for different dyes. For instance, for 9a (5.4 kDa) and 9c (16.1 kDa) the emission wavelength shifts 65 nm, while from 12a (5.6 kDa) to 12c (16.6 kDa) it shifts only 23 nm (Figures 5, 6 and Table 2).
Figure 5.
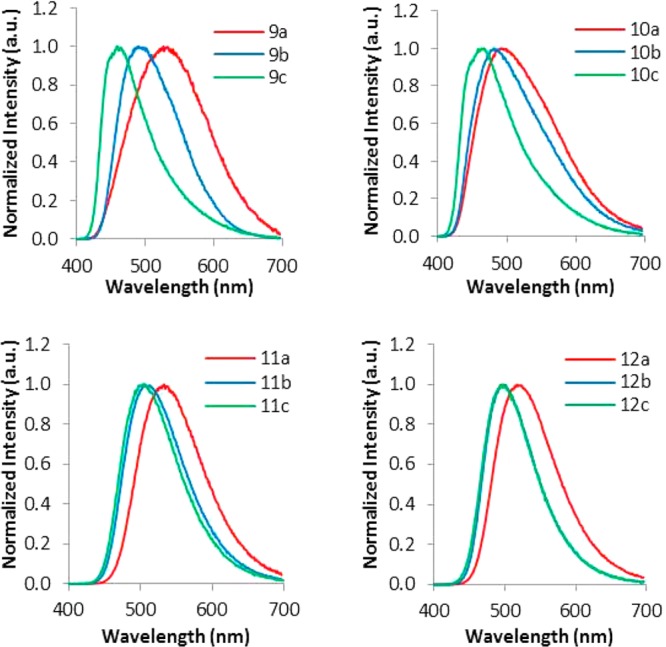
Comparison of the emission spectra of dye–polymer conjugates 9–12 of different molecular weights.
Figure 6.

Images showing emission color changes for polymers 9–12 of different molecular weights under air with UV lamp excitation.
Fluorescence lifetimes were recorded for BF2bdkPLA films under ambient conditions (Tables 2 and Table S7). In all cases, lifetimes fit to triple-exponential decay, which may be ascribed to different fluorophore associations or polymer microenvironments in the solid state.57 As the polymer molecular weight increases, dye–dye interactions decrease, and the pre-exponential weighted lifetimes,58 τpw0, steadily decrease. For the entire molecular weight range examined in this study, dye aggregation may be present in polymer films, because only for 12c do lifetimes approach the value measured for monomeric structures in solution. As expected, the heavy atom has a significant influence on the fluorescence lifetime. For nonhalogenated polymers 9a and 11a lifetimes are ∼13 ns, whereas lifetimes for brominated analogues 10a and 12a decrease to 3.7 and 5.0 ns, respectively59−64 (Table 2).
Phosphorescence measurements for polymer films were performed under a nitrogen atmosphere, and spectra were recorded with a 2 ms delay. Previous studies showed that BF2bdk dyes exhibit room temperature phosphorescence (RTP) in rigid media such as poly(lactic acid) (Tg ∼ 60 °C).10,14,16,17,22,65,66 Here, too, RTP of varying intensities was observed for dye–polymer conjugates 9–12. Phosphorescence data for polymer films is provided in Table 2. The phosphorescence maxima vary little (538–568 nm) for dyes in this series and for different molecular weights (i.e., dye loadings) of a given dye. For example, the RTP for BF2nbmPLA polymers 9a–c is ∼555 nm, and for its brominated derivatives BF2n(Br)bmPLA 10a–d the phosphorescence maxima slightly red-shifted to ∼565 nm. For the BF2bnmPLA polymers 11a–c, the RTP is ∼545 nm, and the −Br derivatives BF2b(Br)mPLA, 12a–c show slightly blue-shifted RTP at ∼538 nm (Figure 7).
Figure 7.
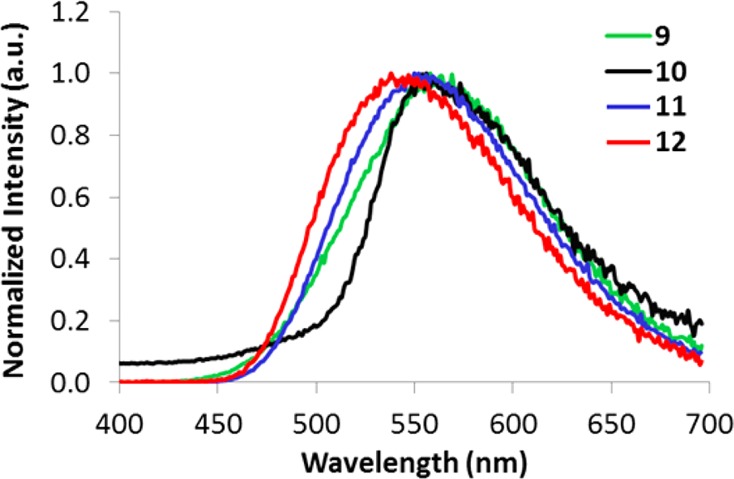
Normalized delayed emission spectra (i.e., phosphorescence + delayed fluorescence) for polymer films 9–12 at room temperature under nitrogen (λex = 369 nm). Phosphorescence maxima range from 538 to 568 nm (∼30 nm range).
The phosphorescence lifetimes were measured under a nitrogen atmosphere. The polymers without a bromide heavy atom usually have longer triplet state lifetimes than their heavy atom derivatives. Heavy atoms increase intersystem crossing (ISC), decrease quantum yields, and also shorten singlet and triplet state lifetimes. For example, the phosphorescence lifetime τP for BF2nbmPLA, 9b (7.7 kDa), is ∼103 ms, and τP for the corresponding brominated derivative, BF2n(Br)bmPLA, 10a (6.8 kDa), is ∼11 ms. When the heavy atom is present on the weaker phenyl donor instead of the naphthyl ring, the effect is not so pronounced.21 For example, polymers BF2bnmPLA 11a (5.2 kDa) and BF2b(Br)nmPLA 12a (5.6 kDa) show comparable phosphorescence lifetimes, τP of ∼20 and ∼26 ms, respectively. This provides further evidence that it is the stronger arene donor that dominates emission properties in these boron β-diketonate systems.
Polymer molecular weight has a significant influence on the fluorescence wavelength. Because the phosphorescence energy remains relatively constant throughout the series, the singlet–triplet state energy gap11,14 increases with polymer molecular weight. Previously for BF2dbm(I)PLA we showed that phosphorescence intensity increases with heavy atom substitution and for lower molecular weights. Nearly full range fluorescence/phosphorescence (F/P) intensity tuning was possible; namely, for high molecular weight polymers, fluorescence was strong and phosphorescence was weak and for low molecular weight polymers, the opposite was true.14 In this study, we explore how fluorescence and phosphorescence peak separations and F/P intensity ratios vary for the naphthyl dye series. This is important for ratiometric oxygen sensing, where the oxygen invariant fluorescence signal serves as the standard and oxygen sensitive phosphorescence serves as the sensor.
A comparison of total emission spectra under air (fluorescence) with emission under nitrogen (fluorescence and phosphorescence) reveals a red shoulder for 9 and slight peak broadening for 11 and 12 under nitrogen (Figure 8). This contrasts with BF2n(Br)bmPLA 10, with a distinct fluorescence peak at λF ∼490 and a more intense phosphorescence peak at ∼565 nm (Figure 9). Total emission spectra also reveal a much stronger heavy atom effect when the bromide is present on the major donor (i.e., naphthalene ring). For 11 and 12, phosphorescence and fluorescence emission maxima are comparable and emission bands effectively overlap for the entire molecular weight range investigated. As expected, for 11 and 12, the overall intensity at the emission band maximum increases under nitrogen (fluorescence + phosphorescence) versus air (fluorescence; phosphorescence is quenched). Though not useful for ratiometric oxygen sensing, materials with small singlet triplet energy gaps are of interest for high efficiency OLED materials. For polymer 9, in contrast, distinct fluorescence and phosphorescence bands are observed for the high molecular weight polymer 9c with maximal peak separation. However, at this lower dye loading the phosphorescence is weak relative to fluorescence, corresponding to a smaller dynamic range for oxygen sensing.
Figure 8.
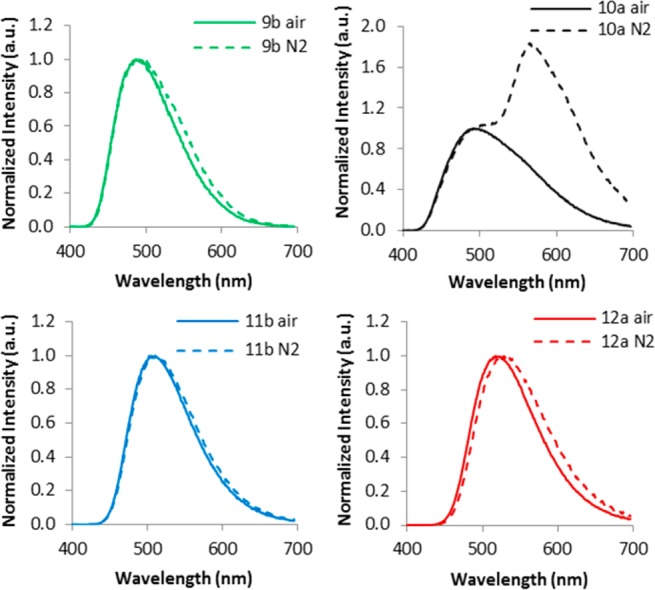
Total emission spectra for polymers 9–12 under air and under a nitrogen atmosphere.
Figure 9.
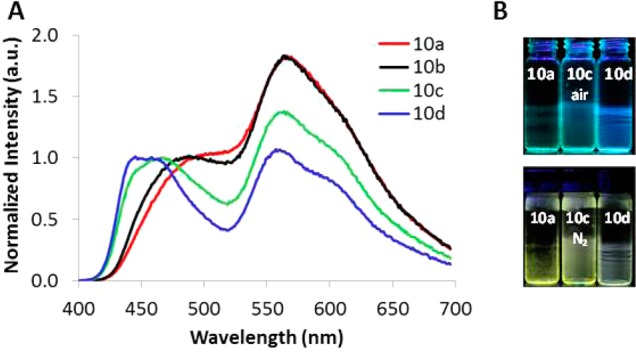
(A) Total emission spectra showing relative fluorescence and phosphorescence intensities for films of BF2n(Br)bmPLA 10a–d of different molecular weights under N2. (B) Images showing emission (excitation by UV lamp): top, under ambient conditions; bottom, under N2.
Polymer 10, BF2n(Br)bmPLA, shows ideal features for ratiometric sensing. Fluorescence and phosphorescence peaks are well separated, and the phosphorescence intensity is strong for all samples 10a–d (Mn = 7–26 kDa) (Figure 9). Even the sample with the lowest dye loading has strong phosphorescence intensity and could be useful for sensing, minimizing the valuable dye reagent and possible toxicity in a biological environment. What is more, the fluorescence and phosphorescence peaks are well aligned with the blue and green channels of RGB CCD cameras, allowing for ratiometric sensing with a simple hand-held imaging modality.
Conclusions
In summary, naphthyl-substituted BF2bdks with and without bromide heavy atoms were prepared for use as initiators to generate polymers with different molecular weights. Compared to previous dbm systems, the naphthyl materials displayed red-shifted absorbance, fluorescence, and phosphorescence.10,11,14 Even when the general structure and the conjugation length of the dye are the same, the polymer attachment site—whether on the naphthyl or phenyl ring—plays an important role in electron density delocalization and optical properties. Computational studies and HOMO and LUMO analysis for the dye initiators 5 (−OCH2CH2OH attached to phenyl ring) and 7 (−OCH2CH2OH attached to the naphthyl ring) revealed that both have ICT character. Nevertheless, the experimentally Stokes shift for 5 is small and for 7 it is large in solution.
In the solid state, fluorescence is sensitive to the position of polymer attachment whereas phosphorescence remains relatively constant. Consequently, fluorescence and phosphorescence peaks are well separated for polymers with blue-shifted fluorescence (e.g., 9) but are poorly separated for those with the most red-shifted fluorescence (11). Aside from altering the dye molecular structure, it is also possible to adjust fluorescence energies and emission lifetimes via polymer molecular weight. Increasing the molecular weight decreases dye–dye interactions and results in blue-shifted fluorescence, decreased fluorescence lifetimes, increased phosphorescence lifetimes, and a larger singlet to triplet energy gap. Achieving fluorescence and phosphorescence peak separation in these ways is important for ratiometric sensing.
Heavy atom substitution and placement also play an important role in optical properties. Halide substitution shifts the HOMO from ICT to π–π* character (e.g., 5 vs 6), as supported by computational studies and radiative lifetimes. The position of bromide substitution also influences luminescence properties. Fluorescence lifetimes and quantum yields decrease significantly upon bromide substitution on the major donor, namely the naphthyl ring (6), whereas the heavy atom effect is considerably weaker for bromide substitution on the minor arene donor (e.g., phenyl ring) (8). These trends extend to the polymer systems.
These fundamental material studies provide important insights for sensing applications. Dyes without heavy atoms such as BF2nbmPLA have longer phosphorescence lifetimes and thus are more sensitive to oxygen. Because phosphorescence intensity is relatively weak, they find application in lifetime sensing. Furthermore, it is possible to fine-tune the lifetime and thus oxygen sensitivity with polymer molecular weight (i.e., dye loading). Dyes such as BF2bnmPLA and BF2b(Br)nmPLA with very small singlet–triplet energy gaps, indistinguishable fluorescence and phosphorescence signals, could be of interest for OLED materials. Finally, BF2n(Br)bmPLA, with red-shifted emission, good fluorescence and phosphorescence peak separation, strong phosphorescence versus fluorescence intensity, and emission wavelengths that are well aligned with the blue and green channels of a CCD camera make this material an excellent candidate for ratiometric oxygen imaging. Further investigation of these materials for oxygen sensing and biomedical imaging is underway.
Acknowledgments
This research was supported by the National Cancer Institute of the National Institutes of Health (R01 CA167250). We gratefully acknowledge the University of Virginia Cancer Center for a fellowship to J.S.-K. through the Farrow Fellowship Fund and the NCI Cancer Center Support Grant, P30 CA44579. We thank Prof. James N. Demas and Prof. Carl O. Trindle for helpful discussions and Rebecca Babski, Tiandong Liu, and Alexander Zestos for their assistance.
Supporting Information Available
Synthesis, characterization, luminescence data, and representative kinetics for 9–12 with different molecular weights; computational data for 5–8. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wang X. D.; Wolfbeis O. S.. Chem. Soc. Rev. 2014, ASAP
- Esipova T. V.; Karagodov A.; Miller J.; Wilson D. F.; Busch T. M.; Vinogradov S. A. Anal. Chem. 2011, 83, 8756–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq J.; Parpaleix A.; Roussakis E.; Ducros M.; Houssen Y. G.; Vinogradov S. A.; Charpak S. Nat. Med. 2011, 17, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier R. J.; Schreml S.; Wang X. D.; Landthaler M.; Babilas P.; Wolfbeis O. S. Angew. Chem., Int. Ed. 2011, 50, 10893–10896. [DOI] [PubMed] [Google Scholar]
- Wang X. D.; Gorris H. H.; Stolwijk J. A.; Meier R. J.; Groegel D. B. M.; Wegener J.; Wolfbeis O. S. Chem. Sci. 2011, 2, 901–906. [Google Scholar]
- Ceroni P.; Lebedev A. Y.; Marchi E.; Yuan M.; Esipova T. V.; Bergamini G.; Wilson D. F.; Busch T. M.; Vinogradov S. A. Photochem. Photobiol. Sci. 2011, 10, 1056–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A. Y.; Cheprakov A. V.; Sakadžić S.; Boas D. A.; Wilson D. F.; Vinogradov S. A. ACS Appl. Mater. Interfaces 2009, 1, 1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo Y. E.; Cao Y.; Kopelman R.; Koo S. M.; Brasuel M.; Philbert M. A. Anal. Chem. 2004, 76, 2498–2505. [DOI] [PubMed] [Google Scholar]
- Lee Y. E.; Ulbrich E. E.; Kim G.; Hah H.; Strollo C.; Fan W.; Gurjar R.; Koo S.; Kopelman R. Anal. Chem. 2010, 82, 8446–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Chen J.; Payne S. J.; Kooi S. E.; Demas J. N.; Fraser C. L. J. Am. Chem. Soc. 2007, 129, 8942–8943. [DOI] [PubMed] [Google Scholar]
- Zhang G. Q.; Kooi S. E.; Demas J. N.; Fraser C. L. Adv. Mater. (Weinheim, Ger.) 2008, 20, 2099–2104. [Google Scholar]
- Koren K.; Borisov S. M.; Saf R.; Klimant I. Eur. J. Inorg. Chem. 2011, 2011, 1531–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Bull B.; Christensen K.; McNeill J. Angew. Chem., Int. Ed. 2009, 48, 2741–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Palmer G. M.; Dewhirst M. W.; Fraser C. L. Nat. Mater. 2009, 8, 747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister A.; Zhang G.; Zareno J.; Horwitz A. F.; Fraser C. L. ACS Nano 2008, 2, 1252–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne S. J.; Zhang G.; Demas J. N.; Fraser C. L.; Degraff B. A. Appl. Spectrosc. 2011, 65, 1321–1324. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Evans R. E.; Campbell K. A.; Fraser C. L. Macromolecules 2009, 42, 8627–8633. [Google Scholar]
- Kersey F. R.; Zhang G.; Palmer G. M.; Dewhirst M. W.; Fraser C. L. ACS Nano 2010, 4, 4989–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Xu S.; Zestos A. G.; Evans R. E.; Lu J.; Fraser C. L. ACS Appl. Mater. Interfaces 2010, 2, 3069–3074. [DOI] [PubMed] [Google Scholar]
- Xu S.; Evans R. E.; Liu T.; Zhang G.; Demas J. N.; Trindle C. O.; Fraser C. L. Inorg. Chem. 2013, 52, 3597–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Zhang G.; Chien A. D.; Evans R. E.; Fraser C. L., unpublished data.
- Zhang X.; Xie T.; Cui M.; Yang L.; Sun X.; Jiang J.; Zhang G. ACS Appl. Mater. Interfaces 2014, 6, 2279–2284. [DOI] [PubMed] [Google Scholar]
- Nagai A.; Chujo Y. Macromolecules 2010, 43, 193–200. [Google Scholar]
- Yoshii R.; Yamane H.; Nagai A.; Tanaka K.; Taka H.; Kita H.; Chujo Y.. Macromolecules 2014, ASAP
- Jäkle F. Chem. Rev. 2010, 110, 3985–4022. [DOI] [PubMed] [Google Scholar]
- Chow Y. L.; Johansson C. I.; Zhang Y. H.; Gautron R.; Yang L.; Rassat A.; Yang S. Z. J. Phys. Org. Chem. 1996, 9, 7–16. [Google Scholar]
- Hudson Z. M.; Liu X. Y.; Wang S. Org. Lett. 2010, 13, 300–303. [DOI] [PubMed] [Google Scholar]
- Rao Y. L.; Amarne H.; Wang S. N. Coord. Chem. Rev. 2012, 256, 759–770. [Google Scholar]
- Li Y.; Kang Y.; Lu J. S.; Wyman I.; Ko S. B.; Wang S. Organometallics 2014, 33, 964–973. [Google Scholar]
- Ko S. B.; Lu J. S.; Wang S. Org. Lett. 2014, 16, 616–619. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Bonder E. M.; Jäkle F. J. Am. Chem. Soc. 2013, 135, 17286–17289. [DOI] [PubMed] [Google Scholar]
- Poon C. T.; Lam W. H.; Wong H. L.; Yam V. W. J. Am. Chem. Soc. 2010, 132, 13992–13993. [DOI] [PubMed] [Google Scholar]
- Kong L.; Wong H. L.; Tam A. Y.; Lam W. H.; Wu L.; Yam V. W. ACS Appl. Mater. Interfaces 2014, 6, 1550–1562. [DOI] [PubMed] [Google Scholar]
- Jiang X. X.; Liu X. H.; Jiang Y. L.; Quan Y. W.; Cheng Y. X.; Zhu C. J. Macromol. Chem. Phys. 2014, 215, 358–364. [Google Scholar]
- Glotzbach C.; Kauscher U.; Voskuhl J.; Kehr N. S.; Stuart M. C.; Frohlich R.; Galla H. J.; Ravoo B. J.; Nagura K.; Saito S.; Yamaguchi S.; Wurthwein E. U. J. Org. Chem. 2013, 78, 4410–4418. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Giebink N. C.; Kanno H.; Ma B.; Thompson M. E.; Forrest S. R. Nature 2006, 440, 908–912. [DOI] [PubMed] [Google Scholar]
- Entwistle C. D.; Marder T. B. Angew. Chem., Int. Ed. 2002, 41, 2927–2931. [DOI] [PubMed] [Google Scholar]
- Williams D. B.; Lawton M. J. Org. Chem. 2010, 75, 8351–8354. [DOI] [PubMed] [Google Scholar]
- Heller C. A.; Henry R. A.; McLaughlin B. A.; Bliss D. E. J. Chem. Eng. Data 1974, 19, 214–219. [Google Scholar]
- Melhuish W. H. J. Chem. Phys. 1961, 65, 229–235. [Google Scholar]
- Rioboo R. J.; Philipp M.; Ramos M. A.; Kruger J. K. Eur. Phys. J. E: Soft Matter 2009, 30, 19–26. [DOI] [PubMed] [Google Scholar]
- Gaussian 09: Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö., Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Phys. Rev. B: Condens. Matter 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- Dennington R.; Keith T.; Millam J. In Shawnee Mission KS; Semichem Inc., 2009; Vol. GaussView Version 5. [Google Scholar]
- Chang K. W.; Chen C. C.; Lee S. Y.; Wang H. E. Drug Chem. Toxicol. 2009, 32, 429–437. [DOI] [PubMed] [Google Scholar]
- Bender J. L.; Corbin P. S.; Fraser C. L.; Metcalf D. H.; Richardson F. S.; Thomas E. L.; Urbas A. M. J. Am. Chem. Soc. 2002, 124, 8526–8527. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Storti G.; Morbidelli M. Ind. Eng. Chem. Res. 2011, 50, 7927–7940. [Google Scholar]
- Yu Y.; Storti G.; Morbidelli M. Macromolecules 2009, 42, 8187–8197. [Google Scholar]
- Penczek S.; Duda A.; Szymanski R. Macromol. Symp. 1998, 132, 441–449. [Google Scholar]
- Gorczynski J. L.; Chen J.; Fraser C. L. J. Am. Chem. Soc. 2005, 127, 14956–14957. [DOI] [PubMed] [Google Scholar]
- Sun X. X.; Zhang X. P.; Li X. Y.; Liu S. Y.; Zhang G. Q. J. Mater. Chem. 2012, 22, 17332–17339. [Google Scholar]
- Yamaguchi Y.; Matsubara Y.; Ochi T.; Wakamiya T.; Yoshida Z. J. Am. Chem. Soc. 2008, 130, 13867–13869. [DOI] [PubMed] [Google Scholar]
- Sahai R.; Hofeldt R. H.; Lin S. H. Trans. Faraday Soc. 1971, 67, 1690–1697. [Google Scholar]
- Nijegorodov N.; Mabbs R. Spectrochim. Acta, Part A 2001, 57, 1449–1462. [DOI] [PubMed] [Google Scholar]
- Grabowski Z. R.; Rotkiewicz K.; Rettig W. Chem. Rev. 2003, 103, 3899–4032. [DOI] [PubMed] [Google Scholar]
- Schwartz B. J. Annu. Rev. Phys. Chem. 2003, 54, 141–172. [DOI] [PubMed] [Google Scholar]
- Carraway E. R.; Demas J. N.; DeGraff B. A.; Bacon J. R. Anal. Chem. 1991, 63, 337–342. [Google Scholar]
- El-Sayed M. A. J. Chem. Phys. 1964, 41, 2462–2467. [Google Scholar]
- Dreeskamp H.; Koch E.; Zander M. Chem. Phys. Lett. 1975, 31, 251–253. [Google Scholar]
- Kasha M. J. Chem. Phys. 1952, 20, 71–74. [Google Scholar]
- McClure D. S. J. Chem. Phys. 1949, 17, 905–913. [Google Scholar]
- Kearvell A.; Wilkinson F. Mol. Cryst. 1968, 4, 69–81. [Google Scholar]
- McGlynn S. P.; Reynolds M. J.; Daigre G. W.; Christodoyleas N. D. J. Chem. Phys. 1962, 66, 2499–2505. [Google Scholar]
- Benabdillah K. M.; Boustta M.; Coudane J.; Vert M. In Polymers from Renewable Resources; American Chemical Society: Washington, DC, 2001; Vol. 764, pp 200–220. [Google Scholar]
- Narladkar A.; Balnois E.; Vignaud G.; Grohens Y. Macromol. Symp. 2008, 273, 146–152. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.