

Abstract
As part of the chromosome-centric human proteome project (C-HPP) initiative, we report our progress on the annotation of chromosome 22. Chromosome 22, spanning 51 million base pairs, was the first chromosome to be sequenced. Gene dosage alterations on this chromosome have been shown to be associated with a number of congenital anomalies. In addition, several rare but aggressive tumors have been associated with this chromosome. A number of important gene families including immunoglobulin lambda locus, Crystallin beta family, and APOBEC gene family are located on this chromosome. On the basis of proteomic profiling of 30 histologically normal tissues and cells using high-resolution mass spectrometry, we show protein evidence of 367 genes on chromosome 22. Importantly, this includes 47 proteins, which are currently annotated as “missing” proteins. We also confirmed the translation start sites of 120 chromosome 22-encoded proteins. Employing a comprehensive proteogenomics analysis pipeline, we provide evidence of novel coding regions on this chromosome which include upstream ORFs and novel exons in addition to correcting existing gene structures. We describe tissue-wise expression of the proteins and the distribution of gene families on this chromosome. These data have been deposited to ProteomeXchange with the identifier PXD000561.
Keywords: human proteome, “missing” proteins, genome annotation, uORF
Introduction
With the near completion of the human genome project, there has been a continued effort to characterize the function of proteins involved in cellular processes. The chromosome-centric human proteome project (C-HPP) initiative led by the Human Proteome Organization (HUPO) attempts to map and annotate all of the estimated 20 300 human protein coding genes across each chromosome.1 The major goal of the C-HPP is to identify and functionally characterize at least one protein product encoded by each gene with a major emphasis on identifying 20% of the proteome that currently lacks proteomic evidence.2 Several groups have adopted a chromosome and are employing mass spectrometry to identify and characterize the proteome encoded.3−5 In addition to mass spectrometry-based analysis, several other strategies including antibody-based detection are being employed.6
To enable characterization of the entire proteome, several challenges need to be overcome. A majority of the proteins that currently lack evidence are likely to have tissue/cell-restricted expression, may be expressed at certain stages of development, or are present in very low abundance. It is becoming more apparent that at least some of the current annotations of the human genome are erroneous, for example, incorrectly annotated translation start sites and missed exons. The global C-HPP initiative of allocating individual chromosomes to teams around the world to identify proteins encoded by genes across all chromosomes will help address these issues. The knowledge base and the technical resources that would be generated will lead the community to apply these findings to a broad array of biomedical problems. In this regard, our team has focused on the annotation of chromosome 22.
Chromosome 22 was the first human chromosome to be fully sequenced,7 and it is the second smallest human autosome, spanning about 51 million base pairs. It accounts for ∼1.8% of the genomic DNA.8 NCBI annotation release 104 lists a total of 884 genes, of which 442 are annotated as protein coding genes. A number of different genetic alterations associated with various diseases and genetic abnormalities on chromosome 22 have been reported. These include some of the well-known as well as rare disorders such as cat eye syndrome, which is caused by extra copies of the proximal region of chromosome 22q,9 DiGeorge syndrome,10 velocardiofacial syndrome (VCFS),11 and 22q13.3 deletion syndrome12 among others. In addition, chromosome 22 is also reported in the formation of the Philadelphia chromosome by reciprocal translocation between chromosome 9 and 22, generating the BCR-ABL oncogene which is found in 95% of chronic myeloid leukemia patients and 25–30% of acute lymphoblastic leukemia patients.13
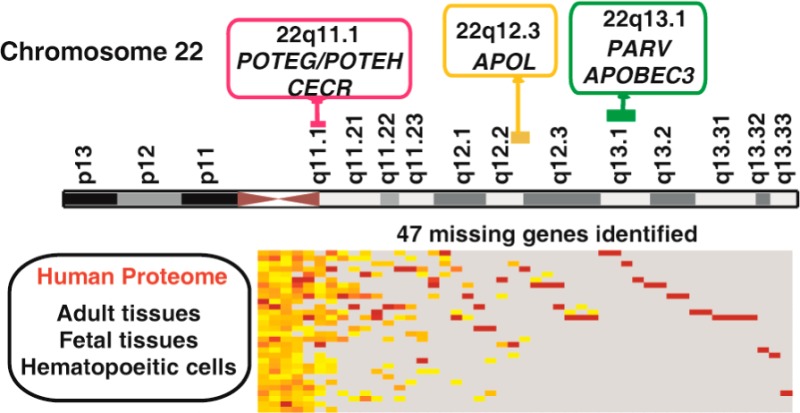
In our previous study, we carried out deep proteomic profiling of 30 different histologically normal tissues and hematopoietic cells, thereby generating a draft map of the human proteome.14 Here, we describe a detailed annotation of the chromosome 22-encoded proteome. Upon mapping our proteomics data to chromosome 22, we identified protein products encoded by 367 genes, including 47 which have been reported to be missing based on C-HPP criteria.2 We also confirm the translation start site for 120 proteins. Tissue-wise expression of several disease-related genes and gene families that are clustered on this chromosome is reported. Finally, employing our proteogenomics strategy, we provide translation evidence for several novel coding regions such as upstream ORFs and novel exons in addition to correction of existing gene structures.
Materials and Methods
Sample Preparation and Fractionation
Histologically normal human organs, fetal tissues, and hematopoietic cells were procured after obtaining institutional ethical clearance. The tissues/cells were lysed in lysis buffer containing 4% SDS, 100 mM DTT, and 100 mM Tris pH 7.5, and were homogenized, sonicated, heated for 10–15 min at 75 °C, cooled, and centrifuged at 2000 rpm for 10 min. The protein concentration of the cleared lysate was estimated using BCA assay, and equal amounts from each of the donors was pooled for further fractionation.
Proteins from SDS lysates were separated on SDS-PAGE, and in-gel digestion was carried out as described earlier.15 Briefly, the protein bands were destained, reduced, alkylated, and subjected to in-gel digestion using trypsin. The peptides were extracted, vacuum-dried, and stored at −80 °C until further analysis.
Four hundred micrograms of proteins from each sample was subjected to in-solution trypsin digestion following reduction and alkylation with DTT and IAA, respectively. The peptide digests were then desalted using Sep-Pak C18 columns (Waters Corporation, Milford, MA) and lyophilized. The lyophilized samples were reconstituted in bRPLC solvent A (10 mM TEABC, pH 8.5) and loaded on an XBridge C18, 5 μm 250 × 4.6 mm column (Waters, Milford, MA). The peptide digest was resolved using a gradient of 0 to 100% solvent B (10 mM TEABC in acetonitrile, pH 8.5) in 50 min. The total fractionation time was 60 min. Ninety-six fractions were collected which were then concatenated to 24 fractions, vacuum-dried, and stored at −80 until further LC-MS/MS analysis.
LC-MS/MS Analysis
Peptide samples from in-gel digestion and bRPLC fractionation were analyzed on LTQ-Orbitrap Velos and LTQ-Orbitrap Elite mass spectrometers (Thermo Electron, Bremen, Germany) interfaced with an Easy-nLC II nanoflow liquid chromatography system (Thermo Scientific, Odense, Southern Denmark). The peptide digests were reconstituted in 0.1% formic acid and loaded onto a trap column (75 μm × 2 cm) packed in-house with Magic C18 AQ (Michrom Bioresources, Inc., Auburn, CA) at a flow rate of 5 μL/min with solvent A. Peptides were resolved on an analytical column (75 μm × 10 cm) at a flow rate of 350 nL/min using a linear gradient of 7–30% solvent B (0.1% formic acid in 95% acetonitrile) over 60 min. Data-dependent acquisition with full scans in the 350–1800 m/z range were carried out using an Orbitrap mass analyzer at a mass resolution of 60 000 in Velos and 120 000 in Elite at 400 m/z, respectively. Twenty of the most intense precursor ions from a survey scan were selected for MS/MS and were fragmented using higher-energy collision dissociation (HCD) with 35% normalized collision energy and detected at a mass resolution of 15 000 and 30 000 at 400 m/z in Velos and Elite, respectively. Dynamic exclusion was set for 30 s with a 10 ppm mass window.
Proteomic Data Analysis
Mass spectrometry data obtained from all LC-MS/MS analysis were searched against Human RefSeq50 database (containing 33 833 entries along with common contaminants) using Sequest and Mascot (version 2.2) search algorithms through Proteome Discoverer 1.3 (Thermo Scientific, Bremen, Germany). Enzyme specificity was set as trypsin with maximum of two missed cleavages allowed. The minimum peptide length was specified as six amino acids. Carbamidomethylation of cysteine was specified as fixed modification and oxidation of methionine, acetylation of protein N-termini and cyclization of N-terminal glutamine were included as variable modifications. The peptide and fragment mass tolerance was set to 10 ppm and 0.05 Da for fragment ions. The data were also searched against a decoy database, and the results were used to estimate q values using the Percolator algorithm16 within Proteome Discoverer. Peptides were considered identified at a q value <0.01 and used for further analysis.
Identification of Novel Peptides Using Alternative Databases Searches
To enable identification of novel peptides and correction of existing gene annotations in the human genome, five alternative databases were used. These databases were generated using in-house python scripts for proteogenomic analysis. The databases used were (1) six-frame translated genome database, (2) three-frame translated RefSeq mRNA sequences from NCBI, (3) three-frame translated pseudogene database with sequences derived from sequences from NCBI and Gerstein’s pseudogenes, (4) three-frame translated non coding RNAs from NONCODE, and (5) N-terminal UTR database of RefSeq mRNA sequences from NCBI. A decoy database was created for each database by reversing the sequences from a target database. Peptide identification was carried out using X!Tandem. The following parameters were common to all searches: (1) precursor mass error set at 10 ppm, (2) fragment mass error set at 0.05 Da, (3) carbamidomethylation of cysteine defined as a fixed modification, (4) oxidation of methionine defined as a variable modification, and (5) only tryptic peptides with up to two missed cleavages were considered. The sequences of common contaminants including trypsin used as protease were appended to the database. Unmatched MS/MS spectra peaklist files were extracted from the protein database search result and searched against these databases using the X!Tandem search engine installed locally.
All the databases were created in-house using python scripts. For the six-frame translated genome database, the human reference genome assembly hg19 was downloaded from NCBI and translated in six reading frames, and peptide sequences greater than six amino acids were retained in the database. The mRNA sequences were downloaded from NCBI RefSeq (RefSeq version 56 containing 33 580 sequences) and translated in three reading frames. Custom pseudogene database from NCBI (11 160 sequences) and Gerstein’s pseudogene database (16 881 sequences from http://pseudogene.org/, version 68) as well as noncoding RNA sequences from NONCODE (91 687 sequences, version 3) were translated in three reading frames.
Peptide sequences identified from each of the alternate database searches were filtered, and only those passing 1% FDR score threshold at the peptide level were considered. Unique lists of genome search specific peptides (GSSPs), pseudogene specific peptides, and transcriptome search specific peptides were generated by comparing the peptides with the protein database. Peptides mapping to multiple regions in the genome and with isoleucine/leucine ambiguity were not considered for further analysis. The final list of the peptides was then manually validated to verify the confidence of spectral assignment.
Gene Ontology Analysis
Molecular functions and primary localization information for all the proteins were obtained from Human Protein Reference Database, HPRD (http://www.hprd.org), containing manually curated protein annotations along with protein–protein interactions related to human proteins.17
Quantitation Based on Spectral Counts
Normalized spectral counts were calculated by summing spectral counts from all the peptides mapping to a given gene per experiment (i.e., SDS-PAGE or bRPLC). Total acquired tandem MS were then used to normalize between experiments, and the resulting spectral counts per gene were averaged across multiple experiments (e.g., bRPLC and SDS-PAGE fractionation) per sample (e.g., esophagus). Final numbers per gene were used for plotting the heat map.
Results and Discussion
Proteomic Evidence for Genes on Chromosome 22
As part of our deep proteomic profiling of 30 different histologically normal human tissues and cell types using high-resolution mass spectrometry, we specifically looked for proteins that are encoded by genes on chromosome 22. Of the 442 RefSeq gene entries, our data provides translation evidence for protein products of 367 genes. Twenty proteins have >90% sequence coverage and 52% of the proteins have sequence coverage >50% (Figure 1a). The distribution of identified proteins based on their subcellular localization and molecular function reveals proteins to be predominantly localized in the cytoplasm followed by the nucleus and the plasma membrane (Figure 1b). In addition to providing translation evidence for many of the chromosome 22-encoded proteins, we also confirmed the start sites of 120 proteins.
Figure 1.

(a) Sequence coverage of proteins encoded by chromosome 22. (b) Distribution of identified chromosome-22-encoded proteins based on subcellular localization. (c) Tissue-wise distribution of protein coding genes encoded by chromosome 22. Distribution of proteins identified from 30 histologically normal tissues and cell lines based on their spectral abundance. The color schema is based on the spectral counts: red blocks represent proteins with relatively higher expression, orange depicts moderate expression, and yellow represents relatively low expression. Proteins not detected in a given tissue are represented in gray.
Figure 1c depicts the relative expression of chromosome 22-encoded proteins identified in this study based on normalized spectral counts. Several proteins identified in our study show tissue restricted expression. For example, proteins encoded by GNAZ, PDXP, PI4KA, SBF1, SYNGR1, and PRODH showed higher expression in adult frontal cortex compared to other tissues, whereas those encoded by PARVG, EP300, THOC5, PARVB, and ADRBK2 showed consistently high expression in the hematopoietic cells compared to fetal and adult tissues. Parvins are actin-binding proteins that form multiprotein complexes with integrin-linked kinases that link integrins to the actin cytoskeleton. γ-Parvin has been reported to be expressed specifically in hematopoietic cells of both myeloid and lymphoid origin and is in agreement with our findings.18 β-Parvin, another member of the parvin family, shows high expression in platelets compared to all other tissues. Interestingly, we identified proteins encoded by ACR, HORMAD2, TSSK2, and TMEM191B, among others, that showed relatively higher expression in adult ovary and testis compared to other tissues sampled in our study.
When we compared our data with neXtProt entries, we found 317 of 356 proteins categorized as PE1 entries present in our data set. We also compared our data with 337 genes from PeptideAtlas and 339 genes from GPMDB.2 We found 63 genes unique to our data set as compared to PeptideAtlas and 56 genes as compared to GPMDB. This was made possible by the deep proteome coverage obtained by combining diverse human samples, different fractionation strategies, and high-resolution mass spectrometers.
“Missing” Proteins Identified in Chromosome 22
To enable identification of “missing proteins”, we compared our list of proteins encoded by chromosome 22 with the list of “missing proteins” derived from proteins that do not have any proteomic evidence in neXtProt, PeptideAtlas, or GPMDB.19 Based on the current metrics2, 108 proteins are reported to be missing or lacking proteomic evidence from chromosome 22. Of these, 21 are currently categorized as PE5 entries. Comparing our list of identified proteins with neXtProt database resulted in the identification of 47 missing proteins, which include 5 of 21 proteins designated as uncertain/dubious (Supplementary Table S1).
On the basis of our analysis, we observed several proteins that showed tissue-restricted expression, whereas only a few showed ubiquitous expression (Figure 2). Proteins encoded by ATP5L2, DDTL, TSPO, and POTEH show ubiquitous expression across all tissues with certain tissues showing higher expression than in others. For example, translocator protein (TSPO), a mitochondrial membrane protein involved in various physiological functions, including immunologic responses,20 showed high expression in monocytes followed by other hematopoietic cells. In the case of tissue-restricted expression, CECR6, one of the dosage-sensitive genes responsible for cat eye syndrome (CES)21 was identified from our study specifically in the brain with higher expression in adult frontal cortex compared to fetal brain. Three RIMBP3 genes—RIMBP3, RIMBP3B, and RIMBP3C—are located on chromosome 22, and each of these is a single exon gene sharing 99% identity at nucleotide level.22 All RIMBP3 genes contain a central SH3 domain, followed by two FN3 domains and two C-terminal SH3 domains. RNASeq data from Illumina BodyMap shows transcript expression in several tissues, with the highest expression in testis. We identified gene-specific peptides only for RIMBP3, with relatively high expression in testis, consistent with the transcriptome data. Another protein, RIBC2 (RIB43A domain with coiled-coils 2) also known as C22orf11, is expressed only in fetal tissues and has the highest expression in fetal heart. RIBC2 is located on the q13.31 locus and consists of two-coiled coil domains. The biological role of this protein has not been determined but it seems to play a role in early developmental stages. In addition, proteins encoded by solute carrier family members located on this chromosome, such as SLC5A4 (adult kidney), SLC35E4, SLC2A11 (high expression in fetal brain), SLC16A8 (adult retina), and SLC7A4 (adult testis), have been identified with tissue-restricted expression pattern. Thus, sampling specific tissue/cell types has enabled us to identify majority of the missing proteins that had been missed earlier.
Figure 2.

Tissue-wise expression of “missing proteins” identified by proteomic study. Distribution of “missing” proteins identified in this study from 30 histologically normal tissues and cell lines based on their spectral abundance. The color schema is based on the spectral counts: red blocks represent proteins with relatively higher expression, orange depicts moderate expression, and yellow represents relatively low expression. Proteins not detected in a given tissue are represented in gray.
Gene Families on Chromosome 22
Several gene families have been reported to be clustered on chromosome 22. Gene families on the same chromosome are presumed to have arisen by tandem gene duplication. Figure 3a depicts some of the gene families on this chromosome. The immunoglobulin λ locus, which encodes the light chain of antibodies, is one such gene family clustered on chromosome 22.23 In addition to the lambda locus, there are other immunoglobulin-related genes on this chromosome such as immunoglobulin λ-like (IGLL) family and immunoglobulin k variable-region pseudogenes. Apart from the immunoglobulin genes, gene families that encode glutathione S-transferases,24 Ret-finger-like proteins,25APOL family,26 and β-crystallins27,28 are present on this chromosome. The γ-glutamyl transferase genes represent a family that is likely to have been duplicated in tandem along with other gene families such as the BCR-like genes that span the 22q11 region. Together these genes form the LCR22 (low-copy repeat 22) repeats. The APOBEC gene family, which is involved in deaminating cytosine to uracil, has 7 of 10 members clustered on chromosome 22.29 These genes are involved in RNA/DNA editing and exhibit diverse physiological functions.30,31APOBEC1, the first member to be discovered, is involved in deaminating apolipoprotein B RNA, thereby creating a premature stop codon and producing a shorter protein.32,33 Tissue-wise distribution of the APOBEC proteins reveals a relatively higher abundance in hematopoietic cells compared to adult and fetal tissues (Figure 3b). Interestingly, our analysis shows APOBEC3H to be restricted in its expression to adult liver tissue. This gene is known to possess antiretroviral activity and inhibits retrovirus replication and retrotransposon mobility by deamination.34
Figure 3.

(a) Selected examples of gene families on chromosome 22. The gene families are shown in boxes with information pertaining to band annotation. Within parentheses, the details of the gene family are provided in the following order: number of genes of a family adjacent to each other/total number of members on the chromosome/total family members in the genome. (b) Panel shows tissue-wise distribution of APOBEC gene family on chromosome 22. 7 of 10 APOBEC gene family members are clustered on chromosome 22.
Cancer-Associated Genes on Chromosome 22
Chromosome 22 is also known to have strong association with cancers and is extensively rearranged in several tumors, including chronic myeloid leukemia, astrocytic tumors,35 and neurofibromatosis type 2a.36 Table 1 lists several known cancer-associated genes located on this chromosome, including some of the prominent genes such as BCR (Philadelphia chromosome – translocation), CRKL (chronic myeloid leukemia), PDGFB (glioma), and genes associated with some rare tumors, including SMARCB1 (rhabdoid tumor), MN1 (meningioma), EWSR1 (Ewing’s sarcoma), and NF2 (schwannoma tumors). A complete list of cancer-associated genes on chromosome 22 has been generated using the method described by Liu et al.37 On the basis of the information related to oncogenes integrated from several websites (Sanger, Waldman, and GeneCards, see legends Table 1), we have identified 24 genes on chromosome 22. Nineteen of the 24 cancer-associated genes are present in regions which are transcriptionally active with gene density above the average on chromosome 22 (i.e., 51.6 genes/Mb). Only five genes occur in a region with <45 genes per Mb.
Table 1. List of Cancer-Associated Genes on Chromosome 22.
| cancer gene list
for chromosome 22 | ||||||
|---|---|---|---|---|---|---|
| position | oncogene | Sanger(S)/Waldman(W)/cancer index(C)a | top Novoseek cancer associationb | GeneCards Novoseek-logPc | adjacent cancer-related genes scored | gene densitye |
| 22q11.21 | BID | C | tumors | 52.3 | 4 | 90 |
| CLTCL1 | S | n/a | n/a | 12 | 100 | |
| COMT | C | breast cancer | 49.4 | 17 | 100 | |
| CRKL | W,C | chronic myeloid leukemia | 77.7 | 0 | 98 | |
| SEPT5 | S | leukemia | 0 | 21 | 100 | |
| 22qll.23 | BCRf | S,W,C | chronic myeloid leukemia | 91.4 | 6 | 130 |
| GSTT1 | C | bladder cancer | 56.9 | 16 | 251 | |
| MIF | C | tumors | 44.9 | 16 | 251 | |
| MMP11 | C | breast carcinoma | 64.3 | 21 | 251 | |
| SMARCB1 | S,C | rhabdoid tumor | 97 | 16 | 251 | |
| 22ql2.1 | CHEK2 | S,C | breast cancer | 54.6 | 13 | 52 |
| MN1 | S,C | meningioma | 68.3 | 12 | 21 | |
| 22ql2.2 | EWSR1 | S,C | Ewing’s sarcoma | 96.3 | 13 | 52 |
| LIF | C | leukemia | 94.9 | 8 | 70 | |
| NF2f | S,W,C | schwannoma tumors | 89.9 | 13 | 52 | |
| PATZ1 | S | colon carcinoma | 35.6 | 0 | ||
| 22ql2.3 | MYH9 | S | leukemia | 17.9 | 6 | 44 |
| TIMP3 | W | tumors | 57.7 | 1 | 23 | |
| 22ql3.1 | MKL1 | S | acute megakaryoblastic leukemia | 78.3 | 5 | 36 |
| PDGFBf | S,W,C | glioma* | 50.4 | 10 | 84 | |
| 22ql3.2 | BIK | C | breast cancer | 14.3 | 3 | 40 |
| CYP2D6 | W,C | breast cancer | 26.4 | 6 | 110 | |
| EP300 | S | leukemia | 60.5 | 5 | 68 | |
| 22ql3.33 | TYMP | W | tumors | 71.4 | 3 | 72 |
Websites that list oncogene information are http://www.sanger.ac.uk/genetics/CGP/Census/, http://waldman.ucsf.edu/GENES/completechroms.html, and http://www.cancerindex.org/geneweb/genes_d.htm.
Top cancer association from GeneCards Novoseek disease relationship table, http://www.genecards.org/.
-logP of top cancer association (b) from GeneCards Novoseek disease relationship table, http://www.genecards.org/.
Information derived from neXtProt and GeneCards. As a way to assess degree of cancer association, we have calculated a score as follows: the adjacent 10 genes on either side of the oncogene (from neXtProt) are scored as oncogene designation +5 points, direct literature association with cancer +3 points, present in cancer data sets +1 point.
The gene density was derived from GeneCards: http://genecards.weizmann.ac.il/geneloc-bin/gene_densities.pl. All values represent the regions with a gene density above the average on chromosome 22 (i.e., 51.6 genes/Mb), except for numbers indicated in bold.
Recognized as driver oncogene.
Italic text indicates known gene mutation associated with cancer.
Bold text and * indicates cancer gene target with therapy from My Cancer Genome, http://www.mycancergenome.org/.
NF2, a gene responsible for neurofibromatosis type 2, encodes a cytoskeletal associating protein, Merlin. Merlin is similar to the ERM (Ezrin–Radixin–Moesin) family of proteins and has been implicated in the regulation of a number of signaling pathways, including Rac1,38 Ras/MAPK,39 mTOR,40 and Hippo pathways.41NF2 has been reported to be a negative regulator of Rac signaling.38 Several studies have reported NF2 as a tumor suppressor protein.42,43 Deletion and mutations in the NF2 gene have been shown to cause autosomal dominant predisposition to tumors in the nervous system and skin.43 In addition, several mutations and breakpoints are reported in the NF2 gene locus located in the 22q12 region of chromosome 22. This gene locus lies in one of the transcriptionally active regions on the chromosome and consists of genes such as LIF and NEFH in addition to several cancer-associated genes, including EWSR1, which contains the Ewing’s sarcoma translocation breakpoint, and other known tumor suppressor genes, GAS2L1 and RASL10A.44 Figure 4 illustrates some of these important genes clustered around NF2 as well as the diversity of protein isoforms of these genes in terms of the number of alternative splice variants (ASVs, red circle) and single nucleotide variants (SNVs, purple circle) resulting from the transcription of missense SNPs. Genes with a significant number of variants in the region include NF2 with 11 ASVs and 90 SNVs, ASCC2 (10, 26), MTMR3 (6, 36), and TBC1D10A (6, 17). RFPL1, RASL10A, UQCR10, ZMAT5, and HORMAD2 are currently categorized as “missing proteins” with no evidence at the proteome level. In our proteomics data, we observed HORMAD2 to be expressed ubiquitously with relatively high expression in adult testis and RFPL1 with restricted expression in fetal heart. This warrants further studies to be performed to functionally characterize these proteins as they may have an impact on the phenotype and could likely contribute to disease progression in NF2 deleted/mutated tumors.
Figure 4.

Genes around NF2. Ten genes adjacent to NF2 have been considered for the analysis. Genes in filled boxes (blue shade) have evidence of PTMs, whereas genes in unfilled boxes have no known PTMs. Red circles within the boxes indicate number of alternative splice variants in Ensembl/neXtProt. Purple circles denote number of proteins in neXtProt.
Proteogenomic Analysis
Evidence for the protein-coding potential of the genome has been largely dependent on gene prediction algorithms and sometimes on the presence of corresponding transcripts or orthologous genes. Currently, about 50% of human transcripts are classified as noncoding. In recent years, mass spectrometry-derived data has been employed to refine/redefine the genome annotation of various organisms.45−48 Employing a proteogenomics strategy established previously by our group, we discovered several potential novel coding regions on chromosome 22. From our large scale proteome analysis, following the protein database search, unmatched spectra were extracted and searched using reference genome translated into six reading frames, RefSeq transcript sequences translated in three reading frames, and custom databases of annotated pseudogenes and conceptually translated protein N-termini databases. Our analysis resulted in the identification of 10 novel events on chromosome 22 including identification of upstream ORFs, evidence for translation of noncoding RNA correction/refinement of gene structure. The summary of the analysis is provided in Table 2. The details of all the proteogenomic annotation on chromosome 22 is provided in Supplementary Table S2.
Table 2. Summary of Novel Findings Identified through Proteogenomics.
| category | no. of cases |
|---|---|
| novel coding region (upstream ORF) | 2 |
| novel coding exons | 2 |
| translation evidence for noncoding RNA | 1 |
| translation evidence for pseudogene | 1 |
| novel N-termini | 3 |
It is estimated that 41–49% of all known human RefSeq transcripts with annotated 5′ UTR contain at least one upstream ORF.49−51 Studies have shown that uAUG codons and associated upstream ORFs in the 5′-UTR region play an important role in translational control of mRNAs. From our analysis, we identified two cases of uORFs. MIEF1 is a mitochondrial membrane protein that regulates mitochondrial membrane dynamics.52,53 Overexpression of MIEF1 was shown to cause extensive mitochondrial fission, recruitment of DRP1 to mitochondria membrane, and formation of long mitochondrial tubules. Conversely, MIEF1 knockdown resulted in mitochondrial fission and fragmentation. A targeted database search against three-frame translated transcript database resulted in the identification of a peptide “YTDRDFYFASIR” in the 5′ UTR region of MIEF1 (Figure 5a). The translated ORF is 70 amino acids in length and is conserved in rabbit and giant panda. Further studies have to be carried out to delineate the role of the uORF in MIEF1 function.
Figure 5.

(a) Identification of novel upstream ORF in the 5′ untranslated region of mitochondrial elongation factor 1 (MIEF1). The figure represents peptide sequence “YTDRDFYFASIR” mapping to 5′UTR of MIEF1 (SMCR7L). In silico translation of the 5′UTR region revealed the presence of a novel ORF of 70 amino acids. Sequence alignment shows conservation of the novel ORF with other mammals. The alignment of the peptide sequence identified in our study has been highlighted in the panel depicting sequence alignment with rabbit and panda. (b) N-terminal extension of tyrosylprotein sulfotransferase (TPST2). The figure represents peptide sequences “ALPADSLGTQAQGELEPR” and “CEPGGLPANLSLKPQK” mapping to 5′UTR of TPST2 just upstream of the currently annotated translation start site. The peptide sequences are translated in the same frame as the existing protein. Sequence alignment with other primate species reveals existence of an alternate translation start site 210 bp upstream of the currently annotated start site. (c) Evidence of translation on noncoding RNA (FAM230B) The figure represents peptide sequence “EDAAQGIANEAADK” mapping to noncoding RNA FAM230B. GENSCAN, a gene prediction algorithm, predicts an ORF in this region. (d) Identification of novel coding exon in the intron of smoothelin (SMTN). We identified “VAVTQAAEVAVATVEPVAR” mapping to the intronic region of SMTN between exon 9 and 10. Our finding is also supported by several ESTs (subset listed in the figure) as well as GENSCAN prediction.
Based on our proteogenomics analysis, we identified two cases of N-terminal extension of proteins including extension of tyrosylprotein sulfotransferase (TPST2) (Figure 5b) and Ran GTPase activating protein 1 (RANGAP1) both with orthologous evidence in primates. We also provide evidence for noncoding RNA, FAM230B (Figure 5c) and translation of intronic region of smoothelin (SMTN), which encodes a structural protein that is found exclusively in contractile smooth muscle cells. It is known to associate with stress fibers and constitutes part of the cytoskeleton. This gene is localized to chromosome 22q12.3 (Figure 5d). The representative MS/MS spectra of the peptides identified are provided in Supplementary Figure 1.
Conclusions
High-resolution mass spectrometry has enabled us to carry out deep proteome profiling resulting in the high confidence identification of the proteome. Additionally, we have also been able to identify 47 proteins that were previously reported to be missing. Future studies have to be aimed at identifying proteins that are still missed using the targeted approach and sampling of tissues and cells. In addition, generation of MRM assays for proteins encoded by chromosome 22 becomes imperative as the disease-associated role of genes on this chromosome is evident. Proteogenomics has enabled redefining the genome annotation in several organisms, and we have demonstrated its ability to identify several novel events that have previously not been reported. The future efforts of C-HPP should enable characterization of these novel events in addition to focusing on the proteome that currently lacks proteomic evidence. Furthermore, a concerted effort is required to map the different post-translational modifications and subproteomes and to identify protein interaction networks of genes encoded by chromosome 22.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository54 with the data set identifier PXD000561.
Acknowledgments
We thank Drs. Lydie Lane and Amos Bairoch for providing us with the list of missing genes. This work was supported by an NIH roadmap grant for Technology Centers of Networks and Pathways (U54GM103520), NCI’s Clinical Proteomic Tumor Analysis Consortium initiative (U24CA160036), a contract (HHSN268201000032C) from the National Heart, Lung and Blood Institute and the Sol Goldman Pancreatic Cancer Research Center. T.S.K.P is supported by DBT Program Support on Neuroproteomics (BT/01/COE/08/05) to IOB. H.G. is a Wellcome Trust-DBT India Alliance Early Career Fellow. S.M.P. is a recipient of Senior Research Fellowship from Council of Scientific and Industrial Research (CSIR), Government of India. S.S.M. is recipient of a Senior Research Fellowship from the University Grants Commission (UGC), India.
Supporting Information Available
Representative MS/MS spectra of peptides identified by proteogenomics analysis, a list of proteins designated “missing proteins” identified by our study, and a summary of novel protein features on chromosome 22 identified through proteogenomic analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Paik Y. K.; Jeong S. K.; Omenn G. S.; Uhlen M.; Hanash S.; Cho S. Y.; Lee H. J.; Na K.; Choi E. Y.; Yan F.; Zhang F.; Zhang Y.; Snyder M.; Cheng Y.; Chen R.; Marko-Varga G.; Deutsch E. W.; Kim H.; Kwon J. Y.; Aebersold R.; Bairoch A.; Taylor A. D.; Kim K. Y.; Lee E. Y.; Hochstrasser D.; Legrain P.; Hancock W. S. The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol. 2012, 303221–223. [DOI] [PubMed] [Google Scholar]
- Lane L.; Bairoch A.; Beavis R. C.; Deutsch E. W.; Gaudet P.; Lundberg E.; Omenn G. S. Metrics for the Human Proteome Project 2013–2014 and strategies for finding missing proteins. J. Proteome Res. 2014, 13115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J.; Jeong S. K.; Na K.; Lee M. J.; Lee S. H.; Lim J. S.; Cha H. J.; Cho J. Y.; Kwon J. Y.; Kim H.; Song S. Y.; Yoo J. S.; Park Y. M.; Hancock W. S.; Paik Y. K. Comprehensive genome-wide proteomic analysis of human placental tissue for the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2013, 1262458–2466. [DOI] [PubMed] [Google Scholar]
- Segura V.; Medina-Aunon J. A.; Mora M. I.; Martinez-Bartolome S.; Abian J.; Aloria K.; Antunez O.; Arizmendi J. M.; Azkargorta M.; Barcelo-Batllori S.; Beaskoetxea J.; Bech-Serra J. J.; Blanco F.; Monteiro M. B.; Caceres D.; Canals F.; Carrascal M.; Casal J. I.; Clemente F.; Colome N.; Dasilva N.; Diaz P.; Elortza F.; Fernandez-Puente P.; Fuentes M.; Gallardo O.; Gharbi S. I.; Gil C.; Gonzalez-Tejedo C.; Hernaez M. L.; Lombardia M.; Lopez-Lucendo M.; Marcilla M.; Mato J. M.; Mendes M.; Oliveira E.; Orera I.; Pascual-Montano A.; Prieto G.; Ruiz-Romero C.; Sanchez Del Pino M. M.; Tabas-Madrid D.; Valero M. L.; Vialas V.; Villanueva J.; Albar J. P.; Corrales F. J. Surfing transcriptomic landscapes. A step beyond the annotation of chromosome 16 proteome. J. Proteome Res. 2013, 131158–172. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Wen B.; Yan G.; Wei J.; Xie L.; Xu S.; Jiang D.; Wang T.; Lin L.; Zi J.; Zhang J.; Zhou R.; Zhao H.; Ren Z.; Qu N.; Lou X.; Sun H.; Du C.; Chen C.; Zhang S.; Tan F.; Xian Y.; Gao Z.; He M.; Chen L.; Zhao X.; Xu P.; Zhu Y.; Yin X.; Shen H.; Zhang Y.; Jiang J.; Zhang C.; Li L.; Chang C.; Ma J.; Yao J.; Lu H.; Ying W.; Zhong F.; He Q. Y.; Liu S. Qualitative and quantitative expression status of the human chromosome 20 genes in cancer tissues and the representative cell lines. J. Proteome Res. 2013, 121151–161. [DOI] [PubMed] [Google Scholar]
- Fagerberg L.; Oksvold P.; Skogs M.; Algenas C.; Lundberg E.; Ponten F.; Sivertsson A.; Odeberg J.; Klevebring D.; Kampf C.; Asplund A.; Sjostedt E.; Al-Khalili Szigyarto C.; Edqvist P. H.; Olsson I.; Rydberg U.; Hudson P.; Ottosson Takanen J.; Berling H.; Bjorling L.; Tegel H.; Rockberg J.; Nilsson P.; Navani S.; Jirstrom K.; Mulder J.; Schwenk J. M.; Zwahlen M.; Hober S.; Forsberg M.; von Feilitzen K.; Uhlen M. Contribution of antibody-based protein profiling to the human Chromosome-Centric Proteome Project (C-HPP). J. Proteome Res. 2013, 1262439–48. [DOI] [PubMed] [Google Scholar]
- Dunham I.; Shimizu N.; Roe B. A.; Chissoe S.; Hunt A. R.; Collins J. E.; Bruskiewich R.; Beare D. M.; Clamp M.; Smink L. J.; Ainscough R.; Almeida J. P.; Babbage A.; Bagguley C.; Bailey J.; Barlow K.; Bates K. N.; Beasley O.; Bird C. P.; Blakey S.; Bridgeman A. M.; Buck D.; Burgess J.; Burrill W. D.; O’Brien K. P.; et al. The DNA sequence of human chromosome 22. Nature 1999, 4026761489–495. [DOI] [PubMed] [Google Scholar]
- Morton N. E. Parameters of the human genome. Proc. Natl. Acad. Sci. U.S.A. 1991, 88177474–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson G. N.; Baker D. L.; Schau J.; Parker J. Cat eye syndrome owing to tetrasomy 22pter leads to q11. J. Med. Genet 1984, 21160–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll D. A.; Budarf M. L.; Emanuel B. S. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am. J. Hum. Genet. 1992, 505924–933. [PMC free article] [PubMed] [Google Scholar]
- Shprintzen R. J.; Goldberg R. B.; Lewin M. L.; Sidoti E. J.; Berkman M. D.; Argamaso R. V.; Young D. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978, 15156–62. [PubMed] [Google Scholar]
- Wong A. C.; Ning Y.; Flint J.; Clark K.; Dumanski J. P.; Ledbetter D. H.; McDermid H. E. Molecular characterization of a 130-kb terminal microdeletion at 22q in a child with mild mental retardation. Am. J. Hum. Genet. 1997, 601113–120. [PMC free article] [PubMed] [Google Scholar]
- Dreazen O.; Klisak I.; Jones G.; Ho W. G.; Sparkes R. S.; Gale R. P. Multiple molecular abnormalities in Ph1 chromosome positive acute lymphoblastic leukaemia. Br. J. Haematol. 1987, 673319–324. [DOI] [PubMed] [Google Scholar]
- Kim M. S.; Pinto S. M.; Getnet D.; Nirujogi R. S.; Manda S. S.; Chaerkady R.; Madugundu A. K.; Kelkar D. S.; Isserlin R.; Jain S.; Thomas J. K.; Muthusamy B.; Leal-Rojas P.; Kumar P.; Sahasrabuddhe N. A.; Balakrishnan L.; Advani J.; George B.; Renuse S.; Selvan L. D. N.; Patil A. H.; Nanjappa V.; Radhakrishnan A.; Prasad S.; Subbannayya T.; Raju R.; Kumar M.; Sreenivasamurthy S. K.; Marimuthu A.; Sathe G. J.; Chavan S.; Datta K. K.; Subbannayya Y.; Sahu A.; Yelamanchi S. D.; Jayaram S.; Rajagopalan P.; Sharma J.; Murthy K. R.; Syed N.; Goel R.; Khan A. K.; Ahmad S.; Dey G.; Mudgal K.; Chatterjee A.; Huang T.; Zhong J.; Wu X.; Shaw P. G.; Freed D.; Zahari M. S.; Mukherjee K. K.; Shankar S.; Mahadevan A.; Lam H.; Mitchell C. J.; Shankar S. K.; Satishchandra P.; Schroeder J. T.; Sirdeshmukh R.; Maitra A.; Leach S. D.; Drake C. G.; Halushka M. K.; Prasad T. S. K.; Hruban R. H.; Kerr C. L.; Bader G. D.; Iacobuzio-Donahue C. H.; Gowda H.; Pandey A.. A draft map of the human proteome. Nature 2014, In press [DOI] [PMC free article] [PubMed]
- Harsha H. C.; Molina H.; Pandey A. Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat. Protoc. 2008, 33505–516. [DOI] [PubMed] [Google Scholar]
- Kall L.; Canterbury J. D.; Weston J.; Noble W. S.; MacCoss M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 411923–925. [DOI] [PubMed] [Google Scholar]
- Keshava Prasad T. S.; Goel R.; Kandasamy K.; Keerthikumar S.; Kumar S.; Mathivanan S.; Telikicherla D.; Raju R.; Shafreen B.; Venugopal A.; Balakrishnan L.; Marimuthu A.; Banerjee S.; Somanathan D. S.; Sebastian A.; Rani S.; Ray S.; Harrys Kishore C. J.; Kanth S.; Ahmed M.; Kashyap M. K.; Mohmood R.; Ramachandra Y. L.; Krishna V.; Rahiman B. A.; Mohan S.; Ranganathan P.; Ramabadran S.; Chaerkady R.; Pandey A. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37Database issueD767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H.; Thievessen I.; Sixt M.; Lammermann T.; Waisman A.; Braun A.; Noegel A. A.; Fassler R. Gamma-Parvin is dispensable for hematopoiesis, leukocyte trafficking, and T-cell-dependent antibody response. Mol. Cell. Biol. 2006, 2651817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko-Varga G.; Omenn G. S.; Paik Y. K.; Hancock W. S. A first step toward completion of a genome-wide characterization of the human proteome. J. Proteome Res. 2013, 1211–5. [DOI] [PubMed] [Google Scholar]
- Veenman L.; Papadopoulos V.; Gavish M. Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des 2007, 13232385–2405. [DOI] [PubMed] [Google Scholar]
- Footz T. K.; Brinkman-Mills P.; Banting G. S.; Maier S. A.; Riazi M. A.; Bridgland L.; Hu S.; Birren B.; Minoshima S.; Shimizu N.; Pan H.; Nguyen T.; Fang F.; Fu Y.; Ray L.; Wu H.; Shaull S.; Phan S.; Yao Z.; Chen F.; Huan A.; Hu P.; Wang Q.; Loh P.; Qi S.; Roe B. A.; McDermid H. E. Analysis of the cat eye syndrome critical region in humans and the region of conserved synteny in mice: a search for candidate genes at or near the human chromosome 22 pericentromere. Genome Res. 2001, 1161053–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelstaedt T.; Schoch S. Structure and evolution of RIM-BP genes: identification of a novel family member. Gene 2007, 4031–270–79. [DOI] [PubMed] [Google Scholar]
- Frippiat J. P.; Williams S. C.; Tomlinson I. M.; Cook G. P.; Cherif D.; Le Paslier D.; Collins J. E.; Dunham I.; Winter G.; Lefranc M. P. Organization of the human immunoglobulin lambda light-chain locus on chromosome 22q11.2. Hum. Mol. Genet. 1995, 46983–991. [DOI] [PubMed] [Google Scholar]
- Morris C.; Courtay C.; Geurts van Kessel A.; ten Hoeve J.; Heisterkamp N.; Groffen J. Localization of a gamma-glutamyl-transferase-related gene family on chromosome 22. Hum. Genet. 1993, 91131–36. [DOI] [PubMed] [Google Scholar]
- Seroussi E.; Kedra D.; Pan H. Q.; Peyrard M.; Schwartz C.; Scambler P.; Donnai D.; Roe B. A.; Dumanski J. P. Duplications on human chromosome 22 reveal a novel Ret Finger Protein-like gene family with sense and endogenous antisense transcripts. Genome Res. 1999, 99803–814. [DOI] [PubMed] [Google Scholar]
- Duchateau P. N.; Pullinger C. R.; Cho M. H.; Eng C.; Kane J. P. Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J. Lipid Res. 2001, 424620–630. [PubMed] [Google Scholar]
- Bijlsma E. K.; Delattre O.; Juyn J. A.; Melot T.; Westerveld A.; Dumanski J. P.; Thomas G.; Hulsebos T. J. Regional fine mapping of the beta Crystallin genes on chromosome 22 excludes these genes as physically linked markers for neurofibromatosis type 2. Genes, Chromosomes Cancer 1993, 82112–118. [DOI] [PubMed] [Google Scholar]
- Hulsebos T. J.; Jenkins N. A.; Gilbert D. J.; Copeland N. G. The beta Crystallin genes on human chromosome 22 define a new region of homology with mouse chromosome 5. Genomics 1995, 252574–576. [DOI] [PubMed] [Google Scholar]
- Jarmuz A.; Chester A.; Bayliss J.; Gisbourne J.; Dunham I.; Scott J.; Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 793285–296. [DOI] [PubMed] [Google Scholar]
- Cullen B. R. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol 2006, 8031067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey L.; Law E. K.; Brown W. L.; Harris R. S. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 2013, 125762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaratnam N.; Morrison J. R.; Bhattacharya S.; Patel D.; Funahashi T.; Giannoni F.; Teng B. B.; Davidson N. O.; Scott J. The p27 catalytic subunit of the apolipoprotein B mRNA editing enzyme is a cytidine deaminase. J. Biol. Chem. 1993, 2682820709–20712. [PubMed] [Google Scholar]
- Teng B.; Burant C. F.; Davidson N. O. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 1993, 26051151816–1819. [DOI] [PubMed] [Google Scholar]
- Dang Y.; Siew L. M.; Wang X.; Han Y.; Lampen R.; Zheng Y. H. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J. Biol. Chem. 2008, 2831711606–11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seng T. J.; Ichimura K.; Liu L.; Tingby O.; Pearson D. M.; Collins V. P. Complex chromosome 22 rearrangements in astrocytic tumors identified using microsatellite and chromosome 22 tile path array analysis. Genes, Chromosomes Cancer 2005, 432181–193. [DOI] [PubMed] [Google Scholar]
- Tsilchorozidou T.; Menko F. H.; Lalloo F.; Kidd A.; De Silva R.; Thomas H.; Smith P.; Malcolmson A.; Dore J.; Madan K.; Brown A.; Yovos J. G.; Tsaligopoulos M.; Vogiatzis N.; Baser M. E.; Wallace A. J.; Evans D. G. Constitutional rearrangements of chromosome 22 as a cause of neurofibromatosis 2. J. Med. Genet 2004, 417529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Im H.; Bairoch A.; Cristofanilli M.; Chen R.; Deutsch E. W.; Dalton S.; Fenyo D.; Fanayan S.; Gates C.; Gaudet P.; Hincapie M.; Hanash S.; Kim H.; Jeong S. K.; Lundberg E.; Mias G.; Menon R.; Mu Z.; Nice E.; Paik Y. K.; Uhlen M.; Wells L.; Wu S. L.; Yan F.; Zhang F.; Zhang Y.; Snyder M.; Omenn G. S.; Beavis R. C.; Hancock W. S. A chromosome-centric human proteome project (C-HPP) to characterize the sets of proteins encoded in chromosome 17. J. Proteome Res. 2013, 12145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R. J.; Paez J. G.; Curto M.; Yaktine A.; Pruitt W. M.; Saotome I.; O’Bryan J. P.; Gupta V.; Ratner N.; Der C. J.; Jacks T.; McClatchey A. I. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev. Cell 2001, 1163–72. [DOI] [PubMed] [Google Scholar]
- Morrison H.; Sperka T.; Manent J.; Giovannini M.; Ponta H.; Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007, 672520–527. [DOI] [PubMed] [Google Scholar]
- James M. F.; Han S.; Polizzano C.; Plotkin S. R.; Manning B. D.; Stemmer-Rachamimov A. O.; Gusella J. F.; Ramesh V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol. Cell. Biol. 2009, 29154250–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F.; Willecke M.; Kango-Singh M.; Nolo R.; Hyun E.; Tao C.; Jafar-Nejad H.; Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006, 8127–36. [DOI] [PubMed] [Google Scholar]
- Rouleau G. A.; Merel P.; Lutchman M.; Sanson M.; Zucman J.; Marineau C.; Hoang-Xuan K.; Demczuk S.; Desmaze C.; Plougastel B.; et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 1993, 3636429515–521. [DOI] [PubMed] [Google Scholar]
- Trofatter J. A.; MacCollin M. M.; Rutter J. L.; Murrell J. R.; Duyao M. P.; Parry D. M.; Eldridge R.; Kley N.; Menon A. G.; Pulaski K.; et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993, 725791–800. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J.; Legoix P.; Thomas G. Identification of new members of the Gas2 and Ras families in the 22q12 chromosome region. Genomics 1996, 383247–254. [DOI] [PubMed] [Google Scholar]
- Pandey A.; Mann M. Proteomics to study genes and genomes. Nature 2000, 4056788837–846. [DOI] [PubMed] [Google Scholar]
- Chaerkady R.; Kelkar D. S.; Muthusamy B.; Kandasamy K.; Dwivedi S. B.; Sahasrabuddhe N. A.; Kim M. S.; Renuse S.; Pinto S. M.; Sharma R.; Pawar H.; Sekhar N. R.; Mohanty A. K.; Getnet D.; Yang Y.; Zhong J.; Dash A. P.; MacCallum R. M.; Delanghe B.; Mlambo G.; Kumar A.; Keshava Prasad T. S.; Okulate M.; Kumar N.; Pandey A. A proteogenomic analysis of Anopheles gambiae using high-resolution Fourier transform mass spectrometry. Genome Res. 2011, 21111872–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar D. S.; Kumar D.; Kumar P.; Balakrishnan L.; Muthusamy B.; Yadav A. K.; Shrivastava P.; Marimuthu A.; Anand S.; Sundaram H.; Kingsbury R.; Harsha H. C.; Nair B.; Prasad T. S.; Chauhan D. S.; Katoch K.; Katoch V. M.; Chaerkady R.; Ramachandran S.; Dash D.; Pandey A. Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry. Mol. Cell Proteomics 2011, 101210.1074/mcp.M111.011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar H.; Sahasrabuddhe N. A.; Renuse S.; Keerthikumar S.; Sharma J.; Kumar G. S.; Venugopal A.; Sekhar N. R.; Kelkar D. S.; Nemade H.; Khobragade S. N.; Muthusamy B.; Kandasamy K.; Harsha H. C.; Chaerkady R.; Patole M. S.; Pandey A. A proteogenomic approach to map the proteome of an unsequenced pathogen - Leishmania donovani. Proteomics 2012, 126832–844. [DOI] [PubMed] [Google Scholar]
- Calvo S. E.; Pagliarini D. J.; Mootha V. K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. U.S.A. 2009, 106187507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri S.; Pandey A. A reassessment of the translation initiation codon in vertebrates. Trends Genet 2001, 1712685–687. [DOI] [PubMed] [Google Scholar]
- Yamashita R.; Suzuki Y.; Nakai K.; Sugano S. Small open reading frames in 5′ untranslated regions of mRNAs. C R Biol. 2003, 32610–11987–991. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Liu T.; Jin S.; Wang X.; Qu M.; Uhlen P.; Tomilin N.; Shupliakov O.; Lendahl U.; Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011, 30142762–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C. S.; Elgass K. D.; Parton R. G.; Osellame L. D.; Stojanovski D.; Ryan M. T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 2883827584–27593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino J. A.; Cote R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; O’Kelly G.; Schoenegger A.; Ovelleiro D.; Perez-Riverol Y.; Reisinger F.; Rios D.; Wang R.; Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013, 41Database issueD1063–D1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository54 with the data set identifier PXD000561.