

Abstract
During infection by picornaviruses, the cellular environment is modified to favour virus replication. This includes the modification of specific host proteins, including the recently discovered viral proteinase cleavage of mRNA decay factor AU-rich binding factor 1 (AUF1). This cellular RNA-binding protein was shown previously to act as a restriction factor during poliovirus, rhinovirus and coxsackievirus infection. During infection by these viruses, AUF1 relocalizes to the cytoplasm and is cleaved by the viral 3C/3CD proteinase. In this study, we demonstrated that replication of encephalomyocarditis virus (EMCV), a picornavirus belonging to the genus Cardiovirus, is AUF1 independent. During EMCV infection, AUF1 relocalized to the cytoplasm; however, unlike what is seen during enterovirus infections, AUF1 was not cleaved to detectable levels, even at late times after infection. This suggests that AUF1 does not act broadly as an inhibitor of picornavirus infections but may instead act as a selective restriction factor targeting members of the genus Enterovirus.
The family Picornaviridae encompasses a wide range of positive-sense RNA viruses that complete their replication cycles in the cytoplasm of infected cells and employ unique mechanisms to alter the cytoplasmic environment to favour virus production. All picornaviruses encode a proteolytically active 3C/3CD protein, which functions in proteolytic processing of the viral polyprotein and in alteration of the cellular environment during infection. Additionally, members of the genus Enterovirus, including poliovirus (PV), coxsackievirus and human rhinovirus (HRV), encode a proteolytically active 2A protein that cleaves specific cellular proteins in infected cells. In contrast, the prototypic member of the genus Cardiovirus, encephalomyocarditis virus (EMCV), encodes a unique 2A protein that is not a proteinase. Despite differences in proteinase-coding capacity, enteroviruses and cardioviruses alter the cellular environment in a similar manner. For example, during the initial stages of infection, modification of host proteins, including the cleavage of eIF4G (for enteroviruses) and dephosphorylation of 4E-BP1 (for EMCV), effectively shuts down cap-dependent translation (Agol & Gmyl, 2010; Castelló et al., 2011; Gingras et al., 1996). Additionally, the cleavage of nucleoporin proteins by the enterovirus 2A proteinase or phosphorylation of nucleoporins by the EMCV leader (L) protein alters the activity of specific nucleocytoplasmic transport pathways (Castelló et al., 2009; Park et al., 2010; Porter & Palmenberg, 2009; Porter et al., 2010; Watters & Palmenberg, 2011). This disruption leads to an increase in cytoplasmic levels of cellular proteins usurped by the virus in its replication cycle, including hnRNP C, SRp20, PTB and La (Back et al., 2002; Fitzgerald & Semler, 2011; Gustin & Sarnow, 2001; Shiroki et al., 1999; reviewed by Castelló et al., 2011).
Restriction factors are important determinants of the outcome of viral infection, and RNA viruses have evolved multiple routes of escape from such limiting factors. To evade the immune response, PV and EMCV degrade the cellular sensors of viral RNA: RIG-I and MDA-5 (Barral et al., 2007, 2009; Papon et al., 2009). Additionally, cleavage of IPS-1 and NF-κB during PV infection and disruption of IRF-3 dimerization by the EMCV L protein act to interrupt IFN signalling (Agol & Gmyl, 2010; Hato et al., 2007; Racaniello, 2010). Recently, the potential disruption of mRNA decay pathways has been discovered for enteroviruses. The decapping enzyme Dcp1a, the exonuclease Xrn1, the deadenylase complex component Pan3 and the mRNA stability factor AU-rich element-binding factor 1 (AUF1) are cleaved or degraded during PV infection (Dougherty et al., 2011; Rozovics et al., 2012), presumably as a viral defence against the degradation of viral RNA during infection.
We recently reported that AUF1, a cellular protein involved in mRNA stability, binds directly to PV stem–loop IV of the 5′ non-coding region (NCR) in vitro and has an inhibitory effect on viral translation and overall viral titres (Cathcart et al., 2013). This AUF1–viral RNA interaction is disrupted by incubation with proteolytically active 3CD, which has been shown to cleave AUF1 in vitro, suggesting the cleavage of AUF1 by 3C/3CD may act as a viral defence against AUF1 and the cellular mRNA decay machinery. Additionally, AUF1 relocalizes to the cytoplasm of PV-infected cells where it partially co-localizes with viral proteins but is excluded from a perinuclear region by peak times of virus replication (Cathcart et al., 2013; Rozovics et al., 2012). The inhibitory effect of AUF1 on viral titre, as well as the relocalization of AUF1 and 3C proteinase cleavage during infection, has been confirmed for other members of the genus Enterovirus, including HRV and coxsackievirus B3 (CVB3) (Cathcart et al., 2013; Rozovics et al., 2012; Spurrell et al., 2005; Wong et al., 2013). In this study, we sought to determine whether AUF1 acts as a broad regulator of picornavirus infections by exploring the potential modification of AUF1 during EMCV infection.
Given the known relocalization of AUF1 to the cytoplasm of enterovirus-infected cells, we first investigated the localization of AUF1 in EMCV-infected cells by confocal microscopy (Fig. 1). HeLa cell monolayers seeded on coverslips were either mock infected (Fig. 1a) or infected with EMCV at an m.o.i. of 20 (Fig. 1b–d) and fixed with formaldehyde at the indicated times post-infection (p.i.). Endogenous AUF1 was visualized using polyclonal anti-AUF1 antibody (Millipore) and Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes). In mock-infected cells, AUF1 was predominantly nuclear (Fig. 1a). By 4 h p.i. (Fig. 1c), AUF1 had partially relocalized to the cytoplasm of infected cells, and this relocalization was more dramatic by 6 h p.i. (Fig. 1d). Although EMCV alters nuclear transport via different mechanisms than enteroviruses, AUF1 does relocalize to the cytoplasm during EMCV infection. Notably, the EMCV L protein and PV 2A proteinase are known to modify the same nucleoporins, including Nup153 and Nup62 (Gustin & Sarnow, 2001; Porter & Palmenberg, 2009; Porter et al., 2010).
Fig. 1.
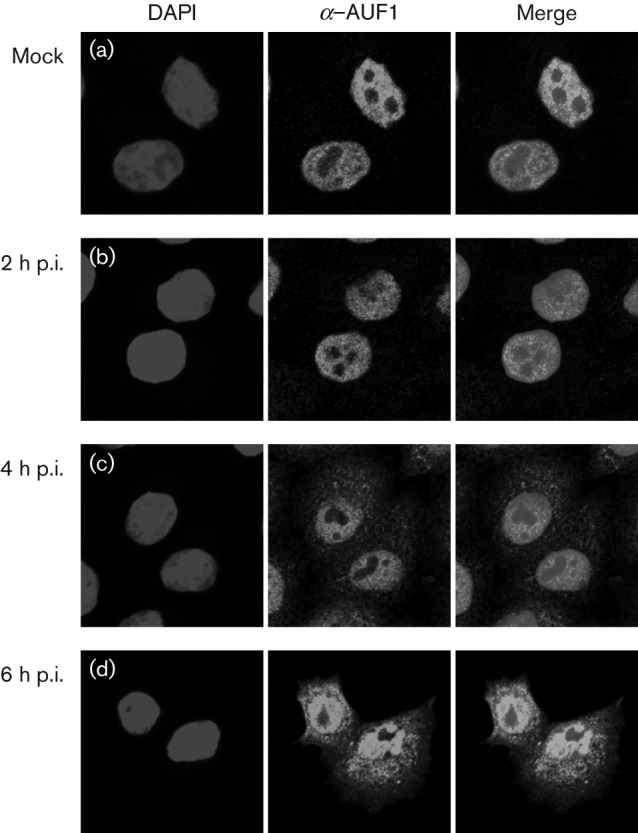
Relocalization of AUF1 in EMCV-infected cells. HeLa cells were seeded on coverslips and either mock infected (a) or infected with EMCV at an m.o.i. of 20. Cells were fixed at 2 (b), 4 (a, c) or 6 (d) h p.i. and localization of AUF1 was examined by confocal microscopy with antibodies against AUF1 (green). DAPI staining (blue) was used to visualize nuclei.
During enterovirus infections, the electrophoretic mobility of AUF1 is modified, due to the cleavage activity of viral 3C/3CD proteinase on AUF1 (Rozovics et al., 2012; Wong et al., 2013). To determine whether AUF1 is modified in a similar manner during cardiovirus infection, Western blot analysis was performed on lysates from HeLa cells infected with PV, CVB3, HRV1a or EMCV (Fig. 2). To generate lysates, cell monolayers were mock infected or infected at an m.o.i. of 20 with the indicated virus. Cells were collected at the indicated times p.i., lysed in buffer containing NP-40 and the proteins resolved by SDS-PAGE followed by Western blotting using polyclonal antibody to AUF1. Whilst cleavage of AUF1 was readily seen for cells infected with PV (Fig. 2, lane 2), CVB3 (Fig. 2, lane 4) or HRV1a (Fig. 2, lane 6) by late times after infection, cleavage of AUF1 was not detected in EMCV-infected cells, even at very late times p.i. (Fig. 2, lane 8). Cleavage products of AUF1 were still not detected when twice the amount of total protein was loaded from lysates generated at 6 or 7 h p.i. with EMCV (data not shown). These results demonstrated that AUF1 is differentially modified during infection by picornaviruses. Notably, HRV1a displayed an alternative cleavage pattern of AUF1 when compared with cleavage of AUF1 during CVB3 or PV infection (Fig. 2) (Rozovics et al., 2012). This is consistent with reports of lower stringency in cleavage-site recognition by the HRV 3C proteinase compared with that of PV (Palmenberg, 1990).
Fig. 2.
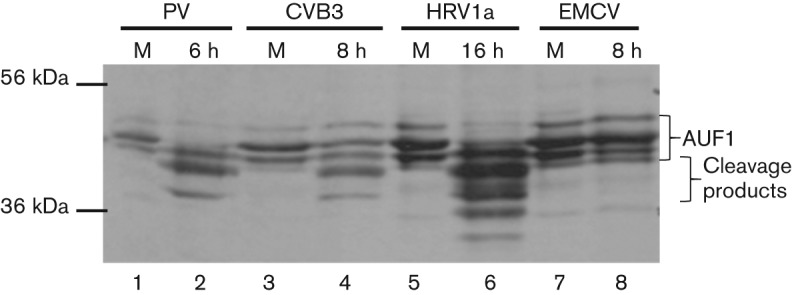
Differential cleavage of AUF1 in picornavirus-infected cells. HeLa cell monolayers were infected with PV (lane 2), CVB3 (lane 4), HRV1a (lane 6) or EMCV (lane 8) and NP-40 lysates were generated at the indicated times p.i. in tandem with mock-infected samples (lanes 1, 3, 5 and 7). The lysates were subjected to SDS-PAGE and Western blot analysis with anti-AUF1 rabbit polyclonal antibody to visualize AUF1. Molecular mass markers are indicated on the left of the gel image and AUF1 and AUF1 cleavage products are indicated with brackets to the right.
To determine the effect of AUF1 on EMCV infection, we used mouse embryonic fibroblast cells genetically deleted for AUF1 (MEF-AUF1−/−) (Lee et al., 2008). For the results shown in Fig. 3(a), MEF-AUF1−/−(black bars) or MEF-AUF1+/+(white bars) monolayers were infected with CVB3, HRV1a or EMCV. As mouse cells lack the PV receptor, PV transcripts corresponding to full-length genomic RNAs were transfected into cells as described by Cathcart et al. (2013). The cells and supernatant were collected at late times p.i., and viral titres were determined by plaque assay on HeLa cell monolayers. As reported previously, PV, CVB3 and HRV1a displayed an increase in viral titre in cells lacking AUF1, demonstrating a negative role for AUF1 in their replication cycle. In contrast, EMCV titres were not significantly different in the presence or absence of AUF1. To confirm this result, a single-cycle growth analysis was performed in HeLa, MEF-AUF1−/− or MEF-AUF1+/+ cell monolayers (Fig. 3b). The titres of EMCV remained similar over the course of infection in both MEF-AUF1−/− and MEF-AUF1+/+ cells (Fig. 3b, 0–8 h p.i.). In contrast to the increase in titre seen in MEF-AUF1−/− cells during enterovirus infection, titres of EMCV were instead slightly lower in MEF cells lacking AUF1 at 10 or 12 h p.i., although not to statistically significant levels (Fig. 3a, b, and data not shown).
Fig. 3.
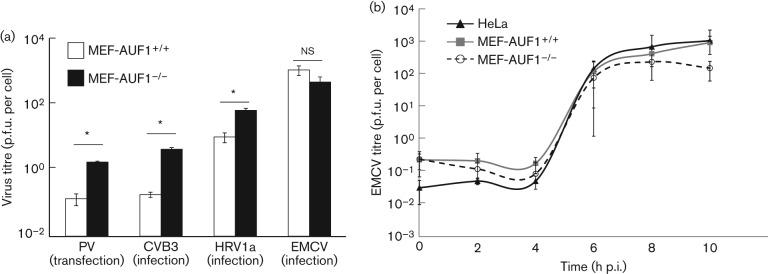
Depletion of AUF1 differentially affects picornavirus infectious cycles. (a) MEF cell monolayers (AUF1+/+ or AUF1−/−) were infected with CVB3 (m.o.i. 100), HRV1a (m.o.i. 10) or EMCV (m.o.i. 20). The cells and supernatant were collected at late times p.i. (CVB3 at 8 h p.i., HRV1a at 20 h p.i. and EMCV at 10 h p.i.) and the titre was determined by plaque assay on HeLa cell monolayers. For PV, 5 µg transcript RNA was transfected into MEF cell monolayers using DEAE-dextran; the cells and supernatant were collected at 8 h post-transfection. Statistical significance is indicated on the graph (*P<0.01, Student’s t-test; ns, not significant). (b) HeLa, MEF-AUF1−/− or MEF-AUF1+/+ cell monolayers were infected with EMCV at an m.o.i. of 20. The cells and supernatant were harvested at different times from 0 up to 10 h p.i., and virus yield was determined by plaque assay on HeLa cell monolayers. Results are shown as means±sd from triplicate samples.
Taken together, these results indicate that, although AUF1 relocalizes to the cytoplasm during EMCV infection, AUF1 is not cleaved to detectable levels in EMCV-infected cells and does not have an effect on overall viral titres. These results suggest a different susceptibility to cellular mRNA decay factors between genera of picornaviruses, despite a similar replication scheme. In accordance with the results reported in this study, translation driven by the EMCV internal ribosome entry site (IRES) has been shown previously to be unaffected by AUF1 in a luciferase reporter assay (Paek et al., 2008). Notably, the secondary structure and IRES trans-acting factor requirements of the EMCV IRES are different than those of enteroviruses (reviewed by Fitzgerald & Semler, 2009). It is possible that these structural differences render EMCV RNA resistant to interaction with AUF1.
The discordance between relocalization of AUF1 during infection and cleavage state raises the question of 3C/3CD proteinase substrate recruitment. Is the difference in the cleavage state of AUF1 during enterovirus and EMCV infection a result of substrate specificity or of intracellular localization? It would be informative to determine whether AUF1 is cleaved in vitro by recombinant EMCV 3C proteinase. Perhaps it is the binding of AUF1 to enterovirus RNA that makes it a target for 3C/3CD proteinase cleavage, and this cleavage acts solely to disrupt the AUF1–viral RNA interaction.
To establish productive infections, viruses must evade cellular antiviral responses, including protecting viral RNA from degradation. As positive-sense RNA viruses, the genomic RNAs of picornaviruses are inherently targets for mRNA decay. As described recently for AUF1 and shown previously for other factors involved in mRNA decay (Xrn1, Dcp1a and PAN3), PV evades, in part, the cellular mRNA decay response by cleavage of essential players in the RNA degradation pathway (Cathcart et al., 2013; Dougherty et al., 2011; Rozovics et al., 2012; Wong et al., 2013). Members of the genus Flavivirus in the family Flaviviridae use a different strategy to avoid host mRNA decay proteins. Their genomic RNAs are capped at the 5′ end but lack a 3′ poly(A) tract. The 3′ NCR of flavivirus RNAs contains a pseudoknot structure that effectively blocks the Xrn1 exoribonuclease, and this stalling has also been linked to the disruption of cellular mRNA degradation (Funk et al., 2010; Moon et al., 2012; Silva et al., 2010). PV has also been shown to contain an RNA element that inhibits RNA decay machinery via its interaction with the IFN-induced RNase L (Townsend et al., 2008). As an alternative to disrupting mRNA decay, viruses may usurp cellular factors to increase viral RNA stability, as has been shown for PV with PCBP2 (Murray et al., 2001). Another AU-rich element-binding protein, HuR, binds to the positive-strand genomic RNA of Sindbis virus (an alphavirus), protecting the viral RNA from deadenylation (Garneau et al., 2008; Sokoloski et al., 2010). Hepatitis C virus, a flavivirus, stabilizes its RNA via binding of the microRNA miR-122 to the 5′ NCR, potentially protecting the 5′ end from exonuclease digestion (Shimakami et al., 2012). EMCV may avoid RNA degradation via an unknown alternative mechanism, perhaps through cleavage of other cellular mRNA decay proteins that are upstream of AUF1 in the mRNA decay pathway, or by usurping stabilizing factors.
In summary, our results highlight the different susceptibilities of RNA viruses to negative regulators produced in infected cells. Such susceptibilities may vary significantly for members of different genera within the family Picornaviridae, as our data have suggested. In addition, the factors determining the sensitivity of picornaviruses to AUF1 may include RNA structure, RNA sequence, subcellular localization, ribonucleoprotein complex composition or other features that, for now, remain to be determined.
Acknowledgements
We thank Sonia Maciejewski for critical comments on the manuscript. We are indebted to Robert Schneider for generously providing the MEF-AUF1+/+ and MEF-AUF1−/− cell lines, Yury Bochkov for the HRV1a virus stock and Ann C. Palmenberg for the plasmid encoding EMCV cDNA. We thank Hung Nguyen and MyPhuong Tran for their expert technical assistance. We also thank Adeeyla Syed at the UCI Optical Biology Core for confocal microscopy training. Confocal microscopy images were generated at the UCI Optical Biology Core facility, which is supported by the Comprehensive Cancer Center award P30CA062203 from the National Cancer Institute. This research was supported by the National Institutes of Health (Public Health Service grant AI026765) and by the California Center for Antiviral Drug Discovery (a Multicampus Research Program Initiative from the University of California).
References
- Agol V. I., Gmyl A. P. (2010). Viral security proteins: counteracting host defences. Nat Rev Microbiol 8, 867–878 10.1038/nrmicro2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back S. H., Kim Y. K., Kim W. J., Cho S., Oh H. R., Kim J. E., Jang S. K. (2002). Translation of polioviral mRNA is inhibited by cleavage of polypyrimidine tract-binding proteins executed by polioviral 3Cpro. J Virol 76, 2529–2542 10.1128/jvi.76.5.2529-2542.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral P. M., Morrison J. M., Drahos J., Gupta P., Sarkar D., Fisher P. B., Racaniello V. R. (2007). MDA-5 is cleaved in poliovirus-infected cells. J Virol 81, 3677–3684 10.1128/JVI.01360-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral P. M., Sarkar D., Fisher P. B., Racaniello V. R. (2009). RIG-I is cleaved during picornavirus infection. Virology 391, 171–176 10.1016/j.virol.2009.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelló A., Izquierdo J. M., Welnowska E., Carrasco L. (2009). RNA nuclear export is blocked by poliovirus 2A protease and is concomitant with nucleoporin cleavage. J Cell Sci 122, 3799–3809 10.1242/jcs.055988 [DOI] [PubMed] [Google Scholar]
- Castelló A., Alvarez E., Carrasco L. (2011). The multifaceted poliovirus 2A protease: regulation of gene expression by picornavirus proteases. J Biomed Biotechnol 2011, 369648. 10.1155/2011/369648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathcart A. L., Rozovics J. M., Semler B. L. (2013). Cellular mRNA decay protein AUF1 negatively regulates enterovirus and human rhinovirus infections. J Virol 87, 10423–10434 10.1128/JVI.01049-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty J. D., White J. P., Lloyd R. E. (2011). Poliovirus-mediated disruption of cytoplasmic processing bodies. J Virol 85, 64–75 10.1128/JVI.01657-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald K. D., Semler B. L. (2009). Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta 1789, 518–528 10.1016/j.bbagrm.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald K. D., Semler B. L. (2011). Re-localization of cellular protein SRp20 during poliovirus infection: bridging a viral IRES to the host cell translation apparatus. PLoS Pathog 7, e1002127. 10.1371/journal.ppat.1002127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk A., Truong K., Nagasaki T., Torres S., Floden N., Balmori Melian E., Edmonds J., Dong H., Shi P. Y., Khromykh A. A. (2010). RNA structures required for production of subgenomic flavivirus RNA. J Virol 84, 11407–11417 10.1128/JVI.01159-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau N. L., Sokoloski K. J., Opyrchal M., Neff C. P., Wilusz C. J., Wilusz J. (2008). The 3′ untranslated region of Sindbis virus represses deadenylation of viral transcripts in mosquito and mammalian cells. J Virol 82, 880–892 10.1128/JVI.01205-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A. C., Svitkin Y., Belsham G. J., Pause A., Sonenberg N. (1996). Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc Natl Acad Sci U S A 93, 5578–5583 10.1073/pnas.93.11.5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin K. E., Sarnow P. (2001). Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J 20, 240–249 10.1093/emboj/20.1.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hato S. V., Ricour C., Schulte B. M., Lanke K. H., de Bruijni M., Zoll J., Melchers W. J., Michiels T., van Kuppeveld F. J. (2007). The mengovirus leader protein blocks interferon-α/β gene transcription and inhibits activation of interferon regulatory factor 3. Cell Microbiol 9, 2921–2930 10.1111/j.1462-5822.2007.01006.x [DOI] [PubMed] [Google Scholar]
- Lee C., Gyorgy A., Maric D., Sadri N., Schneider R. J., Barker J. L., Lawson M., Agoston D. V. (2008). Members of the NuRD chromatin remodeling complex interact with AUF1 in developing cortical neurons. Cereb Cortex 18, 2909–2919 10.1093/cercor/bhn051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S. L., Anderson J. R., Kumagai Y., Wilusz C. J., Akira S., Khromykh A. A., Wilusz J. (2012). A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 18, 2029–2040 10.1261/rna.034330.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray K. E., Roberts A. W., Barton D. J. (2001). Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA 7, 1126–1141 10.1017/S1355838201010044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek K. Y., Kim C. S., Park S. M., Kim J. H., Jang S. K. (2008). RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J Virol 82, 12082–12093 10.1128/JVI.01405-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmenberg A. C. (1990). Proteolytic processing of picornaviral polyprotein. Annu Rev Microbiol 44, 603–623 10.1146/annurev.mi.44.100190.003131 [DOI] [PubMed] [Google Scholar]
- Papon L., Oteiza A., Imaizumi T., Kato H., Brocchi E., Lawson T. G., Akira S., Mechti N. (2009). The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393, 311–318 10.1016/j.virol.2009.08.009 [DOI] [PubMed] [Google Scholar]
- Park N., Skern T., Gustin K. E. (2010). Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. J Biol Chem 285, 28796–28805 10.1074/jbc.M110.143404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter F. W., Palmenberg A. C. (2009). Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition by cardioviruses. J Virol 83, 1941–1951 10.1128/JVI.01752-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter F. W., Brown B., Palmenberg A. C. (2010). Nucleoporin phosphorylation triggered by the encephalomyocarditis virus leader protein is mediated by mitogen-activated protein kinases. J Virol 84, 12538–12548 10.1128/JVI.01484-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaniello V. R. (2010). Innate immune responses. In The Picornaviruses, pp. 287–302 Edited by Ehrenfeld E., Domingo E., Roos R. P. Washington, DC: American Society for Microbiology; 10.1128/ISBN978-1-55581-603-2.18 [DOI] [Google Scholar]
- Rozovics J. M., Chase A. J., Cathcart A. L., Chou W., Gershon P. D., Palusa S., Wilusz J., Semler B. L. (2012). Picornavirus modification of a host mRNA decay protein. MBio 3, e00431–e12 10.1128/mBio.00431-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakami T., Yamane D., Jangra R. K., Kempf B. J., Spaniel C., Barton D. J., Lemon S. M. (2012). Stabilization of hepatitis C virus RNA by an Ago2–miR-122 complex. Proc Natl Acad Sci U S A 109, 941–946 10.1073/pnas.1112263109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiroki K., Isoyama T., Kuge S., Ishii T., Ohmi S., Hata S., Suzuki K., Takasaki Y., Nomoto A. (1999). Intracellular redistribution of truncated La protein produced by poliovirus 3Cpro-mediated cleavage. J Virol 73, 2193–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva P. A., Pereira C. F., Dalebout T. J., Spaan W. J., Bredenbeek P. J. (2010). An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J Virol 84, 11395–11406 10.1128/JVI.01047-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloski K. J., Dickson A. M., Chaskey E. L., Garneau N. L., Wilusz C. J., Wilusz J. (2010). Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 8, 196–207 10.1016/j.chom.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurrell J. C., Wiehler S., Zaheer R. S., Sanders S. P., Proud D. (2005). Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 289, L85–L95 10.1152/ajplung.00397.2004 [DOI] [PubMed] [Google Scholar]
- Townsend H. L., Jha B. K., Han J. Q., Maluf N. K., Silverman R. H., Barton D. J. (2008). A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 14, 1026–1036 10.1261/rna.958908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters K., Palmenberg A. C. (2011). Differential processing of nuclear pore complex proteins by rhinovirus 2A proteases from different species and serotypes. J Virol 85, 10874–10883 10.1128/JVI.00718-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J., Si X., Angeles A., Zhang J., Shi J., Fung G., Jagdeo J., Wang T., Zhong Z. & other authors (2013). Cytoplasmic redistribution and cleavage of AUF1 during coxsackievirus infection enhance the stability of its viral genome. FASEB J 27, 2777–2787 10.1096/fj.12-226498 [DOI] [PubMed] [Google Scholar]