

Abstract
Metoclopramide is a widely used clinical drug in a variety of medical settings with rare acute dystonic events reported. The aim of this study was to assess a previous report of inactivation of CYP2D6 by metoclopramide, to determine the contribution of various CYPs to metoclopramide metabolism, and to identify the mono-oxygenated products of metoclopramide metabolism.
Metoclopramide interacted with CYP2D6 with Type I binding and a Ks value of 9.56 ± 1.09 μM. CYP2D6 was the major metabolizer of metoclopramide and the two major products were N-deethylation of the diethyl amine and N-hydroxylation on the phenyl ring amine. CYPs 1A2, 2C9, 2C19, and 3A4 also metabolized metoclopramide.
While reversible inhibition of CYP2D6 was noted, CYP2D6 inactivation by metoclopramide was not observed under conditions of varying concentration or varying time using Supersomes™ or pool human liver microsomes.
The major metabolites of metoclopramide were N-hydroxylation and N-deethylation formed most efficiently by CYP2D6 but also formed by all CYPs examined. Also, while metoclopramide is metabolized primarily by CYP2D6, it is not a mechanism-based inactivator of CYP2D6 in vitro.
Keywords: CYP2D6, metoclopramide
Introduction
Metoclopramide is a dopamine receptor antagonist with primary function to stimulate gastric contractions. This has lead to the use of metoclopramide as an antiemetic for patients taking chemotherapy, recovering from or preparing for surgery, and other clinical applications. In rare cases, prolonged administration of metoclopramide can lead to acute dystonic reactions that result in permanent disability (DiPalma, 1990).
Previously, metoclopramide was reported to be metabolized to both conjugated and oxidized products by the action of CYPs as well as UDP-glucuronosyltransferases and sulfotransferases (Teng et al., 1977, Argikar et al., 2010). Structurally, metoclopramide has an aromatic ring and basic nitrogens – both characteristic of substrates of CYP2D6. In fact, CYP2D6 has been identified as a metabolizer of metoclopramide, though the role of individual CYPs in metabolism has not been shown (Yu et al., 2006, Desta et al., 2002). One group has also reported that metoclopramide is a mechanism-based inactivator of CYP2D6 in HLMs (Desta et al., 2002).
Identification of chemical agents and moieties that act to inactivate CYP2D6 are of clinical importance in prevention of adverse drug events since CYP2D6 is an important drug metabolizing enzyme responsible for metabolism of ~20% of pharmaceuticals. To date, only a handful of CYP2D6 inactivators have been identified and characterized (Nagy et al., 2011, Livezey et al., 2012, Hutzler et al., 2004, Heydari et al., 2004, Bertelsen et al., 2003, Obach et al., 2007, Hollenberg et al., 2008). Furthermore, CYP2D6 is also a highly polymorphic enzyme and there are reports of correlation between CYP2D6 genotype/phenotype and cases of acute dystonic reactions with prolonged use of metoclopramide (van der Padt et al., 2006). Therefore, increased understanding of interactions between metoclopramide and CYP2D6 are warranted.
Argikar et al. (2010) previously identified 10 principal metabolites of metoclopramide from human urine and in vitro (in HLM and HLC) labeled M1-M10. Of these, five were conjugated to either glucuronide or sulfate at the primary amine of metoclopramide (M1, M2, M6, M7, and M8) while the other five were non-conjugated oxidation products (M3, M4, M5, M9, and M10) (for consistency, we will use the same metabolite labels as used in Argikar et al.). The non-conjugated products were N-deethylation (m/z 272; M3); monooxygenation (m/z 316; M4); phenyl ring N-carboxylation (m/z 344; M9); and phenyl ring N-nitro group addition (m/z 330; M10). The position of monooxygenation on M4 was not determined.
Using in vitro methods, the current study sought to establish the roles of various CYPs in the metabolism of metoclopramide and to examine possible mechanism-based inactivation of CYP2D6 that had previously been reported (Desta et al., 2002). Our studies show that metoclopramide does not behave as an inactivator of CYP2D6. Furthermore, we establish the roles of various drug metabolizing CYPs in metabolism of metoclopramide to primarily two products in vitro. Metabolites observed are also compared to those predicted by site of metabolism software.
Materials and Methods
Chemicals
Metoclopramide (4-amino-5-chloro-N-(2-(diethylamino)ethyl)-2-methoxybenzamide) and deethylated metoclopramide (4-amino-5-chloro-N-[2-(ethylamino)ethyl]-2-methoxybenzamide) were purchased from Sigma-Aldrich (St. Louis, MO) and were reconstituted in water for use in assays described below. Ultra-pure solvents (water, ACN, and methanol) for MS were purchased from EMD Chemicals, Inc. (Gibbstown, NJ). All other solvents were HPLC grade and purchased from Sigma-Aldrich (St. Louis, MO). Potassium phosphate, NADPH, TiCl3, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, NADP+, bufuralol, 1′-hydroxybufuralol, dextromethorphan, dextrorphan, catalase, and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Enzymes
Human CYP enzymes 2D6, 3A4, 2C9, 2C19, and 1A2 all with P450 reductase (Supersomes™) and human liver microsomes (UltraPool™ HLM 150 mixed gender pool) were purchased from BD-Gentest (Woburn, MA). For spectral analysis and binding titrations described below, recombinant human CYP2D6 purified from E. coli as previously described was used (a generous gift from Dr. F. P. Guengerich, Vanderbilt University, Nashville, TN) (Gillam et al., 1995, Hanna et al., 2001).
Spectral Binding Titrations
Spectral binding titration studies were carried out with recombinant purified CYP2D6 (1 μM) in 100 mM potassium phosphate buffer, pH 7.4. The enzyme was evenly divided between two cuvettes and the experiments were performed at room temperature by titrating in aliquots of metoclopramide (0-70 μM) into the sample cuvette with the solvent control added to the reference cuvette. A baseline of the reference cuvette was recorded (350-500 nm) on a Cary-300 dual-beam spectrophotometer (Varian, Walnut Creek, CA). Ligand was subsequently added and the spectra were recorded (350-500 nm) after each addition. The solvent was added to the reference cuvette. The difference in absorbance between the wavelength maximum and minimum was plotted versus the concentration of metoclopramide and the data were analyzed by nonlinear regression methods with KaleidaGraph software (Synergy Software, Reading, PA). The dissociation constant, Ks, was determined using the following quadratic velocity equation or tight-binding equation:
where S is the substrate concentration, E is the total enzyme concentration, and Ks is the spectral dissociation constant for the reaction .
Site of Mechanism Predictions
The software programs SMARTCyp (Rydberg et al., 2010, Rydberg and Olsen, 2012) and RS-Predictor (Zaretzki et al., 2012) were used for prediction of sites of metabolism on metoclopramide by individual CYPs.
Analysis of Metabolites
The role of individual P450s in the metabolism of metoclopramide was examined by use of Supersomes for CYPs 2D6, 2C9, 2C19, 3A4, and 1A2. Each enzyme (40 pmol) was incubated with 25 μM metoclopramide in 100 mM potassium phosphate buffer, pH 7.4 (final volume 200 μL) in the presence of NADPH (1 mM). Reactions were incubated at 37 °C for 20 minutes and then quenched by addition of 30 μL of ACN. Samples were then spiked with 4 μM caffeine as an internal standard and centrifuged. An aliquot of the supernatant (20 μL) was directly injected onto a Kinetex C18 (2.6 μm, 2.1 × 100 mm) column (Phenomenex, Torrance, CA) for chromatographic separation using an Alliance 2690 HPLC system (Waters, Milford, MA) with a solvent system composed of solvent mixtures A (90% water, 10% methanol, and 0.05% TFA) and B (90% ACN, 10% methanol and 0.05% TFA). After 5 min at the initial conditions (5% B), separation of metabolites was achieved by a linear gradient of 5% B to 50% B over 20 minutes then holding at 50% B for 1 min before returning to the initial conditions. The flow rate was 0.1 mL/min. The column was allowed to re-equilibrate for 15 min at initial conditions (95% A/5% B) prior to the next injection. The internal standard, caffeine, eluted at 10 min while the various metoclopramide metabolites eluted between 15-25 min. Mass spectrometry was performed using a Thermo LXQ (Thermo Scientific, West Palm Beach, FL) mass spectrometer with an ESI source in a positive ion mode. The ESI conditions were sheath gas set at 27 arbitrary units, the auxiliary gas was set at 5 arbitrary units, the spray voltage was set at 5 kV, and the capillary temperature was set at 250 °C. For MSn experiments, target ions were captured and collision induced dissociation (CID) produced using 35% collision energy.
Reduction of N-hydroxyl amine
Monooxygenation of metoclopramide at three different positions was apparent by the presence of three distinct m/z 316 ions in the mass spectral analysis (M+1) (vide infra). To determine if monooxygenation was on carbon or nitrogen atoms, titanium (III) trichloride (TiCl3) was used to selectively reduce any hydroxylamines as previously described (Kulanthaivel et al., 2004, Seto and Guengerich, 1993). Briefly, reaction mixtures containing 25 μM metoclopramide and 60 pmol of CYP3A4 Supersomes in 100 mM potassium phosphate buffer, pH 7.4 (final volume 300 μL) were initiated by addition of NADPH (1 mM). Reactions were incubated at 37 °C for 20 minutes and then quenched by addition of 45 μL of ACN. One aliquot (100 μL) of the reaction mixture was treated with 20 μL of a solution of TiCl3 (~10 wt. % in 20-30 wt. % HCl). For controls, one aliquot (100 μL) was treated with HCl alone (20 wt %) and another aliquot (100 μL) was left untreated. Samples were left at room temperature for 1.5 hr to allow for TiCl3 reduction of hydroxylamines to the parent amines. The samples were then centrifuged and the supernatant was analyzed by LC/MS as described for the metabolites above. To confirm findings with TiCl3 and HCl, 4-methylmorpholine N-oxide (contains a hydroxylated tertiary amine) and deferoxamine (contains three hydroxylated amide nitrogens) were used as a controls to show reversal of N-oxidation under our reaction conditions while acetominophen was used as a control to show that C-hydroxylation would not be reversed under our reaction conditions.
Determination of Km and vmax for CYP2D6 metabolism of metoclopramide
Assays contained varying concentrations of metoclopramide (0-50 μM) and 15 pmols CYP2D6 in 100 mM potassium phosphate buffer, pH 7.4 (final volume of 100 μL) were warmed to 37 °C for 3 min prior to initiation by the addition of NADPH generating system (5 mM glucose 6-phosphate, 0.5 mM NADP+, and 0.5 units/mL glucose 6-phosphate dehydrogenase). After 1 min at 37 °C, reactions were quenched by the addition of acetonitrile (15 μL) and analyzed by HPLC. The formation of the N-deethylated metoclopramide product was quantified by HPLC with a Phenomenex Kinetex C18 column (2.6 μm, 4.6 × 100 mm) with a 2487 Dual Wavelength UV detector controlled with Empower software (Waters, Milford, MA). The detector was set at 272 nm and N-deethylated metoclopramide eluted at 4.1 min while unreacted parent metoclopramide eluted at 6.5 min. Peak separation was achieved under isocratic conditions with 85% A (10% methanol, 90% water, 0.05% trifluoroacetic acid) and 15% B (10% methanol, 90% acetonitrile, 0.05% trifluoroacetic acid). All analyses were performed at room temperature with a flow rate of 0.75 mL/min. N-deethylated metoclopramide product peak areas were compared to a standard curve generated from dilutions of N-deethylated metoclopramide to quantitate enzyme velocity.
Time- and concentration-dependent assays with metoclopramide using dilution reactions and substrate addition reactions
Inactivation of CYP2D6 by metoclopramide was examined by two types of analysis of CYP2D6 activity in the presence of metoclopramide. In one type of analysis, primary inactivation assays were followed by 20-fold dilution into secondary reporter assays as previously described (Nagy et al., 2011). Briefly, primary reaction mixtures containing 0-100 μM metoclopramide and 20 pmol of CYP2D6 Supersomes in 100 mM potassium phosphate buffer, pH 7.4 (final volume of 100 μL), were preincubated in a 37 °C shaking water bath. After 3 minutes, the primary reactions were initiated with the addition of NADPH generating system and incubated at 37 °C. The time zero control received an equivalent amount of water. The time dependent inactivation assays were incubated from 0-40 minutes during the inactivation phase as indicated while a 40-minute inactivation phase was used in the metoclopramide concentration dependent inactivation assays. Aliquots of 10 μL were then removed from the primary reactions and added to the secondary reaction mixtures containing 100 μM bufuralol or 50 μM dextromethorphan and NADPH in 100 mM potassium phosphate buffer, pH 7.4, with a final volume of 200 μL (20-fold dilution). The secondary mixtures were incubated for 10 minutes at 37 °C and quenched with 15 μL of 70% perchloric acid.
To be consistent with a previously reported study of metoclopramide inhibition of CYP2D6, a second type of analysis reaction was also used in which the reporter (bufuralol, 100 μM final or dextromethorphan, 50 μM final) was added directly to the inactivation assay following a 0-40 min inactivation phase, as indicated, in time-dependent inhibition assays or following a 40-minute inactivation phase in metoclopramide concentration dependent assays (Desta et al., 2002). These reactions had no dilution of metoclopramide before addition of reporter substrate and are referred to as substrate addition reactions. The analysis was repeated using HLM (100 μg protein) in substrate addition reactions.
All reaction mixtures were centrifuged (2000 × g, 3 minutes) to remove the precipitated enzyme and 20 μL aliquots of the recovered supernatants were directly injected onto a Waters Alliance e2695 HPLC for analysis.
The formation of the 1′-hydroxybufuralol product was quantified by HPLC with a Phenomenex Kinetex C18 column (2.6 μm, 4.6 × 100 mm) with a 474 fluorescence detector controlled with Empower software (Waters, Milford, MA) as previously described (Nagy et al., 2011). Briefly, the detector was set at an excitation wavelength of 252 nm and an emission wavelength of 302 nm. The mobile phase consisted of 30% acetonitrile and 70% 1 mM perchloric acid in water. All analyses were performed at room temperature with a flow rate of 0.75 mL/min.
The formation of the dextrorphan product was quantified by HPLC with a Phenomenex Kinetex C18 column (2.6 μm, 4.6 × 100 mm) with a 474 fluorescence detector controlled with Empower software (Waters, Milford, MA). The detector was set at an excitation wavelength of 280 nm and an emission wavelength of 310 nm. The mobile phase consisted of 30% acetonitrile, 1 % acetic acid, and 0.5% triethylamine. All analyses were performed at room temperature with a flow rate of 0.75 mL/min.
Assays of N-deethylated metoclopramide interaction with CYP2D6
To test possible inhibition of CYP2D6 by a product of metoclopramide metabolism, CYP2D6 dilution activity assays were repeated in the presence of deethylated metoclopramide. Deethylated metoclopramide was dissolved in water for use in assays described. Assays were initiated by addition of NADPH (1 mM, final) to reactions containing deethylated metoclopramide (0-100 μM) and 40 pmols CYP2D6 Supersomes in 100 mM potassium phosphate, pH 7.4 (final volume 100 μL). After 20 minutes, 10 μL of the reaction was removed and added to a secondary reporter reaction containing bufuralol (100 μM) and NADPH (1 mM) in 100 mM potassium phosphate, pH 7.4 (final volume 200 μL). Secondary reporter reactions were quenched after 10 min with 70% perchloric acid (15 μL). Samples were centrifuged and analyzed by HPLC for production of 1′-hydroxybufuralol as described above.
Partition Ratio
Partition ratio was performed as previously described (Nagy et al., 2011). Primary reaction mixtures containing 0-240 μM metoclopramide and 20 pmol of CYP2D6 Supersomes in 100 mM potassium phosphate buffer, pH 7.4 (final volume of 100 μL), were preincubated in a 37 °C shaking water bath. After 5 minutes, the primary reactions were initiated with the addition of NADPH-generating system and incubated at 37 °C. To allow the inactivation to go to completion, inactivation assays were incubated for 60 minutes. Aliquots of 10 μL were then removed from the primary reactions and added to the secondary reaction mixtures containing 100 μM bufuralol and NADPH-generating system in 100 mM potassium phosphate buffer, pH 7.4, with a final volume of 200 μL. The secondary mixtures (in triplicate) were incubated for 10 minutes at 37 °C and quenched with 15 μL of 70% perchloric acid. Reaction mixtures were centrifuged (2000 × g, 5 minutes) to remove the precipitated enzyme and aliquots of the recovered supernatants were directly injected onto a Waters Alliance e2695 HPLC for analysis. The formation of the 1′-hydroxybufuralol product was quantified by HPLC as described above.
To confirm that metoclopramide was behaving as a substrate and not an inactivator or stimulator of CYP2D6 activity, partition ratio experiments were repeated as described above except primary reaction mixtures contained 0-240 μM dextromethorphan, a known substrate for CYP2D6, instead of metoclopramide.
Dixon Analysis
Dixon analysis was performed to determine the inhibition constant, Ki (Dixon, 1953). Reactions contained CYP2D6 Supersomes (2 pmol) in 100 mM potassium phosphate buffer, pH 7.4 (final volume of 200 μL) with one of four concentrations of the reporter substrate bufuralol (5, 10, 50, and 100 μM) and varying concentrations of metoclopramide (0, 5, 30, 100 μM). Reactions were preincubated in a 37 °C shaking water bath prior to initiation with NADPH generating system. After a 20 minute incubation, reactions were quenched with 15 μL of ACN. Samples were analyzed by HPLC for 1′-hydroxybufuralol product formation.
Molecular Modeling and Docking Simulations
AutoDock 4.0 was employed to perform docking simulations and molecular models (http://autodock.scripps.edu) (Morris et al., 1998, Huey et al., 2007). The protein structures used in these studies were CYP2D6 without ligand bound (PDB ID: 2F9Q) and CYP2D6 with prinomastat bound (PDB ID: 3QM4). Ligands and solvent molecules were removed, but the heme was retained. The Fe atom of the heme and the heme nitrogens were set to neutral charge. A water molecule was placed 1.7 Å from the heme iron using COOT (Emsley and Cowtan, 2004) to simulate the electrostatics of Compound I in the mechanism of P450 catalysis (Shahrokh et al., 2012). The PDB 2F9Q of CYP2D6 was modified in Swiss-Deep View at position 374 to the reference amino acid (M374V). Residues within 5 Å of the heme iron were identified and set as flexible residues for computations and included Ala305, Cys443, Thr309, Glu446, Leu444, and Gly445. For each protein structure, charges were calculated by the Gasteiger-Marsili method. The 3D structures of the ligands for docking studies were built in Spartan 4.0 (Wavefunction, Inc., Irvine, CA) with all hydrogen atoms added and energy minimization. The grid maps were calculated using AutoGrid. The dimensions of the grid box were set to 40 × 40 × 40 Å and the grid spacing was set to 0.375 Å. Docking was performed using the Lamarckian genetic algorithm. Each docking experiment was performed 100 times, yielding 100 docked conformations. The consensus binding postures of the molecules were obtained by visual inspection and docking scores.
Results
Spectral Binding
The binding constant for metoclopramide was determined by titration of CYP2D6 with increasing concentrations of metoclopramide (Figure 1A). We found that metoclopramide showed Type I binding typical of substrates with a Ks value of 9.56 ± 1.09 μM (Figure 1B).
Figure 1.

Spectral binding titration of CYP2D6 and determination of spectral binding parameter Ks. (A) Titration of CYP2D6 with metoclopramide and (B) plot of ΔA430-395 (from panel A) vs. concentration of metoclopramide.
Metabolites
In the present study with the individual CYP Supersomes 2D6, 3A4, 2C9, 2C19, and 1A2, we observed four of the previously identified metabolites: M3, M4, M9 and M10 (Figure 2). Our studies show the major products to be M3 and M4. Furthermore, CYP2D6 produced more M3 and M4 than any of the other CYPs based on normalized levels in the mass spectrometry compared to an internal standard of caffeine (Figure 2). We also found that M9 is a non-NADPH dependent product present in all reactions and is not formed by the action of CYPs, and thus is not a true metabolite.
Figure 2.

Structure of metoclopramide and observed metabolites. Metabolite mass to charge values (m/z) for each metabolite are indicated (M+1). Metabolite labels are consistent with Argikar et al. (Argikar et al., 2010). The ratio of product formation as compared to an internal standard for indicated metabolites by each CYP is shown in the bar graphs. For M4, levels of metabolite formed by 3A4, 2C9, and 2C19 are shown as an inset. Caffeine was used as an internal standard for quantification as indicated in the Methods.
We observed three different M4 metabolites with m/z 316 (consistent with monooxygenation). These are most apparent in reactions with CYP3A4 that produces all three (Figure 3). We have designated the metabolites M4a, M4b, and M4c (Figure 3). Using TiCl3 treatment of metabolites as described in the Methods, monooxygenation of two of the three M4 metabolites was shown to be on nitrogen atoms: M4a - N of the phenyl ring amine and M4c - N of the diethylamine. TiCl3 treatment failed to reverse M4b metabolite formation and was therefore presumed to be a product of carbon hydroxylation.
Figure 3.

Identification of m/z 316 isomeric products. Extracted ion chromatograms (m/z 316) of CYP3A4 metabolite samples after (A) no treatment, (B) HCl treatment only, or (C) TiCl3 in HCl treatment. Note that after treatment with HCl and TiCl3, M4b retention time was slightly shifted to ~20 min; however, all fragmentation patterns matched those for M4b eluting at ~21 min in the control sample. (D) Corresponding MS2 for m/z 316 isomeric product with retention time of ~19 min, M4a. (E) Corresponding MS2 for m/z 316 isomeric product with retention time of ~21 min, M4b. (F) Corresponding MS2 for m/z 316 isomeric product with retention time of ~23 min, M4c. (G) Corresponding MS3 for m/z 316 isomeric product M4a with retention time of ~19 min and MS2 of 243. (H) Corresponding MS3 for m/z 316 isomeric product M4b with retention time of ~21 min and MS2 of 243. (I) Corresponding MS3 for m/z 316 isomeric product M4c with retention time of ~23 min and MS2 of 227. Data shown are representative of those found in a minimum of four separate experiments.
M4a eluted at ~19 min and was shown to be hydroxylation of the N of the phenyl ring amine, not a phenyl ring carbon. All CYPs examined produced this product. The position of this hydroxylation has been suggested by others (Yu et al., 2006) and for the first time is confirmed in the present study. TiCl3 selectively reverses hydroxylation of nitrogen, but not carbon (Seto and Guengerich, 1993, Kulanthaivel et al., 2004), and reversed formation of M4a. Furthermore, MS2 (principal fragment of m/z 243) and MS3 data (principal fragment of m/z 226) support N-hydroxylation (Figure 3D & 3G). The m/z 226 peak from MS3 data are consistent with the loss of a hydroxyl group (−OH).
The M4b product eluting at ~21 min was most visible in reactions with CYP3A4 because the M4a peak is smaller with CYP3A4 than with CYP2D6 or CYP1A2 (Figure 3 and data not shown). M4b appears to be a hydroxylation of a phenyl ring carbon since the M4b peak was unaffected by treatment with HCl or TiCl3 in HCl (Figure 3A compared to Figure 3C). The MS2 and MS3 data yield m/z 243 and m/z 228, respectively (Figure 3E and 3H). The m/z 228 peak in the MS3 is consistent with loss of an amine (-NH2) from the m/z 243 fragment when hydroxylation has occurred elsewhere. MS4 data produced a very weak signal but consistently with m/z 184 suggesting hydroxylation has likely occurred on a phenyl ring carbon, most likely between the amine and methoxy groups (data not shown).
CYPs 2D6, 3A4, 2C9, and 2C19 also produce a minor m/z 316 peak at ~23 min elution time, M4c. The MS2 fragment is m/z 227 – the same as the MS2 fragment of the parent metoclopramide, so the M4c peak is the product of oxygenation on the diethylamine end of the molecule that is lost in the first CID (Figure 3F). Since the peak is lost upon treatment of the sample with TiCl3 that reverses N-hydroxylation, this product is oxygenation of the nitrogen of the diethylamine (Figure 3C).
Control reactions treated with or without HCl retain the presence of the m/z 316 peaks in the mass analysis for the M4a, M4b and M4c products (Figure 3B). Samples treated with TiCl3 in HCl show complete loss of only the M4a and M4c peaks (Figure 3C).
For comparison to observed metabolites, the metabolism prediction software programs SMARTCyp and RS Predictor were used. For CYP2D6, SMARTCyp predicted metabolism at the phenyl ring amine nitrogen as the highest ranking site of metabolism (Table 1). This prediction is consistent with our findings regarding the m/z 330 metabolite (M10) and the M4a m/z 316 metabolite and subsequent TiCl3 confirmation of N-hydroxylation (Figure 3). Hydroxylation at the ring carbon between the amine and methoxy groups may occur as predicted by SMARTCyp, though we were unable to definitively identify which of the two phenyl ring carbons is hydroxylated; we note that the one between the amine and methoxy group would be the lower energy product. We saw no evidence of modification of metoclopramide at the other predicted site – the methyl group of the methoxy moiety. Metabolism at the methoxy group, specifically demethylation, would be consistent with known metabolism activity of CYPs, but no m/z 285 (M+1) product was observed. Conversely, the N-deethylated product was a CYP2D6 major product and not predicted to be by SMARTCyp. Furthermore, we also observed monooxygenation on the nitrogen of the diethylamine moiety that was not predicted by SMARTCyp for CYP2D6.
RS Predictor sites of metabolism were also generated. In RS Predictor, sites of metoclopramide metabolism by CYP2D6 were predicted at the phenyl amine - which was observed, the methylene group of the di-ethyl amine – which was also observed, and at the methoxy group - which was not observed. Like SMARTCyp, RS predictor did not suggest metabolism at the nitrogen of the diethylamine though we observed a product of oxygenation at that site (M4c). Sites of metabolism by the other CYPs were also predicted using SMARTCyp and RS Predictor as shown in Table 1 (SMARTCyp only shows predicted sites of metabolism by CYP2D6, CYP2C9, and CYP3A4 while RS Predictor considers possible sites of metabolism by other drug metabolizing CYPs).
Table 1.
Predicted of Sites of Metabolism of Metoclopramide by CYPs.*
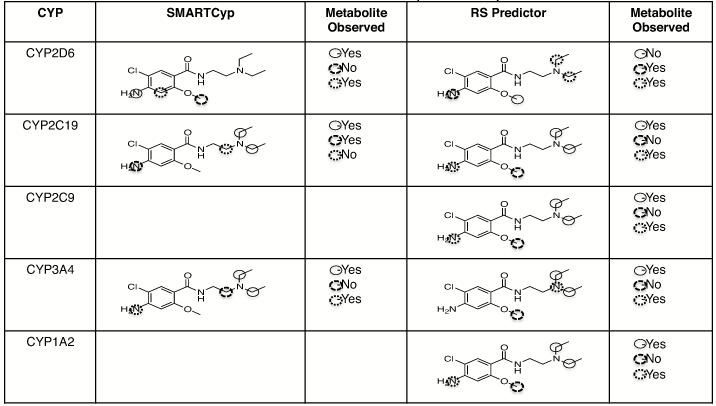
|
Ranked atom sites of probable reac6vity are indicated by circles. Atoms circled with a solid circle  are predicted to be the most probable site of metabolism followed by atoms circled with dashed circle
are predicted to be the most probable site of metabolism followed by atoms circled with dashed circle  and finally atoms circled with dojed circle
and finally atoms circled with dojed circle  . Observed metabolism of any type at the predicted atoms is indicated by “yes” in the “Metabolite Observed” column.
. Observed metabolism of any type at the predicted atoms is indicated by “yes” in the “Metabolite Observed” column.
Molecular Modeling using AutoDock
Metoclopramide bound in two possible binding orientations: one with the phenyl ring pointing toward the heme and the other with the diethylamine pointing toward the heme (Supplemental Figure 2 and data not shown). Both orientations are consistent with metabolite findings. Binding energies for metoclopramide to either structure and in either orientation were similar and around −5 kcal/mol (or −21 kJ/mol). This finding further validates the observation that CYP2D6 oxidizes basic nitrogens on both ends of the drug in roughly equivalent ratios.
Kinetics of CYP2D6 Metabolism of Metoclopramide
Metabolism of metoclopramide by CYP2D6 from Supersomes followed Michaelis-Menten kinetics with Km of 1.20 ± 0.29 μM and vmax of 6.1 ± 0.23 pmol product/min/pmol CYP2D6 (Figure 4).
Figure 4.

Michaelis-Menten Analysis of CYP2D6 Metabolism of Metoclopramide. CYP2D6 Supersomes were used to determine Km and vmax values for CYP2D6 metabolism of metoclopramide to deethylated metoclopramide. Km was 1.20 ± 0.29 μM and vmax was 6.1 ± 0.23 pmol product/min/pmol CYP2D6 with R2 value of 0.95 for the analysis.
Dixon Analysis
We performed a Dixon analysis using CYP2D6 Supersomes and bufuralol as a reporter substrate (Supplemental Figure 3) or dextromethorphan as substrate (data not shown). From the Dixon analysis of both assays, metoclopramide exhibited a Ki = 13.8 μM. This is similar to the Ki value of 4.71 μM reported by Desta et al. using two donor HLM sets and dextromethorphan as the reporter (Desta et al., 2002).
Partition ratio
Given a previous report of mechanism-based inactivation of CYP2D6 by metoclopramide (Desta et al., 2002), we attempted to determine the partition ratio for metoclopramide inactivation of CYP2D6 by incubation of CYP2D6 with various concentrations of metoclopramide over 60 minutes to allow the inactivation to progress to presumed completion. The percentage of the activity remaining was plotted as a function of the molar ratio of metoclopramide to 2D6 as described previously (Silverman, 1988, Nagy et al., 2011). However, the data were not consistent with inactivation and followed the same results seen in experiments where a substrate, dextromethorphan, was used as the “inactivator” in the inactivation phase and bufuralol as reporter as in assays with metoclopramide (Supplemental Figure 4).
Activity Assays
We explored other possible effects of metoclopramide on the activity of CYP2D6 using CYP2D6 from Supersomes and in pooled HLM with either dextromethorphan or bufuralol as the reporter substrate. We used both dilution assays and substrate addition assays for comparison since the previous study reporting inactivation had used dextromethorphan in substrate addition assays only (Desta et al., 2002).
In our time-dependent inhibition substrate addition assays with HLM we observed an ~40% loss of CYP2D6 activity over 40 minutes that was paralleled by an equivalent loss of CYP2D6 activity in control reactions (Supplemental Figure 5A). In concentration dependent substrate addition assays in pooled HLM with dextromethorphan as reporter substrate there was a ~20% loss of CYP2D6 activity at concentrations of metoclopramide from 10 – 75 μM (Supplemental Figure 5B). Thus, in our assays with pooled HLM the IC50 for metoclopramide would be >100 μM.
To confirm our findings in HLM, we also considered inactivation of CYP2D6 in Supersomes by both substrate addition and dilution assays. In time dependent inactivation assays we saw some loss of CYP2D6 activity over time, but as with HLM, the loss in activity in the control reactions was similar indicating that there was no apparent metoclopramide dependent inactivation (Figure 5A and Supplemental Figure 6A). For concentration-dependent assays, CYP2D6 retained ~80% activity at concentrations as high as 50 μM metoclopramide (Figure 5B and Supplemental Figure 6B). In dilution assays, the decrease in CYP2D6 activity at the lowest concentration of metoclopramide (Figure 5B) is due to instability of CYP2D6 Supersomes in the presence of NADPH and low substrate concentrations (below the Kd, Figure 1). This was confirmed when assays were repeated with NADPH included in control reactions with no metoclopramide; the activity of CYP2D6 was much lower and the reactions with metoclopramide showed higher activity than the control (Supplemental Figure 7).
Figure 5.

Dilution Assay for Determination of Metoclopramide (A) Time-dependent and (B) Concentration-dependent Inhibition of CYP2D6. Time-dependent assays contained 20 μM metoclopramide in the inactivation reaction. Concentration-dependent assays were carried out for 40 min. Bufuralol was used as the reporter substrate. Control reactions without metoclopramide and without NADPH are shown as solid circles (●). Control reactions without metoclopramide but with NADPH are shown as solid triangles (▲). Reactions containing both metoclopramide and NADPH are shown as open squares (□). Reactions shown are the average of triplicate experiments.
Since the loss of CYP2D6 activity in the previous study using a single human liver was significant (Desta et al., 2002), we considered that perhaps a metabolite of metoclopramide could be acting as an inhibitor of CYP2D6. We tested the ability of deethylated metoclopramide (M3) to inhibit CYP2D6 in dilution assays. No inhibition of CYP2D6 activity was observed (Supplemental Figure 4).
Discussion
The present study has established that metoclopramide is not an inactivator of CYP2D6*1. The study has further established the metabolites of metoclopramide formed in vitro by the five major drug metabolizing CYPs and the extent of metabolite formation by each CYP. SMARTCyp and RS Predictor software were fairly accurate in prediction as the only site of metabolism projected that was inconsistent with isolated metabolites was the methoxy group of metoclopramide.
Analysis of metabolite formation showed production of M3 (deethylated) and M4 (monooxygenated) as the major metabolites and that each was produced primarily by CYP2D6, though deethylation of the amine nitrogen (M3) was the major product formed by all CYPs examined. In addition, three monooxygenated products of metoclopramide metabolism were observed (M4a, M4b, and M4c). Two were confirmed to be hydroxylation on the phenylic amine and the diethylamine nitrogens (M4a and M4c, respectively). The third hydroxylation product was on one of the carbons on the phenyl ring (M4b). TiCl3 reversal of N-hydroxylation was used to confirm these findings (Figure 3). All three monooxygenation products (M4a, M4b, M4c) were formed by CYP3A4 and CYP2D6 and all three were formed in NADPH dependent reactions. The M4a product if formed in vivo could be a substrate for glucuornidation to form M1 observed in a previous study (Argikar et al., 2010).
Besides identification of metabolites and contributions of specific CYPs to metabolism of metoclopramide, we were interested in a previous report of inactivation of CYP2D6 in HLM by metoclopramide (Desta et al., 2002). Our group has studied inactivation of CYP2D6 by both protein inactivators, e.g. SCH66712 and EMTPP, and heme modifiers, e.g. paroxetine and MDMA (Nagy et al., 2011, Livezey et al., 2012). The chemical moieties in metoclopramide are different from these other inactivators and are not consistent with known structural alerts (Evans et al., 2004, Kalgutkar et al., 2007, Hollenberg et al., 2008), thus we believed this warranted further study.
Our analysis of concentration dependent inhibition of CYP2D6 by metoclopramide showed little or no inhibition of CYP2D6 regardless of enzyme source (HLM or Supersomes) or assay style (substrate addition or dilution) (Figure 5 and Supplemental Figures 5, 6, and 7). Calculated IC50 values would be >>100 μM. Likewise, while there was some loss of activity in time-dependent assays (Figures 5 and Supplemental Figures 5 and 6), the loss was paralleled by equivalent losses in CYP2D6 activity in control reactions. Inclusion of catalase and SOD in reactions did not reduce the loss of activity observed (data not shown).
Desta et al. had previously observed a rapid loss of CYP2D6 activity (IC50 of 1 μM) in the presence of increasing concentrations of metoclopramide using HLM from a single donor and dextromethorphan as reporter (2002). We believe the discrepancy in findings between our study and the previous one are largely be due to the differences in enzyme source since our study used both pooled HLM and Supersomes systems. For example, Argikar et al. found in eight (8) human study subjects as much as a 21-fold difference in metoclopramide metabolite production among study subjects; this suggest that use of a single human liver, as was the case in the Desta et al. study, could lead to non-typical results. CYP2D6 is well recognized for its many and prevalent polymorphic forms in all populations. The genotype/phenotype was not established for the donor used by Desta et al. Metoclopramide could interact differently with some polymorphic forms of CYP2D6 that might have been present in the single HLM used in the previous study. It is intriguing to consider this possibility since in the clinical literature there are reports of several types of extrapyramidal symptoms such as rare acute dystonic reactions to metoclopramide in individuals with CYP2D6 polymorphisms (van der Padt et al., 2006, Silfeler et al., 2012). Alternatively, inactivation of CYP2D6 seen in the previous study could have been due to formation of an inactivating metabolite of metoclopramide formed in the microsomes (there are other polymorphic enzymes, such as monoamine oxidases, glucuronosyltransferases, sulfotransferases, and others, that could contribute to formation of an inactivator in vivo) or increased susceptibility to auto-oxidation of the CYP2D6 in the donor liver.
The typical clinical dose of metoclopramide as an antiemetic is 10 mg four times a day. This would yield peak plasma concentrations of 6-10 μM metoclopramide. Bateman et al. report plasma metoclopramide concentrations in patients at the time of dystonia symptoms (within 6 hours of dosing) to be 0.22 – 1.33 μM (Bateman et al., 1983). Clinical reports of adverse reactions to metoclopramide after usage including several types of extrapyramidal symptoms such as akathisia, parkinsonism, tardive dyskinesia, acute dystonic reactions, and other adverse effects. These reactions occur in as many as 20% of patients prescribed the drug and are believed to be due to inhibition of specific dopamine receptor targets; some of these are related to poor elimination of metoclopramide while others are independent of the elimination rate (Bateman et al., 1983; van der Padt et al., 2006; Silfeler et al., 2012). Understanding the role of CYP2D6 inhibition or loss of activity in these adverse events can aid in appropriate usage of metoclopramide. The Ki for reversible inhibition of CYP2D6 was found to be 13.8 μM – similar to the peak plasma concentration. This suggests that there may be some potential adverse effects of CYP2D6 reversible inhibition at peak plasma concentrations.
Partition ratio analysis to determine the turnover rate of the enzyme in the presence of an inactivator was attempted with metoclopramide since we have previously established partition ratios for other inactivators of CYP2D6 (Livezey et al., 2012). However, the partition ratio analysis did not show inhibition after 60 minutes of inactivation phase (Supplemental Figure 4). Based on these data and the lack of inactivation seen in activity assays, metoclopramide would not be expected to behave as an inactivator. Reports for CYP2D6 activity in high throughput screenings using the substrate 3-[2-(N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin (AMMC) that produces a fluorescent product show metoclopramide to be an inhibitor of recombinant CYP2D6 with IC50 value of ~9 μM (Yu et al., 2006).This is similar to the IC50 value for dextromethorphan inhibition of CYP2D6 with AMMC as a probe (Chauret et al., 2001). Dextromethorphan is often used as a reporter substrate for CYP2D6 activity and is not typically thought of as an inhibitor. We show here that metoclopramide behaves in similar fashion to dextromethorphan in inactivation assays further supporting the claim that metoclopramide is not an inactivator of CYP2D6 (Supplemental Figure 4). We note that both metoclopramide and dextromethorphan are able to stabilize the activity of CYP2D6 Supersomes in the presence of NADPH (Supplemental Figure 4).
Conclusions
Overall, the results of our activity assays suggest that metoclopramide is a reversible, competitive inhibitor of CYP2D6 since there was little or no time-dependent loss in CYP2D6 activity compared to the control. Furthermore, we have established the in vitro products of metoclopramide metabolism by the five major drug-metabolizing enzymes including identification three monooxygenation products and sites of hydroxylation.
Supplementary Material
Acknowledgement
We thank Drs. F. P. Guengerich, P. Hollenberg, E. Johnson, N. Hosea, B. Burke, Z. Desta, and H. Zhang for helpful comments. This work was supported by the National Institutes of Health (L.L.F.) [Grants 1R15GM086767-01, -02 and 3R15BM086767-01S1]; a grant to Kalamazoo College from the Howard Hughes Medical Institute [52006304] through the Precollege and Undergraduate Science Education Program; and the Hutchcroft Fund of Kalamazoo College.
This work was supported by the National Institutes of Health (L. L. F) [Grants: 1R15GM086767-01, -02 and 3R15BM086767-01S1]
Abbreviations used
- CYP
cytochrome P450 enzyme
- HLM
human liver microsomes
- HPLC
high performance liquid chromatography
Footnotes
Declaration of Interest. The authors declare no financial interest or conflict of interest.
Authorship Contributions
Participated in research design: Furge
Conducted experiments: Furge, Bolles, Briggs, Livezey, Nagy, Fujiwara
Performed data analysis: Furge, Bolles, Briggs, Livezey, Nagy
Wrote or contributed to the writing of the manuscript: Furge, Bolles, Briggs, Livezey
Other: Furge acquired funding for the research
References
- Argikar UA, Gomez J, Ung D, Parkman HP, Nagar S. Identification of novel metoclopramide metabolites in humans: in vitro and in vivo studies. Drug Metab Dispos. 2010;38:1295–307. doi: 10.1124/dmd.110.033357. [DOI] [PubMed] [Google Scholar]
- Bateman DN, Craft AW, Nicholson E, Pearson ADJ. Dystonic reactions and the pharmacokinetics of metoclopramide in children. Br J Clin Pharm. 1983;15:557–559. doi: 10.1111/j.1365-2125.1983.tb02090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen KM, Venkatakrishnan K, Von Moltke LL, Obach RS, Greenblatt DJ. Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: comparison with fluoxetine and quinidine. Drug Metab Dispos. 2003;31:289–93. doi: 10.1124/dmd.31.3.289. [DOI] [PubMed] [Google Scholar]
- Chauret N, Dobbs B, Lackman RL, Bateman K, Nicoll-Griffith DA, Stresser DM, Ackermann JM, Turner SD, Miller VP, Crespi CL. The use of 3-[2-(N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin (AMMC) as a specific CYP2D6 probe in human liver microsomes. Drug Metab Dispos. 2001;29:1196–200. [PubMed] [Google Scholar]
- Desta Z, Wu GM, Morocho AM, Flockhart DA. The gastroprokinetic and antiemetic drug metoclopramide is a substrate and inhibitor of cytochrome P450 2D6. Drug Metab Dispos. 2002;30:336–43. doi: 10.1124/dmd.30.3.336. [DOI] [PubMed] [Google Scholar]
- Dipalma JR. Metoclopramide: a dopamine receptor antagonist. Am Fam Physician. 1990;41:919–24. [PubMed] [Google Scholar]
- Dixon M. The determination of enzyme inhibitor constants. Biochemical Journal. 1953;55:170–171. doi: 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans DC, Watt AP, Nicoll-Griffith DA, Baille TA. Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol. 2004;17:3–16. doi: 10.1021/tx034170b. [DOI] [PubMed] [Google Scholar]
- Gillam EMJ, Guo Z, Martin MV, Jenkins CM, Guengerich FP. Expression of cytochrome P450 2D6 in Escherichia coli, purification, and spectral and catalytic characterization. Archives of Biochemistry and Biophysics. 1995;319:540–550. doi: 10.1006/abbi.1995.1329. [DOI] [PubMed] [Google Scholar]
- Hanna IH, Kim MS, Guengerich FP. Heterologous expression of cytochrome P450 2D6 mutants, electron transfer, and catalysis of bufuralol hydroxylation: the role of aspartate 301 in structural integrity. Arch Biochem Biophys. 2001;393:255–61. doi: 10.1006/abbi.2001.2510. [DOI] [PubMed] [Google Scholar]
- Heydari A, Yeo KR, Lennard MS, Ellis SW, Tucker GT, Rostami-Hodjegan A. Mechanism-based inactivation of CYP2D6 by methylenedioxymethamphetamine. Drug Metab Dispos. 2004;32:1213–7. doi: 10.1124/dmd.104.001180. [DOI] [PubMed] [Google Scholar]
- Hollenberg PF, Kent UM, Bumpus NN. Mechanism-based inactivation of human cytochromes p450s: experimental characterization, reactive intermediates, and clinical implications. Chem Res Toxicol. 2008;21:189–205. doi: 10.1021/tx7002504. [DOI] [PubMed] [Google Scholar]
- Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J. Computational Chemistry. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- Hutzler JM, Steenwyk RC, Smith EB, Walker GS, Wienkers LC. Mechanism-based inactivation of cytochrome P450 2D6 by 1-[(2-ethyl-4-methyl-1H-imidazol-5-yl)methyl]-4-[4-(trifluoromethyl)-2-pyridinyl]piperazine: kinetic characterization and evidence for apoprotein adduction. Chem Res Toxicol. 2004;17:174–84. doi: 10.1021/tx034199f. [DOI] [PubMed] [Google Scholar]
- Kalgutkar AS, Obach RS, Maurer TS. Mechanism-based inactivation of cytochrome P450 enzymes: chemical mechanisms, structure-activity relationships and relationship to clinical drug-drug interactions and idiosyncratic adverse drug reactions. Curr Drug Metab. 2007;8:407–47. doi: 10.2174/138920007780866807. [DOI] [PubMed] [Google Scholar]
- Kulanthaivel P, Barbuch RJ, Davidson RS, YI P, Rener GA, Mattiuz EL, Hadden CE, Goodwin LA, Ehlhardt WJ. Selective reduction of N-oxides to amines: application to drug metabolism. Drug Metab Dispos. 2004;32:966–72. [PubMed] [Google Scholar]
- Livezey MR, Nagy LD, Diffenderfer LE, Arthur EJ, Hsi DJ, Holton JM, Furge LL. Molecular analysis and modeling of inactivation of human CYP2D6 by four mechanism based inactivators. Drug Metab Lett. 2012;6 doi: 10.2174/187231212800229318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
- Nagy LD, Mocny CS, Diffenderfer LE, Hsi DJ, Butler BF, Arthur EJ, Fletke KJ, Palamanda JR, Nomeir AA, Furge LL. Substituted imidazole of 5-fluoro-2-[4-[(2-phenyl-1H-imidazol-5-yl)methyl]-1-piperazinyl]pyrimidine Inactivates cytochrome P450 2D6 by protein adduction. Drug Metab Dispos. 2011;39:974–83. doi: 10.1124/dmd.110.037630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatatakrishnan K. Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab Dispos. 2007;35:246–55. doi: 10.1124/dmd.106.012633. [DOI] [PubMed] [Google Scholar]
- Rowland P, Blaney FE, Smyth MG, Jones JJ, Leydon VR, Oxobrow AK, Lewis CJ, Tennant MG, Modi S, Eggleston DS, Chenery RJ, Bridges AM. Crystal structure of human cytochrome P450 2D6. J Biol Chem. 2006;281:7614–22. doi: 10.1074/jbc.M511232200. [DOI] [PubMed] [Google Scholar]
- Rydberg P, Gloriam DE, Olsen L. The SMARTCyp cytochrome P450 metabolism prediction server. Bioinformatics. 2010;26:2988–9. doi: 10.1093/bioinformatics/btq584. [DOI] [PubMed] [Google Scholar]
- Rydberg P, Olsen L. Predicting drug metabolism by cytochrome P450 2C9: comparison with the 2D6 and 3A4 isoforms. ChemMedChem. 2012;7:1202–9. doi: 10.1002/cmdc.201200160. [DOI] [PubMed] [Google Scholar]
- Seto Y, Guengerich FP. Partitioning between N-dealkylation and N-oxygenation in the oxidation of N,N-dialkylarylamines catalyzed by cytochrome P450 2B1. J Biol Chem. 1993;268:9986–97. [PubMed] [Google Scholar]
- Shahrokh K, Orendt A, Yost GS, Cheatham TE., 3RD Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J Comput Chem. 2012;33:119–33. doi: 10.1002/jcc.21922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silfeler I, Arica V, Arica S, Dogan M. Development of acute dystonia in three brothers due to metoclopramide. J Res Med Sci. 2012;17:308–9. [PMC free article] [PubMed] [Google Scholar]
- Silverman RB. Mechanism-based Enzyme Inactivation: Chemistry and Enzymology. CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- Teng L, Bruce RB, Dunning LK. Metoclopramide metabolism and determination by high-pressure liquid chromatography. J Pharm Sci. 1977;66:1615–8. doi: 10.1002/jps.2600661128. [DOI] [PubMed] [Google Scholar]
- Van der Padt A, Van Schaik RH, Sonneveld P. Acute dystonic reaction to metoclopramide in patients carrying homozygous cytochrome P450 2D6 genetic polymorphisms. Neth J Med. 2006;64:160–2. [PubMed] [Google Scholar]
- Wang A, Savas U, Hsu MH, Stout CD, Johnson EF. Crystal structure of human cytochrome P450 2D6 with prinomastat bound. J Biol Chem. 2012;287:10834–43. doi: 10.1074/jbc.M111.307918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Paine MJ, Marechal L,JD, Kemp CA, Ward CJ, Brown S, Sutcliffe MJ, Roberts GC, Rankin EM, Wolf CR. In silico prediction of drug binding to CYP2D6: identification of a new metabolite of metoclopramide. Drug Metab Dispos. 2006;34:1386–92. doi: 10.1124/dmd.106.009852. [DOI] [PubMed] [Google Scholar]
- Yuan R, Madani S, Wei XX, Reynolds K, Huang SM. Evaluation of cytochrome P450 probe substrates commonly used by the pharmaceutical industry to study in vitro drug interactions. Drug Metab Dispos. 2002;30:1311–9. doi: 10.1124/dmd.30.12.1311. [DOI] [PubMed] [Google Scholar]
- Zaretzki J, Rydberg P, Bergeron C, Bennett KP, Olsen L, Breneman CM. RS-Predictor models augmented with SMARTCyp reactivities: robust metabolic regioselectivity predictions for nine CYP isozymes. J Chem Inf Model. 2012;52:1637–59. doi: 10.1021/ci300009z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.