

Abstract
Lipid deposition in the liver is associated with metabolic disorders including fatty liver disease, type II diabetes, and hepatocellular cancer. The enzymes acetyl-CoA carboxylase 1 (ACC1) and ACC2 are powerful regulators of hepatic fat storage; therefore, their inhibition is expected to prevent the development of fatty liver. In this study we generated liver-specific ACC1 and ACC2 double knockout (LDKO) mice to determine how the loss of ACC activity affects liver fat metabolism and whole-body physiology. Characterization of LDKO mice revealed unexpected phenotypes of increased hepatic triglyceride and decreased fat oxidation. We also observed that chronic ACC inhibition led to hyper-acetylation of proteins in the extra-mitochondrial space. In sum, these data reveal the existence of a compensatory pathway that protects hepatic fat stores when ACC enzymes are inhibited. Furthermore, we identified an important role for ACC enzymes in the regulation of protein acetylation in the extra-mitochondrial space.
Keywords: Lipid metabolism, Liver, Steatosis, Acetylation
1. Introduction
The liver plays an important role in the regulation of whole body lipid and carbohydrate homeostasis during fluctuations in nutrient intake [1]. During fasting, the liver maintains euglycemia by releasing glucose into circulation. Hepatocyte glucose output is facilitated by an altered metabolic state wherein fatty acids are preferentially oxidized as an energy source to prevent the catabolism of glucose. In contrast, food intake triggers a rapid switch in hepatocyte metabolism that promotes the oxidation and metabolism of excess carbohydrates to glycogen and fat. With feeding, fat oxidation is repressed to prevent the catabolism of newly made lipid [2]. The acetyl-CoA carboxylase (ACC) enzymes ACC1 and ACC2 are important regulators of these metabolic transitions through their product malonyl-CoA [3,4]. ACC1 is localized in the cytosol and generates malonyl-CoA for de novo lipogenesis, while ACC2 is bound to the mitochondrial outer membrane and produces malonyl-CoA that can be used both as a substrate for lipogenesis [5] and as a negative modulator of mitochondrial fat oxidation by inhibiting carnitine palmitoyltransferase 1a (CPT1a). Thus, ACC-dependent malonyl-CoA production is considered to be a central control point for metabolic flexibility [6].
Both ACC isotypes are susceptible to dysregulation and are commonly over-expressed or over-activated in disease states associated with fatty liver [7–9]. Simple hepatosteatosis alone is thought to have a benign course [10,11], but up to 25% of patients progress to advanced diseases [10]. Fatty liver disease is a risk factor for insulin resistance, diabetes, and hepatocellular cancer. Thus, there is considerable interest in developing small molecule drugs that inhibit ACC enzymes [12,13]. Recently, it was shown that chronic activation of ACC enzymes in mice is sufficient to increase hepatic fat accumulation [14]; however, it remains unclear how the complete and chronic inhibition of ACC activity will impact liver lipid content, whole body metabolic physiology, or the metabolic fate of cytosolic acetyl-CoA in hepatocytes. To investigate these questions, we generated and characterized liver-specific ACC1 and ACC2 double knockout mice.
2. Materials and methods
2.1. Mice and diets
Ozgene Australia (Murdoch, Australia) was contracted to generate Acc1 and Acc2 floxed mice described previously [15,16]. Mice were produced on a pure C57BL/6 background using Bruce4 embryonic stem cells. Acc1 and Acc2 floxed mice were bred with C57BL/6 FLPe mice to delete the neomycin selection cassettes. To generate liver-specific deletions, female Acc1lox/lox/Acc2lox/lox mice were bred with male Acc1lox/lox/Acc2lox/lox mice expressing liver-specific albumin-Cre. Offspring from this cross produced a 1:1 ratio of LDKO (liver-specific double ACC knockout) to flox control offspring. Mice were maintained on a 12 h light–dark cycle and fed normal chow ad libitum (7912 Teklad LM-485 from Harlan Laboratories; 25 kcal% protein, 17 kcal% fat and 58 kcal% carbohydrates). All animal experiments were performed according to standard operating procedures approved by the Institutional Animal Care and Use Committee at University of Virginia.
2.2. Respirometry
Oxygen consumption rate (VO2) and respiratory control ratio (RCR) were measured under consistent environmental temperature (20–22 °C) using an indirect calorimetry system (Oxymax series, Columbus Instruments, Columbus, OH), as described [15]. Studies were commenced after acclimation to the metabolic chamber with airflow of 0.5 L/min. Gas samples were measured at 16-min intervals over a 24 h period with food and water provided ad libitum unless indicated otherwise.
2.3. Cell culture and drug treatment
Primary hepatocytes from control and LDKO mice were isolated based on established protocol [17], seeded in Dulbecco's modified Eagle's medium (DMEM) supplemented with 4.5 g/L glucose, 10% fetal bovine serum, 1 μM dexamethasone and 0.1 μM insulin for 4 h, and cultured overnight prior to experiment in serum-free DMEM with 4.5 g/L glucose, 0.2% bovine serum albumin, 100 nM dexamethasone and 1 nM insulin. For protein acetylation experiments, primary hepatocytes were treated with nicotinamide (NAM) at 5 mM for 24 h. For insulin stimulation experiments, hepatocytes were cultured in serum-free DMEM with 1 g/L glucose, 0.2% bovine serum albumin, 100 nM dexamethasone and no insulin for 3 h prior to addition of insulin for 10 min. DMSO was used as vehicle control.
2.4. Metabolic substrate competition assays and enzyme activity assays
Substrate competition assays were performed on monolayered hepatocytes (20,000 cells/cm2) in Krebs-Ringer Phosphate buffer (1.2 M NaCl, 6 mM Na2HPO4, 60 mM KCl, 4 mM NaH2PO4, 12 mM MgSO4, 125 mM HEPES pH7.4, 10 mM CaCl2) supplemented with non-labeled substrates: 50 μM acetate, 5 mM glucose, 0.5 mM glutamine, 125 μM palmitate and 1 mM carnitine, and one of the following radioactive labeled substrates: 10 μCi/mL 14C-acetate (de novo lipogenesis), 10 μCi/mL 3H-glucose (glycolysis) or 2 μCi/mL 14C-palmitate (fatty acid oxidation). Cells were incubated in sealed wells at 37 °C for 1.5 h. Lipids were extracted by the Folch method [18] and analyzed by scintillation counting or thin-layer chromatography to measure de novo lipogenesis. Palmitate oxidation was determined by acidifying each well with 2 M perchloric acid and trapping carbon dioxide in 2 M NaOH in a small tube placed in the well. Acid soluble 14C-labeled metabolites were extracted by centrifugation to determine incomplete palmitate oxidation [19]. Glycolysis was measured by scintillation counting 3H-glucose that was converted to 3H-H2O using the diffusion equilibrium method. Measurements of the activity of oxidative enzymes involved in metabolism and mitochondrial function were performed as described previously [20].
2.5. Acetyl-CoA measurements
Acetyl-CoA levels were measured by mass spectrometry using methods described in Refs. [15,21]. For quantification in mouse liver, tissue pieces (40 mg) were homogenized in 500 μL ice-cold isolation media (250 mM sucrose, 10 mM Tris–HCl and 1 mM EGTA), and centrifuged at 800 × g for 5 min at 4 °C to pellet cell debris and nuclei. Cleared cell lysate was centrifuged at 10,000 × g for 10 min at 4 °C to obtain a pellet enriched in mitochondria. Pellets were resuspended in 1 mL of 6% perchloric acid, and the supernatant (cytoplasm and microsome) fraction was mixed with an equal volume of 12% perchloric acid. 13C-3-malonyl-CoA (0.5 μM final) was added as a recovery standard. After centrifugation and ultracentrifugation of extract, supernatant was applied to a solid-phase extraction column (Oasis HLB 1 cc–30 mg, Waters) preconditioned with acetonitrile then milliQ water. Bound acyl-CoAs were washed with milliQ water, eluted with acetonitrile, dried under nitrogen gas at 37 °C, and resuspended in 120 μL of solvent A (2% ACN, 10 mM ammonium acetate, 5 mM acetic acid, 10 mM DIPEA) and analyzed by HPLC–MS. Analyses were performed using a triple quadrupole mass spectrometer (AB-Sciex 4000 Q-Trap) coupled to a Shimadzu LC-20AD LC system equipped with a Supelco Discovery C18 column (50 mm × 2.1 mm × 5 μm bead size) integrated with a precolumn (4 × 4 mm). A binary solvent system (total flow 0.25 mL/min) was used that consisted of the following solvents, A: 98.6% H2O, 2% acetonitrile, 5 mM acetic acid, 10 mM N,N-Diisopropylethylamine, 10 mM ammonium acetate; B: 75% acetonitrile, 25% solvent A. Chromatographic runs started at 100% A for 1 min, a linear gradient to reach 100% at 3.5 min, then 100% B for 2 min, and finally 100% solvent A for 2.5 min (8 min total). Column temperature was set to 30 °C and the flow rate was 0.5 mL/min. Measurements were carried out in positive mode using previously published transitions for acetyl-CoA (m/z 857.2 → m/z 350.2) and 13C-malonyl-CoA (m/z 810.4 → m/z 303.2) [21,22] using the following settings (DP, EP, CE, CXP, in volts: 91, 10, 43, 8; 106, 10, 41, 10; 116, 10, 43, 10). Quantification was carried out by measuring peak areas using the software Analyst 1.5.1 that were corrected for recovery using 13C-malonyl-CoA as an internal standard.
2.6. Proteomics sample preparation
Liver tissue was dounce homogenized in 9 M Urea with addition of complete EDTA-free protease inhibitor cocktail (Roche), 10 mM nicotinamide, and 50 mM butyric acid. After sonication on ice for 30 seconds, lysates were centrifuged at 10,000 × g and assayed for protein content using the BCA method. Protein extracts were reduced with 5 mM DTT and alkylated with 15 mM iodoacetamide. A 2.5 mg aliquot of protein from each sample was diluted 5-fold with 50 mM Tris pH 8.2 and digested overnight with trypsin (Promega) at 37 °C. The resulting peptides were acidified to pH 2 with trifluoroacetic acid (TFA) and desalted (but not eluted) using a tC18 SepPak cartridge (Waters) prior to on-column isotopic labeling of primary amines by reductive dimethylation [23]. After desalting, 5 mL of labeling reagent (light: 0.4% CH2O, 60 mM NaCNBH3 in 0.5 M MES pH 5.5, heavy: 0.4% CD2O, 60 mM NaCNBD3 in 0.5 M MES pH 5.5) was passed through the column at approximately 0.5 mL/min. The reaction was quenched by 15 column volumes of 1% TFA, washed with 0.5% acetic acid. Labeled peptides were eluted and mixed in a 1:1 ratio for further analysis.
Immediately after mixing heavy (+34.0689 Da) and light (+28.0313 Da) peptides, a 100 μg aliquot was removed for quantitative analysis of the unmodified proteome and dried by vacuum centrifugation. Peptides were resuspended in 0.1% NH4OH and fractionated in step-wise format with increasing concentrations of acetonitrile on microfilters constructed in-house using pH-resistant SDB-XC reverse phase chromatography material (3M Empore) [24]. Eluates were dried by vacuum centrifugation and stored at −20 °C prior to further analysis. Labeled peptides containing acetylated lysines were enriched after mixing by immunoaffinity purification. Briefly, 4.9 mg of dried peptides were resuspended in 50 mM MOPS-NaOH pH 7.2, 10 mM Na2HPO4, 50 mM NaCl and incubated overnight at 4 °C with pre-conjugated acetyl-lysine antibody (Immunechem). Immunoprecipitates were washed four times and eluted with 0.15% TFA. Acetylated peptides were desalted and fractionated in a step-wise format with increasing concentrations of NH4HCO3 using microfilters constructed in-house using SCX material (3M Empore). Eluates were desalted, dried, and stored at −20 °C prior to further analysis.
2.7. LC–MS/MS and data processing
Samples were subjected to reverse phase liquid chromatography on an EASY nLC (Thermo) equipped with a 40 cm × 75 μm column packed in-house with 1.9 μM Reprosil C18 particles (Dr. Maisch) and online analyzed by tandem mass spectrometry in a Q-Exactive (for unmodified peptides, 90 min gradients) or an LTQ Orbitrap Velos (for acetylated peptides, 120 min gradients). Mass spectra were acquired using a data dependent acquisition method (twenty most intense precursors selected for fragmentation) with dynamic exclusion (30 seconds). Raw spectra were converted to mzXML open data format and searched using Sequest against a concatenated forward and reverse version of the Uniprot mouse protein sequence database (v11/29/2012), digested with trypsin and allowing for up to two missed cleavages. Peptide mass tolerance was 50 ppm. Fragment ion tolerance was 0.36 for LTQ-Orbitrap-Velos data and 0.01 for Q-Exactive data. Carbamidomethylated cysteine (+57.021464), dimethylated lysine (+28.0313), and dimethylated peptide N-terminus (+28.0313) were searched as fixed modifications. Oxidized methionine (+15.994915), heavy dimethylated lysine (+6.03766), and heavy dimethylated peptide N-terminus (+6.03766) were searched as variable modifications in all cases. For acetyl-lysine enriched samples, an additional variable lysine modification was used: +13.97926, which corresponds to the difference between an acetyl group (+42.0105) and the fixed dimethyl group (+28.0313). This accounts for the fact that a lysine can be acetylated or dimethylated, but not both. Identified peptides were filtered to a false discovery rate of <1% and allowing only peptides that were correctly labeled and a minimum of 7 amino acids in length. For protein analysis, peptides were additionally filtered to a protein level FDR of <1%. In general, because acetylation events cause missed cleavages to occur yielding only one possible lysine candidate, site localization was not an issue. Nevertheless, localization scores and site refinement were performed using an in-house implementation of the Ascore algorithm [25] where an Ascore >13 equates to p <0.05. Maximum peak intensities and heavy-to-light (H/L) ratios for identified peptides were calculated using an in-house peptide quantification algorithm. To be considered for quantitation, we required a peptide signal to noise ratio >5. For most peptides (>90%), both light and heavy isotope intensities were measured. If only one isotope was measured, a ratio was calculated between peptide intensity and local noise. When more than one peptide was measured, the average H/L ratio was computed. For protein quantitation, H/L ratios for peptides mapping to the same protein were averaged. Similarly, for acetylation site quantitation, H/L ratios for peptides mapping to the same site were averaged. Resulting datasets were log2 transformed. To control for mixing error, both the acetylation site data and protein data were normalized to the median protein ratio. Finally, acetylation site quantifications were individually corrected for changes in its respective protein by subtracting the log2 ratio of the parent protein from the acetylation site ratio.
2.8. Bioinformatics
Statistical analysis was performed in R version 2.15.2. For subcellular compartment analysis, protein identifications were mapped to Gene Ontology Cellular Component terms using gene sets from the Molecular Signatures Database MsigDB [26]. For mitochondrial protein analysis, the mouse MitoCarta database [27] was converted to Uniprot identifiers and used to assign high confidence mitochondrial proteins to the datasets. Metabolic pathways and proteins were manually curated from IUBMB-Nicholson metabolic pathway diagrams (http://www.iubmb-nicholson.org), and plotted according to log2 fold change using Cytoscape v2.8.3 [28]. Functional enrichment analysis was performed using DAVID bioinformatics resources v6.7 [29]. Acetylated proteins were queried for enrichment against a background containing all proteins found in the proteome dataset.
2.9. Triglyceride production and oral triglyceride tolerance assays
To measure hepatic triglyceride production rate, female mice (12 wks of age) were injected i.p. with 1 g/kg body weight Poloxamer 407 [30] and serum triglyceride levels were measured over 24 h. Mice were fasted from 9 am–1 pm, and tail vein whole blood was sampled prior to injection then at 1, 2, 6, and 24 h after injection. To determine oral triglyceride tolerance, mice were fasted for 4 h (8 am–12 pm) prior to receiving safflower oil by oral gavage (10 μL/g body weight). Tail vein blood was sampled prior to gavage then hourly for 6 h. Serum was isolated and stored at −20 °C until triglyceride assay (Pointe Scientific).
2.10. Tissue and serum metabolites
Submandibular whole blood was sampled from 16-week-old mice at fed (9 pm) then 12 h fasted states. Serum was isolated by centrifugation at 2,000 × g for 15 min at 4 °C. Metabolites were determined according to manufacturers' protocols: Triglyceride assay (Pointe Scientific); Cholesterol assay (Infinity Cholesterol Liquid Reagent, Thermo Scientific); Free fatty acid assay (BioVision); Insulin ELISA (Crystal Chem Inc); Ketone assay (Cayman); NAD+ assay (BioAssay System).
2.11. Histology
Liver samples were fixed in 10% neutral buffered formalin and paraffin-embedded for microtome sectioning (5 μm thick) and hematoxylin–eosin staining. Frozen-sections from OCT-embedded liver samples were used for Oil-Red-O staining as previous described [31]. Microscopy was performed and analyzed on a ScanScope.
2.12. Tolerance tests
Glucose, insulin and pyruvate tolerance tests were performed 6 h after the removal of food (8 am–2 pm). Glucose (1.5 g/kg), insulin (1 U/kg) or pyruvate (2 g/kg) tolerance tests were performed via i.p. injection. Blood glucose was measured with an AccuChek II glucometer (Roche).
2.13. Western blotting
Liver ACC proteins were analyzed using avidin-pulldown as described previously [15]. For acetyl-lysine immunoblotting, liver tissue or cells were lysed in RIPA buffer (50 mM TRIS pH 8.0, 100 mM NaCl, 2 M EGTA pH 7.0, 0.4% v/v Triton X-100, 10 mM nicotinamide), with protease inhibitors cocktail (Roche) and phosphatase inhibitors (2 mM sodium orthovanadate, 1 mM sodium pyrophosphate, 10 mM sodium fluoride, 250 nM microcystin LR), and immunoblotted with an anti-acetyl-lysine antibody (Cell Signaling 9441). Other antibodies used in this study: phospho-Akt (S473) (587F11; Cell Signaling 4051), pan Akt (C67E7, Cell Signaling 4691), pan 14-3-3 (H-8, Santa Cruz, sc-1657), Mitomix rodent OXPHOS cocktail (Mitosciences MS604), CPT1 (H-40, Santa Cruz, sc-98834) and alpha-tubulin (H-300, Santa Cruz, sc-5546). Line scan analysis was performed using Image J.
2.14. Reverse transcription realtime PCR
Liver (2 μg, Trizol) or cells (1 μg, Direct-zol) total RNA was semi-quantitated by standard two-step RT-PCR (High Capacity cDNA synthesis kit, Roche; Sensifast SYBR Green mix, Bioline) using gene-specific primers (Integrated DNA Technologies). Acaca-sense, 5′-CATCACCATCAGCCTGGTTACA-3′, Acaca-antisense 5′-ACTGTGTACGCTCTTCGGCAT-3′; Acacb-sense, 5′-GTTTGGGCACTGCTTCTCCT-3′, Acacb-antisense, 5′-CACACACCACCCCAAGCAT-3′; Acadm-sense 5′-GCAGGTTTCAAGATCGCAATG-3′, Acadm-antisense 5′-TGAAACTCCTTGGTGCTCCACT-3′; Ppargc1b-sense 5′-GACGAGCTTTCACTGCTACAGA-3′, Ppargc1b-antisense, 5′-TGCCATCCACCTTGACACA-3′; Esrrg-sense, 5′-GAGGACGATTCAAGGTAACATAGAG-3′, Esrrg-antisense, 5′-GACGGACCCCTTCTTTCAGC-3′. Other genes of interest were performed using primer pairs established in previous publications: Ppara, Fgf21, and Pdk4 [32]; Acox1 and Hmgcs2 [33]; Cpt1a [34]; Pck1 and G6pc [35]; Ppargc1a and Esrra [36]; Ppifa (housekeeping gene) [37].
2.15. Statistical analyses
Data are expressed as means ± standard error of the mean (SEM). p-values were calculated by Mann–Whitney test or one-way ANOVA with Fisher's PLSD post-hoc test, unless otherwise stated. Statistical significance was set at p <0.05.
3. Results
3.1. Increased hepatic triglyceride accumulation in the absence of ACC activity
Other groups have reported single gene knockout of either ACC1 or ACC2 individually in mice [5,15,38,39]. However, no studies have genetically knocked out both ACC enzymes specifically in mouse liver. In this study we generated liver-specific ACC1 and ACC2 double knockout (LDKO) mice, as follows. ACC1 and ACC2 floxed mice were bred with mice expressing Cre under the control of a liver-specific albumin promoter. Knockout was confirmed at the mRNA and protein levels in liver tissue (Figures 1A and B). To verify that ACC enzymatic activity was deficient in the liver, we isolated primary hepatocytes from LDKO and flox control mice and tested their capacity to utilize 14C-acetate (a cell-permeable precursor to cytosolic acetyl-CoA) for lipogenesis. Hepatocytes from LDKO mice were completely deficient in lipogenic conversion of acetate into lipid, but were fully capable of synthesizing sterols from this substrate (Figure 1C). The utilization of 14C-acetate for sterol synthesis was expected because this process does not require ACC activity. Compared to flox controls, LDKO mice had similar body weight and adiposity (Supplemental Figure S1), and had normal energy expenditure and respiratory quotient in both fed and fasted states (Supplemental Figures S2 and S3). The LDKO mice also demonstrated normal responses to metabolic challenges with a bolus injection of glucose, pyruvate, or insulin (Figure 1D–F). LDKO mice had similar serum glucose, insulin, free fatty acid, and triglyceride levels to flox controls in both the fed and fasted states, but had 22% less (p < 0.05) serum cholesterol in the fasted state (Table 1). Hepatocytes and liver tissue demonstrated normal insulin-stimulation of Akt (Supplemental Figure S4). Together, these data demonstrate that loss of ACC activity in the liver does not adversely affect whole body nutrient handling or insulin sensitivity. However, we did find that LDKO liver mass was increased (Figure 1G) concomitant with increased triglyceride content (Figure 1H). Histological analysis of liver tissue by Oil-red-O and H&E staining revealed that LDKO hepatocytes accumulated microvesicular fat (Figure 1I), a phenotype normally associated with a defect in fat oxidation [40]. ACC enzymes normally function to promote fat storage; therefore, the finding that ACC deletion increases basal liver fat accumulation was unexpected.
Figure 1.
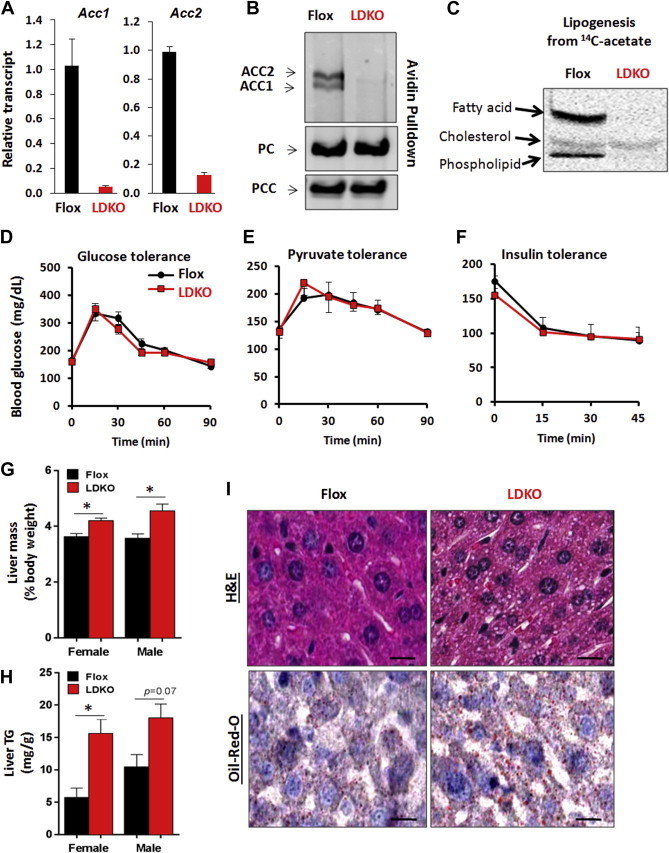
Loss of hepatic ACC activity increases fat storage. (A) Liver Acc1 and Acc2 mRNA transcripts quantified by qPCR in flox control and LDKO mice (p < 0.0001, n = 10, Mann–Whitney). (B) Avidin-bead pulldown with immunoblotting for ACC enzymes in liver lysates from flox and liver-specific ACC1 and ACC2 knockout (LDKO) mice, with pyruvate carboxylase (PC) and propionyl-CoA carboxylase (PCC) enzymes serving as protein loading controls. (C) Incorporation of 14C-acetate into fatty acids and sterols in primary hepatocytes. (D–F) Glucose, pyruvate and insulin tolerance tests (D and E, n = 5; F, n = 3). (G) Liver to body weight percentage and (H) triglyceride content of flox and LDKO mice (female LDKO (n = 8) and Flox (n = 5), male LDKO (n = 5) and Flox (n = 3); *p < 0.05, Mann–Whitney.). Data expressed as mean ± SEM. (I) Representative liver sections stained with hematoxylin and eosin (H&E) and Oil-Red-O; Scale bars equal 100 μm.
Table 1.
Serum parameters of fed or 12 h-fasted LDKO and flox mice.
| Fed |
Fasted |
|||
|---|---|---|---|---|
| Flox | LDKO | Flox | LDKO | |
| Triglyceride (mg/dL) | 204.5 ± 28.8 (9) | 233.8 ± 27.0 (10) | 156.0 ± 12.9 (9) | 136.3 ± 5.0 (11) |
| Cholesterol (mg/dL) | 91.4 ± 6.7 (9) | 71.9 ± 5.4 (10) | 93.6 ± 6.0 (9) | 73.5 ± 3.7* (11) |
| Free fatty acid (mM) | 0.92 ± 0.11 (9) | 0.87 ± 0.09 (10) | 1.40 ± 0.15 (9) | 1.15 ± 0.12 (11) |
| Glucose (mg/dL) | 170.8 ± 18.5 (6) | 171.3 ± 22.9 (4) | 124.9 ± 7.6 (17) | 144.5 ± 7.9 (17) |
| Insulin (ng/ml) | 0.97 ± 0.12 | 1.34 ± 0.27 | 0.42 ± 0.08 | 0.51 ± 0.11 |
Data expressed as mean ± SEM. * Significant difference (p < 0.05) between Flox vs. LDKO of same condition (fed or fasted). Parentheses indicate the number of mice per group.
3.2. ACC inhibition paradoxically suppresses fatty acid oxidation
Hepatic fat accumulation can be caused by numerous mechanisms including defective lipid export, increased lipid intake, increased lipid synthesis, or decreased fat oxidation. Lipid synthesis can be ruled out (Figure 1C); therefore, we measured triglyceride export from the liver, the clearance of triglyceride after a bolus oral gavage of safflower oil, and the oxidation of the saturated fatty acid palmitate (Figure 2A–C). The rate of lipid efflux from the liver was measured by treating LDKO and flox mice with the lipoprotein lipase inhibitor, Poloxamer 407, and monitoring serum triglyceride accumulation over time (Figure 2A). LDKO mice had greater rates of triglyceride export from the liver compared to flox control mice (Figure 2A), thus indicating the increase in hepatic fat in LDKO mice was not due to impaired lipid efflux from the liver. We next challenged LDKO and flox mice with an oral bolus of safflower oil and measured triglyceride appearance and clearance from the serum. This experiment showed that LDKO mice had normal uptake and clearance of lipid (Figure 2B). Finally, we measured fatty acid oxidation in LDKO and flox primary hepatocytes using 14C-palmitate. This experiment revealed that LDKO hepatocytes had a significant decrease in fatty acid oxidation compared to flox controls (48% decrease, p < 10−4, Figure 2C).
Figure 2.
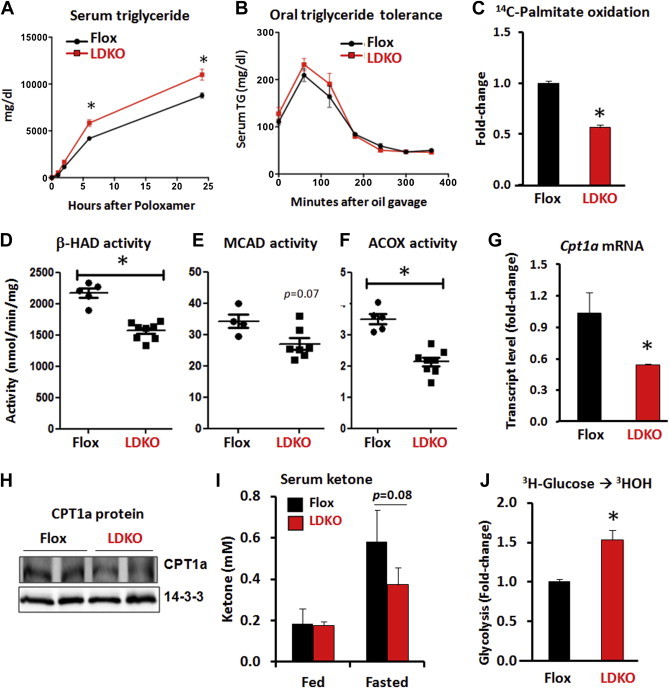
The loss of ACC activity decreases hepatic fat oxidation. (A) Hepatic triglyceride production assessed by serum triglyceride levels at time points after i.p. treatment of 1 g/kg lipoprotein lipase inhibitor, Poloxamer 407 (n = 5 LDKO vs. n = 4 flox). (B) Oral triglyceride tolerance assessed by serum triglyceride levels at various time points after a bolus of safflower oil (n = 5). Data expressed as mean ± SEM; *p < 0.0001, multiple unpaired t-test with correction using Holm-Sidak method. (C) Rate of palmitate oxidation in isolated primary hepatocytes (*p < 0.001, Mann–Whitney, seven independent experiments). Enzyme activity assays of (D) 3-hydroxyacyl-CoA dehydrogenase (β-HAD), (E) media-chain acyl-CoA dehydrogenase (MCAD), and (F) peroxisomal acyl-CoA oxidase (ACOX) in liver tissue (*p < 0.05, Mann–Whitney, n = 7 LDKO vs. n = 5 flox). (G and H) Hepatic CPT1a transcript levels and protein expression.14-3-3 serves as a protein loading control. (I) Serum ketone levels at fed and fasted states (Mann–Whitney; n = 5 flox vs. n = 10 LDKO). (J) Rate of glycolysis in isolated primary hepatocytes (*p < 0.01, Mann–Whitney, five independent experiments). Data expressed as mean ± SEM.
The mechanism(s) of reduced fat oxidation were investigated in more detail by assaying the activity of key enzymes in the mitochondrial and peroxisomal fatty acid oxidation pathways in liver lysates. Compared to flox control tissue, LDKO liver lysates had decreased activity of mitochondrial 3-hydroxyacyl-CoA dehydrogenase (β-HAD, p = 0.002), mitochondrial medium-chain acyl-CoA dehydrogenase (MCAD, p = 0.07), and peroxisomal acyl-CoA oxidase (ACOX, p = 0.002) (Figure 2D–F). The LDKO liver tissue also had lower mRNA and protein expression of CPT1a; the rate-limiting enzyme in mitochondrial fat oxidation (Figure 2G and H). Furthermore, LDKO mice showed a trend of impaired fasting-induced ketone production; an indirect marker of hepatic fat oxidation in vivo (Figure 2I). To determine whether the decrease in fat oxidation was compensated by altered glucose utilization, as predicted by the Randle cycle [2], we measured the rate of glycolysis in isolated primary hepatocytes from LDKO and flox mice. These data revealed a 40% increase in glycolysis in LDKO hepatocytes compared to flox controls (Figure 2J). Collectively, these data identify that inhibition of ACC enzymes leads to reprogramming of hepatic glucose and fatty acid metabolism that resembles the chronic fed state.
Peroxisomal proliferator-activation receptor alpha (PPARα) regulates the expression of several hepatic genes involved in fat oxidation, including Cpt1a. Therefore, we investigated transcriptional changes in pathways involved in nutrient metabolism including 5 other PPARα-regulated genes: Pdk4, Fgf21, Acox1, Hmgcs2 and Ppara. In contrast to Cpt1a, no other PPARα-regulated genes were down regulated and both Pdk4 and Hmgcs2 were upregulated in liver tissue from LDKO mice compared to controls (Figure 3A). Furthermore, there were no statistically significant transcriptional changes in other glucose or fatty acid metabolic genes regulated by SREBP1c, PGC-1α or PGC-1β (Figure 3B–D). These data also show that the decreases in ACOX and MCAD activity in LDKO liver tissue (Figure 2D–F) was not due to alterations in transcription of their respective genes, Acox1 and Acadm (Figure 3A and D).
Figure 3.
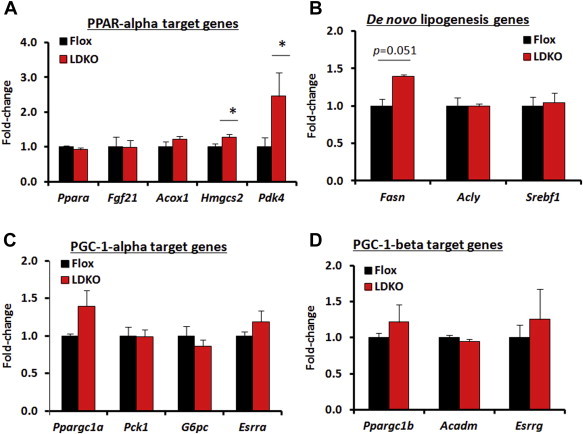
PPARα, SREBP1c, and PGC-1 target gene transcription in LDKO vs. flox liver. (A–D) qPCR analysis of target genes regulated by PPARα, SREBP1c, PGC-1α and PGC-1β. Data expressed as mean ± SEM (n = 5, *p < 0.05 LDKO vs. Flox).
3.3. Liver ACC inhibition disrupted acetyl-CoA homeostasis and altered protein acetylation
Acetyl-CoA is a versatile cellular metabolite utilized for ATP production, cholesterol and lipid biosynthesis, and protein acetylation. Given the high flux of acetyl-CoA through ACC enzymes for lipogenesis, we investigated whether the perturbation of ACC activity could impact acetyl-CoA utilization for protein acetylation. Primary hepatocytes and liver tissue from LDKO and flox control mice were immunoblotted with an antibody that recognizes acetylated lysine residues (Figure 4A and B). Compared to flox controls, the LDKO tissues and cells had increased immunoblotting signals across a broad range of molecular weights (Figure 4A and B). Since deacetylase enzymes may also regulate protein acetylation, we measured NAD+ levels and compared the acetylation pattern of ACC-deficient cells with the pattern caused by the NAD+-dependent deacetylase inhibitor, nicotinamide. NAD+ levels were similar in flox and LDKO liver lysates (Figure S5A), and NAM treatment induced a different pattern of protein acetylation than was observed with ACC inhibition (Figure 4A). These data reveal that ACC inhibition promotes global protein acetylation, and indicate that NAD+-dependent deacetylase inhibition cannot account for the changes in lysine acetylation.
Figure 4.
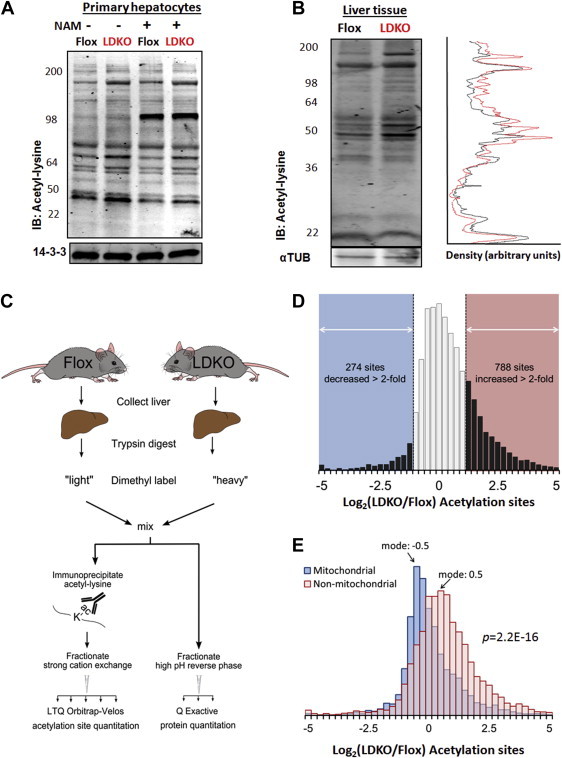
ACC inhibition alters the liver acetyl-lysine proteome. (A) Acetyl-lysine immunoblotting of whole cell lysates of primary hepatocytes treated with or without 5 mM nicotinamide, isolated from LDKO and flox mice. (B) Acetyl-lysine immunoblotting of liver tissue from LDKO and flox mice with line scan analysis. (C) Work flow of quantitative acetylproteomics experiments on flox and LDKO livers. (D) Distribution of relative abundances in acetylation sites between LDKO and flox mice, expressed in logarithmic scale (log2) for the entire proteome and (E) mitochondrial vs. non-mitochondrial proteins according to Mitocarta classification (n = 3, p = 2.2e-16, Mann–Whitney).
3.4. Subcellular compartmentalization of ACC-dependent changes in protein acetylation
It was recently shown in Saccharomyces cerevisiae that ACC1 inhibition increases nuclear histone acetylation [41]; however, the Western blots demonstrated lysine hyper-acetylation across a broad range of protein molecular weights that are not indicative of histones. To identify the proteins that were hyper-acetylated, we enriched for acetyl-peptides by an anti-acetyl-lysine antibody pulldown and quantified them by proteomics (Figure 4C). This approach resulted in the quantification of 26,843 acetylated peptides corresponding to 3586 unique acetylation sites on 1151 proteins (Supplementary Dataset S1). After normalization to protein abundance (Supplementary Dataset S2), the distribution of acetylation site relative abundances were positively biased (mean Log2(LDKO/flox) = 0.30), signifying global hyper-acetylation in the LDKO liver. Specifically, 788 acetylation sites were increased by greater than 2-fold (22% of the unique sites identified) (Figure 4D). Curiously however, 274 acetylation sites (8% of the unique sites identified) were decreased by more than 2-fold compared to floxed controls. Since ACC enzymes only have access to cytoplasmic acetyl-CoA, we investigated whether subcellular protein distribution correlated with lysine acetylation. These analyses identified a significant enrichment in the acetylation of proteins in the extra-mitochondrial space and hypo-acetylation of proteins located in mitochondria (Figures 4E and 5A).
Figure 5.
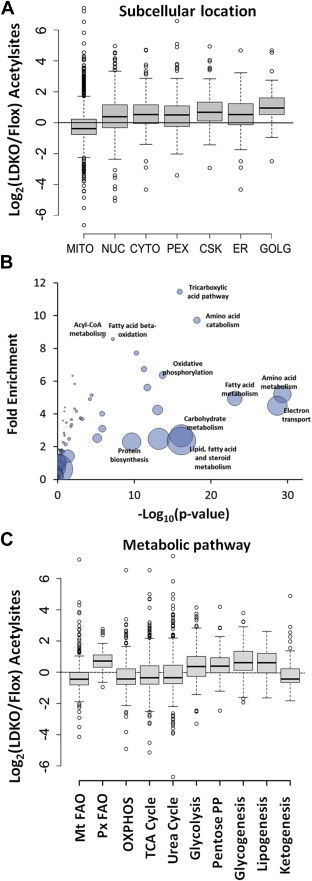
ACC-mediated alterations in protein acetylation are enriched for metabolic networks involved in intermediary nutrient metabolism. (A) Boxplot distribution of acetylation site of proteins based on their annotated subcellular localizations. (B) Functional enrichment analysis of acetylated proteins identifies that metabolic networks are significantly affected. Bubble size represents the number of proteins in each cluster. (C) Boxplot distribution of protein acetylation sites based on their metabolic process.
We next evaluated whether altered protein acetylation patterns caused by ACC inhibition were biased toward particular biological or metabolic pathways. Functional enrichment analysis identified that proteins involved in intermediary nutrient metabolism were highly acetylated in the liver tissue of LDKO mice compared to flox controls (eight out of the ten most acetylated pathways, Figure 5B). In livers from LDKO mice, glycolytic and peroxisomal fatty acid metabolic enzymes were generally hyper-acetylated, while mitochondrial proteins involved in fat oxidation and the tricarboxylic acid cycle (TCA) were generally unaffected or hypo-acetylated compared to floxed controls (Figures 5C and 6). To determine whether protein acetylation corresponded with acetyl-CoA levels, we measured acetyl-CoA in mitochondrial and cytoplasmic fractions of flox control and LDKO liver tissues. Curiously, both mitochondrial and cytoplasmic acetyl-CoA levels were similar between flox and LDKO liver tissues (Supplementary Figure S5B). These data suggest that alterations in metabolic substrate flux or increased protein acetylation balance cellular acetyl-CoA levels independently of functional ACC enzymes.
Figure 6.
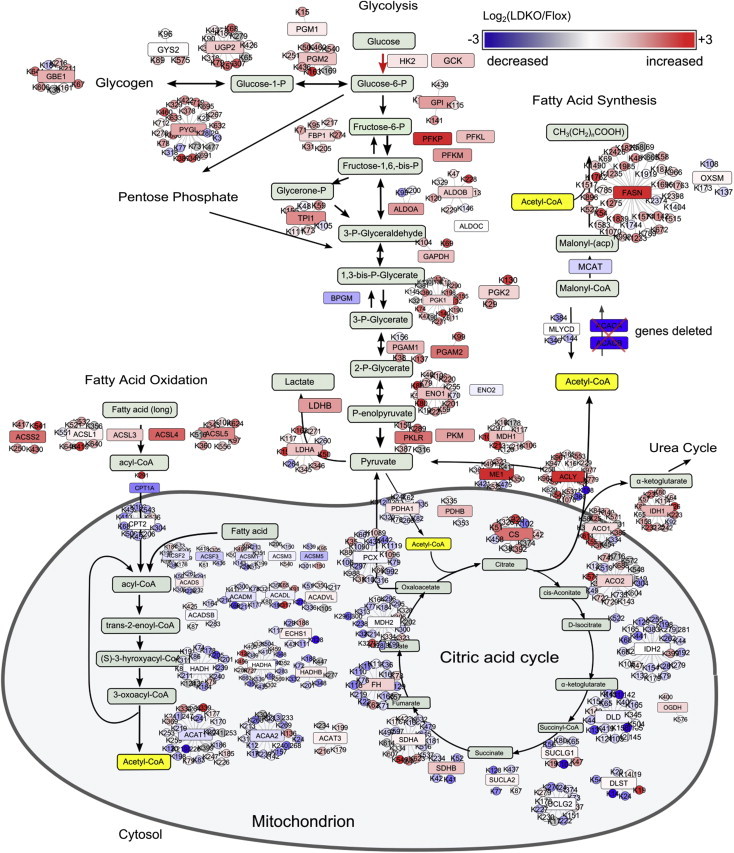
Schematic showing the protein acetylation sites in metabolic pathways that are most differentially acetylated in LDKO/flox liver. Pathway color scheme: Red, increased in LDKO vs. flox; blue, decreased; red or blue box, enzymes; circle, acetylation sites on each enzyme.
Since mitochondria were generally hypo-acetylated, we investigated the activity and acetylation status of complexes I, II and IV of the electron transport chain (ETC) and citrate synthase of the tricarboxylic acid cycle (TCA). The expression of all ETC complexes was similar between flox and LDKO livers as determined by Western blot using antibodies against complex I subunit NDUFB8, complex II subunit of 30 kDa, complex III core protein 2, complex IV subunit I and complex V alpha subunit (Figure 7A). However, proteomics analysis of acetylated ETC components revealed variable differences in the expression and acetylation of other individual components of the ETC (Figure 7B). To determine whether these changes affected complex activity, we measured their activity (Figure 7C). LDKO liver lysates had increased activity of ETC complex I, lower activity of complex II, and similar activity of complex IV compared to control (Figure 7C). In addition, citrate synthase enzyme activity was increased in LDKO tissue lysates (Figure 7C).
Figure 7.
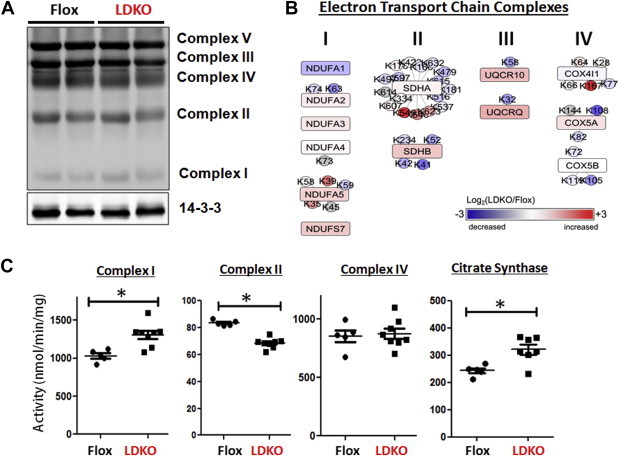
Protein expression and activity of electron transport chain complexes. (A) Western blot analysis of a liver lysates from LDKO and flox mice separated by SDS-PAGE and immunoblotted with five monoclonal antibodies recognizing a representative protein in each complex involved in mitochondrial oxidative phosphorylation. (B) Changes in protein expression (boxes) and acetylation (circles) of electron transport chain complexes I, II, III, and IV. (C) Enzyme activity of complexes I, II, and IV, and citrate synthase in tissue lysates from LDKO and flox control liver (n = 5 flox, n = 8 LDKO, *p < 0.05, Mann–Whitney, n = 5 flox, n = 8 LDKO). Data expressed as mean ± SEM.
4. Discussion and conclusions
ACC enzymes are attractive drug targets for fatty liver diseases because inhibition of their activity is predicted to decrease lipogenesis, increase fat oxidation, and thereby reduce fat storage. Our observation that the deletion of ACC activity in the liver promotes excess fat accumulation is entirely unexpected based on previous work in this area. The only study to genetically target both ACC enzymes in vivo utilized an antisense oligonucleotide (ASO) in rats. The ASO markedly decreased ACC expression in both adipose tissue and liver, and resulted in a decrease in hepatic fat and improved insulin sensitivity when rats were fed a high-fat diet [42]. The phenotype of the LDKO mouse and ASO treatment are not directly comparable for many reasons including differences in the species of rodent, the diet, the duration of inhibition, and the different tissues targeted. Regardless of these discrepancies, the LDKO mouse described herein has allowed us to evaluate the consequences of hepatic ACC inhibition on liver fat metabolism and whole animal physiology. Importantly, the increase in liver fat storage in LDKO mice reveals a gap in knowledge concerning the role of ACC enzymes in liver fat metabolism.
The increase in liver fat in LDKO hepatocytes is likely caused by reduced fatty acid oxidation, as supported by our findings demonstrating decreased CPT1a expression; decreased fasting ketone production; accumulation of microvesicular fat; and reduced activities of peroxisomal acyl-CoA oxidase, mitochondrial βHAD and mitochondrial MCAD enzymes in the LDKO liver compared to floxed controls. Furthermore, since complex II is involved in FADH2 oxidation that is driven by fatty acid catabolism, the reduced activity of this complex may also contribute to impaired fatty acid oxidation in LDKO hepatocytes. These data indicate that ACC inhibition triggers multiple mechanisms to repress fatty acid oxidation.
It has been suggested that newly synthesized lipids are ligands for PPARα [33,43]. However, our data indicate that liver-derived lipogenesis is not required for PPARα-mediated gene transcription since only CPT1a was decreased in LKDO liver tissue compared to flox controls, while five other PPARα regulated genes were unaltered or upregulated (Pdk4 and Hmgcs2). Furthermore, we did not observe significant effects on other metabolic genes regulated by SREBP1c, PGC-1α or PGC-1β in LDKO mice. These data reveal the existence of a very precise, but unknown, mechanism whereby ACC inhibition specifically targets CPT1a at the mRNA level to decrease fatty acid oxidation without disrupting other closely regulated genes involved in carbohydrate and mitochondrial metabolism.
In organelles such as the nucleus, the regulation of protein acetylation is fairly well-characterized with respect to the roles of histone acetyltransferases and deacetylases. By contrast, the regulation of protein acetylation in the cytoplasm is insufficiently understood. In recent years, several reports have demonstrated that protein acetylation, particularly in cytoplasmic proteins, plays an important role in liver metabolism [44,45]. In LDKO liver, protein hyper-acetylation was observed in all organelles except mitochondria, indicating an important regulatory role for ACC enzymes in the control of protein acetylation in the extra-mitochondrial space. It is likely that the metabolic phenotype of reduced fat oxidation and increased glycolysis observed in LKDO liver is related to the alterations in protein acetylation. For example, hyper-acetylation of glycolytic enzymes is known to promote glycolysis [44,46,47].
The mechanisms that underlie the compartment-specific changes in acetylation caused by ACC inhibition require further investigation, and several scenarios are possible. First, the loss of ACC activity in the extra-mitochondrial space may increase the availability of acetyl-CoA for protein acetyltransferases. Several acetyltransferases are known to localize in the cytoplasmic compartment including GCN5 and PCAF [48–50]; however, it remains unclear whether these acetyltransferases have the broad substrate specificity or subcellular distribution necessary to mediate the hyper-acetylation observed in LDKO liver tissue. Another possibility is that the increase in protein hyper-acetylation occurs non-enzymatically, as has been demonstrated previously [51–53]. Studies in yeast demonstrate that ACC gene inhibition is sufficient to promote histone acetylation due to increased availability of acetyl-CoA [41]. Similarly, inhibition of AMPK in yeast activated ACC and led to reduced histone acetylation [54]. Therefore, a similar regulation of protein acetylation by ACC enzymes may also exist in the mammalian hepatocyte on non-histone proteins. Although we were unable to detect an increase in acetyl-CoA levels in LDKO liver, it remains possible that certain nutritional or hormone-activated states are required to observe acetyl-CoA accumulation. It is less likely that decreased NAD+-dependent deacetylase activity drives global protein acetylation since NAD+ levels were unchanged in LDKO tissue and nicotinamide did not increase protein acetylation with a similar pattern. Finally, it was recently shown that decreased activity of mitochondrial ETC complex I is associated with increased mitochondrial protein acetylation [55]. The LDKO liver had significantly higher complex I activity concomitant with a decrease in protein acetylation in mitochondria, thus it is possible that mitochondrial hypo-acetylation may be secondary to increased complex I activity.
In summary, we observe that complete inhibition of hepatocyte ACC enzymes triggers the activation of a compensatory pathway that preserves fat storage in the liver. We hypothesize that the mechanism linking the loss of ACC activity to increased fat storage is due to increased acetylation of key metabolic enzymes and transcriptional regulatory sensors. These sensors interpret hyper-acetylation as a nutrient replete state and coordinate the feeding response to decrease fatty acid oxidation; an appropriate response to nutrient excess. Future mutational studies are required to test this hypothesis and to define how specific acetyl-lysine sites identified in this study affect protein function. Collectively, this new information advances our understanding of the role of ACC enzymes in hepatic nutrient metabolism and protein acetylation, and also reveals the existence of a therapeutic window for drug discovery efforts targeting ACC. One possibility is that an ideal ACC inhibitor would impair lipogenesis but maintain enough residual activity in specific subcellular compartments to prevent protein hyper-acetylation and avoid the compensatory inhibition of fat oxidation. These data also implicate acetyl-CoA, like malonyl-CoA, as a potent regulator of hepatic metabolic flexibility.
Acknowledgments
We thank Lars Löfgren and Nick Oakes (AstraZeneca) for valuable advice regarding acyl-CoA measurements. Drs Deborah Muoio and Michael Davies (Duke) provided helpful advice regarding reagents for detecting protein acetylation. This work was funded primarily through start-up funds and Research and Development funds from the University of Virginia (to KLH). KLH is supported by an American Diabetes Association Junior Faculty Award and JV is supported by an Ellison Medical Foundation New Scholar Award. We thank Eric Ward, Steve Breazeale, and Rich Anderson at Cropsolutions for the provision of reagents. Dr. Ivan K. H. Poon provided scientific advice as well as experimental and editorial assistance.
Footnotes
Jenny D.Y. Chow and Robert T. Lawrence contributed equally to this work.
Contributor Information
Judit Villén, Email: jvillen@uw.edu.
Kyle L. Hoehn, Email: klh8st@virginia.edu.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data related to this article:
Lysine acetylome data comparing LDKO vs. flox livers.
Global proteomic data comparing LDKO vs. flox livers.
References
- 1.Strable M.S., Ntambi J.M. Genetic control of de novo lipogenesis: role in diet-induced obesity. Critical Reviews in Biochemistry and Molecular Biology. 2010;45:199–214. doi: 10.3109/10409231003667500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Randle P.J., Garland P.B., Hales L.N., Newsholme E.A. The glucose fatty acid cycle, its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;281:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 3.McGarry J.D., Takabayashi Y., Foster D.W. The role of malonyl-CoA in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. Journal of Biological Chemistry. 1978;253:8294–8300. [PubMed] [Google Scholar]
- 4.Xue B., Kahn B.B. Ampk integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. Journal of Physiology. 2006;574:73–83. doi: 10.1113/jphysiol.2006.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harada N., Oda Z., Hara Y., Fujinami K., Okawa M., Ohbuchi K. Hepatic de novo lipogenesis is present in liver-specific acc1-deficient mice. Molecular and Cellular Biology. 2007;27:1881–1888. doi: 10.1128/MCB.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGarry J.D., Mannaerts G.P., Foster D.W. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. Journal of Clinical Investigation. 1977;60:265–270. doi: 10.1172/JCI108764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvisi D.F., Wang C., Ho C., Ladu S., Lee S.A., Mattu S. Increased lipogenesis, induced by akt-mtorc1-rps6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140 doi: 10.1053/j.gastro.2010.12.006. 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohjima M., Enjoji M., Higuchi N., Kato M., Kotoh K., Yoshimoto T. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. International Journal of Molecular Medicine. 2007;20:351–358. [PubMed] [Google Scholar]
- 9.Yahagi N., Shimano H., Hasegawa K., Ohashi K., Matsuzaka T., Najima Y. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. European Journal of Cancer. 2005;41:1316–1322. doi: 10.1016/j.ejca.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 10.Angulo P. Nonalcoholic fatty liver disease. New England Journal of Medicine. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 11.Sun Z., Lazar M.A. Dissociating fatty liver and diabetes. Trends in Endocrinology and Metabolism. 2013;24:4–12. doi: 10.1016/j.tem.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corbett J.W. Review of recent acetyl-CoA carboxylase inhibitor patents: mid-2007–2008. Expert Opinion on Therapeutic Patents. 2009;19:943–956. doi: 10.1517/13543770902862180. [DOI] [PubMed] [Google Scholar]
- 13.Tong L., Harwood H.J., Jr. Acetyl-coenzyme a carboxylases: versatile targets for drug discovery. Journal of Cellular Biochemistry. 2006;99:1476–1488. doi: 10.1002/jcb.21077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fullerton M.D., Galic S., Marcinko K., Sikkema S., Pulinilkunnil T., Chen Z.P. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nature Medicine. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoehn K.L., Turner N., Swarbrick M.M., Wilks D., Preston E., Phua Y. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cellular Metabolism. 2010;11:70–76. doi: 10.1016/j.cmet.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J., Walsh M.C., Hoehn K.L., James D.E., Wherry E.J., Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. Journal of Immunology. 2014;192:3190–3199. doi: 10.4049/jimmunol.1302985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Severgnini M., Sherman J., Sehgal A., Jayaprakash N.K., Aubin J., Wang G. A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology. 2012;64:187–195. doi: 10.1007/s10616-011-9407-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Folch J., Lees M., Sloane Stanley G.H. A simple method for the isolation and purification of total lipides from animal tissues. Journal of Biological Chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- 19.Hirschey, M.D., Verdin, E., 2010. Measuring fatty acid oxidation in tissue homogenates.
- 20.Taddeo E.P., Laker R.C., Breen D.S., Akhtar Y.N., Kenwood B.M., Liao J.A. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Molecular Metabolism. 2013;3:124–134. doi: 10.1016/j.molmet.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bengtsson C., Blaho S., Saitton D.B., Brickmann K., Broddefalk J., Davidsson O. Design of small molecule inhibitors of acetyl-CoA carboxylase 1 and 2 showing reduction of hepatic malonyl-CoA levels in vivo in obese zucker rats. Bioorganic & Medicinal Chemistry. 2011;19:3039–3053. doi: 10.1016/j.bmc.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Gao L., Chiou W., Tang H., Cheng X., Camp H.S., Burns D.J. Simultaneous quantification of malonyl-CoA and several other short-chain acyl-CoAs in animal tissues by ion-pairing reversed-phase HPLC/MS. Journal of Chromatography B, Analytical Technologies in the Biomedical and Life Sciences. 2007;853:303–313. doi: 10.1016/j.jchromb.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 23.Boersema P.J., Raijmakers R., Lemeer S., Mohammed S., Heck A.J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nature Protocols. 2009;4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 24.Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using stagetips. Nature Protocols. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 25.Beausoleil S.A., Villen J., Gerber S.A., Rush J., Gygi S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nature Biotechnology. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 26.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdottir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Millar J.S., Cromley D.A., McCoy M.G., Rader D.J., Billheimer J.T. Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR-1339. Journal of Lipid Research. 2005;46:2023–2028. doi: 10.1194/jlr.D500019-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Chow J.D., Jones M.E., Prelle K., Simpson E.R., Boon W.C. A selective estrogen receptor alpha agonist ameliorates hepatic steatosis in the male aromatase knockout mouse. Journal of Endocrinology. 2011;210:323–334. doi: 10.1530/JOE-10-0462. [DOI] [PubMed] [Google Scholar]
- 32.Oishi K., Uchida D., Ishida N. Circadian expression of FGF21 is induced by PPARalpha activation in the mouse liver. FEBS Letters. 2008;582:3639–3642. doi: 10.1016/j.febslet.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 33.Chakravarthy M.V., Pan Z., Zhu Y., Tordjman K., Schneider J.G., Coleman T. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cellular Metabolism. 2005;1:309–322. doi: 10.1016/j.cmet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 34.Gao X.F., Chen W., Kong X.P., Xu A.M., Wang Z.G., Sweeney G. Enhanced susceptibility of Cpt1c knockout mice to glucose intolerance induced by a high-fat diet involves elevated hepatic gluconeogenesis and decreased skeletal muscle glucose uptake. Diabetologia. 2009;52:912–920. doi: 10.1007/s00125-009-1284-0. [DOI] [PubMed] [Google Scholar]
- 35.Bai P., Canto C., Brunyanszki A., Huber A., Szanto M., Cen Y. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cellular Metabolism. 2011;13:450–460. doi: 10.1016/j.cmet.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhalla K., Hwang B.J., Dewi R.E., Ou L., Twaddel W., Fang H.B. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Research. 2011;71:6888–6898. doi: 10.1158/0008-5472.CAN-11-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Breevoort S.R., Angdisen J., Fu M., Schmidt D.R., Holmstrom S.R. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. Journal of Clinical Investigation. 2012;122:1688–1699. doi: 10.1172/JCI59817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olson D.P., Pulinilkunnil T., Cline G.W., Shulman G.I., Lowell B.B. Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7598–7603. doi: 10.1073/pnas.0913492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao J., DeMayo F.J., Li H., Abu-Elheiga L., Gu Z., Shaikenov T.E. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8552–8557. doi: 10.1073/pnas.0603115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy J.K., Rao M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2006;290:G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 41.Galdieri L., Vancura A. Acetyl-CoA carboxylase regulates global histone acetylation. Journal of Biological Chemistry. 2012;287:23865–23876. doi: 10.1074/jbc.M112.380519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savage D.B., Choi C.S., Samuel V.T., Liu Z.X., Zhang D., Wang A. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. Journal of Clinical Investigation. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakravarthy M.V., Lodhi I.J., Yin L., Malapaka R.R., Xu H.E., Turk J. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138:476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L., Vaitheesvaran B., Hartil K., Robinson A.J., Hoopmann M.R., Eng J.K. The fasted/fed mouse metabolic acetylome: N6-acetylation differences suggest acetylation coordinates organ-specific fuel switching. Journal of Proteome Research. 2011;10:4134–4149. doi: 10.1021/pr200313x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hallows W.C., Yu W., Denu J.M. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by SIRT1 protein-mediated deacetylation. Journal of Biological Chemistry. 2012;287:3850–3858. doi: 10.1074/jbc.M111.317404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sterner D.E., Berger S.L. Acetylation of histones and transcription-related factors. Microbiology and Molecular Biology Reviews. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roth S.Y., Denu J.M., Allis C.D. Histone acetyltransferases. Annual Review of Biochemistry. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 50.Lerin C., Rodgers J.T., Kalume D.E., Kim S.H., Pandey A., Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cellular Metabolism. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 51.Cai L., Tu B.P. On acetyl-CoA as a gauge of cellular metabolic state. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:195–202. doi: 10.1101/sqb.2011.76.010769. [DOI] [PubMed] [Google Scholar]
- 52.Paik W.K., Pearson D., Lee H.W., Kim S. Nonenzymatic acetylation of histones with acetyl-CoA. Biochimica et Biophysica Acta. 1970;213:513–522. doi: 10.1016/0005-2787(70)90058-4. [DOI] [PubMed] [Google Scholar]
- 53.Nohara H., Takahashi T., Ogata K. Enzymatic acetylation of histones and some chemical characters of their acetyl groups. Biochimica et Biophysica Acta. 1968;154:529–539. doi: 10.1016/0005-2795(68)90013-5. [DOI] [PubMed] [Google Scholar]
- 54.Zhang M., Galdieri L., Vancura A. The yeast AMPK homolog SNF1 regulates acetyl coenzyme a homeostasis and histone acetylation. Molecular and Cellular Biology. 2013;33:4701–4717. doi: 10.1128/MCB.00198-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karamanlidis G., Lee C.F., Garcia-Menendez L., Kolwicz S.C., Jr., Suthammarak W., Gong G. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cellular Metabolism. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lysine acetylome data comparing LDKO vs. flox livers.
Global proteomic data comparing LDKO vs. flox livers.