

Abstract
Immune-driven inflammation plays an important part in atherogenesis and is therefore believed to be key to the development of cardiovascular disease (CVD), which is currently the leading cause of death in the Western world. By fulfilling some of the Koch postulates, atherogenesis has even been proposed to be considered as an autoimmune disease, raising the hope that CVD could be prevented by immunomodulation. Nevertheless, the role of the immune system and autoimmune reactions in atherosclerosis appear to be a double edged-sword, with both pro-atherogenic and anti-atherogenic attributes. Hence, if immunomodulation is to become a therapeutic option for atherosclerosis and CVD, it will be crucial to correctly identify patients who might benefit from targeted suppression of deleterious autoimmune responses. This could be achieved, for example, by the detection of disease-associated autoantibodies. In this work, we will review the currently available clinical, in vitro, and animal studies dedicated to autoantibodies against apolipoprotein A-1 (anti-apoA-1 IgG), the major proteic fraction of high density lipoprotein. Current clinical studies indicate that high levels of anti-apoA-1 IgG are associated with a worse cardiovascular prognosis. In addition, in vitro and animal studies indicate a pro-inflammatory and pro-atherogenic role, supporting the hypothesis that these autoantibodies may play a direct causal role in CVD, and furthermore that they could potentially represent a therapeutic target for CVD in the future.
Keywords: Autoantibodies, Cardiovascular disease, Atherosclerosis, Apolipoprotein A-1, Autoimmunity, Biomarkers
Core tip: This review provides a comprehensive and critical analysis of the most recent basic research articles and clinical trials on the role of autoantibodies to apolipoprotein A-1 as biomarkers and potential mediators of cardiovascular diseases (CVD). Evidence from both in vitro and in vivo studies showed that anti-apolipoprotein A-1 IgG might have critical pro-atherosclerotic activities by activating immune cells to release pro-inflammatory mediators and proteases. In addition, these autoantibodies might increase heart rate and arrhythmias both in humans and animal models. These studies suggest a causal role of anti-apolipoprotein A-1 immunoglobulins of G class in CVD, indicating that those autoantibodies could potentially represent an emerging therapeutic target to better fight CVD.
INTRODUCTION
Current epidemiology of cardiovascular diseases and preventive strategies
Despite increasing public awareness and major therapeutic progress, cardiovascular disease (CVD) remains the leading cause of morbidity and mortality worldwide. In the United States, CVD prevalence in the general population is expected to reach 40%, with direct related costs set to reach 800 billion dollars per year in the next two decades[1].
In Europe, CVD causes 47% of all deaths (Figure 1), accounting for 4 million fatalities each year, and costing 196 billion euros a year. Roughly half of these costs (54%) have been attributed to direct health care costs, and the other half (46%) to indirect losses (Heart Network: www.ehnheart.org).
Figure 1.
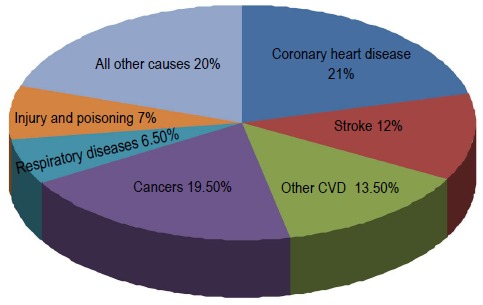
Deaths by cause in Europe for the latest available year, and by gender. Adapted from European Heart Network (www.ehnheart.org). CVD: cardiovascular disease.
Because the disease progresses asymptomatically, the first indication that an individual has atherosclerosis is often a severe cardiovascular event. According to statistics obtained in the United States during the last two decades, the first indicator of atherosclerosis for 30%-50% of patients was an acute, and in many cases fatal, myocardial infarction (MI)[2]. Current guidelines address this problem by identifying high-risk individuals according to the cumulative presence of different Framingham risk factors (smoking, obesity, diabetes, dyslipidemia, and hypertension), with the decision to go forward into preventive treatment made according to the estimated risk. Based upon these clinically-based cardiovascular (CV) risk stratification tools, individuals identified as at-risk for atherosclerosis and CVD are subjected to treatment that directly addresses the established risk factors, combining lifestyle modification (e.g., smoking, exercise, diet) with anti-platelet therapy (aspirin), and medication to reduce both blood pressure (anti-hypertensive agents) and levels of circulating cholesterol (statins).
While this strategy has undoubtedly made some impact, current CV risk stratification tools only have the power to segregate very high-risk individuals from very low-risk individuals, and lack sensitivity and specificity in persons deemed to be at intermediate risk[3]. As a consequence, up to 60% of CV events occur in primary prevention (i.e., in patients with asymptomatic CVD), affecting subjects deemed at low or intermediate risk of CVD (false negative)[4,5]. At the same time, other patients are unnecessarily given lifelong prevention medication (false positives) (reviewed in[2,6]).
For this reason, strong calls have been made to exploit existing knowledge and technology to improve the sensitivity and specificity of risk stratification approaches used to guide preventive therapy[2]. To be effective as public health measures, new approaches would have to be not only sensitive and specific, but also low-cost, non-invasive and adaptable to scale-up and commercialization for widespread use[2,6]. While solutions involving imaging technologies such as ultrasound, chest computed tomography (CT) and magnetic resonance imaging have been proposed[2], their implementation at population level in primary care is currently difficult to envisage mainly for economic reasons, and also because of health hazards related to radiation exposure.
As a more viable alternative strategy with respect to costs and health issues, much attention has been drawn to CV biomarkers that allow, on the basis of a simple blood sample measurement, to quantify either the amount of myocardial necrosis, such as cardiac troponins[7], the degree of myocardial stretch, such as natriuretic peptides[8], or the amount of systemic inflammation, such as high sensitive C-reactive protein (hs-CRP)[9], to only quote the “usual suspects” in the field. The complete list of candidate CV biomarkers is much longer, reflecting the numerous studies published in the field (Figure 2), but only a few of these candidates, notably those shown to be causally involved in the disease, are likely to make their way into clinical practice. For this reason it is hoped that improved knowledge of the pathogenesis of atherosclerosis will lead to the identification and validation of biomarkers for atherosclerosis and CVD, enabling the development of new risk stratification approaches[6].
Figure 2.
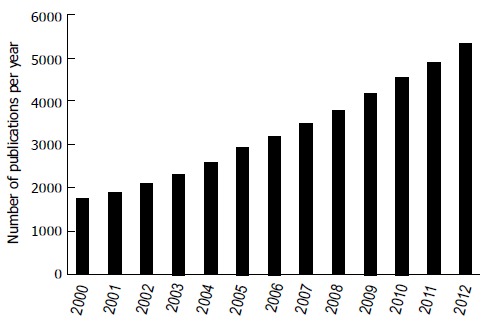
Annual evolution of publications on cardiac biomarkers since 2000. This graphic represents the number of publications per year indexed and retrieved in Pubmed between 2000 and 2012 when the key words “cardiovascular biomarkers” are entered. Entry date: 22nd of January 2014.
Pathogenesis of atherosclerosis and cardiovascular disease
CVD is causally linked to atherosclerosis, the swelling of artery walls due to the formation of plaque lesions. Plaques are made up of leukocytes, smooth muscle cells and lipid deposits, with the surface of the plaque in contact with the arterial lumen covered with a fibrous connective tissue cap. Although atherosclerosis accumulates gradually and asymptomatically from childhood, it is accelerated by a number of established risk factors, including Framingham risk factors. Atherosclerotic plaques may remain stable as they grow, gradually reducing arterial blood flow as the lumen becomes increasingly obstructed, or may become prone to rupture. When plaque rupture occurs, the highly thrombogenic interior of the plaque is revealed, leading to atherothrombosis. The resulting ischemia is what causes CVD morbidity and mortality. Depending on the location of the affected artery the outcome can be myocardial infarction, stroke, or peripheral artery disease[4].
Atheroslcerosis as an immune-mediated disease
Evidence linking high blood cholesterol to atherosclerosis, together with the presence of lipid deposits within atherosclerotic plaques led to the prevailing view that atherosclerosis was a lipid-related disease. This view was held until the 1990’s, when a series of discoveries led to a paradigm shift in the understanding of atherosclerosis, shifting emphasis from lipid metabolism and transport to inflammation[10-12]. Inflammatory responses are now believed to underlie all of the key steps in atherosclerotic pathogenesis, from the initial modification of healthy arterial endothelium to thrombus formation at the site of plaque rupture.
According to this current paradigm (reviewed in[2,10-12]), atherosclerosis is initiated by inflammatory activation of arterial wall endothelial cells, allowing adhesion of circulating leukocytes. Expression of inflammatory chemokines leads to the migration of these leukocytes, predominantly circulating monocytes, across the endothelium and into the tunica intima. At this site the monocytes mature, acquiring a macrophage phenotype and the capacity to ingest native and modified low-density lipoprotein (LDL) particles that exit the blood and permeate the activated arterial endothelium. Following extensive lipid ingestion, these macrophages become “foam cells”, which are the main constituents of an early atherosclerotic lesion. Foam cells release a broad range of cytokines and serve to amplify the inflammatory response, as well as inducing the proliferation of resident smooth muscle cells and promoting local angiogenesis. Chronic inflammation leads to the formation of an advanced atherosclerotic plaque, comprising a mass of foam cells surrounding a “necrotic core” of lipids released by dead and dying cells, capped by a fibrous layer made up of smooth muscle cells and extracellular matrix. Inflammatory responses also play a key role in atherothrombosis, which is recognized to account for up to 80% of acute CV manifestations[13]. Inflammation influences the local extracellular matrix composition through a complex interplay between different matrix-metalloproteinases (MMPs) determining the propensity of the fibrous cap to rupture[14-17]. Furthermore, a pro-inflammatory micro-environment also promotes thrombus formation via the activation of coagulation factors, leading to acute vessel occlusion[6].
Detailed analysis of the content of atherosclerotic plaques, together with the advent of a wide range of genetically modified mouse strains, has enabled further elucidation of the inflammatory pathogenesis of atherosclerosis[18]. The identification of autoantibodies as well as autoreactive T cells in atherosclerotic plaques[19], and the correlation established in clinical studies between their detection and disease severity provided a clear indication that adaptive immunity plays a role in atherosclerosis (reviewed in[20]). This role was underlined in a number of studies in which ApoE-/- knockout mice, which are predisposed to hypercholesterolemia and atherosclerosis, were crossed with different mouse strains deficient in specific arms of the adaptive immune system. These studies revealed a key pro-atherogenic role for the Th1 subset of CD4 T cells, and an anti-atherogenic role for the regulatory T cell subset (reviewed in[20]), as well as both pro- and anti-atherogenic roles for different B cell subsets[21]. In addition, they highlighted the importance in atherogenesis of signaling through pattern recognition receptors (PRR) of the innate immune system, such as Toll-like receptors (TLR) (reviewed in[18,20]).
Atherosclerosis as an autoimmune disease?
Grounded on the fact that atherogenesis fulfills several of “Koch” postulates (Table 1), atherosclerosis has even been proposed to be of autoimmune etiology[22,23]. This hypothesis is based on the following evidence. Firstly, atherosclerotic plaques are infiltrated by both T cells and antibodies specific for various autoantigens[20], patients suffering from autoimmune disease, such as systemic lupus erythematosus (SLE), anti-phospholipid syndrome (APS) and rheumatoid arthritis (RA) display an increased CV risk, independently of traditional CV risk factors[24-26]. Secondly, as reviewed elsewhere, in patients without autoimmune diseases, but established CVD, levels of antibodies directed against various and numerous endogenous epitopes, such as modified LDL, heat-shock proteins (HSP), and cardiolipin, have been shown to independently predict CV outcome[27]. Thirdly, in vivo and in vitro evidence demonstrated that some autoantibodies might directly influence atherogenesis and atherosclerotic plaque vulnerability, mostly by activating innate immune receptors, thereby supporting a causal role of humoral autoimmunity in atherosclerosis[28-31].
Table 1.
Koch posultates applied to the role of autoimmunity in atherosclerosis
| Basic Koch postulates | Koch postulates transposed to the role of autoimmunity in atherosclerosis | Koch postulates met ? |
| Pathogens must be detected in the diseased host at every stage of the disease | Autoantibodies and auto-reactive T cells can be detected in atherosclerotic plaques and serum of patients in primary or secondary prevention of CVD | Yes |
| Pathogens must be isolated from the diseased host and grown in culture | Autoreactive T-cells can be isolated and cultivated from diseased host presenting experimental atherosclerosis | Yes |
| When inocculated in healthy animals, the pathogens from pure culture must induce the disease | Passive or active immunization drastically affect the course of atherogenesis in animal models | Yes |
| The pathogen must be re-isolated from the diseased animal and must correspond to the primary pathogen in pure culture | Protective autoantibodies of expected specificity can be isolated from animals exposed to active immunization | Partly |
To establish a causality link between a mircoorganism and an infection, the four Koch postulates must be fulfilled. When applied to the role of autoimmunity in atherosclerosis, the Koch postulates support a causal role between autoimmunity, atherosclerosis and cardiovascular disease (CVD). Adapted from references[20-28].
Nevertheless, the relationship between autoantibodies and CVD is debated, because some of them have been shown to be anti-atherogenic, while others act as pro-atherogenic molecules[27,28]. The reason for such duality is still elusive and will not be further discussed in the present work.
Another unresolved question concerns the mechanisms by which tolerance is broken to generate autoimmunity. Certain lines of evidence point to pathogen molecular mimicry, i.e., cross-reactivity between microbial antigens and components of host structures, including modified LDL and HSP[32,33]. In addition, modification of proteins by oxidation can generate new epitopes that are recognized as non-self by the adaptive immune system[32]. However, the presence of a non-self-epitope is not normally sufficient to drive an autoimmune response, since in order to effectively prime T cells, antigen presenting cells must concomitantly receive “danger signals” through their PRR. In the case of pathogen molecular mimicry, the PRR ligands are provided by the pathogen in the form of pathogen-associated molecular patterns (PAMPs). In the absence of a pathogen, “sterile inflammation” can be induced when antigen presenting cells are stimulated via their PRR by an analogous set of structures called damage-associated molecular patterns (DAMPs), which are typically released by stressed or necrotic cells[31-33].
Hence, both pathogen molecular mimicry, as a consequence of infection with, e.g., Chlamydia pneumoniae or Helicobacter pylori, or DAMP-mediated sterile inflammation represent mechanisms by which autoantibodies targeting antigens implicated in atherosclerosis can emerge (reviewed in[12]).
Autoantibodies as CV risk stratification tools?
As mentioned previously, there is a clear need for new biomarkers to improve current CV risk stratification[6,34]. Driven by the paradigm shift of atherogenesis moving from a lipid-centered to inflammatory-centered etiology, the quest for new potential cardiovascular risk markers to better assess global cardiovascular vulnerability was principally oriented on inflammatory biomarkers, including autoantibodies[27-29].
Among the advantages identified for some autoantibodies is that they meet the current benchmark specifications requested for novel CV biomarkers[35,36]. Firstly, their association with CV outcomes has not only been shown to be independent of traditional CV risk factors (reviewed in[27,28]), but could also provide incremental predictive information over current CV risk stratification tools. Secondly, the stability provided by their long half-life place them as good candidates for long-term prognosis when compared to biomarkers with a shorter half-life. Thirdly, their measurement is typically simple, accurate, robust, and achievable at moderate costs.
Autoantibodies as potential therapeutic targets?
Providing that some autoantibodies have been shown to modulate atherogenic processes in antagonistic ways (reviewed in[27,28]), attempts to induce atheroprotective immunity through active immunization raised the hope that vaccination against different specific antigens (wide variety of modified LDL, HSP, etc.) could lead to lifelong protection against atherosclerosis and CVD. This hypothesis is currently under active investigation in humans[37,38].
On the other hand, neutralizing the deleterious effects of pro-atherogenic autoantibodies represents another interesting therapeutic modality which could currently be achieved through passive immunization with intravenous immunoglobulins (IVIG). In this respect, data concerning IVIG administration in humans after MI yield rather contradictory results[38,39], and although data restricted to animal models do support an anti-atherogenic role of IVIG[40-43], the costs related to IVIG therapy may well prohibit widespread administration of IVIG to all MI or CVD patients in the long-term, even if it proves to be effective. One solution might be to identify specific autoantibodies that could then be selectively neutralized by anti-idiotypic molecules rather than IVIG. Accordingly, an approach based on the detection of specific autoantibodies would enable the identification of a subset of CVD patients that could benefit either from immunomodulation (passive or active immunization) or from a specific mimetic peptide-based therapy. Such a strategy could represent an affordable step forward toward personalized medicine in the field of CVD, allowing a more targeted therapeutic intervention.
HIGH-DENSITY LIPOPROTEIN, APOLIPOPROTEIN A-1, AND ITS RELATED AUTOANTIBODIES
Human apolipoprotein A-1 (apoA-1) is a 28-kD protein with 243 amino acid residues encoded by the apolipoprotein multigene superfamily located on chromosome 11q23[44]. The protein is synthesized as a 24 amino-acid-longer prepro sequence of apoA-1, primarily by hepatocytes in the liver and also by enterocytes. Mature apoA-1 constitutes the principal protein fraction of high density lipoprotein (HDL) whose protective role in the CV system derives, to a great extent, from the inverse association of HDL-cholesterol and apoA-1 plasma concentrations with the risk of myocardial infarction. The atheroprotective role of HDL in the cardiovascular system has been attributed to the pleiotropic effects of HDL, including reverse cholesterol transport from resident arterial wall macrophages to the biliary tract for elimination, vasodilatation, anti-thrombotic, anti-coagulant and anti-inflammatory effects[45,46]. Mirroring those versatile properties, mass spectrometry analyses revealed that HDL encompasses very heterogeneous macromolecular complexes of lipids and proteins. Only one third of the up to 80 different proteins identified in HDL is dedicated to lipid transport. The remaining proteins being either acute-phase proteins, proteases, anti-oxidant, anti-thrombotic enzymes or proteins involved in complement regulation[45,46].
In addition to being the principal protein fraction of HDL and a limiting factor for HDL formation, apoA-1 per se has many of the HDL-related atheroprotective properties, such as inhibition of immune cell trans-endothelial migration, inhibition of monocyte activation, inhibition of cytokine production induced by T-cell contact, inhibition of lipid peroxidation, and interference with innate immune receptors pro-inflammatory signaling[45]. There is also a growing body of evidence indicating that both acute and chronic inflammatory conditions induce post-translational modifications of apoA-1 transforming HDL and apoA-1 into pro-inflammatory molecules[46]. Furthermore, as reviewed in the next paragraphs, recent data suggest that that humoral autoimmunity to apoA-1 and HDL could be new possible biomarkers for CVD, and possibly a mediator of inflammation, atherosclerosis, and CVD.
ANTI-APOA-1 IGG IN AUTOIMMUNE DISEASES
Anti-apoA-1 IgG in SLE and APS patients
In 1995, using early phage display technology, Merrill et al[47] reported that sera derived from SLE patients were immunoreactive against a protein displaying 82% DNA sequence homology with human apoA-1, followed by the confirmation that those sera were indeed reactive to human apoA-1 when coated on gamma-irradiated enzyme-linked immunosorbent assay plates. Further understanding of anti-apoA-1 autoantibody architecture was provided by the same group in 1998 who reported that high levels of anti-apoA-1 IgG were retrieved in a significant subset of SLE (32.5%) and primary APS patients (22.9%)[35]. Those autoantibodies were found to be associated with the presence of anti-beta2glycoprotoein I (β2GPI) antibodies, and to display an optimal affinity for mature HDLs[48]. In 2001, Abe and colleagues characterized six different monoclonal anti-apoA-1 antibodies (derived from two SLE patients) displaying a low specificity, as reflected by their broad cross-reactivity to single strand DNA, thrombin, cardiolipin (CL), and to HDL[48,49]. Because of the latter observation, anti-apoA-1 IgG were considered a possible subgroup of anti-HDL antibodies[49]. The first insight regarding the potential pathogenicity of this class of autoantibodies in atherogenesis was demonstrated in 2003 by Delgado Alves and colleagues, who showed an inverse correlation between anti-HDL IgG and paraoxonase-1 (PON-1) activity, and with the total antioxidant capacity of the corresponding sera[50]. More specifically, those initial results suggested that anti-HDL, and later anti-apoA-1 IgG[51], could be related to atherogenesis, through HDL dysfunction[52,53], whose pathophysiological role in atherogenesis was starting to be recognized[54].
Anti-apoA-1 IgG in rheumatoid arthritis patients
In 2010, we demonstrated in a case-control study that anti-apoA-1 IgG levels were higher in patients suffering from rheumatoid arthritis (RA) than in matched-controls (17% vs 2%, P = 0.01)[55]. In this study, those autoantibodies were associated with higher oxidized LDL levels and were significantly associated with anamnestic CVD. Nevertheless, no association was found with the RA disease activity score[55]. Concomitantly, in a longitudinal prospective study which will be described in detail in the following paragraph entitled “anti-apoA-1 IgG as independent predictors of CV risk“, we confirmed that those autoantibodies were predictive of CVD in RA patients, and were associated with a pro-inflammatory cytokine profile[56,57].
ANTI-APOA-1 IGG IN OTHER NON-AUTOIMMUNE POPULATIONS
If high levels of anti-apoA-1 IgG are initially described as raised in patients with autoimmune diseases associated with an increased risk of CVD, high levels of those autoantibodies can also be detected in patients without autoimmune disease, but with CVD, such as acute coronary syndrome[58-62], and severe carotid stenosis[63,64].
In addition, the existence of elevated levels of anti-apoA-1 IgG was demonstrated in patients with periodontitis[65], and patients under hemodialysis[66], two clinical conditions known to be associated with increased CVD risk[67,68]. Finally, the existence of high levels of anti-apoA-1 IgG was recently reported in obese, but otherwise healthy subjects[69].
In those different settings the prevalence of a high titer of anti-apoA-1 IgG varies between 10% and 20%, against 0% to 6.5% in healthy blood donors or controls[56,59,65]. The clinical relevance of such findings will be discussed later.
ANTI-APOA-1 IGG AS A MARKER AND POSSIBLE MEDIATOR OF INFLAMMATION AND ATHEROGENESIS
Anti-apoA-1 IgG are associated with a pro-inflammatory and pro-atherogenic cytokine profile in humans
Most studies published to date have reported significant associations between high levels of anti-apoA-1 IgG levels and markers of oxidative stress, inflammation and endothelial dysfunction related to atherogenesis and atherosclerotic plaque rupture.
In SLE patients, anti-apoA-1 IgG levels were found to be positively correlated with nitric oxide (r = 0.37, P = 0.007), inversely related to PON-1 activity (r = -0.31, P = 0.006), and the total anti-oxidant capacity of the sera (r = -0.47, P < 0.0001) suggesting that those autoantibodies could interfere with the anti-oxidant properties of HDL, giving rise to a pro-oxidative micro-environment facilitating atherogenesis[70]. Similarly, RA patients tested positive for those autoantibodies were shown in two different studies to have higher plasma levels of oxLDL levels[55,56], considered a major player in all stages of atherogenesis[2,10,11]. Furthermore, RA patients tested positive for anti-apoA-1 antibodies were found to have higher levels of interleukin-8 (IL-8) and MMP-9[55], two inflammatory mediators known to be associated with atherogenesis, and atherosclerotic plaque vulnerability in humans[14,71].
In a retrospective study involving MI patients, we reported a positive association between anti-apoA-1 IgG and serum amyloid A (SAA) protein levels (r = 0.76, P = 0.006), a multifunctional protein located at the crossroad of inflammation and cholesterol homeostasis[58].
Subsequently, in a prospective cohort study involving MI patients (n = 127), we noted the same relationship between anti-apoA-1 IgG and oxLDL levels as had been documented in RA patients[59]. MI patients considered as positive for anti-apoA-1 IgG had significantly higher median levels of oxLDL when compared to patients tested negative for those autoantibodies (226.5 U/L vs 47.7 U/L, P < 0.0001), and a positive correlation between oxLDL and anti-apoA-1 IgG was observed (Spearman r = 0.28, P < 0.05)[59]. On the other hand, no association with PON-1 activity was observed in this study.
In a prospective study enrolling 221 MI patients, we demonstrated that patients tested positive for anti-apoA-1 antibodies had higher circulating levels of IL-6, TNF-α, and MMP-9, and lower MMP-3 levels[72], a cytokine constellation known to be associated with increased atherosclerotic plaque vulnerability, and worse CV prognosis[73,74]. This increase in MMP-9 levels retrieved in anti-apoA-1 IgG positive patients was associated with an increase in MMP-9 activity[63].
Furthermore, in our periodontitis study, we observed a positive correlation between anti-apoA-1 IgG and ADMA levels (Spearman; r = 0.20, P = 0.02)[65], a marker of endothelial-dependent dysfunction with strong CV prognostic value[75,76].
Among other associations observed between anti-apoA-1 IgG CV relevant prognostic features was an association with basal heart rate. In one of our prospective MI cohort studies[60], we demonstrated that when compared to those tested negative for anti-apoA-1 IgG, patients tested positive for those antibodies had a higher basal heart rate upon discharge, a well-established CV prognostic feature after MI[77-79]. The possible impact of those autoantibodies on nervous autonomic dysfunction will be presented in the paragraph entitled “Anti-apoA-1 IgG elicits a positive chronotropic effect on cardiomyocytes”.
In conclusion, the most consistent associations observed so far between anti-apoA-1 IgG and CV-relevant markers of inflammation concern mostly oxLDLs and MMP-9. Although no causal link can be inferred based on such statistical associations, they were nevertheless instrumental in orienting the subsequent in vitro and animal studies described in the next paragraph.
Anti-apoA-1 IgG as active mediators of atherosclerosis and atherosclerotic plaque vulnerability in vitro and in vivo
Experiments carried out in cellular and animal models indicated that certain autoantibodies contribute directly to the induction of atherogenesis and atherosclerotic plaque vulnerability through their capacity to signal through innate immune receptors, notably TLR-2 ([29-31]; reviewed in[27]). By analogy, we investigated whether anti-apoA-1 autoantibodies could act through innate immune receptors signaling to elicit a pro-inflammatory response.
In this respect, we recently showed that lipopolysaccharide-free anti-apoA-1 IgG dose-dependently induced the production of a range pro-inflammatory cytokines, such as IL-8, MMP-9, IL-6, TNF-α, and MCP-1 in human monocyte-derived macrophages[55,63,72], and that this process was mediated by the TLR2/CD14 complex[72]. In addition, our in silico modeling studies revealed evidence of structural homology between apoA-1 and part of the extracellular domain of TLR2, suggesting a molecular mechanism for this cross-reactivity[72]. Our current understanding on how anti-apoA-1 IgG promotes sterile inflammation through the activation of TLR2/CD14 complex is summarized in Figure 3.
Figure 3.
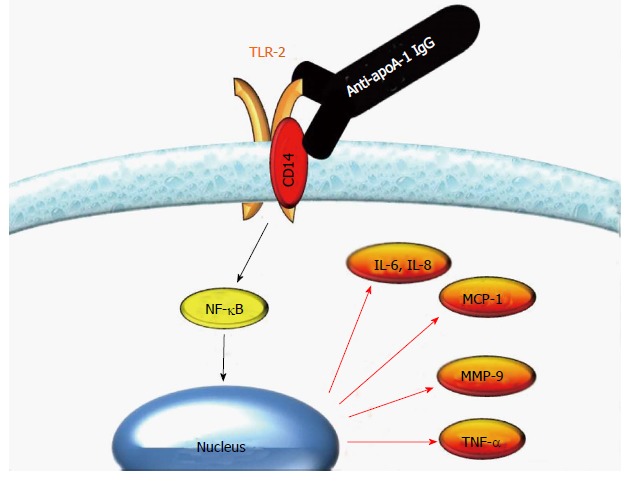
Autoantibodies against apolipoprotein A-1 IgG elicit a pro-inflammatory response through Toll-like receptor 2/CD14 complex on human macrophages. Autoantibodies against apolipoprotein A-1 (anti-apoA-1) IgG specifically bind to Toll-like receptor (TLR)2 due to conformational homology between apoA-1 and TLR2. In the presence of CD14, the binding of anti-apoA-1 IgG to TLR2 induces a nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-dependent production of pro-inflammatory cytokines. MMP-9: Matrix-metalloproteinases; IL-8: Interleukin-8.
Anti-apoA-1 IgG elicits a positive chronotropic effect on cardiomyocytes
We have recently demonstrated that there is a positive association between levels of anti-apoA-1 IgG and resting heart rate following myocardial infarction, a well-established parameter for CVD prognosis in secondary prevention[60,79]. In the same study, we showed that in the presence of aldosterone, anti-apoA-1 IgG elicits a dose-dependent increase in the spontaneous contraction rate of neonatal rat ventricular cardiomyocytes[60]. Using patch-clamp electrophysiology combined with a pharmacological approach, we subsequently showed that this positive chronotropic effect was mediated by L-type calcium channel activation, itself induced by the concomitant activation of both the mineralocorticoid receptor-dependent phosphatidyl 3-kinase pathway and the protein kinase A pathway[80]. In support of an activation mechanism involving aldosterone and antibody, we demonstrated that the chronotropic effect can be abrogated by addition of eplerenone, an aldosterone antagonist, and by intravenous immunoglobulins[60,80]. Hence, there is compelling evidence in support of a role for anti-apoA-1 IgG in the induction of a positive chronotropic effect in cardiomyocytes, but further work will be required to define (1) whether this is a direct or indirect effect; and (2) if anti-apoA-1 IgG acts directly on cardiomyocytes, which receptor does it engage to activate the protein kinase A pathway (Figure 4)? At the present time, there is no indication suggesting that this chronotropic effect could be mediated by interference with the activity of the autonomous nervous system; further work will be required to address this question.
Figure 4.
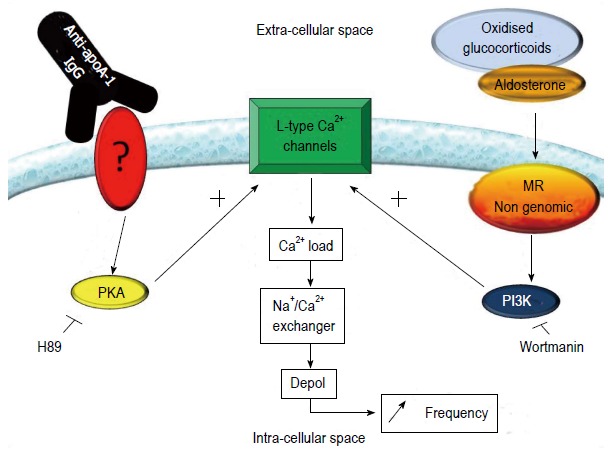
Current understanding of the mechanism by which autoantibodies against apolipoprotein A-1 IgG elicit chronotropic responses in nenonatal rat cardiomyocytes. Stimulation of the mineralocorticoid receptor (MR), either by aldosterone or oxidized glucocorticoids, induces the downstream activation of PI3K, which in turn activates L-type calcium channels. Anti-apoA-1 IgG has been shown to sensitize the L-type calcium channel in a protein kinase A (PKA)-dependent manner. The PI3K and PKA activated pathways alone are not sufficient to induce an increase in basal contraction rate, when simultaneously activated L-type calcium channels are activated, leading to an increase in intracellular Ca2+. This signal is amplified by the Na+/Ca2+ exchanger, leading to an increase of the prepotential slope of the cells, which ultimately translates into an increased contraction rate.
Anti-apoA-1 IgG induces atherosclerosis and death in apoE-/- mice
Animal studies that we have performed provided direct evidence that anti-apoA-1 IgG was sufficient to induce atherosclerosis. Passive immunization of atherosclerosis-prone apoE-/- mice with anti-apoA-1 IgG increased both atherosclerotic lesion size and histological features of atherosclerotic plaque vulnerability[63]. In a lupus-prone mouse model, Srivastava and colleagues demonstrated that the presence of anti-apoA-1 antibodies was associated with a decrease in the anti-oxidant properties of HDL which inferred a decrease in PON-1 activity, leading to an increase in pro-inflammatory reactive oxygen species[81]. These results support the hypothesis that anti-apoA-1 IgG and HDL dysfunction are two related phenomena. Although a causal link between anti-apoA-1 IgG and HDL dysfunction remains elusive, these results are consistent with clinical observations reported previously[49-52].
ANTI-APOA-1 IGG AS INDEPENDENT PREDICTORS OF CV RISK
In 2010, we demonstrated that anti-apoA-1 IgG positivity assessed in samples taken within the first 24 h of patient admission for MI was a significant and independent predictor of MACE during 1 year follow-up[60]. The presence of high anti-apoA-1 IgG levels on admission increased the subsequent risk of MACE by 4-fold, independently of Framingham risk factors [adjusted OR = 4.3, 95%CI: 1.46-12.6, P = 0.007][60]. Cox regression analysis demonstrated that for each arbitrary unit increase in anti-apoA-1 IgG, there was a concomitant 3% increase in MACE risk (P = 0.0003). All 221 patients tested negative for anti-nuclear antibodies and no association with any other autoantibodies (rheumatoid factor, anti-β2GPI and anti-cardiolipin antibodies) was observed[60].
These findings were extended in an ancillary study derived from the same cohort of patients aimed at comparing, in a “head to head” fashion, the prognostic accuracies of other autoantibodies described as potentially relevant for CV event prediction. Among those, we measured antibodies to β2GPI domain I and IV, cardiolipin, heat-shock protein 60 (anti-HSP-60), and phosphorylcholine (anti-PC IgM)[61]. In this study, autoantibodies to apoA-1 were found to be the only autoantibodies to significantly predict subsequent MACE occurrence, although a non-significant trend was observed for anti-cardiolipin (P = 0.05), and anti-HSP60 antibodies P = 0.07). In this study, the prognostic accuracy measured by the area under the curve (AUC) was rather modest (AUC: 0.65, P = 0.007)[61], and of the same order of magnitude as the 10-year global Framingham risk score. Risk analyses demonstrated that anti-apoA-1 IgG positivity increased the risk of MACE by 4-fold, independently of the 10-year global Framingham risk score (adjusted hazard ratio = 3.8, P = 0.002)[61]. Those preliminary results pointed to anti-apoA-1 IgG as a promising humoral autoimmune candidate for MACE prediction in secondary prevention settings.
Furthermore, in a single center prospective study involving 138 patients presenting to the emergency room with acute chest pain, we demonstrated that anti-apoA-1 IgG values assessed on the first sample available had a relatively good diagnostic accuracy for non-ST elevation myocardial infarction (NSTEMI) with an AUC of 0.75 (P < 0.0001) that could be increased up to 0.88 when combined with anti-PC IgM and the NSTEMI-TIMI score to generate a clinical antibody ratio (CABR) score[62]. Also, anti-apoA-1 IgG was found to be a good predictor (AUC = 0.80, P < 0.0001) of subsequent troponin I elevation when the first sample tested negative, which was the secondary endpoint of this study. Risk analyses indicated that in the presence of high anti-apoA-1 IgG levels, the risk of subsequent NSTEMI diagnosis was increased by 6-fold after the adjustment for NSTEMI-TIMI score (OR: 6.4, 95%CI: 1.72-24.2). At the pre-specified cut-off, this test displayed an interesting negative predictive value of 88% and 95% for the primary and secondary study endpoints, respectively. To summarize, in ACS patients, the predictive accuracy according to ROC curve analysis revealed AUC values ranging between 0.65 and 0.75[61,62]. If these AUC values are relatively modest (they should ideally be above 0.80[36]), they are still in the same range as those reported for the Framingham risk score, which currently determines patient management[3].
Furthermore, we demonstrated that anti-apoA-1 IgG were also predictors of MACE at one-year after elective surgery for severe carotid stenosis with an AUC of 0.74 (95%CI: 0.59-0.90, P = 0.01)[64], and that its combined used with myeloperoxidase could improve the predictive accuracy of the model[64]. In this study, high levels of anti-apoA-1 IgG were associated with a 5-fold increase in MACE during follow-up (exact OR = 5.29, 95%CI: 1.08-34.02, P = 0.04), which remained significant after adjustment for the 10-year Framingham risk score according to conventional logistic regression, but not when the exact logistic regression model was applied[64].
In a longitudinal prospective study involving 133 RA patients followed-up for a median duration of 9 years[56], we demonstrated that high levels of anti-apoA-1 IgG was associated with a 4-fold increase in MACE during follow-up, independently of Framingham risk factors and RA disease duration (HR = 4.2, 95%CI: 1.5-12.1). In this study, ROC curve analyses indicated that those autoantibodies were the strongest predictors of MACE with an AUC of 0.73 (P = 0.0008), a specificity of 50%, and a sensitivity of 90% at the predefined cut-off[56]. In addition to their independency of traditional CV risk factors to predict poor CV outcome, we also demonstrated that anti-apoA-1 IgG provides incremental prognostic information over traditional cardiovascular risk factors in ACS, in severe carotid stenosis, and in RA patients. When compared to current risk stratification tools (NSTEMI-TIMI score in acute chest pain patients, or the 10-year global Framingham risk score in ACS, RA, or severe carotid stenosis patients), it significantly improved the patient risk reclassification with significant integrated discrimination index values ranging between 1.8% and 175%[57,62,64].
Anti-apoA-1 IgG as a biomarker predictive of atherosclerosis and atherosclerotic plaque vulnerability
Of clinical relevance, we have also demonstrated that anti-apoA-1 IgG is also detectable in a proportion of healthy subjects without autoimmune disease and CVD (0%-6.5%), albeit at lower levels than seen in patient cohorts[56,59,65]. Significantly, in a small case-control study on healthy subjects[69], we demonstrated that anti-apoA-1 IgG levels in the obese subgroup were raised to levels previously described in CVD patients, with high levels of anti-apoA-1 IgG being a significant predictor of coronary artery calcifications visualized by chest computed tomography. Because coronary artery calcifications are a major predictor of subsequent cardiovascular events in asymptomatic subjects[82], the results of this preliminary study suggest that anti-apoA-1 IgG may be a valuable biomarker for use in primary prevention to screen for the presence of coronary artery lesions. Indeed in this setting, anti-apoA-1 IgG testing had a negative predictive value of 94% to detect the presence of coronary artery calcification, with an AUC of 0.83[69]. Similarly, we demonstrated in patients with periodontitis younger than 50 years old that anti-apoA-1 IgG was the only predictor of a pathological ankle brachial index[65], a measure used to detect peripheral artery disease and known to reflect the global atherosclerosis burden[83,84].
Extending those results, we also reported that the presence of anti-apoA-1 antibodies in patients with severe carotid stenosis was associated with histological features of atherosclerotic plaque vulnerability determined on surgical biopsy specimens[63]. Indeed, in this study, we demonstrated that circulating levels of anti-apoA-1 IgG were positively correlated with intraplaque macrophages (r = 0.33, P = 0.002), MMP-9 expression (r = 0.43, P = 0.0001) and neutrophils (r = 0.42, P = 0.0001), and inversely correlated with total collagen content (r = -0.29, P = 0.008). Furthermore, patients deemed as positive for anti-apoA-1 IgG had significantly higher levels of macrophages, MMP-9 expression and neutrophils within their atherosclerotic lesions, and lower levels of total collagen when compared to patients tested negative for those autoantibodies[63]. Interestingly, those findings were mimicked in apoE-/- mice exposed to passive immunization with anti-apoA-1 IgG when compared to the CTL group[63]. Taken together those results indicate that assessing anti-apoA-1 IgG levels could not only be a possible biomarker of atherosclerosis, but could also be used to detect the presence of atherosclerotic plaque vulnerability. Because assessing atherosclerotic plaque vulnerability is currently an unmet clinical need, the possibility of using anti-apoA-1 IgG detection as a simple and affordable surrogate biomarker of atherosclerotic plaque fragility is of patent clinical interest.
FUTURE PERSPECTIVES
Because current in vitro and in vivo results indicate that anti-apoA-1 IgG could well be active mediators of atherogenesis, those autoantibodies may represent an emergent therapeutic target. In other words, we speculate that measuring circulating levels of anti-apoA-1 IgG would enable the identification of a subset of patients who would benefit from specific therapy aimed at reversing the deleterious effect of those autoantibodies. In this respect, we have demonstrated that the chronotropic effect of those autoantibodies could be reversed by existing therapeutic compounds such as IVIG and eplerenone, a selective MR antagonist[80].
In parallel, we will pursue our work aimed at defining the exact CV-relevant epitope(s) targeted by those autoantibodies. Once determined, those epitopes could be useful both for the detection of anti-apoA-1 IgG by occupying binding sites, and for neutralizing the pathogenic effects of the antibodies (pro-arrythmogenic and pro-inflammatory effects), which hopefully would translate in a reduction of atherogenesis-related complications in humans.
CONCLUSION
To summarize, recent studies demonstrate that IgG autoantibodies against apoA-1 are raised in many diseases associated with a high cardiovascular risk, such as SLE, ACS, RA, severe carotid stenosis, and end-stage renal disease. To date, high levels of anti-apoA-1 IgG have been shown to be an independent prognostic marker of poor CV outcome in MI, RA and carotid stenosis patients, to display clinically relevant properties for NSTEMI diagnosis in acute chest pain patients, to be associated with atherosclerotic plaque vulnerability in patients with severe carotid stenosis, and to predict coronary artery lesions in obese, but otherwise healthy subjects. In most studies reported so far, high levels of anti-apoA-1 IgG are associated with a pro-inflammatory cytokine profile, and in SLE/APS, those autoantibodies have been shown to be associated with the presence of dysfunctional HDLs.
Concomitantly, in vitro data tend to indicate that anti-apoA-1 IgG are active modulators of atherogenesis by (1) promoting a sterile inflammation through the TLR2/CD14 complex; and (2) eliciting specific neutrophil chemotaxis. Furthermore, in vitro experiments suggest that those autoantibodies could act as pro-arrhythmogenic molecules through an aldosterone-dependent L-type calcium channel activation that can be reversed using existing therapeutic compounds. In parallel, work in mouse models demonstrated that passive immunization with anti-apoA-1 IgG increases atherogenesis, atherosclerotic plaque vulnerability, death rate, and decreases the anti-oxidant properties of HDL by inhibiting PON-1 activity. The preliminary clinical results need to be replicated in larger multicenter cohorts and further basic science studies will be required to gain a better understanding of the pathophysiological involvement of anti-apoA-1 IgG in atherogenesis. Nevertheless, the current converging in vitro and animal observations lend weight to the hypothesis that anti-apoA-1 IgG are active mediators of atherogenesis rather than innocent bystanders. Hence, these autoantibodies, could in the future, represent a new possible therapeutic target, whose deleterious effect could be abrogated by therapeutic synthetic apoA-1 mimetic peptides. In this context, anti-apoA-1 IgG appears to be a promising biomarker of pathological cardiovascular autoimmunity, allowing the identification of a subset of CVD patients who could benefit from specific immunomodulation in the future, substantially contributing to the development of personalized medicine in the field of CVD.
Footnotes
P- Reviewers: Cheng XW, Sun ZH S- Editor: Gou SX L- Editor: Webster JR E- Editor: Wu HL
Supported by Swiss National Science Foundation Grants to Dr. Vuilleumier N No. 310030_140736; and to Dr. Montecucco F No. 32003B_134963/1; a grant from the Foundation “Gustave and Simone Prévot” to Dr. Montecucco F
References
- 1.Eyre H, Kahn R, Robertson RM, Clark NG, Doyle C, Hong Y, Gansler T, Glynn T, Smith RA, Taubert K, et al. Preventing cancer, cardiovascular disease, and diabetes: a common agenda for the American Cancer Society, the American Diabetes Association, and the American Heart Association. Circulation. 2004;109:3244–3255. doi: 10.1161/01.CIR.0000133321.00456.00. [DOI] [PubMed] [Google Scholar]
- 2.Naghavi M, Falk E, Hecht HS, Jamieson MJ, Kaul S, Berman D, Fayad Z, Budoff MJ, Rumberger J, Naqvi TZ, et al. From vulnerable plaque to vulnerable patient--Part III: Executive summary of the Screening for Heart Attack Prevention and Education (SHAPE) Task Force report. Am J Cardiol. 2006;98:2H–15H. doi: 10.1016/j.amjcard.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Murphy TP, Dhangana R, Pencina MJ, Zafar AM, D’Agostino RB. Performance of current guidelines for coronary heart disease prevention: optimal use of the Framingham-based risk assessment. Atherosclerosis. 2011;216:452–457. doi: 10.1016/j.atherosclerosis.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 4.Nasir K, Michos ED, Blumenthal RS, Raggi P. Detection of high-risk young adults and women by coronary calcium and National Cholesterol Education Program Panel III guidelines. J Am Coll Cardiol. 2005;46:1931–1936. doi: 10.1016/j.jacc.2005.07.052. [DOI] [PubMed] [Google Scholar]
- 5.Brindle P, Emberson J, Lampe F, Walker M, Whincup P, Fahey T, Ebrahim S. Predictive accuracy of the Framingham coronary risk score in British men: prospective cohort study. BMJ. 2003;327:1267. doi: 10.1136/bmj.327.7426.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 7.Thygesen K, Alpert JS, White HD. Universal definition of myocardial infarction. J Am Coll Cardiol. 2007;50:2173–2195. doi: 10.1016/j.jacc.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Tang WH, Francis GS, Morrow DA, Newby LK, Cannon CP, Jesse RL, Storrow AB, Christenson RH, Apple FS, Ravkilde J, et al. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: Clinical utilization of cardiac biomarker testing in heart failure. Circulation. 2007;116:e99–e109. doi: 10.1161/CIRCULATIONAHA.107.185267. [DOI] [PubMed] [Google Scholar]
- 9.Sabatine MS, Morrow DA, Jablonski KA, Rice MM, Warnica JW, Domanski MJ, Hsia J, Gersh BJ, Rifai N, Ridker PM, et al. Prognostic significance of the Centers for Disease Control/American Heart Association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease. Circulation. 2007;115:1528–1536. doi: 10.1161/CIRCULATIONAHA.106.649939. [DOI] [PubMed] [Google Scholar]
- 10.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 11.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 12.Hansson GK, Nilsson J. Introduction: atherosclerosis as inflammation: a controversial concept becomes accepted. J Intern Med. 2008;263:462–463. doi: 10.1111/j.1365-2796.2008.01959.x. [DOI] [PubMed] [Google Scholar]
- 13.Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276–1282. doi: 10.1056/NEJM199705013361802. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705–713. doi: 10.1161/01.ATV.0000261709.34878.20. [DOI] [PubMed] [Google Scholar]
- 15.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci USA. 2005;102:15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samnegård A, Silveira A, Tornvall P, Hamsten A, Ericsson CG, Eriksson P. Lower serum concentration of matrix metalloproteinase-3 in the acute stage of myocardial infarction. J Intern Med. 2006;259:530–536. doi: 10.1111/j.1365-2796.2006.01632.x. [DOI] [PubMed] [Google Scholar]
- 17.Loftus IM, Naylor AR, Goodall S, Crowther M, Jones L, Bell PR, Thompson MM. Increased matrix metalloproteinase-9 activity in unstable carotid plaques. A potential role in acute plaque disruption. Stroke. 2000;31:40–47. doi: 10.1161/01.str.31.1.40. [DOI] [PubMed] [Google Scholar]
- 18.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 19.Hansson GK, Holm J, Jonasson L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol. 1989;135:169–175. [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis. Clin Immunol. 2010;134:33–46. doi: 10.1016/j.clim.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Perry HM, McNamara CA. Refining the role of B cells in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1548–1549. doi: 10.1161/ATVBAHA.112.249235. [DOI] [PubMed] [Google Scholar]
- 22.Blasi C. The autoimmune origin of atherosclerosis. Atherosclerosis. 2008;201:17–32. doi: 10.1016/j.atherosclerosis.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 23.Pereira IA, Borba EF. The role of inflammation, humoral and cell mediated autoimmunity in the pathogenesis of atherosclerosis. Swiss Med Wkly. 2008;138:534–539. doi: 10.4414/smw.2008.12287. [DOI] [PubMed] [Google Scholar]
- 24.Skaggs BJ, Hahn BH, McMahon M. Accelerated atherosclerosis in patients with SLE--mechanisms and management. Nat Rev Rheumatol. 2012;8:214–223. doi: 10.1038/nrrheum.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeller CB, Appenzeller S. Cardiovascular disease in systemic lupus erythematosus: the role of traditional and lupus related risk factors. Curr Cardiol Rev. 2008;4:116–122. doi: 10.2174/157340308784245775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan MJ. Cardiovascular complications of rheumatoid arthritis: assessment, prevention, and treatment. Rheum Dis Clin North Am. 2010;36:405–426. doi: 10.1016/j.rdc.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roux-Lombard P, Pagano S, Montecucco F, Satta N, Vuilleumier N. Auto-antibodies as emergent prognostic markers and possible mediators of ischemic cardiovascular diseases. Clin Rev Allergy Immunol. 2013;44:84–97. doi: 10.1007/s12016-010-8233-z. [DOI] [PubMed] [Google Scholar]
- 28.Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F. Evidence on the pathogenic role of auto-antibodies in acute cardiovascular diseases. Thromb Haemost. 2013;109:854–868. doi: 10.1160/TH12-10-0768. [DOI] [PubMed] [Google Scholar]
- 29.Satta N, Dunoyer-Geindre S, Reber G, Fish RJ, Boehlen F, Kruithof EK, de Moerloose P. The role of TLR2 in the inflammatory activation of mouse fibroblasts by human antiphospholipid antibodies. Blood. 2007;109:1507–1514. doi: 10.1182/blood-2005-03-024463. [DOI] [PubMed] [Google Scholar]
- 30.Satta N, Kruithof EK, Fickentscher C, Dunoyer-Geindre S, Boehlen F, Reber G, Burger D, de Moerloose P. Toll-like receptor 2 mediates the activation of human monocytes and endothelial cells by antiphospholipid antibodies. Blood. 2011;117:5523–5531. doi: 10.1182/blood-2010-11-316158. [DOI] [PubMed] [Google Scholar]
- 31.Yokota S, Minota S, Fujii N. Anti-HSP auto-antibodies enhance HSP-induced pro-inflammatory cytokine production in human monocytic cells via Toll-like receptors. Int Immunol. 2006;18:573–580. doi: 10.1093/intimm/dxh399. [DOI] [PubMed] [Google Scholar]
- 32.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, Boullier A, Gonen A, Diehl CJ, Que X, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epstein SE, Zhu J, Burnett MS, Zhou YF, Vercellotti G, Hajjar D. Infection and atherosclerosis: potential roles of pathogen burden and molecular mimicry. Arterioscler Thromb Vasc Biol. 2000;20:1417–1420. doi: 10.1161/01.atv.20.6.1417. [DOI] [PubMed] [Google Scholar]
- 34.Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54:24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 35.Morrow DA, de Lemos JA. Benchmarks for the assessment of novel cardiovascular biomarkers. Circulation. 2007;115:949–952. doi: 10.1161/CIRCULATIONAHA.106.683110. [DOI] [PubMed] [Google Scholar]
- 36.Pencina MJ, D’Agostino RB, D’Agostino RB, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172; discussion 207-212. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 37.Hansson GK, Nilsson J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin Immunopathol. 2009;31:95–101. doi: 10.1007/s00281-009-0151-x. [DOI] [PubMed] [Google Scholar]
- 38.de Jager SC, Kuiper J. Vaccination strategies in atherosclerosis. Thromb Haemost. 2011;106:796–803. doi: 10.1160/TH11-05-0369. [DOI] [PubMed] [Google Scholar]
- 39.Gullestad L, Aass H, Fjeld JG, Wikeby L, Andreassen AK, Ihlen H, Simonsen S, Kjekshus J, Nitter-Hauge S, Ueland T, et al. Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation. 2001;103:220–225. doi: 10.1161/01.cir.103.2.220. [DOI] [PubMed] [Google Scholar]
- 40.Gullestad L, Orn S, Dickstein K, Eek C, Edvardsen T, Aakhus S, Askevold ET, Michelsen A, Bendz B, Skårdal R, et al. Intravenous immunoglobulin does not reduce left ventricular remodeling in patients with myocardial dysfunction during hospitalization after acute myocardial infarction. Int J Cardiol. 2013;168:212–218. doi: 10.1016/j.ijcard.2012.09.092. [DOI] [PubMed] [Google Scholar]
- 41.Matsuura E, Kobayashi K, Inoue K, Shoenfeld Y. Intravenous immunoglobulin and atherosclerosis. Clin Rev Allergy Immunol. 2005;29:311–319. doi: 10.1385/CRIAI:29:3:311. [DOI] [PubMed] [Google Scholar]
- 42.Persson L, Borén J, Nicoletti A, Hansson GK, Pekna M. Immunoglobulin treatment reduces atherosclerosis in apolipoprotein E-/- low-density lipoprotein receptor-/- mice via the complement system. Clin Exp Immunol. 2005;142:441–445. doi: 10.1111/j.1365-2249.2005.02954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okabe TA, Kishimoto C, Shimada K, Murayama T, Yokode M, Kita T. Effects of late administration of immunoglobulin on experimental atherosclerosis in apolipoprotein E-deficient mice. Circ J. 2005;69:1543–1546. doi: 10.1253/circj.69.1543. [DOI] [PubMed] [Google Scholar]
- 44.Gordon SM, Hofmann S, Askew DS, Davidson WS. High density lipoprotein: it’s not just about lipid transport anymore. Trends Endocrinol Metab. 2011;22:9–15. doi: 10.1016/j.tem.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Besler C, Lüscher TF, Landmesser U. Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med. 2012;4:251–268. doi: 10.1002/emmm.201200224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- 47.Merrill JT, Rivkin E, Shen C, Lahita RG. Selection of a gene for apolipoprotein A1 using autoantibodies from a patient with systemic lupus erythematosus. Arthritis Rheum. 1995;38:1655–1659. doi: 10.1002/art.1780381118. [DOI] [PubMed] [Google Scholar]
- 48.Dinu AR, Merrill JT, Shen C, Antonov IV, Myones BL, Lahita RG. Frequency of antibodies to the cholesterol transport protein apolipoprotein A1 in patients with SLE. Lupus. 1998;7:355–360. doi: 10.1191/096120398678920262. [DOI] [PubMed] [Google Scholar]
- 49.Abe H, Tsuboi N, Suzuki S, Sakuraba H, Takanashi H, Tahara K, Tonozuka N, Hayashi T, Umeda M. Anti-apolipoprotein A-I autoantibody: characterization of monoclonal autoantibodies from patients with systemic lupus erythematosus. J Rheumatol. 2001;28:990–995. [PubMed] [Google Scholar]
- 50.Delgado Alves J, Kumar S, Isenberg DA. Cross-reactivity between anti-cardiolipin, anti-high-density lipoprotein and anti-apolipoprotein A-I IgG antibodies in patients with systemic lupus erythematosus and primary antiphospholipid syndrome. Rheumatology (Oxford) 2003;42:893–899. doi: 10.1093/rheumatology/keg248. [DOI] [PubMed] [Google Scholar]
- 51.Delgado Alves J, Ames PR, Donohue S, Stanyer L, Nourooz-Zadeh J, Ravirajan C, Isenberg DA. Antibodies to high-density lipoprotein and beta2-glycoprotein I are inversely correlated with paraoxonase activity in systemic lupus erythematosus and primary antiphospholipid syndrome. Arthritis Rheum. 2002;46:2686–2694. doi: 10.1002/art.10542. [DOI] [PubMed] [Google Scholar]
- 52.Batuca JR, Ames PR, Isenberg DA, Alves JD. Antibodies toward high-density lipoprotein components inhibit paraoxonase activity in patients with systemic lupus erythematosus. Ann N Y Acad Sci. 2007;1108:137–146. doi: 10.1196/annals.1422.016. [DOI] [PubMed] [Google Scholar]
- 53.Ames PR, Matsuura E, Batuca JR, Ciampa A, Lopez LL, Ferrara F, Iannaccone L, Alves JD. High-density lipoprotein inversely relates to its specific autoantibody favoring oxidation in thrombotic primary antiphospholipid syndrome. Lupus. 2010;19:711–716. doi: 10.1177/0961203309357765. [DOI] [PubMed] [Google Scholar]
- 54.Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, La Du BN, Fogelman AM, Navab M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995;96:2758–2767. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vuilleumier N, Bratt J, Alizadeh R, Jogestrand T, Hafström I, Frostegård J. Anti-apoA-1 IgG and oxidized LDL are raised in rheumatoid arthritis (RA): potential associations with cardiovascular disease and RA disease activity. Scand J Rheumatol. 2010;39:447–453. doi: 10.3109/03009741003742755. [DOI] [PubMed] [Google Scholar]
- 56.Vuilleumier N, Bas S, Pagano S, Montecucco F, Guerne PA, Finckh A, Lovis C, Mach F, Hochstrasser D, Roux-Lombard P, et al. Anti-apolipoprotein A-1 IgG predicts major cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum. 2010;62:2640–2650. doi: 10.1002/art.27546. [DOI] [PubMed] [Google Scholar]
- 57.Finckh A, Courvoisier DS, Pagano S, Bas S, Chevallier-Ruggeri P, Hochstrasser D, Roux-Lombard P, Gabay C, Vuilleumier N. Evaluation of cardiovascular risk in patients with rheumatoid arthritis: do cardiovascular biomarkers offer added predictive ability over established clinical risk scores? Arthritis Care Res (Hoboken) 2012;64:817–825. doi: 10.1002/acr.21631. [DOI] [PubMed] [Google Scholar]
- 58.Vuilleumier N, Reber G, James R, Burger D, de Moerloose P, Dayer JM, Roux-Lombard P. Presence of autoantibodies to apolipoprotein A-1 in patients with acute coronary syndrome further links autoimmunity to cardiovascular disease. J Autoimmun. 2004;23:353–360. doi: 10.1016/j.jaut.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 59.Vuilleumier N, Charbonney E, Fontao L, Alvarez M, Turck N, Sanchez JC, Burkhard PR, Mensi N, Righini M, Reber G, et al. Anti-(apolipoprotein A-1) IgGs are associated with high levels of oxidized low-density lipoprotein in acute coronary syndrome. Clin Sci (Lond) 2008;115:25–33. doi: 10.1042/CS20070325. [DOI] [PubMed] [Google Scholar]
- 60.Vuilleumier N, Rossier MF, Pagano S, Python M, Charbonney E, Nkoulou R, James R, Reber G, Mach F, Roux-Lombard P. Anti-apolipoprotein A-1 IgG as an independent cardiovascular prognostic marker affecting basal heart rate in myocardial infarction. Eur Heart J. 2010;31:815–823. doi: 10.1093/eurheartj/ehq055. [DOI] [PubMed] [Google Scholar]
- 61.Vuilleumier N, Pagano S, Lahlou K, Antoine P, Charbonney E, Norman GL, Mach F, Roux-Lombard P. Head-to-Head Comparison of Auto-Antibodies for Cardiovascular Outcome Prediction after Myocardial Infarction: a Prospective Study. J Clinic Experiment Cardiol. 2011;2:169. [Google Scholar]
- 62.Keller PF, Pagano S, Roux-Lombard P, Sigaud P, Rutschmann OT, Mach F, Hochstrasser D, Vuilleumier N. Autoantibodies against apolipoprotein A-1 and phosphorylcholine for diagnosis of non-ST-segment elevation myocardial infarction. J Intern Med. 2012;271:451–462. doi: 10.1111/j.1365-2796.2011.02479.x. [DOI] [PubMed] [Google Scholar]
- 63.Montecucco F, Vuilleumier N, Pagano S, Lenglet S, Bertolotto M, Braunersreuther V, Pelli G, Kovari E, Pane B, Spinella G, et al. Anti-Apolipoprotein A-1 auto-antibodies are active mediators of atherosclerotic plaque vulnerability. Eur Heart J. 2011;32:412–421. doi: 10.1093/eurheartj/ehq521. [DOI] [PubMed] [Google Scholar]
- 64.Vuilleumier N, Montecucco F, Spinella G, Pagano S, Bertolotto M, Pane B, Pende A, Galan K, Roux-Lombard P, Combescure C, et al. Serum levels of anti-apolipoprotein A-1 auto-antibodies and myeloperoxidase as predictors of major adverse cardiovascular events after carotid endarterectomy. Thromb Haemost. 2013;109:706–715. doi: 10.1160/TH12-10-0714. [DOI] [PubMed] [Google Scholar]
- 65.Wick PA, Mombelli A, Pagano S, Moren X, Giannopoulou C, Mach F, Roux-Lombard P, Vuilleumier N. Anti-apolipoprotein A-1 autoantibodies as biomarker for atherosclerosis burden in patients with periodontitis. J Periodontal Res. 2013;48:350–356. doi: 10.1111/jre.12014. [DOI] [PubMed] [Google Scholar]
- 66.Pruijm M, Schmidtko J, Aho A, Pagano S, Roux-Lombard P, Teta D, Burnier M, Vuilleumier N. High prevalence of anti-apolipoprotein/A-1 autoantibodies in maintenance hemodialysis and association with dialysis vintage. Ther Apher Dial. 2012;16:588–594. doi: 10.1111/j.1744-9987.2012.01102.x. [DOI] [PubMed] [Google Scholar]
- 67.Dietrich T, Jimenez M, Krall Kaye EA, Vokonas PS, Garcia RI. Age-dependent associations between chronic periodontitis/edentulism and risk of coronary heart disease. Circulation. 2008;117:1668–1674. doi: 10.1161/CIRCULATIONAHA.107.711507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension. 2003;42:1050–1065. doi: 10.1161/01.HYP.0000102971.85504.7c. [DOI] [PubMed] [Google Scholar]
- 69.Quercioli A, Montecucco F, Galan K, Ratib O, Roux-Lombard P, Pagano S, Mach F, Schindler TH, Vuilleumier N. Anti-apolipoprotein A-1 IgG levels predict coronary artery calcification in obese but otherwise healthy individuals. Mediators Inflamm. 2012;2012:243158. doi: 10.1155/2012/243158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Batuca JR, Ames PR, Amaral M, Favas C, Isenberg DA, Delgado Alves J. Anti-atherogenic and anti-inflammatory properties of high-density lipoprotein are affected by specific antibodies in systemic lupus erythematosus. Rheumatology (Oxford) 2009;48:26–31. doi: 10.1093/rheumatology/ken397. [DOI] [PubMed] [Google Scholar]
- 71.Wahlgren CM, Zheng W, Shaalan W, Tang J, Bassiouny HS. Human carotid plaque calcification and vulnerability. Relationship between degree of plaque calcification, fibrous cap inflammatory gene expression and symptomatology. Cerebrovasc Dis. 2009;27:193–200. doi: 10.1159/000189204. [DOI] [PubMed] [Google Scholar]
- 72.Pagano S, Satta N, Werling D, Offord V, de Moerloose P, Charbonney E, Hochstrasser D, Roux-Lombard P, Vuilleumier N. Anti-apolipoprotein A-1 IgG in patients with myocardial infarction promotes inflammation through TLR2/CD14 complex. J Intern Med. 2012;272:344–357. doi: 10.1111/j.1365-2796.2012.02530.x. [DOI] [PubMed] [Google Scholar]
- 73.Maier W, Altwegg LA, Corti R, Gay S, Hersberger M, Maly FE, Sütsch G, Roffi M, Neidhart M, Eberli FR, et al. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation. 2005;111:1355–1361. doi: 10.1161/01.CIR.0000158479.58589.0A. [DOI] [PubMed] [Google Scholar]
- 74.Biasucci LM, Liuzzo G, Fantuzzi G, Caligiuri G, Rebuzzi AG, Ginnetti F, Dinarello CA, Maseri A. Increasing levels of interleukin (IL)-1Ra and IL-6 during the first 2 days of hospitalization in unstable angina are associated with increased risk of in-hospital coronary events. Circulation. 1999;99:2079–2084. doi: 10.1161/01.cir.99.16.2079. [DOI] [PubMed] [Google Scholar]
- 75.Anderssohn M, Schwedhelm E, Lüneburg N, Vasan RS, Böger RH. Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: an intriguing interaction with diabetes mellitus. Diab Vasc Dis Res. 2010;7:105–118. doi: 10.1177/1479164110366053. [DOI] [PubMed] [Google Scholar]
- 76.Böger RH, Maas R, Schulze F, Schwedhelm E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality--an update on patient populations with a wide range of cardiovascular risk. Pharmacol Res. 2009;60:481–487. doi: 10.1016/j.phrs.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 77.Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372:817–821. doi: 10.1016/S0140-6736(08)61171-X. [DOI] [PubMed] [Google Scholar]
- 78.Kannel WB, Kannel C, Paffenbarger RS, Cupples LA. Heart rate and cardiovascular mortality: the Framingham Study. Am Heart J. 1987;113:1489–1494. doi: 10.1016/0002-8703(87)90666-1. [DOI] [PubMed] [Google Scholar]
- 79.Diaz A, Bourassa MG, Guertin MC, Tardif JC. Long-term prognostic value of resting heart rate in patients with suspected or proven coronary artery disease. Eur Heart J. 2005;26:967–974. doi: 10.1093/eurheartj/ehi190. [DOI] [PubMed] [Google Scholar]
- 80.Rossier MF, Pagano S, Python M, Maturana AD, James RW, Mach F, Roux-Lombard P, Vuilleumier N. Antiapolipoprotein A-1 IgG chronotropic effects require nongenomic action of aldosterone on L-type calcium channels. Endocrinology. 2012;153:1269–1278. doi: 10.1210/en.2011-1835. [DOI] [PubMed] [Google Scholar]
- 81.Srivastava R, Yu S, Parks BW, Black LL, Kabarowski JH. Autoimmune-mediated reduction of high-density lipoprotein-cholesterol and paraoxonase 1 activity in systemic lupus erythematosus-prone gld mice. Arthritis Rheum. 2011;63:201–211. doi: 10.1002/art.27764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polonsky TS, McClelland RL, Jorgensen NW, Bild DE, Burke GL, Guerci AD, Greenland P. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA. 2010;303:1610–1616. doi: 10.1001/jama.2010.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fowkes FG, Murray GD, Butcher I, Heald CL, Lee RJ, Chambless LE, Folsom AR, Hirsch AT, Dramaix M, deBacker G, et al. Ankle brachial index combined with Framingham Risk Score to predict cardiovascular events and mortality: a meta-analysis. JAMA. 2008;300:197–208. doi: 10.1001/jama.300.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tziomalos K, Athyros VG, Karagiannis A, Mikhailidis DP. The role of ankle brachial index and carotid intima-media thickness in vascular risk stratification. Curr Opin Cardiol. 2010;25:394–398. doi: 10.1097/hco.0b013e328338c109. [DOI] [PubMed] [Google Scholar]