

Abstract
The peripherally restricted fatty acid amide hydrolase (FAAH) inhibitor URB937 (3, cyclohexylcarbamic acid 3’-carbamoyl-6-hydroxybiphenyl-3-yl ester) is extruded from the brain and spinal cord by the Abcg2 efflux transporter. Despite its inability to enter the central nervous system (CNS), 3 exerts profound antinociceptive effects in mice and rats, which result from the inhibition of FAAH in peripheral tissues and the consequent enhancement of anandamide signaling at CB1 cannabinoid receptors localized on sensory nerve endings. In the present study, we examined the structure-activity relationships (SAR) for the biphenyl region of compound 3, focusing on the carbamoyl and hydroxyl groups in the distal and proximal phenyl rings. Our SAR studies generated a new series of peripherally restricted FAAH inhibitors and identified compound 35 (cyclohexylcarbamic acid 3’-carbamoyl-5-hydroxybiphenyl-3-yl ester) as the most potent brain-impermeant FAAH inhibitor disclosed to date.
Keywords: FAAH, O-biphenyl-3-yl carbamates, URB937, SAR, peripheral FAAH inhibitors
INTRODUCTION
The therapeutic exploitation of the endocannabinoid system with exogenous agonists is limited by the undesired side effects caused by indiscriminate activation of cannabinoid type-1 (CB1) receptors, particularly in the brain.1 An alternative strategy to direct CB1 receptor targeting is to upregulate the signaling activity of the endogenous cannabinoid ligands, arachidonoylethanolamide (anandamide)2 and 2-arachidonoyl-sn-glycerol (2-AG),3 by blocking their intracellular degradation. Anandamide is released on demand by stimulated neurons2 and inhibitors of the enzyme responsible for its hydrolytic cleavage, fatty acid amide hydrolase (FAAH), have been shown to increase anandamide levels and activate central and peripheral CB1 receptors without causing signs of cannabinoid intoxication.4,5
Outside the central nervous system (CNS), CB1 receptors are found in organs such as liver, kidney and intestine, as well as in peripheral sensory terminals and dorsal root ganglia (DRG) neurons.6 Evidence suggests that therapeutic gain devoid of central liability can be achieved in conditions such as pain and metabolic syndrome by targeting these peripheral receptors.7,8 Thus, developing pharmacological agents that do not cross the blood-brain barrier (BBB) provides a potential approach to identify endocannabinoid-based therapies that are both safe and effective. Indeed, synthetic efforts aimed at creating CB1 receptor agonists and antagonists with restricted access to the CNS have been reported.9,10,11,12 In those studies, the main strategies adopted to limit access of compounds of interest to the CNS were either to increase the compounds’ topological polar surface area (TPSA)11 or to exploit the recognition by drug transporters present in the blood-brain barrier (BBB).12
Selective activation of peripheral CB1 receptors by endogenously produced anandamide was first achieved with compound URB937 (3, cyclohexylcarbamic acid 3’-carbamoyl-6-hydroxybiphenyl-3-yl ester, Figure 1), which blocks FAAH activity only outside the CNS through an irreversible mechanism.13 Compound 3 inhibits visceral and inflammatory pain responses in rodents by reducing nociceptive inputs to the spinal cord.13 Genetic and pharmacological studies have shown that this compound is extruded from the CNS by the ATP-binding cassette transporter Abcg2 (ABCG2 in humans).14 Furthermore, Abcg2 limits the access of 3 not only to the CNS, but also to the feto-placental unit of pregnant rodents.15 Recognition by Abcg2 may be exploited, therefore, as a novel strategy to develop therapeutic agents devoid of CNS-mediated effects and, possibly, safe to be used during pregnancy.16
Figure 1.

Design of peripherally restricted FAAH inhibitors.
We have previously shown that introduction of a hydroxyl group in the para position of the proximal phenyl ring of cyclohexylcarbamic acid 3’-carbamoylbiphenyl-3-yl ester (1, URB597, Figure 1) generates the peripherally restricted derivative 3 (Figure 1).13 Conversely, removing the carbamoyl moiety from the distal phenyl ring yields cyclohexylcarbamic acid 6-hydroxybiphenyl-3-yl ester, a compound that readily enters the brain (2, URB694, Figure 1).17,18 These observations suggest that the R1 and R2 regions (Figure 1) are essential to limit the penetration of O-biphenyl-3-yl carbamate FAAH inhibitors into the CNS. To elucidate the substitutions in R1 and R2 that best combine inhibitory potency and lack of brain penetration, we progressively modified the R1 and R2 regions in the scaffold of 3 and tested the new compounds for their ability to inhibit FAAH activity in vitro and in vivo. Dose-response exploration studies allowed us to establish a hierarchy among the different substituents based on their access to the brain. Furthermore, the involvement of Abcg2 in the peripheral distribution of the new compounds was tested using the selective Abcg2 inhibitor, Ko-143.19 These studies allowed us to identify a series of novel peripherally restricted FAAH inhibitors, including the highly potent compound 35, and obtain new insights on the structural requirements underpinning the impaired access of these agents to the CNS.
CHEMISTRY
Different synthetic approaches were utilized for the preparation of compounds 7a–g and 11a– c, bearing structural modifications on the distal phenyl ring (Scheme 1 and 2).
Scheme 1.

Reagents and conditions: a’) BBr3, DCM, –78 °C, 2 h, 97%; b’) ArB(OH)2, CsOAc, PdCl2dppf, dioxane, 80 °C, 5–12 h, 16–62%; a”) ArB(OR)2 (5d,e) or ArB(OH)2 (5f,g), CsOAc, PdCl2dppf, dioxane, 80 °C, 3–12 h, 19–77%; b”) cyclohexene, 10% Pd/C, EtOH, 60 °C, 2 h (7d,f,g) or BBr3, DCM, –78 °C to rt, 2 h (7e), 56–90%.
Scheme 2.
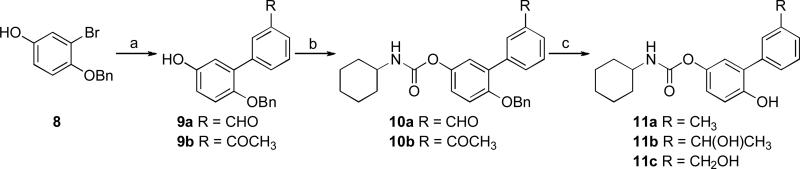
Reagents and conditions: a) ArB(OH)2, Na2CO3, Pd(PPh3)4, toluene/H2O, reflux, 12 h, 64–78%; b) c-C6H11NCO, Et3N, CH3CN, reflux, 5 h, 55–61%; c) H2 (4 atm), 10% Pd/C, EtOH/EtOAc (11b) or EtOH (11a,c), 50 °C, 4 h, 19–50%.
Compounds 7a–c were prepared by the introduction of the targeted structural variations at the last step of the synthesis via a Suzuki cross-coupling reaction directly on the common intermediate 6, obtained from 4 20 through an O-debenzylation by using boron tribromide in dichloromethane (Scheme 1). Alternatively, compounds 7d–g were prepared using a synthetic methodology recently reported for the preparation of multigram scale of 3.20 The intermediates 5d–g were obtained from 4 through a Suzuki cross-coupling reaction and converted into the final compounds via a O-debenzylation reaction employing either cyclohexene and Pd/C in dioxane or boron tribromide in dichloromethane (Scheme 1).
Compounds 11a–c were obtained in three steps starting from 8 13,21, through a Suzuki cross-coupling reaction followed by carbamoylation and Pd/C catalyzed hydrogenative deprotection (Scheme 2).
Compound 15 13 was obtained in a three-step synthetic procedure starting from the commercially available aldehyde 12 which was converted into phenol derivative 13 through a Dakin oxidation 22 followed by a Suzuki cross-coupling reaction to give 14 that was then treated with cyclohexyl isocyanate in acetonitrile to afford the carbamate 15 (Scheme 3).
Scheme 3.
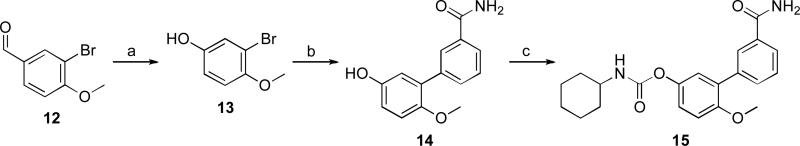
Reagents and conditions: a) i) m-CPBA, DCM, 40 °C, 72 h; ii) NaOCH3, EtOH, rt, 1 h, 60%; b) 3-carbamoylphenylboronic acid, Na2CO3, Pd(PPh3)4, toluene/H2O, reflux, 12 h, 81%; d) c-C6H11NCO, Et3N, CH3CN, reflux, 2 h, 77%.
The synthetic procedure for the preparation of compound 21 is reported in Scheme 4. Starting from the commercially available aldehyde 16, which was protected as acetal 17,23 a two-step sequence was employed to obtain the O-benzylated bromide 18 24 via a nucleophilic aromatic substitution with benzyl alcohol in the presence of potassium tert-butoxide, followed by hydrolysis of the acetal group under acidic condition. The intermediate 18 was efficiently converted into the biphenyl aldehyde 19 under ligand-free Suzuki cross-coupling conditions 25 by reacting with 3-carbamoylphenylboronic acid in aqueous potassium carbonate in the presence of palladium acetate. Compound 19 was then reduced under standard conditions to the corresponding alcohol 20 that was O-debenzylated and selectively converted with cyclohexyl isocyanate into the final compound 21.
Scheme 4.

Reagents and conditions: a) ethylene glycol, p-TSA, toluene, reflux, 12h, 93%; b) t-BuOK, BnOH, dry dioxane, 85 °C, 1 h; c) 2 N HCl, rt, 2 h, 75% (over 2 steps); d) ArB(OH)2, K2CO3, Pd(OAc)2, EGME/H2O (3:1), rt, 40 min, 95%; e) NaBH4, EtOH, 0 °C to rt, 2 h, 81%; f) 10% Pd/C, γ-terpinene, dioxane, reflux, 1 h; g) c-C6H11NCO, Et3N, CH3CN/EtOH, rt, 12h, 44% (over 2 steps).
The synthetic procedure for the preparation of compound 29 is described in Scheme 5. Starting from 22, a Friedel-Crafts acylation produced compound 23 which was oxidized to carboxylic acid 24 by a two-step sequence consisting in a Claisen condensation with diethyl oxalate followed by an oxidative cleavage with potassium peroxymonosulfate.26 The conversion of 24 into 25 was carried out using boron tribromide in dichloromethane. Compound 25 was then chemoselectively transformed into the O-benzylester 26 27 that, after a Suzuki cross-coupling reaction, gave 27. Conversion of the phenol derivative 27 into the carboxylic acid 29 was carried out through a carbamoylation reaction by using cyclohexyl isocyanate in acetonitrile/ethanol followed by Pd/C catalysed hydrogenolysis with cyclohexene in dioxane.
Scheme 5.

Reagents and conditions: a) AcCl, ZrCl4, DCM, 0 °C to rt, 1 h, 55%; b) diethyl oxalate, t-BuONa, THF, rt, 30 min, 77%; c) Oxone®, acetone, 0 °C, 2 h, 61%; d) BBr3, DCM, 0 °C to rt, 12h, 61%; e) BnBr, KHCO3, DMF, rt, 12h; f) 3-carbamoylphenylboronic acid, PdCl2dppf, K2CO3, dioxane/H2O, 90 °C, 1 h, 57%; g) c-C6H11NCO, Et3N, CH3CN/EtOH, 45 °C, 12h, 76%; h) 10% Pd/C, cyclohexene, dioxane, 80 °C, 2 h, 44%.
Scheme 6 reports the synthesis of compound 35 from 31 that was converted by a nucleophilic aromatic substitution in 32.28 A ligand-free Suzuki cross coupling reaction was utilized 29 to afford the corresponding biphenyl derivative 33 that was quantitatively debenzylated to obtain compound 34. Attempts to selectively mono-carbamoylate 34 in the desired carbamate 35 were troublesome. Compound 34 was treated with cyclohexyl isocyanate (1.1 equiv.) in presence of triethylamine or 4-(dimethylamino)-pyridine (DMAP) in acetonitrile at room temperature for 12h to afford mixtures containing the di-carbamoylated derivative 36 as the major side product. In the specific, we observed the formation of (0.6: 1.3: 1.0) and (1.3: 1.0: 2.3) mixtures of compounds (34: 35: 36) by using triethylamine (1.1 equiv.) and DMAP (0.1 equiv.) respectively. Eventually, the use of cyclohexyl isocyanate in N,N-dimethylformamide at room temperature in presence of copper chloride as promoter 30 yielded a (1:2:1) mixture of compounds (34: 35: 36), which were separated by chromatographic purification. Alternatively, attempts to carry out selective mono-protection of compound 34 with tert-butyldimethylsilyl chloride (TBSCl) or triisopropylsilyl chloride (TIPSCl) also failed.
Scheme 6.

Reagents and conditions: a) t-BuONa, BnOH, dry DMF, 90 °C, 3 h, 72%; b) 3-carbamoylphenylboronic acid, K2CO3, Pd(OAc)2, EGME/H2O (3:1), 60 °C, 20 min, 70%; c) 10% Pd/C, cyclohexene, dioxane, 80 °C, 2 h, 100%; d) c-C6H11NCO, CuCl, DMF, rt, 30 min, 29%.
Compound 41 was obtained in a four-step synthetic procedure starting from the commercially available phenol 37, that was converted in the corresponding triflate 38 31 and directly used in the Suzuki cross coupling reaction to afford 39. Compound 39 was selectively mono-demethylated to 40 using boron tribromide in dichloromethane and then converted to the carbamate 41 under standard conditions (Scheme 7).
Scheme 7.

Reagents and conditions: a) (CF3SO2)2O, DMAP, DCM, 0 °C to rt, 15 min; b) 3-carbamoylphenylboronic acid, Na2CO3, PdCl2, EtOH, reflux, 12 h, 45%; c) BBr3, DCM, 0 °C to rt, 30 min, 40%; d) c-C6H11NCO, Et3N, CH3CN, reflux, 2 h, 65%.
RESULTS
Previous studies have shown that compound 3 inhibits FAAH activity in liver and other peripheral tissues of mice with a median effective dose (ED50) of 0.2 mg/kg (intraperitoneal, i.p.), which is 200 times lower than the ED50 for FAAH inhibition in the brain (40 mg/kg).13 The present study evaluated the effects that structural modifications at the meta position of the distal phenyl ring (R1 region) and the para or meta positions of the proximal phenyl ring (R2 region) exert on the inhibitory potency and systemic distribution of 3. Median concentrations to inhibit FAAH activity (IC50) were determined in vitro using rat brain homogenates. FAAH inhibition was also measured ex vivo in liver and brain tissue of mice 1 h after systemic administration of test compounds (1 mg/kg, i.p.). Since O-biphenyl-3-yl carbamates inhibit FAAH through a covalent, irreversible mechanism,32 the degree of FAAH inhibition measured ex vivo provides a useful estimate of the amount of compound reaching that tissue.14,15
Analogues of 3 bearing different substituents on the meta- position of the distal phenyl ring
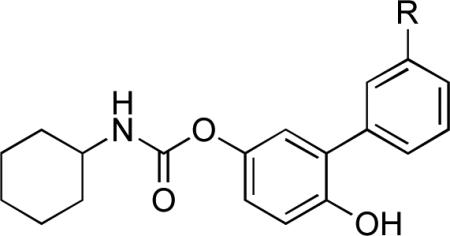
The results of explorative chemistry targeting the meta position of the distal phenyl ring (R1 region) of 3 are summarized in Table 1. Substituting the carbamoyl group with a methyl (11a), 1-hydroxyethyl (11b) or hydroxymethyl (11c) group yielded compounds that retained FAAH inhibitory activity in vitro, but readily accessed the CNS in vivo: 11a–c produced similar inhibitory effects on FAAH activity in brain and liver. Likewise, the methylketone derivative 7a displayed good brain penetration in vivo, along with increased in vitro potency on FAAH (Table 1). These results confirm that the carbamoyl functionality in the R1 region of 3 plays a key role in the peripheral distribution of this compound. This idea was further tested by preparing new compounds in which such functionality was progressively alkylated to a secondary (7d) or tertiary (7e) amide. When administered in vivo (1 mg/kg, i.p.), both 7d and 7e displayed impaired access to the brain, but maintained the ability to block FAAH activity in the liver (Table 1). A dose–exploration study revealed that 7d and 7e gain access to the brain at higher dosages, similarly to what previously found for 3,13 possibly by saturating the mechanism that mediates their extrusion from the brain (Figure 2a). Interestingly, the ED50 values of these compounds for brain FAAH inhibition ex vivo (3, 40 mg/kg; 7d, 15 mg/kg; 7e, 3.5 mg/kg) progressively decreased as the number of methyl substituents linked to the carbamoyl moiety was increased (Figure 2a). The reverse amide 7c displayed significantly higher brain penetration relative to 3, 7d and 7e at all doses tested (Figure 2a). The involvement of the Abcg2 transporter in restricting the access of 7d and 7e to the brain was tested by pre-administration of the selective inhibitor Ko-143 (15 mg/kg, i.p.). Pharmacological blockade of Abcg2 allowed sub-effective doses of 3 and 7d to inhibit FAAH activity in the brain, but failed to do so with 7e (Figure 2c). We interpret these findings to indicate that a primary (3) or secondary (7d) carbamoyl moiety is a key determinant for the interaction of O-biphenyl-3-yl carbamate FAAH inhibitors with Abcg2 in vivo. By contrast, the asymmetric tissue distribution of 7e at low doses appears to be independent of Abcg2.
Table 1.
Inhibitory Potency (IC50) and Systemic Distribution of 3’-Substituted O-Biphenyl-3-yl Carbamates.
| |||||
|---|---|---|---|---|---|
| R | In vitro IC50 (nM)a | FAAH inhibition in liver (%)b | FAAH inhibition in brain (%)b | PSA (Å2)d | |
| 3 | CONH2 | 2.0 | 91.7±0.7 | −3.0±8.0 | 83 |
| 7a | COCH3 | 0.3 | 88.1±0.8 | 89.2±0.6 | 62 |
| 7b | COOH | 32 | 30.0±2.6 | 2.8±1.3 | 77 |
| 66.4±4.1c | −6.8±2.7c | ||||
| 7c | NHCOCH3 | 15 | 76.0±1.6 | 31.6±1.5 | 72 |
| 7d | CONHCH3 | 6.0 | 87.2±2.4 | 0.9±3.7 | 73 |
| 7e | CON(CH3)2 | 1.5 | 84.2±2.5 | 22.6±7.8 | 65 |
| 7f | SO2NH2 | 2.7 | 81.1±1.3 | 7.0±1.7 | 99 |
| 7g | SO2CH3 | 6.3 | 73.9±4.6 | 6.6±2.3 | 78 |
| 11a | CH3 | 2.5 | 72.1±4.9 | 85.4±0.5 | 49 |
| 11b | CH(OH)CH3 | 2.0 | 80.9±0.9 | 89.6±0.6 | 65 |
| 11c | CH2OH | 1.6 | 86.6±0.6 | 88.6±0.7 | 66 |
IC50 measured in membrane preparations of Wistar rat brain
FAAH inhibition measured ex vivo 1 h after injection in Swiss Webster mice (1 mg/kg, intraperitoneal, n = 3)
FAAH inhibition measured ex vivo 1 h after injection in Swiss Webster mice (3 mg/kg, intraperitoneal, n = 3)
PSA values were calculated using ICM version 3.7 (Molsoft LLC, San Diego, CA).
Figure 2.

Inhibition of brain FAAH activity by analogues of 3 bearing different substituents on the meta- position of the distal phenyl ring; a) Dose-dependent effects of secondary (7c), tertiary (7d) or reverse (7e) amide derivatives of compound 3 in Swiss Webster mice; doses were 0.3–40 mg/kg (subcutaneous); FAAH activity was measured ex vivo 1 h after injection; b) Dose-dependent effects of sulfonamide (7f) and methylsulfone (7g) derivatives of compound 3 in Swiss Webster mice; doses were 0.3–75 mg/kg (s.c.); c) Effects of pharmacological blockade of the Abcg2 transporter (Ko-143, 15 mg/kg, i.p., closed bars) on brain inhibition of FAAH activity by a sub-effective dose (selected from the dose-response study: 3 (25); 7d (10); 7e (1); 7f (40); 7g (1) in mg/kg, s.c., open bars) of analogues of compound 3 bearing different functionalities on the meta- position of the distal phenyl ring. Results are expressed as mean ± s.e.m. (n = 3-4). *** P< 0.001 vs non-Ko-143 treated group.
To further test the relevance of the carbamoyl group in the distal ring of 3, and probe its possible role in the interaction with Abcg2 acting as a H-bond donor, we synthesized and tested the carboxylic acid derivative 7b (Table 1). The compound displayed impaired access to the brain, but failed to fully inhibit liver FAAH activity at 1 mg/kg (30% inhibition, Table 1). By increasing the dosage to 3 mg/kg, we were able to improve the blockade of liver FAAH activity (66% inhibition, Table 1), still without affecting brain FAAH activity. These findings suggest that 7b has restricted access to the CNS. However, we did not further pursue the characterization of this compound due to its relatively low potency both in vitro and in vivo. On the other hand, the corresponding bioisosteric sulphonamide (7f) and methylsulphone (7g) derivatives were potent, single-digit nanomolar FAAH inhibitors in vitro and were effective at inhibiting liver FAAH activity in vivo (1mg/kg, i.p.), while having significantly reduced brain penetration (Table 1). In agreement with the results obtained with the primary and secondary carbamoyl derivatives 3 and 7d, we found that 7f displayed a more strongly restricted access to the CNS compared to 7g, with brain ED50 values of 75 and 3 mg/kg, respectively (Figure 2b). However, pharmacological blockade of Abcg2 with Ko-143 did not increase the access of a sub-effective dose of 7f (40 mg/kg) or 7g (1mg/kg) to the brain (Figure 2c), indicating that these compounds are excluded from the CNS by a mechanism that is independent of Abcg2.
Analogues of 3 with different substituents on the meta- or para- position of the proximal phenyl ring
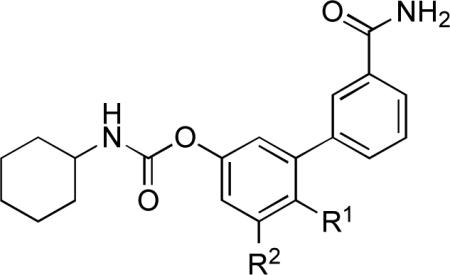
Next, we turned our attention to the SAR exploration of the R2 region of compound 3. The results are summarized in Table 2. We hypothesized that the hydroxyl group in the para position of the proximal phenyl ring, which differentiates 3 from the globally active inhibitor 1 (Figure 1), might be a key element in the peripheral distribution of 3. Supporting this idea, we previously showed that the p-methoxy derivative 15 readily accesses the brain following systemic administration.13 In agreement with this finding, hydroxyl-containing substituents such as the hydroxymethyl 21 and the carboxylic acid 29 were also peripherally restricted when administered at the dosage of 1 mg/kg (Table 2). Compound 21 behaved similarly to 3 and 7d, in that it inhibited brain FAAH when given at high doses (ED50 = 15 mg/kg, Figure 3a) and gained access to the brain when co-administered with the Abcg2 inhibitor Ko-143 (Figure 3b). By contrast, 29 failed to enter the brain even at the highest dose tested (75 mg/kg, Figure 3a). A similar behavior was displayed by the sulfate derivative 30 (Table 2, Figure 3a). Neither 29 nor 30 entered the brain, despite pre-treatment with Ko-143 (Figure 3b), indicating that Abcg2 is not involved in restricting their access to the CNS. It is likely that the presence of absolute charges on the surface of 29 and 30 hinders their diffusion across the BBB.
Table 2.
Inhibitory Potency (IC50) and Systemic Distribution of 5-(or 6-)Substituted 3’-carbamoyl-O-biphenyl-3-yl Carbamates.
| ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | In vitro IC50 (nM)a | FAAH inhibition in liver (%)b | FAAH inhibition in brain (%)b | PSA (Å2)d | |
| 3 | OH | H | 2.0 | 91.7±0.7 | −3.0±8.0 | 83 |
| 15 | OCH3 | H | 0.5 | 94.6±0.7 | 86.4±2.1 | 74 |
| 21 | CH2OH | H | 1.2 | 91.5±1.1 | 10.5±1.5 | 83 |
| 29 | COOH | H | 2100 | 86.3±1.3 | −2.1±0.5 | 94 |
| 30 c | OSO3NH4 | H | 34 | 84.0±1.2 | −11.7±2.6 | 117 |
| 35 | H | OH | 0.5 | 89.5±1.1 | −4.2±2.5 | 84 |
| 41 | H | OCH3 | 2.0 | 85.6±2.4 | 82.6±0.4 | 74 |
IC50 measured in membrane preparations of Wistar rat brain
FAAH inhibition measured ex vivo 1 h after injection in Swiss Webster mice (1 mg/kg, intraperitoneal, n = 3)
30 was obtained from 3 upon treatment with SO3-DMF complex in dry DCM (see experimental section for further details)
PSA values were calculated using ICM version 3.7 (Molsoft LLC, San Diego, CA).
Figure 3.

Inhibition of brain FAAH activity by analogues of compound 3 bearing different substituents on the meta- or para- position of the proximal phenyl ring; a) Dose-dependent inhibition of brain FAAH activity by p-hydroxymethyl (21), p-carboxyl (29), p-sulfate (30) and m-hydroxy (35) derivatives of compound 3 in Swiss Webster mice; doses were 0.3–75 mg/kg (s.c.); b) Effects of pharmacological blockade of the Abcg2 transporter (Ko-143, 15 mg/kg, i.p., closed bars) on brain inhibition of FAAH activity caused by a sub-effective dose (selected from the dose-response study: 3 (25); 21 (10); 29 (40); 30 (75); 35 (40) in mg/kg, s.c., open bars) of analogues of compound 3 bearing different functionalities on the meta- position of the distal phenyl ring. Results are expressed as mean ± s.e.m. (n = 3-4). *** P< 0.001, ** P< 0.01 vs non-Ko-143 treated group.
Lastly, we asked whether relocation of the hydroxyl group of 3 from the para- to the meta-position on the proximal phenyl ring affects FAAH activity and brain penetration. The mhydroxy derivative 35 retained both a strong inhibitory potency toward FAAH in vitro (IC50 = 0.5 mg/kg) and a marked peripheral distribution (Table 2). A dose exploration study revealed that 35 had a markedly restricted access to the brain (ED50 = 75 mg/kg, Figure 3a). For comparison, compound 3 inhibited brain FAAH with an ED50 of 40 mg/kg. The inability of 35 to access the brain appears to require Abcg2, since pharmacological blockade of this transporter allowed a high dose of 35 (40 mg/kg) to inhibit central FAAH activity (Figure 3b). Similarly to what found for 15, the asymmetric tissue distribution of 35 was lost in the corresponding mmethoxy derivative 41, which inhibited FAAH in liver and brain to a similar extent after systemic administration (1 mg/kg, Table 2).
CONCLUSIONS
Previous studies have identified compound 3 as a potent and selective FAAH inhibitor, whose passage through the blood-tissue barriers of the CNS and feto-placental unit is restricted at by the Abcg2 transporter.14,15 Despite this restricted systemic distribution, 3 exhibits marked anti-hyperalgesic and anti-allodynic properties in mice and rats,13 suggesting that peripherally restricted FAAH inhibitors might represent a novel class of clinically relevant analgesics. 21 The primary objective of the present study was to identify new brain-impermeant FAAH inhibitors, which could be used both as tools to understand the role of anandamide in peripheral tissues and as potential candidates for preclinical development. The identification of brain-impermeant drugs is often based on the use of cellular efflux transport assays. However, these in vitro systems do not realistically capture the complexity of the BBB. For this reason, we opted for testing the newly synthesized compounds in vivo, using inhibition of FAAH activity in brain and liver tissues as a measure of central and peripheral exposure to the drug. The contribution of Abcg2 was examined using Ko-143, a selective inhibitor of this transporter. Our SAR exploration of two pharmacophoric regions in the scaffold of 3 allowed us to identify several novel FAAH inhibitors with restricted access to the brain. Among them, the m-hydroxyl derivative 35 showed the greatest inhibitory potency in vitro (IC50 = 0.5 nM) and the lowest brain penetration in vivo (ED50 = 75 mg/kg). This compound thus provides a valuable addition to our still limited armamentarium of peripherally restricted FAAH inhibitors.
The present study also offered several insights on the molecular mechanism responsible for the peripheral distribution of O-biphenyl-3-yl carbamate FAAH inhibitors. By showing that progressive alkylation of the carboxamide group in the distal phenyl ring of 3 (R1 region) leads to a higher degree of brain penetration, our results confirm a key role for this moiety in determining the peripheral distribution of 3. Contrary to what was found for the primary and secondary amides (3 and 7d), the restricted access to the brain of the tertiary amide 7e was not mediated by Abcg2. A plausible interpretation of this finding is that the carboxamide hydrogen bond donors may be necessary for 3 and 7d to interact with Abcg2, possibly through H-bonding.
We were able to confirm the key role played by the hydroxyl substituent of the proximal ring of 3 in the peripheral distribution of O-biphenyl-3-yl carbamate FAAH inhibitors. Some flexibility is allowed around this motif, however, since both the p- and m-hydroxy (3 and 35), as well as the p-hydroxymethyl derivative 21 behaved as substrates for Abcg2 in vivo, gaining access to the brain after pharmacological blockade of the transporter by Ko-143. By contrast, compounds 29 and 30 did not enter the brain, even at the highest dosage tested, and their distribution was not influenced by Ko-143 treatment. The case of the sulfate derivative 30 is particularly significant. This compound was initially synthesized to test the putative role of phenolsulfotransferases (PSTs) in the peripheralization of 3. Sulfate conjugation is known to increase the affinity of Abcg2 for its substrates,33 and PSTs to co-localize with the transporter in brain, intestinal epithelium and other tissues.34 However, the transfer of the sulfate group is expected to occur in the cytosol of epithelial cells, where PSTs and the substrate-binding site of Abcg2 are located, once the compound has diffused through the apical side of the membrane. The present findings suggest that the higher PSA of these two compounds might preclude their diffusion through cellular membranes and therefore their access to the CNS.
Even though further experiments are required to fully characterize the mechanisms that preclude O-biphenyl-3-yl carbamates from entering the brain, our results may be relevant to the design of new pharmacological agents with restricted access to the CNS. In the case of the Abcg2/ABCG2 transporter, the structure of which has not been yet resolved, the information gleamed from the present SAR studies may be useful to understand the interaction between the pharmacophoric sites of 35 and amino acid residues of the transporter involved in substrate recognition.
EXPERIMENTAL SECTION
Animals
Adult male Swiss-Webster mice (25-30 g) were kept in a temperature-controlled environment with a 12-h light/12-h dark cycle receiving standard chow and water ad libitum. All procedures met the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Drug administration
FAAH inhibitors were dissolved in warm saline/PEG400/Tween80 (18:1:1) under sonication, and were administered by i.p. or subcutaneous injection between the shoulder blades. Ko-143 (Tocris, Ellisville, MO) was dissolved in the same vehicle containing 30% DMSO (Sigma, St. Louis, MO) and administered by i.p. injection 20 min prior to FAAH inhibitors.
Tissue processing
Mice were slightly anesthetized with isofluorane and killed by decapitation 1 hour after drug injections. Brain and liver were immediately removed and frozen in liquid N2. Samples were weighed and homogenized in 10 volumes of ice-cold Tris–HCl (50 mM, 5–9 vol., pH 7.5) containing 0.32M sucrose. Homogenates were centrifuged at 1000×g for 10 min at 4°C and supernatants were collected and tested for protein concentration using a bicinchoninic acid (BCA) assay kit (Pierce, Rockford, IL).
Ex vivo FAAH activity assay
FAAH activity was measured at 37°C for 30 min in 0.5mL of Tris buffer (50 mM, pH 7.5) containing fatty acid-free bovine serum albumin (BSA) (0.05%, w/v), protein from tissue homogenates (50 μg from rat brain, 10 μg from liver), non-radioactive anandamide (10 μM) and anandamide[ethanolamine-3H] (10,000 cpm, specific activity 60 Ci/mmol, ARC, St. Louis, MO) as substrate. Reactions were stopped with chloroform/methanol (1:1, 1 mL) and radioactivity was measured in the aqueous layer by liquid scintillation counting. For in vitro IC50 determination, homogenates (50 μg from rat brain) were pre-incubated with inhibitors for 20 min at 37°C prior to substrate addition.
Chemicals, materials and methods
Solvents and reagents were obtained from commercial suppliers and were used without further purification. NMR experiments were run on a Bruker AC 200 spectrometer (200.07 MHz for 1H, and 50.31 MHz for 13C) and on a Bruker Avance III 400 system (400.13 MHz for 1H, and 100.62 MHz for 13C), equipped with a BBI probe and Z-gradients. Spectra were acquired at 300 K, using deuterated dimethylsulfoxide (DMSO-d6) or deuterated chloroform (chloroform-d) as solvents. Chemical shifts (d) for 1H and 13C spectra are reported in parts per million (ppm) using the residual non-deuterated solvent resonance as the internal standard (for chloroform-d: 7.26 ppm, 1H and 77.16 ppm, 13C; for DMSO-d6: 2.50 ppm, 1H; 39.52 ppm, 13C). Data are reported as follows: chemical shift (sorted in descending order), multiplicity (indicated as: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet and combinations thereof), coupling constants (J) in Hertz (Hz) and integration. UPLC/MS analyses were run on a Waters ACQUITY UPLC/MS system consisting of a Single Quadropole Detector (SQD) Mass Spectrometer (MS) equipped with an Electrospray Ionization (ESI) interface and a Photodiode Array (PDA) Detector. PDA range was 210-400 nm. ESI in positive and negative mode was applied. Mobile phases: (A) 10mM NH4OAc in H2O, pH 5; (B) 10mM NH4OAc in CH3CN/H2O (95:5) pH 5. Analyses were performed either with method A, B or C. Method A: gradient 5 to 95% B over 3 min; flow rate 0.5 mL/min; temperature 40 °C. Pre column: Vanguard BEH C18 (1.7µm 2.1x5mm). Column: BEH C18 (1.7μm 2.1×50mm). Method B: gradient 0 to 50% B over 3 min; flow rate 0.5 mL/min; temperature 40 °C. Pre column: VanGuard HSS T3 C18 (1.7μm 2.1×5 mm). Column HSS T3 (1.8μm 2.1 × 50mm). Method C: gradient: 50 to 100% B over 3 min, flow rate 0.5 mL/min; temperature 40 °C. Pre column: Vanguard BEH C18 (1.7μm 2.1×5mm). Column: BEH C18 (1.7μm 2.1×50mm). Flash column chromatography was performed automatically on Teledyne ISCO apparatus (CombiFlash® Rf) with pre-packed silica gel columns of different sizes (Redisep) or manually on silica gel (Kieselgel60, 0.040-0.063 mm, Merck). TLC analyses were performed on precoated silica gel on aluminum sheets (Kieselgel 60 F254, Merck). Purifications by preparative HPLC/MS were run on a Waters Autopurification system consisting of a 3100 Single Quadropole Detector (SQD) Mass Spectrometer (MS) equipped with an Electrospray Ionization (ESI) interface and a 2998 Photodiode Array (PDA) Detector. HPLC system included a 2747 Sample Manager, 2545 Binary Gradient Module, System Fluidic Organizer and 515 HPLC Pump. PDA range was 210-400 nm. Purifications were performed on a XBridgeTM Prep C18 OBD column (100x19mmID, particle size 5μm) with a XBridgeTM Prep C18 (10×19 mmID, particle size 5 μm) Guard Cartridge. Mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in CH3CN -H2O (95:5) at pH 5 (B). ESI in positive and negative mode was used. All final compounds displayed ≥ 95% purity as determined by UPLC analysis.
General Procedure for the Synthesis of Carbamates 5d–g and 7a–c (Procedure A)
A mixture of compound 4 (or 6) (1.0 equiv.), the appropriate aryl boronic acid (or aryl boronic ester) (1.5 equiv.), CsOAc (2.0 equiv.) in dioxane (0.1 M) was degassed with a stream of N2 for 30 min. PdCl2dppf (0.05 equiv.) was added and the reaction mixture was heated at 80 °C until UPLC-MS analysis revealed completion of the reaction. The reaction mixture was cooled down to room temperature and a saturated aqueous NH4Cl solution was added (3 mL). The aqueous phase was separated and extracted with EtOAc (2 × 15 mL). The combined organic phases were washed with brine and dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (SiO2) eluting with a gradient of EtOAc/ cyclohexane or MeOH/ DCM.
General procedure for the Synthesis of Carbamates 7d–g (Procedure B)
Compound 5d (5e, or 5f, or 5g) (1.0 equiv.) was heated in a 1:5 mixture of cyclohexene/EtOH (0.2 M) at 60 °C in presence of 10% Pd/C (catalyst loading: 2.5% w/w) until UPLC-MS analysis revealed completion of the reaction. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo. The residue was purified by flash chromatography (SiO2) eluting with a gradient of cyclohexane/ EtOAc or MeOH/ DCM.
General procedure for the Synthesis of Phenols 9a,b and 14 (Procedure C)
A mixture of 8 (or 13) (1.0 equiv.), the appropriate boronic acid (1.2 equiv.) and Na2CO3 (5 equiv., 10% aqueous solution) in toluene (0.2 M) was degassed with a stream of N2 for 30 min. Pd(PPh3)4 (0.05 equiv.) was added and the reaction mixture was stirred at reflux for 12 h, cooled down to room temperature, and filtered through a pad of Celite. 2N HCl (5 mL) was added and the mixture was extracted with EtOAc. The combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (SiO2) eluting with cyclohexane/ EtOAc or MeOH/ DCM.
General procedure for the Synthesis of Carbamates 10a,b and 15 (Procedure D)
To a solution of 9a (or 9b, or 14) (1.0 equiv.) in CH3CN (0.4 M), c-hexyl-isocyanate (1.1 equiv.) and Et3N (1.1 equiv.) were added. The mixture was stirred for 5 h at reflux, cooled down to room temperature and concentrated in vacuo. The residue was purified by flash chromatography (SiO2) eluting with cyclohexane /EtOAc.
General procedure for the Synthesis of Carbamates 11a–c (Procedure E)
Compound 10a (or 10b) (1.0 equiv.) was heated in EtOAc/EtOH (1:1, 0.1 M) (for 10a) or EtOH (0.1 M) (for 10b) at 50 °C under H2 atmosphere (4 atm) in presence of 10% Pd/C (catalyst loading: 10% w/w) for 4h. The mixture was then cooled down to room temperature and filtered through a pad of Celite. After evaporation of the solvent, the residue was purified by flash chromatography (SiO2) eluting with cyclohexane/EtOAc.
Cyclohexylcarbamic Acid 3’-Acetamido-6-benzyloxybiphenyl-3-yl Ester (5d)
The title compound 5d was prepared according to general procedure A using compound 4 (0.202 g, 0.50 mmol), PdCl2dppf (18.3 mg, 0.025 mmol), CsOAc (192 mg, 1.00 mmol) and N-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzamide (195 mg, 0.75 mmol); reaction time: 2 h. The residue was purified by flash chromatography (0 to 50% EtOAc in cyclohexane) to afford 5d as a white solid. 43 mg, 19%. 1H NMR (400 MHz, chloroform-d) δ 7.85 (s, 1H), 7.77–7.72 (m, 1H), 7.65 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.34–7.26 (m, 5H), 7.12 (d, J = 2.7 Hz, 1H), 7.05 (dd, J = 2.7, 8.8 Hz, 1H), 6.98 (d, J = 8.8 Hz, 1H), 6.19 (d, J = 4.8 Hz, 1H), 5.04– 4.99 (m, 1H), 4.99 (s, 2H), 3.59–3.51 (m, 1H), 2.90 (d, J = 4.8 Hz, 3H), 2.03–1.96 (m, 2H), 1.77–1.69 (m, 2H), 1.65–1.58 (m, 1H), 1.41–1.30 (m, 2H), 1.28–1.14 (m, 3H). MS (ES) C28H30N2O4 requires m/z 458, found 459 [M+H]+.
Cyclohexylcarbamic Acid 6-Benzyloxy-3’-dimethylcarbamoylbiphenyl-3-yl Ester (5e)
The title compound 5e was prepared according to general procedure A using compound 4 (0.404 g, 1.0 mmol), PdCl2dppf (36.6 mg, 0.05 mmol), CsOAc (0.384 mg, 2.0 mmol) and N,N-dimethyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzamide (0.413 g, 1.5 mmol); reaction time: 6 h. The residue was purified by flash chromatography (0 to 50% EtOAc in cyclohexane) to afford 5e as a colourless solid. 229 mg, 48%. 1H NMR (400 MHz, chloroform-d) δ 7.65–7.55 (m, 2H), 7.47–7.35 (m, 2H), 7.35–7.27 (m, 5H), 7.17–7.10 (m, 1H), 7.07 (d, J = 8.81 Hz, 1H), 7.00 (d, J = 8.82 Hz, 1H), 5.03 (s, 2H), 4.94–4.85 (m, 1H), 3.64–3.51 (m, 1H), 2.95 (s, 6H), 2.09–1.95 (m, 2H), 1.80–1.67 (m, 2H), 1.67–1.56 (m, 1H), 1.46–1.30 (m, 2H), 1.30–1.12 (m, 3H). MS (ES) C29H32N2O4 requires m/z 472, found 473 [M+H]+.
Cyclohexylcarbamic Acid 6-Benzyloxy-3’-sulfamoylbiphenyl-3-yl Ester (5f)
The title compound 5f was prepared according to general procedure A using compound 4 (162 mg, 0.4 mmol), PdCl2dppf (14.6 mg, 0.02 mmol), CsOAc (154 mg, 0.80 mmol) and (3-sulfamoylphenyl)boronic acid (201 mg, 0.60 mmol); reaction time: 8 h. The residue was purified by flash chromatography (0 to 30% EtOAc in cyclohexane) to afford 5f as a white solid. 105 mg, 55%. 1H NMR (400 MHz, chloroform-d) δ 8.17 (s, 1H), 7.93 (d, J = 7.4 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.75 (dd, J = 7.4, 7.9 Hz, 1H), 7.37–7.28 (m, 5H), 7.16 (d, J = 2.7 Hz, 1H), 7.11 (dd, J = 2.7, 8.8 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H), 5.04 (s, 2H), 4.93 (d, J = 7.7 Hz, 1H), 4.61 (s, 2H), 3.61–3.52 (m, 1H), 2.05–1.98 (m, 2H), 1.78–1.70 (m, 2H), 1.66–1.60 (m, 1H), 1.41–1.31 (m, 2H), 1.31–1.15 (m, 3H). MS (ESI): C26H28N2O5S requires m/z 480, found 481 [M+H]+.
Cyclohexylcarbamic Acid 6-Benzyloxy-3’-methylsulfonylbiphenyl-3-yl Ester (5g)
The title compound 5g was prepared according to general procedure A using compound 4 (162 mg, 0.4 mmol), PdCl2dppf (14.6 mg, 0.02 mmol), CsOAc (154 mg, 0.80 mmol) and (3-methylsulfonylphenyl)boronic acid (200 mg, 0.60 mmol); reaction time: 4 h. The residue was purified by flash chromatography (0 to 30% EtOAc in cyclohexane) to afford 5g as a colourless solid. 148 mg, 77%. 1H NMR (400 MHz, chloroform-d) δ 8.19 (s, 1H), 7.86 (d, J = 7.9 Hz, 1H), 7.82 (d, J = 7.7 Hz, 1H), 7.56 (dd, J = 7.7, 7.9 Hz, 1H), 7.35–7.27 (m, 5H), 7.16 (d, J = 2.7 Hz, 1H), 7.12 (dd, J = 2.7, 8.8 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 5.05 (s, 2H), 4.95 (d, J = 7.86 Hz, 1H), 3.61–3.52 (m, 1H), 2.89 (s, 3H), 2.06–1.98 (m, 2H), 1.78–1.70 (m, 2H), 1.66–1.60 (m, 1H), 1.44–1.32 (m, 2H), 1.23 (m, 3H). MS (ESI) C27H29NO5S requires m/z 479, found 480 [M+H]+.
Cyclohexylcarbamic Acid 3-Bromo-4-hydroxyphenyl Ester (6)
To a solution of 4 (4.04 g, 10.0 mmol) in dry DCM (50 mL) at –78 °C, BBr3 (20.0 mL, 1.0 M solution in DCM) was slowly added under Ar atmosphere and the reaction mixture stirred at -78 °C for 2 h and then quenched with saturated aqueous NH4Cl solution. The aqueous layer was extracted with DCM (3 x 50 mL) and the combined organic phases were washed with brine and dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (0 to 20% EtOAc in cyclohexane) to afford 6 as a white solid. 3.06 g, 97%. 1H NMR (400 MHz, chloroform-d) δ 7.27 (d, J = 2.5 Hz, 1H), 6.97 (dd, J = 2.5, 8.8 Hz, 1H), 6.93 (d, J = 8.8 Hz, 1H), 5.59 (s, 1H), 4.89 (d, J = 6.9 Hz, 1H), 3.59–3.50 (m, 1H), 2.04–1.96 (m, 2H), 1.78–1.70 (m, 2H), 1.66–1.59 (m, 1H), 1.43–1.31 (m, 2H), 1.27–1.14 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 154.1, 151.8, 143.9, 126.4, 122.5, 116.4, 108.8, 50.2, 33.0, 25.6, 25.0. MS (ESI) C13H16BrNO3 requires m/z 313, 315, found 314, 316 [M+H]+.
Cyclohexylcarbamic Acid 3’-Acetyl-6-hydroxybiphenyl-3-yl Ester (7a)
The title compound 7a was prepared according to general procedure A using compound 6 (0.157 g, 0.5 mmol), PdCl2dppf (18.3 mg, 0.025 mmol), CsOAc (192 mg, 1.00 mmol) and 3-methoxyphenylboronic acid (114 mg, 0.75 mmol); reaction time: 5 h. The crude was purified by flash chromatography (0 to 40% EtOAc in cyclohexane) to afford 7a as a colourless solid. 109 mg, 62%. 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.11 (s, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.62–7.52 (m, 2H), 7.04 (s, 1H), 6.96–6.91 (m, 2H), 3.35–3.28 (m, 1H), 2.62 (s, 3H), 1.86–1.79 (m, 2H), 1.74–1.66 (m, 2H), 1.60–1.52 (m, 1H), 1.33–1.19 (m, 4H), 1.15–1.07 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 198.4, 154.4, 151.8, 144.1, 138.6, 137.2, 134.2, 129.1, 128.9, 127.4, 127.1, 123.6, 122.6, 116.8, 50.2, 33.0, 27.3, 25.6, 25.0. MS (ES) C21H23NO4 requires m/z 353, found 354 [M+H]+.
Cyclohexylcarbamic Acid 3’-Carboxy-6-hydroxybiphenyl-3-yl Ester (7b)
The title compound 7b was prepared according to general procedure A using compound 6 (0.157 g, 0.5 mmol), PdCl2dppf (18.3 mg, 0.025 mmol), CsOAc (192 mg, 1.00 mmol) and 3-phenylboronic acid (124 mg, 0.75 mmol); reaction time: 12 h. The crude was purified by flash chromatography (0 to 10% MeOH in DCM) to afford 7b as a yellow solid. 28 mg, 16%. 1H NMR (400 MHz, DMSO-d6) δ 12.90 (s, 1H), 9.62 (s, 1H), 8.12 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.53 (t, J = 7.7 Hz, 1H), 6.99 (s, 1H), 6.92 (s, 2H), 3.34–3.30 (m, 1H), 1.83–1.79 (m, 2H), 1.72–1.67 (m, 2H), 1.57–1.53 (m, 1H), 1.30–1.18 (m, 4H), 1.16– 1.05 (m, 1H). MS (ESI) C20H21NO5 requires m/z 355, found 356 [M+H]+.
Cyclohexylcarbamic Acid 3’-Acetamido-6-hydroxybiphenyl-3-yl Ester (7c)
The title compound 7c was prepared according to general procedure A using compound 6 (157 mg, 0.50 mmol), PdCl2dppf (18.3 mg, 0.025 mmol), CsOAc (192 mg, 1.00 mmol) and (3-acetamidophenyl)boronic acid (179 mg, 0.75 mmol); reaction time: 6 h. The crude was purified by flash chromatography (0 to 50% EtOAc in cyclohexane) to afford 7c as an off-white solid. 110 mg, 60%. 1H NMR (400 MHz, DMSO-d6) δ 9.94 (s, 1H), 9.47 (s, 1H), 7.71 (s, 1H), 7.57 (d, J = 7.9 Hz, 2H), 7.30 (t, J = 7.9 Hz, 1H), 7.20 (d, J = 7.8 Hz, 1H), 6.89 (s, 2H), 3.29 (s, 1H), 2.04 (s, 3H), 1.81 (d, J = 9.5 Hz, 2H), 1.75–1.65 (m, 3H), 1.55 (d, J = 12.5 Hz, 1H), 1.33–1.17 (m, 4H), 1.18–1.04 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.7, 154.4, 151.7, 144.0, 139.5, 138.7, 128.7, 128.2, 124.3, 123.5, 122.0, 120.2, 118.0, 116.7, 50.2, 33.0, 25.6, 25.0, 24.5. MS (ES) C21H24N2O4 requires m/z 368, found 369 [M+H]+.
Cyclohexylcarbamic Acid 6-Hydroxy-3’-methylcarbamoylbiphenyl-3-yl Ester (7d)
The title compound 7d was prepared according to general procedure B using compound 5d (41.3 mg, 0.09 mmol) and 10% Pd/C (16.5 mg); reaction time: 2 h. The residue was purified by flash chromatography (0 to 2% MeOH in DCM) to afford 7d as a white solid. 23 mg, 56%. 1H NMR (400 MHz, chloroform-d) δ 7.65 (d, J = 7.7 Hz, 1H), 7.58 (s, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.35 (dd, J = 7.7, 7.7 Hz, 1H), 6.90–6.83 (m, 3H), 6.73 (d, J = 8.7 Hz, 1H), 6.67 (d, J = 4.8 Hz, 1H), 5.08 (bd, J = 8.1 Hz, 1H), 3.56–3.51 (m, 1H), 2.90 (d, J = 4.8 Hz, 3H), 2.01–1.96 (m, 2H), 1.76– 1.70 (m, 2H), 1.65–1.59 (m, 1H), 1.41–1.30 (m, 2H), 1.27–1.16 (m, 3H). 13C NMR (101 MHz, chloroform-d) δ 171.3, 168.7, 155.0, 150.9, 144.2, 137.7, 134.8, 132.2, 128.8, 127.4, 126.4, 123.5, 122.2, 117.2, 60.5, 50.4, 33.3, 25.6, 24.9. MS (ES) C21H24N2O4 requires m/z 368, found 369 [M+H]+.
Cyclohexylcarbamic Acid 3’-dimethylcarbamoyl-6-hydroxybiphenyl-3-yl Ester (7e)
To a solution of 5e (227 mg, 0.48 mmol) in dry DCM (5 mL) at -78 °C, BBr3 (0.96 mL, 1.0 M solution in DCM) was slowly added under Ar atmosphere. The reaction was warmed to room temperature, stirred for 1 h and then quenched with saturated aqueous NH4Cl solution. The aqueous solution was extracted with EtOAc (3 × 20 mL) and the organic phase dried (Na2SO4). After evaporation of solvent, the residue was purified by flash chromatography (0 to 4% MeOH in DCM) to afford 5e as a colourless solid. 133 mg, 72%. 1H NMR (400 MHz, DMSO-d6) δ 9.59 (s, 1H), 7.64–7.54 (m, 3H), 7.50–7.41 (m, 1H), 7.33 (d, J = 7.5 Hz, 1H), 7.00 (s, 1H), 6.97–6.89 (m, 2H), 3.33–3.28 (m, 1H), 3.01–2.93 (m, 6H), 1.85–1.78 (m, 2H), 1.71–1.66 (m, 2H), 1.59– 1.51 (m, 1H), 1.33–1.17 (m, 4H), 1.16–1.04 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 154.4, 151.8, 144.1, 138.2, 136.6, 130.3, 128.5, 127.9, 127.5, 125.9, 123.6, 122.4, 116.8, 55.3, 50.2, 33.0, 25.6, 25.0. MS (ES) C22H26N2O4 requires m/z 382, found 383 [M+H]+.
Cyclohexylcarbamic Acid 6-Hydroxy-3’-sulfamoylbiphenyl-3-yl Ester (7f)
The title compound 7f was prepared according to general procedure B using compound 5f (106 mg, 0.22 mmol), and 10% Pd/C (42.4 mg); reaction time: 2 h. The residue was purified by flash chromatography (0 to 50% EtOAc in cyclohexane) to afford 7f as a white solid. 75 mg, 87%. 1H NMR (400 MHz, DMSO-d6) δ 9.72–9.68 (m, 1H), 8.01 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.77–7.73 (m, 1H), 7.62–7.56 (m, 2H), 7.35 (s, 2H), 7.01 (s, 1H), 6.96–6.93 (m, 2H), 3.39–3.26 (m, 1H), 1.85–1.77 (m, 2H), 1.74–1.66 (m, 2H), 1.59–1.52 (m, 1H), 1.32–1.19 (m, 4H), 1.18–1.08 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 154.4, 151.8, 144.4, 144.1, 138.9, 132.7, 129.07, 126.8, 126.5, 124.3, 123.6, 122.9, 116.9, 50.2, 33.0, 25.6, 25.0. MS (ESI) C19H22N2O5S requires m/z 390, found 391 [M+H]+.
Cyclohexylcarbamic Acid 6-Hydroxy-3’-methylsulfonylbiphenyl-3-yl Ester (7g)
The title compound 7g was prepared according to general procedure B using compound 5g (149 mg, 0.31 mmol), and 10% Pd/C (59.6 mg); reaction time: 2 h. The residue was purified by flash chromatography (0 to 50% EtOAc in cyclohexane) to afford 7g as a colourless solid. 109 mg, 90%. 1H NMR (400 MHz, DMSO-d6) δ 9.75 (s, 1H), 8.09–8.07 (m, 1H), 7.94–7.90 (m, 1H), 7.88–7.84 (m, 1H), 7.71–7.65 (m, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.11–7.08 (m, 1H), 6.98–6.92 (m, 2H), 3.34–3.27 (m, 1H), 3.25 (s, 3H), 1.85–1.77 (m, 2H), 1.74–1.66 (m, 2H), 1.59–1.52 (m, 1H), 1.32–1.16 (m, 4H), 1.16–1.06 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 153.9, 151.3, 143.7, 140.7, 138.8, 134.0, 129.1, 127.2, 125.9, 125.2, 123.2, 122.7, 116.4, 49.7, 43.6, 32.6, 25.1, 24.6. MS (ESI) C20H23NO5S requires m/z 389, found 390 [M+H]+.
3-(2-Benzyloxy-5-hydroxyphenyl)benzaldehyde (9a)
The title compound 9a was prepared according to general procedure C using compound 8 (278 mg, 1.0 mmol), 3-formylphenylboronic acid (0.174 g, 1.2 mmol), Na2CO3 (0.53 g, 5 mmol), Pd(PPh3)4 (0.058 g, 0.05 mmol). The crude was purified by flash chromatography (cyclohexane/EtOAc 85:15) to afford 9a as amber oil. 0.194 g, 64%. 1H NMR (200 MHz, DMSO-d6) δ 10.04 (s, 1H), 9.14 (s, 1H), 8.05 (t, J = 1.5 Hz, 1H), 7.88–7.81 (m, 2H), 7.62 (t, J = 7.6 Hz 1H), 7.33–7.22 (m, 5H), 7.06 (d, J = 9.0 Hz 1H), 6.81–6.74 (m, 2H), 5.00 (s, 2H). MS (ESI) C20H16O3 requires m/z 304, found 303 [M-H]-.
1-[3-(2-Benzyloxy-5-hydroxyphenyl)phenyl]ethanone (9b)
The title compound 9b was prepared according to general procedure C using compound 8 (0.278 g, 1.0 mmol), 3-acetylphenylboronic acid (0.196 g, 1.2 mmol), Na2CO3 (0.53 g, 5 mmol), Pd(PPh3)4 (0.058 g, 0.05 mmol). The crude was purified by flash chromatography (cyclohexane/EtOAc 75:25 and then DCM/MeOH 99:1) to afford 9b as white solid after crystallization from EtOH. 0.248 g, 78%. 1H NMR (200 MHz, DMSO-d6) δ 9.12 (br, 1H), 8.09 (t, J = 1.6 Hz, 1H), 7.89 (dt, J = 7.7 Hz, J = 1.4 Hz ,1H), 7.75 (dt, 7.7 Hz, J = 1.4 Hz, 1H), 7.54 (t, J = 7.7 Hz, J = 1.4 Hz, 1H), 7.33– 7.28 (m, 5H), 7.06 (d, 8.8 Hz, 1H), 6.80– 6.73 (m, 2H), 4.99 (s, 2H), 2.55 (s, 3H). MS (ESI) C21H18O3 requires m/z 318, found 319 [M+H]+, 317 [M–H]-.
Cyclohexylcarbamic Acid 6-Benzyloxy-3’-formylbiphenyl-3-yl Ester (10a)
The title compound 10a was prepared according to general procedure D using compound 9a (0.304 g, 1.0 mmol), c-C6H11NCO (0.137 mg, 1.1 mmol), Et3N (0.11 g, 0.154 mL, 1.1 mmol). The crude was purified by flash chromatography (cyclohexane/EtOAc 75:25) to afford 10a as amber oil. 0.236 g, 55%. 1H NMR (200 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.10 (t, J = 1.5 Hz 1H), 7.91–7.85 (m, 2H), 7.67–7.59 (m, 2H), 7.39–7.08 (m, 8H), 5.14 (s, 2H), 3.37–3.33 (m, 1H), 1.83–1.54 (m, 5H), 1.24–1.19 (m, 5H). MS (ESI) C27H27NO4 requires m/z 429, found 430 [M+H]+.
Cyclohexylcarbamic Acid 3’-Acetyl-6-benzyloxybiphenyl-3-yl Ester (10b)
The title compound 10b was prepared according to general procedure D using compound 9b (0.318 g, 1.0 mmol), c-C6H11NCO (0.137 mg, 1.1 mmol), Et3N (0.11 g, 0.154 mL, 1.1 mmol). The crude was purified by flash chromatography (cyclohexane/EtOAc 70:30) to afford 10b as white solid, after crystallization from EtOH. 0.270 g, 61%. 1H NMR (200 MHz, DMSO-d6) δ 8.12 (t, J = 1.5 Hz, 1H), 7.90 (dt, J = 8.0 Hz, J = 1.5 Hz, 1H), 7.79 (dt, J = 8.0 Hz, J = 1.5 Hz 1H), 7.64–7.52 (m, 2H), 7.40–7.20 (m, 6H), 7.12–7.07 (m, 2H), 5.13 (s, 2H), 3.38–3.31 (m, 1H), 2.55 (s, 3H), 1.83– 1.53 (m, 5H), 1.23–1.06 (m, 5H). MS (ESI) C28H29NO4 requires m/z 443, found 444 [M+H]+.
Cyclohexylcarbamic Acid 6-Hydroxy-3’-methylbiphenyl-3-yl Ester (11a)
The title compound 11a was prepared according to general procedure E using compound 10a (0.429 mg, 1 mmol) and 10% Pd/C (0.044 g). The crude was purified by flash chromatography (cyclohexane/EtOAc 40:60) to afford 11a as a white solid, after crystallization from Et2O. 0.061 g, 19%. 1H NMR (200 MHz, chloroform-d) δ 7.39–7.18 (m, 4H), 7.01–6.84 (m, 3H), 5.60 (s, 1H), 4.95 (d, J = 8.0 Hz, 1H), 3.68–3.48 (m, 1H), 2.40 (s, 3H), 2.04–1.99 (d, J = 10.4 Hz, 2H), 1.78–1.61 (m, 3H), 1.48–1.11 (m, 5H). 13C NMR (50 MHz, chloroform-d) δ 154.2, 149.9, 144.3, 139.0, 136.47, 129.7, 129.1, 128.7, 128.6, 126.0, 123.0, 122.0, 116.3, 50.1, 33.3, 25.4, 24.7, 21.5. MS (ESI) C20H23NO3 requires m/z 325, found 326 [M+H]+.
Cyclohexylcarbamic Acid 6-Hydroxy-3’-(1-hydroxyethyl)biphenyl-3-yl Ester (11b)
The title compound 11b was prepared according to general procedure E using compound 10b (0.443 g, 1.0 mmol) and 10% Pd/C (0.044 g). The crude was purified by flash chromatography (cyclohexane/EtOAc 40:60) to afford 11b as a white amorphous solid. 0.176 g, 50%. 1H NMR (200 MHz, chloroform-d) δ 7.37–7.27 (m, 4H), 6.96–6.61 (m, 3H), 5.11 (d, J = 8.0 Hz, 1H), 4.81–4.71 (m, 1H), 3.55–3.51 (m, 1H), 2.82 (br, 1H), 2.05–1.96 (m, 2H), 1.69–1.59 (m, 3H), 1.42 (d, J = 6.4 Hz, 3H), 1.33–1.15 (m, 6H). 13C NMR (50 MHz, chloroform-d) δ 181.4, 154.7, 150.4, 146.1, 144.0, 137.2, 128.7, 128.1, 126.3, 124.6, 123.2, 121.8, 116.8, 70.2, 50.2, 33.2, 25.4, 25.0, 24.7. MS (ESI) C21H25NO4 requires m/z 355, found 354 [M–H]-, 356 [M+H]+.
Cyclohexylcarbamic Acid 6-Hydroxy-3’-hydroxymethylbiphenyl-3-yl Ester (11c)
The title compound 11c was prepared according to general procedure E using compound 10a (0.429 g, 1 mmol) and 10% Pd/C (0.044 g). The crude was purified by flash chromatography (cyclohexane/EtOAc 40:60) to afford 11c as a white amorphous solid. 0.112 g, 30%. 1H NMR (200 MHz, chloroform-d) δ 7.44–7.34 (m, 4H), 7.00–6.83 (m, 3H), 5.60 (s, 1H), 4.93 (d, J = 8.2 Hz,1H), 4.69 (s, 2H), 3.57–3.43 (m, 1H), 2.05–1.97 (m, 3H), 1.77–1.70 (m, 3H), 1.42–1.06 (m, 4H). 13C NMR (50 MHz, chloroform-d) δ 154.5, 150.2, 144.1, 141.5, 137.1, 129.0, 128.6, 128.2, 127.7, 126.3, 123.2, 121.9, 116.7, 66.0, 50.2, 33.2, 25.4, 24.7. MS (ESI) C20H23NO4 requires m/z 341, found 342 [M+H]+.
3-Bromo-4-methoxyphenol (13)
To a solution of 12 (214 mg, 1 mmol) in DCM (5 mL), m-CPBA (0.173 g, 1 mmol) was added. The mixture was stirred for 72 h at 40 °C, and then washed with a saturated aqueous Na2S2O3 solution (5 mL) and with a saturated aqueous NaHCO3 solution (5 mL). The combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was dissolved in EtOH (5 mL), NaOCH3 (0.108 g, 2 mmol) was added and the mixture was stirred for 1 h at room temperature, concentrated in vacuo and acidified with 2 N HCl (5 mL), and extracted with DCM (5 × 3 mL). The combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (cyclohexane/DCM = 20:80) to afford 13. 0.121 g, 60%. MS and 1H NMR are according to the literature. 35
3-(5-Hydroxy-2-methoxyphenyl)benzamide (14)
The title compound 14 was prepared according to general procedure C using compound 13 (0.202 g, 1 mmol), 3’-carbamoylphenylboronic acid (0.198 g, 1.2 mmol), Na2CO3 (0.52 g, 5 mmol), and Pd(PPh3)4 (0.020 mg). The crude was purified by flash chromatography (DCM/MeOH = 94:6) to afford 14 as a white solid. 0.197 g, 81%. 1H NMR (200 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.03 (s, 1H), 7.93–7.91 (m, 1H), 7.82–7.78 (m, 1H), 7.61–7.57 (m, 1H), 7.49–7.45 (m, 1H), 7.41–7.39 (m, 1 H), 6.96–9.91 (m, 1H), 6.77–6.71 (m, 2H), 3.65 (s, 3H). MS (ESI) C14H13NO3 requires m/z 243, found 242 [M–H]-.
Cyclohexylcarbamic Acid 3’-Carbamoyl-6-methoxybiphenyl-3-yl Ester (15)
The title compound 15 was prepared according to general procedure D using compound 14 (0.243 g, 1.0 mmol), c-C6H11NCO (0.137 mg, 1.1 mmol), Et3N (0.11 g, 0.154 mL, 1.1 mmol). The crude was purified by flash chromatography (cyclohexane/EtOAc 30:70) to afford 15 as a white solid. 0.284 g, 77%. 1H NMR (200 MHz, chloroform-d) δ 7.93–7.91 (m, 1H), 7.82–7.77 (m, 1H), 7.72–7.68 (m, 1H), 7.52–7.44 (m, 1H), 7.14–7.07 (m, 2H), 6.97–6.92 (m, 1H), 6.26 –5.73 (m, 2H), 4.98–4.82 (m, 1H), 3.80 (s, 3H), 3.58–3.54 (m, 1H), 1.13–2.04 (m, 10H). 13C NMR (50 MHz, chloroform-d) δ 169.4, 154.1, 153.7, 144.6, 138.2, 133.2, 133.1, 130.1, 128.3, 126.3, 124.0, 121.9, 111.8, 56.0, 50.2, 33.3, 25.4, 24.7. MS (ESI) C21H24N2O4 requires m/z 368, found 369 [M+H]+.
2-(2-Bromo-4-fluorophenyl)-1,3-dioxolane (17)23
To a solution of 16 (4.0 g, 19.7 mmol) in dry toluene (30 mL) ethylene glycol (5.56 mL, 98.5 mmol) and p-TSA (187 mg, 1 mmol) were added and the reaction mixture was heated at reflux for 12h. The mixture was cooled down to room temperature and then poured into saturated aqueous NH4Cl solution (50 mL). The two phases were separated and the organic solution was washed with brine and dried (Na2SO4). Evaporation of the solvent gave 17 as light yellow oil that was used in the next step without further purification. 4.5 g. 1H NMR (400 MHz, chloroform-d) δ 7.62 (dd, J = 8.3, 6.1 Hz, 1H), 7.34 (dd, J = 8.3, 2.6 Hz, 1H), 7.08 (td, J = 8.3, 2.6 Hz, 1H), 6.07 (s, 1H), 4.46–3.85 (m, 4H).
4-Benzyloxy-2-bromobenzaldehyde (18)
To a solution of 17 (4.0 g, 16.19 mmol) in dry dioxane (60 mL), BnOH (6.27 mL, 64.78 mmol) and t-BuOK (7.27 g, 64.78 mmol) were added and then the mixture was heated at 85 °C for 1 h. The mixture was cooled down to room temperature and then poured into H2O (150 mL) and EtOAc (200 mL) and the two phases were separated. To the organic solution 2 N HCl (150 mL) was added and stirring was continued for 2 h at room temperature. The two phases were then separated and the organic layer was dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (0 to 10% EtOAc in cyclohexane) to yield 18 as a white solid. 3.53 g, 75%. 1H NMR (400 MHz, chloroform-d) δ 10.25 (d, J = 0.8 Hz, 1H), 7.92 (d, J = 8.8 Hz, 1H), 7.48–7.34 (m, 5H), 7.25 (d, J = 2.5 Hz, 1H), 7.03 (dd, J = 8.8, 2.5 Hz, 1H), 5.16 (s, 2H). MS (ESI): no ionization.
3-(5-Benzyloxy-2-formylphenyl)benzamide (19)
To a solution of 18 (1.41g, 4.85 mmol) in ethyleneglycol monomethyl ether (EGME) (30 mL), H2O was slowly added (8 mL) followed by the addition of K2CO3 (1.34 g, 9.69 mmol), 3-carbamoylbenzeneboronic acid (1.2 g, 7.27 mmol), and Pd(OAc)2 (10.8 mg, 0.049 mmol). The mixture was stirred at room temperature for 40 min until the mixture became dark and a precipitate was formed. H2O (40 mL) was then added and the solid was filtered and washed with H2O (15 mL) to afford 19 as a whitish solid. 1.53 g, 95%. 1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.08 (bs, 1H), 8.00–7.92 (m, 3H), 7.65–7.55 (m, 2H), 7.55–7.33 (m, 6H), 7.25 (dd, J = 8.6, 2.5 Hz, 1H), 7.15 (d, J = 2.5 Hz, 1H), 5.30 (s, 2H). MS (ESI) C21H17NO3 requires m/z 331, found 332 [M+H]+
3-[5-Benzyloxy-2-hydroxymethylphenyl]benzamide (20)
To a suspension of 19 (1.6 g, 4.83 mmol) in EtOH (20 mL), NaBH4 (365 mg, 9.67 mmol) was added slowly at 0 °C and the mixture was stirred for 2 h. The reaction was diluted with DCM (50 mL) and quenched by the addition of saturated aqueous Na2CO3 solution (20 mL) and H2O (20 mL) to afford a precipitate that was filtered to give 20 as a grayish solid. 1.3 g, 81%. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (bs, 1H), 7.95–7.83 (m, 2H), 7.67–7.27 (m, 9H), 7.06 (dd, J = 8.5, 2.7 Hz, 1H), 6.92 (d, J = 2.7 Hz, 1H), 5.16 (s, 2H), 5.02 (t, J = 5.2 Hz, 1H), 4.32 (d, J = 5.2 Hz, 2H). MS (ESI) C21H19NO3 requires m/z 333, found 316 [M-H2O+H]+
Cyclohexylcarbamic Acid 3’-Carbamoyl-6-hydroxymethylbiphenyl-3-yl Ester (21)
A suspension of 20 (1.3 g, 3.89 mmol) in MeOH (80 mL) under N2 atmosphere was heated at reflux until complete dissolution and then 10% Pd/C (700 mg) were rapidly added followed by the addition of γ-terpinene (6.2 mL, 38.9 mmol). The mixture was heated at reflux for additional 1 h then was cooled down to room temperature, filtered through Celite and washed with MeOH (15 mL). The filtrate was concentrated to dryness affording a colorless solid, which was dissolved in a 1:1 mixture of CH3CN /EtOH (20 mL). To this solution, Et3N (0.32 mL, 2.35 mmol) and c-C6H11NCO (0.5 mL, 3.89 mmol) were added and the mixture was stirred at room temperature for 12h. The reaction was quenched by the addition of EtOAc (80 mL) and 2 N HCl (80 mL) that, after vigorously stirring, were then separated. The aqueous solution was extracted with EtOAc (40 mL) and then the combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (100% EtOAc) and then crystallized from EtOH/H2O to afford 21 as a white solid. 630 mg, 44%. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (bs, 1H), 7.94–7.80 (m, 2H), 7.71 (d, J = 7.9 Hz, 1H), 7.64–7.47 (m, 3H), 7.39 (bs, 1H), 7.13 (dd, J = 8.4, 2.5 Hz, 1H), 6.99 (d, J = 2.5 Hz, 1H), 5.15 (t, J = 5.1 Hz, 1H), 4.38 (d, J = 5.1 Hz, 2H), 3.42–3.22 (m, 1H), 2.03–1.49 (m, 5H), 1.37–0.99 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 168.2, 153.9, 150.3, 140.9, 140.0, 136.3, 134.9, 132.2, 129.7, 128.6, 128.3, 127.0, 122.9, 121.3, 60.8, 50.3, 33.0, 25.6, 25.0. MS (ESI) m/z C21H24N2O4 requires m/z 368, found 386 [M+NH4]+, 737 [2M+H]+.
1-(2-Bromo-4-methoxyphenyl)ethanone (23)
To a suspension of ZrCl4 (37.63 g, 0.16 mol) in dry DCM (500 mL), 22 (16.78 mL, 0.17 mol) was added at -10 °C under N2 atmosphere followed by the addition of AcCl in 15 mL of DCM dropwise. The orange turbid solution was stirred at -10 °C for 1 h, then the reaction mixture was carefully poured into a 3 L flask containing of 2 N HCl (500 mL) and of DCM (150 mL), and stirred for 40 min. The phases were separated and the milky organic phase dried (Na2SO4). The residue was dissolved in MTBE (300 mL) and the mixture was filtered through a pad of Celite to give a clear colorless solution. After evaporation of the solvent, the residue was purified by flash chromatography (0 to 20% EtOAc in cyclohexane) to afford 23 as a light yellow oil. 17 g, 55%. 1H NMR (400 MHz, chloroform-d) δ 7.61 (d, J = 8.7 Hz, 1H), 7.18 (d, J = 2.5 Hz, 1H), 6.90 (dd, J = 8.7, 2.5 Hz, 1H), 3.87 (s, 3H), 2.65 (s, 3H). MS (ESI) C9H9BrO2 requires m/z 228, 230, found 229, 231 [M+H]+.
2-Bromo-4-methoxybenzoic acid (24)
To a suspension of t-BuONa (2.77 g, 28.8 mmol) in dry THF (50 mL), diethyl oxalate (6.06 mL, 39.3 mmol) was carefully added under N2 atmosphere and the yellow reaction mixture was stirred at room temperature for 30 min. A solution of 23 (3.0 g, 13.10 mmol) in dry THF (15 mL) was then added dropwise and the mixture was stirred at room temperature for additional 30 min. The reaction mixture was carefully poured into a mixture of 1 N HCl (200 mL) and EtOAc (200 mL), then the phases were separated and the organic layer was concentrated in vacuo to give a yellow oil. It was dissolved in a 5:3 mixture of acetone/H2O (160 mL) then NaHCO3 was added (11.0 g, 131.0 mmol) and cooled down to 0 °C. To this mixture, Oxone® (20.1 g, 32.8 mmol) was added very carefully and an evolution of gas was immediately observed. The mixture was stirred at 0 °C for 2 h then the solids were filtered off. To the filtrate, solid Na2S2O3 was added under stirring until disappearance of oxidant (KI solution test). The mixture was concentrated to remove residual acetone and then 2N HCl (20 mL) was added until acidic pH while a white precipitate immediately was formed which was filtered and washed with H2O (20 mL) to afford 24 as a white solid. 2.3 g, 77%. 1H NMR (400 MHz, DMSO-d6) δ 13.00 (bs, 1H), 7.82 (d, J = 8.7 Hz, 1H), 7.27 (d, J = 2.5 Hz, 1H), 7.04 (dd, J = 8.7, 2.5 Hz, 1H), 3.84 (s, 3H). MS (ESI) C8H7BrO3 requires m/z 230, 232, found 231, 233 [M+H]+.
2-Bromo-4-hydroxybenzoic acid (25)
To a suspension of 24 (3.0 g, 13.0 mmol) in dry DCM (50 mL) BBr3 (39 mL, 1 M solution in DCM) was added dropwise at 0 °C under N2 atmosphere for 30 min until complete dissolution and then left under stirring at room temperature for 12h while a precipitate was formed. The reaction mixture was quenched at 0 °C by a careful addition of 5 N NaOH (10 mL) until pH >11. H2O (50 mL) and DCM (50 mL) were then added and the mixture was stirred for additional 2 h. The two phases were separated and the organic layer was washed with H2O (30 mL). The aqueous phase was carefully acidified with 37% HCl until pH 1. NaCl (10 g) was added portionwise and a white precipitate immediately was formed. The mixture was stirred at 0 °C for 1.5 h and then filtered to give 25 as a white solid. 1.7 g, 61%. 1H NMR (400 MHz, DMSO-d6) δ 12.78 (bs, 1H), 10.54 (bs, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.08 (d, J = 2.4 Hz, 1H), 6.84 (dd, J = 8.6, 2.4 Hz, 1H). MS (ESI) C7H5BrO3 requires m/z 216, 217, found 215, 217 [M-H]-.
Benzyl 2-bromo-4-hydroxybenzoate (26)
To a solution of 25 (3.0 g, 13.8 mmol) in DMF (30 mL), KHCO3 (2.1 g, 20.7 mmol) was added under vigorous stirring followed by the addition of BnBr (1.47 mL, 12.44 mmol). The yellow mixture was stirred for 12h at room temperature and then poured into a mixture of 1 N HCl (200 mL) and MTBE (200 mL) under stirring. The two phases were separated and the organic layer was dried (Na2SO4). Evaporation of solvent gave 26 as a yellow oil that was used in the next step without further purification. 4.1 g. 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 7.80 (d, J = 8.7 Hz, 1H), 7.51–7.45 (m, 2H), 7.45–7.33 (m, 3H), 7.12 (d, J = 2.4 Hz, 1H), 6.87 (dd, J = 8.7, 2.4 Hz, 1H), 5.30 (s, 2H). MS (ESI) C14H11BrO3 requires m/z 306, 308, found 307, 309 [M+H]+.
Benzyl 2-(3-carbamoylphenyl)-4-hydroxybenzoate (27)
To a solution of 26 (4.2 g, 13.68 mmol) in dioxane (100 mL), H2O (80 mL) was added followed by the addition of Na2CO3 (2.9 g, 27.36 mmol) and 3-carbamoylphenylboronic acid (3.4 g, 20.52 mmol). To this solution, PdCl2dppf (500 mg, 0.034 mmol) was added and then the mixture was heated at 90 °C for 1.5 h under N2 atmosphere. The mixture was cooled down to room temperature and then poured into 1 N HCl (200 mL) and EtOAc (200 mL) under stirring. After 30 min, the two phases were separated and the aqueous phase was extracted with EtOAc (100 mL). The combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (40 to 100% EtOAc in DCM) to afford 27 as a whitish solid. 2.73 g, 57%. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (bs, 1H), 8.02 (bs, 1H), 7.92–7.76 (m, 3H), 7.51–7.33 (m, 3H), 7.30–7.23 (m, 3H), 7.11–7.01 (m, 2H), 6.89 (dd, J = 8.6, 2.5 Hz, 1H), 6.75 (d, J = 2.5 Hz, 1H), 5.02 (s, 2H). MS (ESI) C21H17NO4 requires m/z 347, found 348 [M+H]+.
Cyclohexylcarbamic Acid 6-Benzyloxycarbonyl-3’-carbamoylbiphenyl-3-yl Ester (28)
A suspension of 27 (2.7g, 7.78 mmol) in dioxane (100 mL) was heated at 50 °C until a yellow solution was formed. The reaction mixture was cooled down to room temperature and DMAP (250 mg, 2.04 mmol) and c-C6H11CNO (1.2 mL, 9.33 mmol) were added and the mixture was heated at 45 °C for 12h, then cooled down to room temperature and poured into 1 N HCl (200 mL) and EtOAc (200 mL) under stirring. The two phases were separated and the aqueous layer was extracted with EtOAc (100 mL). The combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (20 to 50% EtOAc in DCM) to afford 28 as a white fluffy solid. 2.8 g, 76%. 1H NMR (400 MHz, DMSO-d6) δ 8.05 (bs, 1H), 7.95–7.82 (m, 4H), 7.52–7.36 (m, 2H), 7.33–7.24 (m, 4H), 7.21 (d, J = 2.4 Hz, 1H), 7.13–7.00 (m, 2H), 5.08 (s, 2H), 3.32 (m, 1H), 1.91–1.48 (m, 5H), 1.38–1.01 (m, 6H). MS (ESI) C28H28N2O5 requires m/z 472, found 473 [M+H]+.
Cyclohexylcarbamic Acid 3’-Carbamoyl-6-carboxybiphenyl-3-yl Ester (29)
To a solution of 28 (2.7 g, 5.72 mmol) in dioxane (200 mL), cyclohexene (50 mL) and 10% Pd/C (2 g) were added. The mixture was heated at 85 °C for 2 h, then was cooled down to room temperature, added of activated carbon (2 g), and filtered through a pad of Celite. Evaporation of solvent gave 29 as white solid. 960 mg, 44%. 1H NMR (400 MHz, DMSO-d6) δ 12.61 (bs, 1H), 8.04 (bs, 1H), 7.95–7.73 (m, 4H), 7.55–7.43 (m, 2H), 7.38 (s, 1H), 7.23 (dd, J = 8.5, 2.4 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 3.33 (m, 1H), 2.00–1.48 (m, 5H), 1.39–0.99 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 169.0, 168.6, 153.3, 153.3, 142.9, 140.7, 134.6, 131.6, 131.5, 128.7, 128.4, 127.8, 127.0, 124.0, 121.1, 50.4, 32.9, 25.6, 25.0. MS (ESI) C21H22N2O5 requires m/z 382, found 383 [M+H]+.
Ammonium Cyclohexylcarbamic Acid 3’-Carbamoyl-6-sulfatebiphenyl-3-yl Ester (30)
To a suspension of 3 (200 mg, 0.62 mmol) in dry DCM (5 mL), SO3-DMF complex (593 mg, 3.73 mmol) was added. After stirring at room temperature for 1 h, pyridine (2 mL) was added and the reaction mixture was concentrated in vacuo to give a colorless oil that was purified by preparative HPLC (column, C18), using the following eluent conditions: 20% B for 0.5 min then 20% to 60% B in 7 min; Rt: 4.5 min to afford 30 as a white solid. 125 mg, 44%. 1H NMR (400 MHz, DMSO-d6) δ 8.08–8.01 (m, 1H), 7.91 (bs, 1H), 7.87–7.76 (m, 2H), 7.69 (d, J = 7.9 Hz, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.36 (bs, 1H), 7.11 (d, J = 2.9 Hz, 1H), 7.05 (dd, J = 8.9, 2.9 Hz, 1H), 3.50 (bs, 4H), 3.41–3.22 (m, 1H), 1.89–1.78 (m, 2H), 1.78–1.66 (m, 2H), 1.63–1.49 (m, 1H), 1.43–0.95 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 154.1, 147.8, 147.1, 137.7, 134.7, 133.2, 132.7, 128.6, 128.3, 126.7, 123.4, 122.3, 121.9, 50.2, 33.0, 25.6, 25.0. MS (ESI) C20H22N2O7S requires m/z 434, found 433 [M-H]- .
1,3-Dibenzyloxy-5-bromobenzene (32)
To a solution of t-BuONa (19.9 g, 207.3 mmol) and BnOH (21.3 mL, 207.3 mmol) in dry DMF (200 mL), 1-bromo-3,5-difluorobenzene 31 (4.8 mL, 41.5 mmol) was added under N2 atmosphere. The reaction mixture was stirred at 90 °C for 3 h. The dark yellow mixture was cooled down to room temperature and, under stirring, slowly transferred in a 3 L flask containing H2O (600 mL) and of MTBE (500 mL). After 30 min, the organic phase was separated, washed with H2O (400 mL) and dried (Na2SO4). Evaporation of the solvent gave 32 as yellow oil that crystallized after 12h upon cooling at -19 °C. The solid was treated with 180 mL of MeOH then filtered and washed with cold MeOH (30 mL). 11 g, 72%. 1H NMR (400 MHz, chloroform-d) δ 7.52–7.31 (m, 10H), 6.80 (d, J= 2.2 Hz, 2H), 6.57 (t, J= 2.2 Hz, 1H), 5.03 (s, 4H). MS (ESI) C20H17BrO2 requires m/z 368, found 367 (M-H)-.
3-(3,5-Dibenzyloxyphenyl)benzamide (33)
To a solution of 32 (11.0 g, 29.8 mmol) in EGME (152 mL), H2O (54 mL) was added dropwise, followed by the addition of K2CO3 (8.2 g, 59.6 mmol), 3-carbamoylphenylboronic acid (7.4 g, 44.7 mmol), and Pd(OAc)2 (80.3 mg 0.36 mmol). The reaction mixture was stirred at 60 °C for 20 min. Then, H2O (100 mL) were added and a precipitate was formed which was filtered and washed with cold H2O (50 mL). The solid was recrystallized from MeOH/THF (2.5:1, 350 mL) to give 33 as a light grey solid. 8.5 g, 70%. 1H NMR (400 MHz, DMSO-d6) δ 8.15 (t, J= 1.8 Hz, 1H), 8.12 (bs, 1H), 7.87 (d, J= 7.8 Hz, 1H), 7.83 (d, J= 7.8 Hz, 1H), 7.61–7.30 (m, 12H), 7.00 (d, J= 2.2 Hz, 2H), 6.73 (t, J= 2.2 Hz, 1H), 5.19 (s, 4H). MS (ESI) C27H23NO3 requires m/z 409, found 410 (M+H)+.
3-(3,5-Dihydroxyphenyl)benzamide (34)
To a suspension of 33 (8.5 g, 20.8 mmol) in dioxane (260 mL), cyclohexene (80 mL) was added and the mixture was heated at 50 °C for 15 min until complete dissolution, cooled down to room temperature and 10% Pd/C (2 g) was added. The reaction mixture was heated at 80 °C for 2 h and an additional amount of 10% Pd/C (2 g) was then added. After additional 2h, the mixture was cooled down to room temperature and filtered through a pad of Celite, washed with dioxane (100 mL) and of absolute EtOH (100 mL). The clear solution was concentrated in vacuo to afford 34 as a light yellow solid. 4.8 g, 100%. 1H NMR (400 MHz, DMSO-d6) δ 9.38 (s, 2H), 8.10 (bs, 1H), 8.07–8.03 (m, 1H), 7.83 (d, J= 7.8 Hz, 1H), 7.68 (d, J= 7.8 Hz, 1H), 7.50 (t, J= 7.7 Hz, 1H), 7.38 (bs, 1H), 6.55 (d, J= 2.1 Hz, 2H), 6.27 (t, J= 2.1 Hz, 1H). MS (ESI) C13H11NO3 requires m/z 229, found 230 (M+H)+.
Cyclohexylcarbamic Acid 3’-Carbamoyl-5-hydroxybiphenyl-3-yl Ester (35)
To a solution of 34 (2.6 g, 11.4 mmol) in dry DMF (30 mL), CuCl (1.1 g, 11.4 mmol) was added and the reaction mixture turned rapidly to a brown color. c-C6H11CNO (1.45 mL, 11.4 mmol) was then added and the mixture was stirred at room temperature for 30 min. To this solution, a mixture of 3% aqueous citric acid solution (200 mL) and EtOAc (100 mL) were then added. The organic phase was separated and dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (50 to 100% EtOAc in cyclohexane) to afford 35 as a white solid. The solid was dissolved in a 6.5:2.0:1.5 mixture of H2O: acetone: EtOH (75 mL). To this solution, H2O (30 mL) were then added and a precipitate was formed which was filtered to afford 35 as a white solid. 1.17 g, 29%. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.13 (bs, 1H), 8.11– 8.09 (m, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.75 (d, J= 7.7 Hz, 1H), 7.70 (d, J= 7.7 Hz, 1H), 7.53 (t, J= 7.7 Hz, 1H), 7.41 (bs, 1H), 6.95 (t, J= 1.9 Hz, 1H), 6.89 (t, J= 1.9 Hz, 1H), 6.53 (d, J= 1.9 Hz, 1H), 3.46–3.32 (m, 1H), 1.99–1.46 (m, 6H), 1.46–0.99 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 168.2, 158.9, 153.8, 153.0, 141.8, 139.9, 135.4, 129.7, 129.4, 127.4, 126.0, 111.4, 110.8, 108.8, 50.2, 33.0, 25.6, 25.0. MS (ESI) C20H22N2O4 requires m/z 354, found 355 (M+H)+.
3,5-Dimethoxyphenyl trifluoromethanesulfonate (38)
To a solution of 37 (0.154 g, 1.0 mmol) in DCM (3 mL), DMAP (0.183 g, 1.5 mmol) and (CF3SO2)2O (0.366 g, 0.22 mL, 1.3 mmol) were added at 0 °C. The mixture was stirred for 15 min at room temperature and concentrated. The residue was purified by flash chromatography (cyclohexane/EtOAc 3:7) to afford 38, which was used directly in the next step without characterization.
3-(3,5-Dimethoxyphenyl)benzamide (39)
To a solution of (n-BuN)4Br (0.332 g, 1.0 mmol) and Na2CO3 (0.265 g, 2.5 mmol) in EtOH (5 mL), 38 (0.286, 1.0 mmol), 3-carbamoylphenylboronic acid (0.165 g, 1.0 mmol) and PdCl2 (18 mg, 0.1 mmol) were added. The mixture was refluxed for 12 h, filtered on a plug of Celite, acidified with 2 N HCl (5 mL), and extracted with EtOAc (5×3 mL). The combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (DCM/MeOH 95:5 and then cyclohexane/EtOAc 30:70) gave 39 as a white solid. 0.115 g, 45%. 1H NMR (200 MHz, DMSO-d6) δ 8.12 (t, J = 1.6 Hz, 1H), 8.08 (br, 1H), 7.88–7.80 (m, 2H), 7.52 (t, J = 7.7 Hz, 1H), 7.39 (br, 1H), 6.85 (d, J = 2.4 Hz, 2H), 6.53 (t, J = 2.2 Hz, 1H), 3.82 (s, 6H). MS (ESI) C15H15NO3 requires m/z 257, found 258 [M+H]+.
3-(3-Hydroxy-5-methoxyphenyl)benzamide (40)
To a solution of 39 (0.257 g, 1.0 mmol) in dry DCM (9 mL), BBr3 (5.0 mL, 1.0 M solution in DCM, 5 mmol) was added at 0 °C. The mixture was stirred for 30 min at room temperature, then H2O (5 mL) was added and the mixture was extracted with DCM (5 mL) and EtOAc (5×2 mL). The combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by flash chromatography (DCM/MeOH 95:5) to afford 40 as a white amorphous solid. 97 mg, 40%. 1H NMR (200 MHz, DMSO-d6) δ 7.87–7.81 (m, 3H), 7.59–7.42 (m, 4H), 5.80 (br, 3H), 1.57 (s, 3H). MS (ESI) C14H13NO3 requires m/z 243, found 244 [M+H]+.
Cyclohexylcarbamic Acid 3’-Carbamoyl-5-metoxybiphenyl-3-yl Ester (41)
To a solution of 40 (0.243 g, 1 mmol) in CH3CN (25 mL), c-C6H11NCO (0.137 mg, 1.1 mmol), Et3N (0.11 g, 0.154 mL, 1.1 mmol) were added. The mixture was refluxed for 2 h and concentrated. The residue was purified by flash chromatography (DCM/EtOAc 70:30) to afford 41 as a white solid after crystallization from EtOAc/petroleum ether. 0.240 g, 65%. 1H NMR (200 MHz, DMSO-d6) δ 8.14–8.09 (m, 2H), 7.80–7.79 (m, 2H), 7.70 (d, J = 8.0 Hz, 1H), 7.53 (t, J = 7.7 Hz,1H), 7.39 (s, 1H), 7.13 (m, 1H), 7.05 (t, J = 1.7 Hz, 1H), 6.71 (t, J = 2.1 Hz, 1H), 3.84 (s, 3H), 3.41–3.30 (m, 1H), 1.85–1.55 (m, 5H), 1.25–1.21 (m, 5H). 13C NMR (50 MHz, DMSO-d6) δ 168.2, 160.9, 153.7, 153.1, 141.8, 139.7, 135.4, 129.9, 129.4, 127.6, 126.1, 113.1, 109.6, 107.7, 56.0, 50.3, 33.0, 25.6, 25.0. MS (ESI) C21H24N2O4 requires m/z 368, found 369 [M+H]+.
ACKNOWLEDGMENT
The authors wish to thank Sine Mandrup Bertozzi for reverse phase HPLC purifications, Luca Goldoni for NMR technical support, Silvia Venzano for compounds handling and Masih A. Babagoli for experimental assistance.
Funding Sources
Funding by NIH grant DA-012423 (to D.P.) is gratefully acknowledged. Universities of Parma and Urbino “Carlo Bo” also supported the work.
ABBREVIATIONS
- FAAH
fatty-acid amide hydrolase
- ABCG2
ATP-binding cassette transporter G2
- BBB
blood-brain barrier
- CB1
cannabinoid type-1 receptor
- CNS
central nervous system
- ED50
median effective dose
- IC50
median inhibitory dose
- Oxone®
potassium peroxymonosulfate
- PSA
polar surface area
- PST
phenol sulfotransferase
- SAR
structure-activity relationship
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- EGME
ethyleneglycol monomethyl ether
- MTBE
methyl tert-butyl ether
- SiO2
silica gel
- TBSCI
tert-butyldimethylchlorosilane
- TIPSCI
triisopropylchlorosilane
Footnotes
The authors declare the following competing financial interests: patents protecting the class of compounds disclosed in this paper were filed by the University of California Irvine, Istituto Italiano di Tecnologia, Università degli Studi di Urbino “Carlo Bo”, and Università degli Studi di Parma on behalf of following individuals: Piomelli, Daniele; Moreno-Sanz, Guillermo; Bandiera, Tiziano; Mor, Marco, and Tarzia, Giorgio.
REFERENCES
- 1.Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol. 2013;64:21–47. doi: 10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- 2.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 3.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 4.Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisogno T, Maccarrone M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin Drug Discov. 2013;8:509–522. doi: 10.1517/17460441.2013.780021. [DOI] [PubMed] [Google Scholar]
- 6.Kunos G, Osei-Hyiaman D, Batkai S, Sharkey KA, Makriyannis A. Should peripheral CB(1) cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol Sci. 2009;30:1–7. doi: 10.1016/j.tips.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, Kuner R. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong WI, Batkai S, Marsicano G, Lutz B, Buettner C, Kunos G. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118:3160–3169. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LoVerme J, Duranti A, Tontini A, Spadoni G, Mor M, Rivara S, Stella N, Xu C, Tarzia G, Piomelli D. Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg Med Chem Lett. 2009;19:639–643. doi: 10.1016/j.bmcl.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plowright AT, Nilsson K, Antonsson M, Amin K, Broddefalk J, Jensen J, Lehmann A, Jin S, St-Onge S, Tomaszewski MJ, Tremblay M, Walpole C, Wei Z, Yang H, Ulander J. Discovery of agonists of cannabinoid receptor 1 with restricted central nervous system penetration aimed for treatment of gastroesophageal reflux disease. J Med Chem. 2013;56:220–240. doi: 10.1021/jm301511h. [DOI] [PubMed] [Google Scholar]
- 11.Fulp A, Bortoff K, Seltzman H, Zhang Y, Mathews J, Snyder R, Fennell T, Maitra R. Design and synthesis of cannabinoid receptor 1 antagonists for peripheral selectivity. J Med Chem. 2012;55:2820–2834. doi: 10.1021/jm201731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wittgen HG, Greupink R, van den Heuvel JJ, van den Broek PH, Dinter-Heidorn H, Koenderink JB, Russel FG. Exploiting transport activity of p-glycoprotein at the blood-brain barrier for the development of peripheral cannabinoid type 1 receptor antagonists. Mol Pharm. 2012;9:1351–1360. doi: 10.1021/mp200617z. [DOI] [PubMed] [Google Scholar]
- 13.Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreno-Sanz G, Barrera B, Guijarro A, d'Elia I, Otero JA, Alvarez AI, Bandiera T, Merino G, Piomelli D. The ABC membrane transporter ABCG2 prevents access of FAAH inhibitor URB937 to the central nervous system. Pharmacol Res. 2011;64:359–363. doi: 10.1016/j.phrs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreno-Sanz G, Sasso O, Guijarro A, Oluyemi O, Bertorelli R, Reggiani A, Piomelli D. Pharmacological characterization of the peripheral FAAH inhibitor URB937 in female rodents: interaction with the Abcg2 transporter in the blood-placenta barrier. Br J Pharmacol. 2012;167:1620–1628. doi: 10.1111/j.1476-5381.2012.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robey RW, Ierano C, Zhan Z, Bates SE. The challenge of exploiting ABCG2 in the clinic. Curr Pharm Biotechnol. 2011;12:595–608. doi: 10.2174/138920111795163913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vacondio F, Silva C, Lodola A, Fioni A, Rivara S, Duranti A, Tontini A, Sanchini S, Clapper JR, Piomelli D, Mor M, Tarzia G. Structure-property relationships of a class of carbamate-based fatty acid amide hydrolase (FAAH) inhibitors: chemical and biological stability. ChemMedChem. 2009;4:1495–1504. doi: 10.1002/cmdc.200900120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, Sanchini S, Tarzia G, Mor M, Piomelli D. A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem. 2009;4:1505–1513. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, Schellens JH, Koomen GJ, Schinkel AH. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1:417–425. [PubMed] [Google Scholar]
- 20.Fiorelli CS, R., Piomelli D, Bandiera T. Development of a Multigram Synthesis of URB937, a Peripherally Restricted FAAH Inhibitor. Org Process Res Dev. 2013;17:359–367. [Google Scholar]
- 21.Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, Moreno-Sanz G, Reggiani A, Piomelli D. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res. 2012;65:553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kürti L, Czakó B. Strategic Applications of Named Reactions in Organic Synthesis. Elsevier Inc.; 2005. pp. 118–119. [Google Scholar]
- 23.Ding D, Zhao Y, Meng Q, Xie D, Nare B, Chen D, Bacchi CJ, Yarlett N, Zhang Y-K, Hernandez V, Hernandez V, Xia Y, Freund Y, Abdulla M, Ang K-H, Ratnam J, McKerrow JH, Jacobs RT, Zhou H, Plattner JJ. Discovery of novel benzoxaborole-based potent antitrypanosomal agents. ACS Med. Chem. Lett. 2010;1:165–169. doi: 10.1021/ml100013s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couture AD, E., Lebrun S, Hoarau C, Grandclaudon P. An expeditious synthesis of goniothalactam. Nat. Prod. Lett. 1999;13:33–40. [Google Scholar]
- 25.Ding D, Meng Q, Gao G, Zhao Y, Wang Q, Nare B, Jacobs R, Rock F, Alley MR, Plattner JJ, Chen G, Li D, Zhou H. Design, synthesis, and structure-activity relationship of Trypanosoma brucei leucyl-tRNA synthetase inhibitors as antitrypanosomal agents. J Med Chem. 2011;54:1276–1287. doi: 10.1021/jm101225g. [DOI] [PubMed] [Google Scholar]
- 26.Ashford SW, Grega KC. Oxidative cleavage of 1,3-dicarbonyls to carboxylic acids with oxone. J Org Chem. 2001;66:1523–1534. doi: 10.1021/jo001579m. [DOI] [PubMed] [Google Scholar]
- 27.Guo W, Li J, Fan N, Wu W, Zhou P, Xia C. A simple and effective method for chemoselective esterification of phenolic acids. Synth. Commun. 2005;35:145–152. [Google Scholar]
- 28.Huang S, Petersen TB, Lipshutz BH. Total Synthesis of (+)-Korupensamine B via an Atropselective Intermolecular Biaryl Coupling. JACS. 2010;132:14021–14023. doi: 10.1021/ja1065202. [DOI] [PubMed] [Google Scholar]
- 29.Del Zotto A, Amoroso F, Baratta W, Rigo P. Very Fast Suzuki-Miyaura Reaction Catalyzed by Pd(OAc)(2) under Aerobic Conditions at Room Temperature in EGME/H2O. Eur J Org Chem. 2009;1:110–116. [Google Scholar]
- 30.Duggan ME, Imagire JS. Copper(I) Chloride Catalyzed Addition of Alcohols to Alkyl lsocyanates. A Mild and Expedient Method for Alkyl Carbamate Formation. Synthesis. 1989;2:131–132. [Google Scholar]
- 31.Subramanian LR, Hanack M, Chang LWK, Imhoff MA, Schleyer P. v. R., Effenberger F, Kurtz W, Stang PJ, Dueber TE. On attempts at solvolytic generation of aryl cations. J. Org. Chem. 1976;41:4099–4103. [Google Scholar]
- 32.Lodola A, Mor M, Rivara S, Christov C, Tarzia G, Piomelli D, Mulholland AJ. Identification of productive inhibitor binding orientation in fatty acid amide hydrolase (FAAH) by QM/MM mechanistic modelling. Chem Commun (Camb) 2008;2:214–216. doi: 10.1039/b714136j. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki M, Suzuki H, Sugimoto Y, Sugiyama Y. ABCG2 transports sulfated conjugates of steroids and xenobiotics. J Biol Chem. 2003;278:22644–22649. doi: 10.1074/jbc.M212399200. [DOI] [PubMed] [Google Scholar]
- 34.Enokizono J, Kusuhara H, Ose A, Schinkel AH, Sugiyama Y. Quantitative investigation of the role of breast cancer resistance protein (Bcrp/Abcg2) in limiting brain and testis penetration of xenobiotic compounds. Drug Metab Dispos. 2008;36:995–1002. doi: 10.1124/dmd.107.019257. [DOI] [PubMed] [Google Scholar]
- 35.Henton DRA, K., Manning MJ, Swenton JS. Chemistry of quinone derivatives. Quinone monoketals via hydrolysis of electrochemically derived quinone bisketals. J. Org. Chem. 1980;45:3422–3433. [Google Scholar]