

Abstract
Despite the remarkable versatility displayed by flavin-dependent monooxygenases (FMOs) in natural product biosynthesis, one notably missing activity is the oxidative generation of carbonate functional groups. We describe a multifunctional Baeyer-Villiger monooxygenase CcsB, which catalyzes the formation of an in-line carbonate in the macrocyclic portion of cytochalasin E. This study expands the repertoire of activities of FMOs and provides a possible synthetic strategy for transformation of ketones into carbonates.
Flavin-dependent monooxygenases (FMOs) catalyze an enormous variety of substrate oxidations and reductions in both primary and secondary metabolism1. During natural product biosynthesis, following the assembly of the structural framework by upstream enzymes, FMOs play essential roles in introducing structural complexity and biological activity2. FMOs are versatile enzymes that can catalyze the formation of different types of C-O bonds3-5; absent from the FMO product portfolio is the carbonate moiety. To date, no enzymatic oxidation of a ketone or an ester to the corresponding carbonate has been described, although there are abundant examples of oxidation of ketones to esters catalyzed by BV monooxygenases (BVMOs)6, 7. Non-enzymatic transformation of an ester to a carbonate is a similarly challenging synthetic transformation. The difficulties in generation of the carbonate by synthetic and enzymatic BV mechanisms are similar, and include increased electron density of the ester carbonyl that deters a second peroxide attack, and the unlikelihood that the resulting Criegee complex will collapse via C-C bond migration to form an additional C-O bond.
The carbonate functionality is therefore very rarely found in natural products8. Remarkably, several members of the cytochalasin family of fungal natural products contain an in-line carbonate moiety in the macrocycle portion of the molecules (Fig.1 and Supplementary Results, Supplementary Fig.1). Both cytochalasins E (1) and K (2) are polyketides produced by Aspergillus clavatus. Related compounds such as phenochalasin B (3)9 and scoparasin A (4)10 also contain a vinyl carbonate. Other members of the large cytochalasin family are less oxidized at the corresponding carbonate carbon than 1-4, including esters such as rosellichalasin (5) and ketones as seen in cytochalasin G (6) (Fig. 1 and Supplementary Figs. 2-3)11.
Figure 1.

Cytochalasins with different oxidation outcomes in the macrocyclic portion. Compounds 1-4 contain a vinyl carbonate group of interest at C21 within the thirteen-membered macrocycle that is fused to an isoindolone bicyclic scaffold. Other members of the large cytochalasin family are less oxidized at the corresponding carbonate carbon than 1-4, including esters such as rosellichalasin (5) and ketones as in cytochalasin G (6).
To study the enzymatic basis of the carbonate group in 1, we first investigated the biosynthetic origin of the oxygen atoms in 1 by growing A. clavatus either in media supplemented with sodium [1-13C, 1-18O2]acetate or in a closed system in which consumed oxygen was replaced by 18O2. A slight upfield shift (Δδc ∼ 0.05 ppm) for 13C connected to 18O was used as an indicator of the source of oxygen atoms in 112. Results showed that the carbonyl oxygen at C21 of 1 is derived from acetate during polyketide assembly. In contrast, both carbonate oxygen atoms attached to C21 are derived from molecular oxygen, thereby pointing to an insertion pathway catalysed by an oxygenase (Supplementary Figs. 4-5).
The ccs biosynthetic gene cluster for 1 and 2 from A. clavatus is centered on a PKS-NRPS megasynthetase CcsA13. CcsB (ACLA_078650) is the only predicted FMO in the gene cluster, with moderate sequence identity to well-characterized type I BVMOs7 (Supplementary Figs. 6-7). CcsB contains the conserved fingerprint motif FXGXXXHXXXW14 and the strictly conserved active site arginine (Arg421) that stabilizes the Fl-4a-OO- anion through electrostatic interactions15. The two remaining oxygenases encoded in the gene cluster, CcsG and CcsD, are both P450 monooxygenases and are possibly involved in the oxidation of other sites in 1 as reported for the related chaetoglobosin16. We therefore propose CcsB is involved in generation of the carbonate group in 1 starting from a ketone precursor, via a mechanism previously not observed among BVMOs.
To investigate the role of CcsB, we sought to inactivate the gene ccsB in A. clavatus. First, the positive regulatory gene ccsR was overexpressed in a Δlig4 modified A. clavatus strain to improve the titer of 1 and 213. We also detected and structurally verified the presence of 5 in the culture extract (Fig.2a, Supplementary Fig.8), which is consistent with other co-isolation reports of 1 and 5 in fungal strains that can produce 117. Using the overproducing strain (OE::ccsR, Δlig4), the ccsB gene was deleted with one of the desired mutants (ΔccsB-37) confirmed by PCR (Supplementary Fig.9). Deletion of the ccsB gene abolished the production of 1, 2 and 5, and led to the production of 1,5 diketone-containing compound 7 (m/z 454[M+Na]+) (Fig. 2a). The structure of 7 (named ketocytochalasin, Fig. 2b) was characterized by UV, MS, and NMR techniques (Supplementary Note 1, Supplementary Table 1 and Supplementary Figs. 10-13), and confirmed by X-ray diffraction (Supplementary Fig.12, Supplementary Data Sets 2 and 4, CCDC 970431). The abolishment of 1, 2 and 5 upon ccsB inactivation and the recovery of 7 strongly indicates CcsB is the enzyme involved in the oxidation reactions at C21. The structure of 7 also points to a relatively early action of CcsB in the tailoring of the cytochalasin scaffold en route to 1 and 2. Additional modifications, such as C18 hydroxylation, and C6-C7 epoxidation, should take place subsequent to the oxidative ring expansion steps (Supplementary Fig.14).
Figure 2.
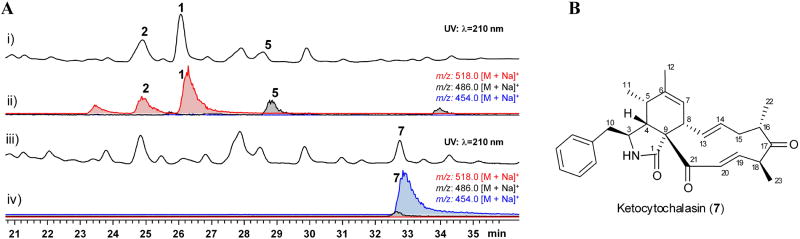
Genetic confirmation of CcsB activity. A) i) and iii): HPLC analysis (λ= 210 nm) of metabolites extracted from OE::ccsR Δlig4 strain and from ΔccsB-37 mutant, respectively; ii) and iv): Selected ion monitoring of m/z: 518 (1, 2), 486 (5), and 454 (7) of traces i) and iii), respectively. All m/z values are [M+Na]+. The metabolites are extracted from the 4th day cultures. The experiments were performed three times and a representative result is shown here. B) Chemical structure of 7.
To examine whether CcsB can generate the ester or carbonate product starting from 7, recombinant CcsB was cloned, expressed and purified to homogeneity from Escherichia coli (Supplementary Fig.15). CcsB was purified with a light yellow hue and its UV spectrum showed the characteristic absorption of FAD (Supplementary Fig.16). LC-MS analysis of the supernatant of denatured protein confirmed the presence of FAD at ∼ 10% of CcsB concentration, which could be reconstituted with addition of FAD (Supplementary Figs.16-17). We then performed an in vitro reaction with CcsB and 7 in the presence of NADPH, FAD, and SsuE (Online Methods). Whereas the control reaction without CcsB did not lead to new products, CcsB catalysed the conversion of 7 into two new products 8 and 9 as evident in the time course analysis (Fig. 3A). The mass of 8 (m/z 448 [M+H]+) and 9 (m/z 464 [M+H]+) are consistent with that of 7 containing one and two additional oxygen atoms, respectively. Removal of NADPH or SsuE from the reaction abolished the conversion of 7. The ratio of 9 to 8 formed in the reaction strongly correlated to the molar ratio of NADPH to 7 in the reaction (Supplementary Fig.18).
Figure 3.

Reactions catalysed by CcsB and chemical complementation of ΔccsB-37. A) HPLC analysis of products of the CcsB reaction (λ= 210 nm). In vitro reaction conditions are 50 mM potassium phosphate buffer (pH 7.0), 4 mM NADPH, 20 μM FAD, 6 μM SsuE, 10 μM CcsB, and 0.4 mM 7. Trace i: Control reaction without CcsB; traces ii-v: time course analysis of the conversion of ketone 7 to ester 8 and carbonate 9; trace vi: reaction with the CcsB mutant R421A; and trace vii: hexane extract of E. coli used for biotransformation of 7 to 8 and 9. B) Chemical structures of compounds 8-11. Compound 11 was not isolated in this study due to its isomerization to 8. C) HPLC analysis (λ=210 nm) of extract from the chemical complementation of 8 and 9 to A. clavatus ΔccsB-37. Traces i-iv: standards of 1, 2, 5, 7, and 10; trace v: A. clavatus ΔccsB-37; trace vi and vii: A. clavatus ΔccsB-37 complemented with 9 or 8 in growth media, respectively. 9 is a precursor to 1 and 2, while 8 is a precursor to 5 and 10. All experiments were performed three times and a representative result is shown here.
Complete NMR and X-ray characterization (Supplementary Note 1, Supplementary Table 2 and Supplementary Figs.19-20, Supplementary Data Sets 1 and 3, CCDC 970432) of 9 (Fig.3B) verified the compound to be indeed the carbonate-containing cytochalasin Z16 (ref 18). The identity of 9 confirmed that CcsB is the only enzyme required to convert the ketone 7 into carbonate 9, an unprecedented example in FMO chemistry. Mutation of the Fl-4a-OO--stabilizing Arg421 to Ala (Supplementary Fig. 21) completely abolished the conversion of 7 to 8 and 9 (Fig. 3A and Supplementary Fig. 15b), thereby confirming the catalytic role of CcsB in the reaction. Feeding of pure 9 to ΔccsB-37 strain led to the restored production of 1 and 2 (but not 5) (Fig.3C, trace vi and Supplementary Fig.22), which verified 9 is indeed an on-pathway intermediate in the ccs biosynthetic pathway.
Initially we were expecting that 8 would be an ester corresponding to structure 11. However, structural elucidation revealed a double bond shift to iso-precytochalasin (Fig. 3B, Supplementary Table 3, Supplementary Fig.23-24, and Supplementary Note 1) No conversion to 9 was observed when pure 8 was incubated with CcsB (Supplementary Fig.25), suggesting that β-γ unsaturated ester 8 may be a shunt product formed from the spontaneous isomerization of the true intermediate 11 (Fig. 3B). Feeding of 8 to A. clavatus ΔccsB-37 strain led to the restored production of 5 along with a new product cytochalasin Z17 (10)18 (Fig.3C, trace vii). Structurally, 5 and 10 represent the variations observed at C6-C7 in 1 and 2, respectively. The simultaneous production of both 8 and 9 from the CcsB assay therefore rationalizes the co-isolation of 5 and 1 in all fungal producers (Supplementary Fig. 14).
The proposed isomerization of 11 to yield 8 unveils a reactive vinylogous β-diketo type system facilitated through deprotonation of the distal α-carbon C18, followed by C20 proton abstraction from solution. As expected, when the CcsB reaction was performed in D2O buffers, we observed the increase in the molecular weight of 8 by 1 mu (Supplementary Fig. 26). Under aqueous conditions, conjugation of the β-diketo system is expected to be favoured for the ketone (8) over the ester (11). Indeed, density functional theory calculations (DFT, Supplementary Note 2) indicate that 8 is roughly 6 kcal mol−1 (ΔG) more stable than 11 (Supplementary Fig. 27). At shorter reaction times and lower NADPH concentrations, we did detect an unstable compound with the same molecular weight and UV absorbance as 8 (Supplementary Fig. 28). However, attempts to purify this compound resulted in rapid conversion to 8, thereby indicating the possible identity as 11 (Supplementary Fig. 29).
Examination of known cytochalasin compounds revealed the vinylogous 1,5-diketo system is absolutely required for carbonate formation (Supplementary Fig. 1). Only esters or ketone compounds are found when such configuration is altered (Supplementary Fig. 2-3). Our experiments here also support that both an α,β-unsaturated ester and an acidic proton in the γ-position (α to a distal carbonyl) are all required for this unique reactivity. Testing CcsB with numerous cytochalasin analogs lacking one or more of these features, including cytochalasin B and dehydrozygrosporin19 (Supplementary Note 3, Supplementary Table 4 and Supplementary Fig. 30), did not lead to formation of carbonate products (Supplementary Fig.25). These observations, combined with the lack of precedent for a second BV oxidation of an ester moiety in both synthetic and biosynthetic literature, suggest alternative mechanisms to generate the carbonate 9 (Supplementary Figs 31-33). The first oxygen insertion step that oxidizes 7 to 11 follows that of the classic BVMO mechanism and is in line with the rules of migratory aptitude. We propose that insertion of the second oxygen into 11 to form the carbonate 9 could be initiated through a Michael addition from the peroxy-flavin anion (Fl-4a-OO-) on the β-carbon of the vinyl ester to yield an α-β epoxide. (Supplementary Figs. 31). Epoxide opening facilitated by abstraction of C18 α-proton leads to an a-alcohol intermediate, which can form the epoxy alkoxide20 and readily rearrange across the 1,5 diketone system to afford 9. This last step is analogous to the reported Lewis-acid-promoted conversion of an α-hydroxy β-dicarbonyl compound to a carbonate 21, 22. Alternatively, direct addition of Fl-4a-OO-at C21 of 11 may also take place (Supplementary Fig. 32-33). Direct testing of these proposals, including trapping of the conjugated flavin intermediate, or testing of molecules containing similarly-tuned functional groups, will be needed to conclusively define the mechanism.
Sequence analysis of CcsB revealed that it is unremarkably grouped with well-characterized BVMOs (Supplementary Fig. 7). An intriguing question is therefore how intrinsically different CcsB is from the canonical BMVO in catalytic function. Close homologs of CcsB are widely found in sequenced fungal genome databases, with several highly homologous hits embedded in gene clusters that resemble the ccs pathway. It is also expected that for all the ester-containing cytochalasins such as cytochalasin B, a CcsB-like enzyme must be present to perform the first BV oxidation. Such activities were demonstrated in the classical feeding studies of deoxaphomin (ketone version of cytochalasin B) to Phoma sp. and the recovery of cytochalasin B23. We therefore propose that formation of the carbonate product by CcsB is the fortuitous result of the perfectly arranged functional groups near the ketone site in 7 and 11. The only comparable BVMO examples are those that can convert an acyl-carrier protein (ACP) bound thioester into a thiocarbonate for subsequent product release, such as that catalyzed by FR9H-Ox in the FR901464 pathway24, 25.
In conclusion, we identified the enzymatic basis for forming the in-line carbonate moiety in cytochalasins isolated from fungi. The conversion of a ketone to a carbonate is unprecedented in natural product biosynthesis and is a completely new type of transformation discovered for the large, well-characterized FMO family.
Onlinemethods
Strains and culture condition
The Aspergillus clavatus strain was obtained from Agriculture Research Service (NRRL 1) Culture Collection and was used as the parental strain in our study. Both the wild type and the mutant strains were grown on malt extract peptone agar (MEPA) (30 g/L malt extract, 3 g/L papaic digest of soybean meal and 15 g/L agar) at 25°C. Escherichia coli strain XL1-Blue (Stratagene) and E coli TOPO10 (Invitrogen) were used for cloning. E coli strain BL21(DE3) (Novagen) was used for protein expression.
DNA manipulation and construction of plasmids
Genomic DNA of A. clavatus NRRL1 was isolated from mycelium grown in stationary liquid culture supplied with malt extract peptone for 48 hours at 25°C. The mycelia collected were lyophilized overnight and were ground to a fine powder using toothpicks. Then, 700 μL LETS buffer (10 mM Tris-HCl pH 8.0, 20 mM EDTA, 0.5% SDS, 0.1 M HCl) was added and the samples were mixed by using the toothpick as well as inverting the tubes several times before leaving the sample on the bench for 5 min. After that, 700 μL of Phenol: chloroform: isoamyl alcohol (25: 24: 1) was added and mixed by inverting 10-15 times before centrifuging at 13,200 rpm for 10 minutes. The supernatant was then transferred into a new 1.5 μL centrifuge tube and equal volume of Phenol: chloroform: isoamyl alcohol (25: 24: 1) was added and mixed by inverting 10-15 times. The mixture was centrifuged at 13,200 rpm for 10 minutes and the supernatant was then transferred into a new 1.5 μL centrifuge tube and 1mL 95% EtOH(ice cold) was added. DNA was pelleted by centrifugation at 4°C (13,200 rpm, 10 min), the supernatant was removed and the pellet was washed twice with ice-cold 70% ethanol, centrifuging briefly and discarding supernatant after each wash. The resulting pellet was allowed to air-dry for 10 minutes before resuspending in 40 μL of 10 mM Tris buffer (pH 8.0). 0.5 μL RNase (10mg/mL stock) was added and incubated at 50°C for 30 min before using for molecular cloning. Platinum Pfx DNA polymerase (Invitrogen) was used to perform PCR reactions from genomic DNA. Sequence of PCR products were confirmed by DNA sequencing (Retrogen, CA). The primers for cloning ccsB and for construction of knockout cassette are shown Supplementary Table 4. The gene encoding ccsB was amplified and inserted into pET23 to yield pYC01. The ccsB gene was fused to an N-terminal FLAG peptide for protein purification. The first 36 residues at N-terminus of CcsB were removed in this construct as these were predicted to be a membrane signal peptide. To perform site-directed mutagenesis of ccsB, PCR directed mutagenesis was used. To construct the knockout cassette for ccsB, the selection marker bar gene with trpC promoter was amplified from the plasmid pBARKS1. This marker was then flanked by two fragments of ccsB using fusion PCR and inserted into the pCR-Blunt (Invitrogen) vector. PCR was then used to amplify up to 10 μg of the entire knockout cassette for fungal transformation.
Fungal Transformation and Genetic manipulation in Aspergillus clavatus
Preparation of A clavatus protoplasts and polyethylene glycol–mediated transformation of A. clavatus was performed as described previously26 with some modification. In detail, A. clavatus spores were collected from 2-3 days culture and inoculated in 50 mL sterile liquid minimal medium and incubated at 28°C (280 rpm) for 14 hours. The culture was harvested by centrifuging at 4°C (3750 rpm) for 10 min. The supernatant was decanted and the aggregated germilings was transferred to a 250 mL flask containing 3 mg/mL lysing enzymes (Sigma-Aldrich) and 2 mg/mL yatalase (Sigma-Aldrich) in Osmotic medium (1.2 M MgSO4, 10 mM Sodium Phosphate buffer) and incubated at 25°C (80 rpm) for 12 hours. Cells were poured directly in a sterile 30 mL glass Corex tube and overlayed very gently with 10 mL of trapping buffer (0.6 M Sorbitol, 0.1 M Tris-HCI, pH 7.0) before centrifuging at 4°C (5000 rpm) for 15 min. Protoplasts at the buffer interface were removed with pipet and diluted with 1 volume of STC buffer (1.2 M sorbitol/10 mM Tris HCl, pH 7.5/10 mM CaCl2), before centrifuging at 4°C (6000 rpm) for 5 min. The supernatant was then decanted and the protoplasts were diluted with 1 volume of STC buffer before using for transformation. Then 100 μL of PEG solution at pH 8.0 (400 mg/mL polyethylene glycol 4000, 50 mM calcium chloride and 10 mM Tris-HCl) was added to the protoplast suspension. The mixture was subsequently combined with 10 μg of the DNA fragment with which the cells were to be transformed. The mixture was incubated at 4°C for 20 min to allow the transformation to proceed. After incubation on ice, 1 ml of the PEG solution was added to the reaction mixture, and the mixture was incubated at room temperature for additional 5 min. The resulting cells were plated on a SMMT agar medium((NH4)2C4H4O6 1.84 g/L, KCl 0.52 g/L, KH2PO4 1.52 g/L, MgSO4·7H2O 0.52 g/L, glucose 10 g/L, sorbitol 218.6 g/L, and agar 18 g/L, supplemented with a mixture comprised of ZnSO4·7H2O 22.0 mg/L, H3BO3 11.0 mg/L, MnCl2-4H2O 5 mg/L, FeSO4·7H2O 5 mg/L, CoCl2·5H2O 1.6 mg/L, CuSO4·7H2O 1.6 mg/L, (NH4)6Mo7O24·4H2O 1.1 mg/L Na2EDTA 50.0 mg/L, and adjusted to pH 6.5 with 1 N KOH) with a suitable selection agent, and overlaid with a SMMT soft agar medium. The transformants were transferred to fresh SMMT agar plates and grown at 25°C for another 7 days. For selection of transformants, zeocin (200 μg/mL), hygromycin (100 μg/mL) or glufosinate (8 mg/mL) was used as the selection marker for ccsR overexpression integration, lig4 knockout or ccsB knockout, respectively. Correct insertion of the bar marker in the ccsB gene among transformants were screened using PCR as shown in Supplementary Fig. 9. ΔccsB-37 was found to be the correct mutant (Supplementary Table 6)
Protein Expression and Purification
The expression plasmid for CcsB pYC01 (Supplementary Table 7) was transformed into E. coli strain BL21 (DE3) for expression of CcsB. Luria Broth media (1 L) supplemented with ampicillin (100 mg/L) inoculated with BL21(DE3)/pYC-1 was grown to an OD600 of 0.6. Protein expression was then induced with 0.12 mM of isopropylthio-β-d-galactoside (IPTG, Sigma-Aldrich), followed by further incubation with shaking at 250 rpm at 16°C for 16 hours. All enzyme purification steps were conducted at 4°C. E. coli cells were harvested by centrifugation (3750 rpm, 15 minutes, 4°C), resuspended in 20 mL TBS buffer and lysed with sonication on ice. Cellular debris was removed by centrifugation (14000 g, 0.5 h, 4°C). FLAG-tagged proteins were purified by using ANTI-FLAG®M1 Agarose Affinity Gel (Sigma-Aldrich), following the supplied protocols. The cleared cell lysate was applied onto a gravity flow column with packed ANTI-FLAG Agarose Affinity Gel. After extensive washing steps, CcsB was eluted with the FLAG peptide elution buffer (100 μg/mL FLAG peptide, 50 mM Tris-HCl, pH 7.4, 100 mM NaCl). Purified proteins were concentrated and buffered exchanged into 50 mM potassium phosphate buffer (pH 7.0) +20% glycerol, concentrated, aliquoted and flash frozen. Protein concentrations were determined using the Bradford dye-binding assay (Biorad).
In vitro activity test for CcsB
CcsB activity was assayed by monitoring the conversion of substrates into products as analyzed by LC-MS. A typical 100 μL assay solution contained 50 mM potassium phosphate buffer (pH 7.0), 4 mM NADPH (Sigma-Aldrich), 20 μM FAD (Sigma-Aldrich), 6 μM SsuE and 10 μM CcsB. The in vitro reaction was initiated by adding 0.4 mM of substrate, such as rosellichalasin (5), cytochalasin B (6, Sigma-Aldrich), ketocytochalasin (7), iso-precytochalasin (8), or cytochalasin D derivative (12). The reactions were performed at 25°C for 3h, 7h, 11h, and 24h. At each time point, the reaction was terminated by the addition of an equal volume of MeOH. Protein precipitate was removed by centrifugation. The substrates and products were extracted with 200 μL hexane twice, dried and redissovled in 60 μL methanol. A 20 μL sample was then analyzed by liquid chromatography mass spectrometry (LC-MS). LC-MS was conducted with a Shimadzu 2010 EV liquid chromatography mass spectrometer by using both positive and negative electrospray ionization and a Phenomenex Luna 5 μm, 2.0 mm×100 mm C18 reverse-phase column. Samples were separated on a linear gradient of 5% to 95% acetonitrile (CH3CN) (v/v) in H2O at a flow rate of 0.1 mL/min. To examine the effect of the relative concentrations of NADPH and 7 on the distribution of products 8 and 9, separate assays containing 7 (0.4 mM) and NADPH of varying concentrations (3.2 mM, 2.0 mM, 1.2 mM, 0.8 mM, 0.4 mM, and 0.2 mM) were performed and analyzed. The products were extracted and monitored by LC-MS after 1h, 3h, 7h, and 12h. To perform the reaction in D2O, all buffers and stock solutions were prepared with D2O. The remaining conditions were kept the same as above.
Chemical analysis and characterization of compounds from A. clavatus and A. clavatus ΔccsB-37 mutant
For small scale analysis, A. clavatus strains were grown in stationary liquid surface culture as a surface mat on 100×15 mm2 Petri dishes with 15 mL malt extract peptone (MEP) media for 4 days at 25°C. The cultures were extracted with the same volume of ethyl acetate (EtOAc) and evaporated to dryness. The dried extract was dissolved in methanol and analyzed by LC-MS (for conditions see above). To purify intermediate 7, A. clavatus ΔccsB-37 mutant strain was grown in stationary liquid surface culture condition in 4 liters MEP liquid medium divided into 120 large 150×15 mm2 Petri dishes for 5 days at 25°C. The resulting mycelia were extracted two times with equal volumes of acetone, while the culture media were treated with XAD16 resin. Absorbed compounds on XAD16 were eluted by washing with acetone. The organic extracts were combined and evaporated by Rotovap. The dried extract was partitioned between methanol and hexane. The hexane fraction was evaporated to dryness, and purified by ISCO-CombiFlash® Rf 200 (Teledyne Isco, Inc) with a gradient of hexane and acetone (linear gradient of 0% to 30% acetone over 30 min at 10 mL/min) on normal phase silica gel. The fractions contained compound 7 were combined and further purified by reverse-phase HPLC (Phenyl-Hexyl 5μm, 250×10 mm) on a linear gradient of 60% to 90% CH3CN (v/v, free of acid) over 30 min at a flow rate of 2.5 mL/min, to give a pure white solid (7, tR = 16.2 min, 10 mg/12 L). 1H, 13C and 2D NMR spectra were obtained using CDCl3 as solvent on Bruker AV500 spectrometer with a 5 mm dual cryoprobe at the UCLA Molecular Instrumentation Center. Pure 7 was crystallized from MeOH/CHCl3 (5:1) with an initial concentration of 2 mg/mL. Ketocytochalasin (7), colorless crystal, [α]d-148° (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) and 13C NMR(125 MHz, CDCl3) see Supplementary Table 1 and attached spectra. ESI-MS m/z 432 [M + H]+ and 454 [M + Na]+. HRESIMS m/z 432.25183 [M + H]+ (for 432.25387, C28H34NO3); 430.23815[M - H]-(calcd for 430.23877, C28H32NO3).
Biotransformation of 7 to yield 8 and 9
To obtain sufficient amount of 8 and 9 for structural characterization, in vivo biotransformation using E coli that overproduces CcsB was performed. The E coli BL21(DE3)/pYC01 strain was grown in 0.5 L of LB medium supplemented with ampicillin (100 mg/L) to an OD600 = 0.6 at 37°C and 250 rpm. Expression was induced with 0.12 mM IPTG. After induction, the culture was shaken for an additional 16 h at 16°C, and then concentrated 20-fold to 25 mL. To initiate the biotransformation, 2 mg/L of 7 was added to the concentrated culture and the culture was shaken at 250 rpm for an additional 24 h at 16°C. To monitor the conversion of 7, 1 mL aliquots were removed at 3 h, 7 h, 13 h and 24 h and extracted with EtOAc for LC-MS analysis. After 24 hours, the culture was extracted with 25 mL of hexane twice, dried and redissolved in 1 mL of MeOH. The crude extract was then purified by reverse-phase HPLC (Phenyl-Hexyl 5μm, 250×10 mm) on a linear gradient of 60% to 90% CH3CN (v/v, free of acid) over 30 min at a flow rate of 2.5 mL/min, to give 8 (tR = 16.7 min, 0.3 mg) and 9 (tR = 17.7 min, 1.5 mg). Small needles of 9 were obtained from MeOH/CHCl3 (10:1) by slow evaporation with an initial concentration of 1 mg/mL. Iso-precytochalasin (8), white solid, [α]d+83° (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) see Supplementary Table 3 and attached spectra. ESI-MS m/z 448 [M + H]+ and 470 [M + Na]+. HRESIMS m/z 448.24674 [M + H]+ (calcd for 448.24878, C28H34NO4,); 446.23307[M - H]- (calcd for 446.23368, C28H32NO4,). Cytochalasin Z16 (9), colourless needle, [a]d+37° (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) and 13C NMR(125 MHz, CDCl3) see Supplementary Table 2 and attached spectra. ESI-MS m/z 464 [M + H]+ and 486 [M + Na]+.
Isotopic labeling study--Feed with 1-13C, 1-18O2 acetate or 18O2 gas
To test the origin of oxygen at C17 in cytochalasin E (1), doubly labeled acetate (1-13C and 18O2) was fed to A. clavatus grown in potato-dextrose (PD) liquid stationary cultures. Labeled 1 was isolated by extraction with EtOAc, purified by SiO2 gel column chromatography (97-3, CH2Cl2-MeOH), and characterized by 1H and 13C-NMR. To confirm the molecular origin of oxygen at C17, 1 was isolated from A clavatus grown in a closed system in which the oxygen consumed was replaced by 18O2 (apparatus see Supplementary Fig. 4a). A. clavatus is inoculated in fresh PD medium (4×50 mL), and incubated in a closed system at 30°C. The closed system is connected via Tygon tubing to a condensation trap, a stirring 3 M KOH trap (to remove excess CO2) and an aquarium pump (controlled by a Variac, 3-5 V). A pressure-equalizing burette (containing 0.5 g/L CuSO4) is filled with either 16O2 or 18O2, to create a slightly over-pressurized system. As the oxygen is consumed, more is added. In the first 24 h of growth 50 mL residual 16O2 is consumed. In the following 49 h, 1130 mL 18O2 is consumed, and finally, the system is purged with 170 mL 12O2 in the final 14 h. Labeled 1 is isolated and characterized by 1H and 13C-NMR.
Crystallographic data of 7 and 9 were deposited at The Cambridge Crystallographic Data Centre, and allocated the deposition numbers CCDC 970431 and CCDC 970432, respectively. The data can be obtained free of charge via www.ccdc.cam.ac.uk/products/csd
Supplementary Material
Acknowledgments
This work was supported by the US NIH (1R01GM085128 and 1DP1GM106413) to Y.T.; Natural Sciences & Engineering Research Council of Canada (NSERC) and Canada Research Chair in Bioorganic & Medicinal Chemistry to J. C. V; US NSF CHE-1059084 to K.N.H. A.P. thanks the Chemistry Biology Interface program (T32GM008496) for support. NMR instrumentation was supported by the NSF equipment grant CHE-1048804. We thank Dr. Yit-Heng Chooi and Prof. N. K. Garg for helpful discussions. We thank Dr. Saeed I. Khan at the UCLA Department of Chemistry and Biochemistry crystallography facility for solving the X-ray structures. We thank Prof. Dehai Li at Chinese Ocean University for providing the standards for 5, 9 and 10. We acknowledge the Extreme Science and Engineering Design Environment (XSEDE) program (TG- CHE040013N) for providing high-performance computing resources.
Footnotes
Contributions: Y. H., D. D., J. C.V. and Y. T. developed the hypothesis and designed the study. D. D. performed the isotopic labeling studies in A. clavatus, J. T. and D. D. prepared selected substrate analogs. Y. H performed the compound isolation and characterization. Y. H. performed the in vitro analysis of CcsB functions. A.P and K.N.H performed and interpreted the density functional theory (DFT) calculations. All authors analyzed and discussed the results. Y. H., J. C. V. and Y. T. prepared the manuscript.
Competing Financial Interests: The authors declare no competing financial interests.
Contributor Information
John C. Vederas, Email: john.vederas@ualberta.ca.
Yi Tang, Email: yitang@ucla.edu.
References
- 1.Huijbers MM, Montersino S, Westphal AH, Tischler D, van Berkel WJ. Flavin dependent monooxygenases. Arch Biochem Biophys. 2014;544:2–17. doi: 10.1016/j.abb.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CT, Wencewicz TA. Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat Prod Rep. 2013;30:175–200. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teufel R, et al. Flavin-mediated dual oxidation controls an enzymatic Favorskii-type rearrangement. Nature. 2013;503:552–556. doi: 10.1038/nature12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsunematsu Y, et al. Distinct mechanisms for spiro-carbon formation reveal biosynthetic pathway crosstalk. Nat Chem Biol. 2013;9:818–825. doi: 10.1038/nchembio.1366. [DOI] [PubMed] [Google Scholar]
- 5.Francisco WA, AbuSoud HM, Topgi R, Baldwin TO, Raushel FM. Interaction of bacterial luciferase with 8-substituted flavin mononucleotide derivatives. J Biol Chem. 1996;271:104–110. doi: 10.1074/jbc.271.1.104. [DOI] [PubMed] [Google Scholar]
- 6.de Gonzalo G, Mihovilovic MD, Fraaije MW. Recent Developments in the Application of Baeyer-Villiger Monooxygenases as Biocatalysts. Chembiochem. 2010;11:2208–2231. doi: 10.1002/cbic.201000395. [DOI] [PubMed] [Google Scholar]
- 7.Leisch H, Morley K, Lau PCK. Baeyer-Villiger Monooxygenases: More Than Just Green Chemistry. Chem Rev. 2011;111:4165–4222. doi: 10.1021/cr1003437. [DOI] [PubMed] [Google Scholar]
- 8.Zhang H, Liu HB, Yue JM. Organic Carbonates from Natural Sources. Chem Rev. 2014;114:883–898. doi: 10.1021/cr300430e. [DOI] [PubMed] [Google Scholar]
- 9.Tomoda H, et al. Phenochalasins, inhibitors of lipid droplet formation in mouse macrophages, produced by Phomopsis sp FT-0211. J Antibiot. 1999;52:851–856. doi: 10.7164/antibiotics.52.851. [DOI] [PubMed] [Google Scholar]
- 10.Pongcharoen W, Rukachaisirikul V, Phongpaichit S, Rungjindamai N, Sakayaroj J. Pimarane diterpene and cytochalasin derivatives from the endophytic fungus Eutypella scoparia PSU-D44. J Nat Prod. 2006;69:856–858. doi: 10.1021/np0600649. [DOI] [PubMed] [Google Scholar]
- 11.Scherlach K, Boettger D, Remme N, Hertweck C. The chemistry and biology of cytochalasans. Nat Prod Rep. 2010;27:869–886. doi: 10.1039/b903913a. [DOI] [PubMed] [Google Scholar]
- 12.Vederas JC. Structural Dependence of O-18-Isotope Shifts in C-13-Nmr Spectra. J Am Chem Soc. 1980;102:374–376. [Google Scholar]
- 13.Qiao KJ, Chooi YH, Tang Y. Identification and engineering of the cytochalasin gene cluster from Aspergillus clavatus NRRL 1. Metab Eng. 2011;13:723–732. doi: 10.1016/j.ymben.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirza IA, et al. Crystal Structures of Cyclohexanone Monooxygenase Reveal Complex Domain Movements and a Sliding Cofactor. J Am Chem Soc. 2009;131:8848–8854. doi: 10.1021/ja9010578. [DOI] [PubMed] [Google Scholar]
- 15.Malito E, Alfieri A, Fraaije MW, Mattevi A. Crystal structure of a Baeyer-Villiger monooxygenase. Proc Natl Acad Sci U S A. 2004;101:13157–13162. doi: 10.1073/pnas.0404538101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishiuchi K, et al. Combinatorial Generation of Complexity by Redox Enzymes in the Chaetoglobosin A Biosynthesis. J Am Chem Soc. 2013;135:7371–7377. doi: 10.1021/ja402828w. [DOI] [PubMed] [Google Scholar]
- 17.Kimura Y, Nakajima H, Hamasaki T. Structure of Rosellichalasin, a New Metabolite Produced by Rosellinia-Necatrix. Agr Biol Chem Tokyo. 1989;53:1699–1701. [Google Scholar]
- 18.Lin ZJ, et al. Bioactive Cytochalasins from Aspergillus flavipes, an Endophytic Fungus Associated with the Mangrove Plant Acanthus ilicifolius. Helv Chim Acta. 2009;92:1538–1544. [Google Scholar]
- 19.Minato H, Matsumot M. Studies on Metabolites of Zygosporium-Masonii .1. Structure of Zygosporin-A. J Chem Soc C. 1970:38-&. doi: 10.1039/j39700000038. [DOI] [PubMed] [Google Scholar]
- 20.Rubin MB, Inbar S. Equilibria among Anions of Alpha-Hydroxy Beta-Diketones and Alpha-Ketol Esters. J Org Chem. 1988;53:3355–3358. [Google Scholar]
- 21.Hong XC, Mejia-Oneto JM, Padwa A. Lewis acid-promoted alpha-hydroxy beta-dicarbonyl to alpha-ketol ester rearrangement. Tetrahedron Lett. 2006;47:8387–8390. doi: 10.1016/j.tetlet.2006.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mejia-Oneto JM, Padwa A. Total synthesis of the alkaloid (+/-)-aspidophytine based on carbonyl ylide cycloaddition chemistry. Helv Chim Acta. 2008;91:285–302. [Google Scholar]
- 23.Robert JL, Tamm C. Biosynthesis of Cytochalasans .5. Incorporation of Deoxaphomin into Cytochalasin B (Phomin) Helv Chim Acta. 1975;58:2501–2504. doi: 10.1002/hlca.19750580830. [DOI] [PubMed] [Google Scholar]
- 24.Frank B, et al. From genetic diversity to metabolic unity: Studies on the biosynthesis of aurafurones and aurafuron-like structures in myxobacteria and streptomycetes. J Mol Biol. 2007;374:24–38. doi: 10.1016/j.jmb.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Tang MC, He HY, Zhang F, Tang GL. Baeyer-Villiger Oxidation of Acyl Carrier Protein-Tethered Thioester to Acyl Carrier Protein-Linked Thiocarbonate Catalyzed by a Monooxygenase Domain in FR901464 Biosynthesis. Acs Catal. 2013;3:444–447. [Google Scholar]
- 26.Yelton MM, Hamer JE, Timberlake WE. Transformation of Aspergillus-Nidulans by Using a Trpc Plasmid. Proc Natl Acad Sci U S A. 1984;81:1470–1474. doi: 10.1073/pnas.81.5.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.