

Abstract
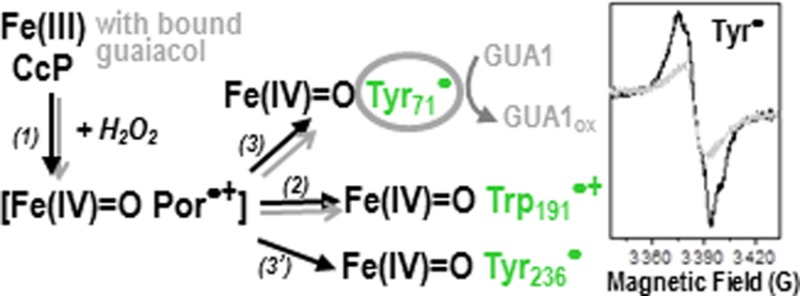
The location of the Trp radical and the catalytic function of the [Fe(IV)=O Trp191•+] intermediate in cytochrome c peroxidase (CcP) are well-established; however, the unambiguous identification of the site(s) for the formation of tyrosyl radical(s) and their possible biological roles remain elusive. We have now performed a systematic investigation of the location and reactivity of the Tyr radical(s) using multifrequency Electron Paramagnetic Resonance (EPR) spectroscopy combined with multiple-site Trp/Tyr mutations in CcP. Two tyrosines, Tyr71 and Tyr236, were identified as those contributing primarily to the EPR spectrum of the tyrosyl radical, recorded at 9 and 285 GHz. The EPR characterization also showed that the heme distal-side Trp51 is involved in the intramolecular electron transfer between Tyr71 and the heme and that formation of Tyr71• and Tyr236• is independent of the [Fe(IV)=O Trp191•+] intermediate. Tyr71 is located in an optimal position to mediate the oxidation of substrates binding at a site, more than 20 Å from the heme, which has been reported recently in the crystal structures of CcP with bound guaicol and phenol [Murphy, E. J., et al. (2012) FEBS J. 279, 1632–1639]. The possibility of discriminating the radical intermediates by their EPR spectra allowed us to identify Tyr71• as the reactive species with the guaiacol substrate. Our assignment of the surface-exposed Tyr236 as the other radical site agrees well with previous studies based on MNP labeling and protein cross-linking [Tsaprailis, G., and English, A. M. (2003) JBIC, J. Biol. Inorg. Chem. 8, 248–255] and on its covalent modification upon reaction of W191G CcP with 2-aminotriazole [Musah, R. A., and Goodin, D. B. (1997) Biochemistry 36, 11665–11674]. Accordingly, while Tyr71 acts as a true reactive intermediate for the oxidation of certain small substrates that bind at a site remote from the heme, the surface-exposed Tyr236 would be more likely related to oxidative stress signaling, as previously proposed. Our findings reinforce the view that CcP is the monofunctional peroxidase that most closely resembles its ancestor enzymes, the catalase-peroxidases, in terms of the higher complexity of the peroxidase reaction [Colin, J., et al. (2009) J. Am. Chem. Soc. 131, 8557–8563]. The strategy used to identify the elusive Tyr radical sites in CcP may be applied to other heme enzymes containing a large number of Tyr and Trp residues and for which Tyr (or Trp) radicals have been proposed to be involved in their peroxidase or peroxidase-like reaction.
Protein-based radicals are involved in the redox chemistry of various metalloproteins.1 Tryptophans and tyrosines can play a crucial role in facilitating electron transfer between redox centers, when behaving as transient radicals,2 and also can be true intermediates in the catalytic cycle of enzymes, when reacting in a concerted way with the metal active site.1 Cytochrome c peroxidase (CcP) and its catalytic reaction have been extensively characterized for more than two decades (reviewed in refs (3−5)). For a long time, CcP was the sole heme peroxidase in which a Trp-based radical intermediate, the [Fe(IV)=O Trp191•+] species,6−8 was identified as the reactive intermediate for the oxidation of an unusual substrate, cytochrome c(9) (for a recent review, see ref (10)). At variance with the typical heme-edge reaction of peroxidases with small substrates (for a recent review, see ref (11)), the reaction of CcP with the surface-bound substrate cyt c occurs via a well-defined electron transfer pathway between Trp191 and the heme site in cyt c(12) (for a recent review, see ref (4)).
EPR and ENDOR spectroscopic characterization unequivocally showed that the broad EPR spectrum at 4 K of the Trp radical in CcP originates from the distinct magnetic properties of the [Fe(IV)=O Trp191•+] intermediate, due to the exchange coupling interaction between the radical and the heme iron moiety.13−16 The contribution of a narrow radical signal to the EPR spectrum of the high-valent intermediate in CcP was also reported.13,17,18 The signal, assigned to a Tyr radical, could possibly reflect different pathways in radical translocation, from the heme to surface tyrosines, related to an antioxidant role in CcP under conditions of excess hydrogen peroxide.18 A number of studies using either direct measurement of the radical spectra by EPR techniques13,16,19 or indirect detection by spin traps and/or spin-labels and mass spectrometry18,20,21 of the tyrosyl radicals resulted in multiple assignments. Accordingly, the unambiguous identification of the site(s) for the formation of tyrosyl radical(s) in CcP has been elusive, as has been their biological role in substrate oxidation.
Interestingly, CcP (and the more recently reported peroxidase from Leishmania major(10)) appears to be the only member(s) of the monofunctional peroxidase family allowing the formation of a high-valent ferryl intermediate comprising a radical delocalized on the heme proximal-side tryptophan (Trp191 in CcP), as in the case of their ancestor enzymes KatGs22,23 (Trp330 in BpKatG and Trp321 in MtKatG). It could then be anticipated that CcP might also exhibit a protein-based radical mechanism related to the oxidation of remote substrates, similar to the case of Burkholderia pseudomallei KatG.24 A recent report of the crystal structure of a CcP–guaiacol complex obtained by soaking the CcP crystals in a substrate solution25 showed that guaiacol does not bind at the typical heme-edge binding site of monofunctional peroxidases (reviewed in ref (11)). One of the two binding sites described for guaiacol is located more than 20 Å from the heme and from Trp191. On the basis of the long distance between this substrate binding site and the heme, and assuming a heme-edge typical peroxidase reaction, Raven and co-workers concluded that the remote binding site should be catalytically inactive, despite the fact that both guaiacol and phenol were found to bind there and not at the typical δ-heme edge of peroxidases.25
To explain the reported efficient reaction of CcP with guaiacol as the substrate, we hypothesized the involvement of a Tyr as redox-active amino acid in the peroxidase reaction, similar to the cases of the Tyr•-mediated oxidation of ABTS substrate in lactoperoxidase,26 and the Trp•-mediated oxidation of veratryl alcohol in lignin peroxidase.27 Therefore, in this work, we conducted a systematic investigation of the localization and putative role in guaiacol oxidation of the Tyr residues formed in CcP using multiple-site Trp/Tyr variants and multifrequency EPR spectroscopy. We have now identified Tyr71 and Tyr236 as those residues contributing primarily to the EPR Tyr• signal in CcP. Our findings confirm the previous assignment of Tyr236, a surface-exposed amino acid, that was based on indirect detection using MNP labeling and protein cross-linking,18 as well as covalent modification upon reaction of W191G CcP with 2-aminotriazole.19 In contrast, the radical on Tyr71 has never been detected probably because the site is not readily accessible to spin-labels or cross-linking reactions. More importantly, we show that [Fe(IV)=O Tyr71•] is the reactive intermediate in the oxidation of guaiacol binding at the site remote from the heme, and that Trp51 is involved in the electron transfer pathway between Tyr71 and the heme.
Experimental Procedures
Sample Preparation
Construction, expression, and purification of site-directed mutant proteins of CcP were performed as described previously.28 The CcP-MI protein used as a template for site-directed mutagenesis reported in this work differs from the yeast wild-type CcP protein at positions 53 and 152 (T53I and D152G) and contains a Met-Ile pair preceding the first amino acid. The mutations were screened by restriction digestion and confirmed by DNA sequencing and mass spectrometry of the purified proteins. Guaiacol (extra pure) was purchased from Fisher Scientific and hydrogen peroxide from Fluka Analytical.
EPR Spectroscopy
The 9 GHz EPR spectra were recorded on a Bruker EleXsys E500 spectrometer equipped with a standard Bruker ER 4102 X-band resonator and a liquid helium cryostat (Oxford Instruments, ESR 900). The home-built high-field EPR spectrometer (95–285 GHz) has been described previously.29 EPR quartz tubes with an external diameter of 4 mm were used for measurements on both EPR spectrometers. Initial (heme) concentrations of 1.5 mM of the CcP samples were used for recording the 9 and 285 GHz EPR spectra. All EPR samples were prepared by mixing, manually and directly in the EPR tubes, the ferric CcP [100 mM potassium phosphate (pH 6.0)] with a 2-fold excess of hydrogen peroxide [100 mM potassium phosphate (pH 6.0)] in equal volumes. The mixing was conducted on ice for 10 s, and then the EPR tube was flash-frozen in liquid nitrogen. Little difference in the type and yield of the EPR signals of the Trp and Tyr radicals was observed for the reaction of the CcP-MI samples with hydrogen peroxide when using mixing times of 5, 15, and 60 s, in contrast to the case of CcP-MKT previously reported.16 The 9 GHz EPR experiments on the reaction of CcP (wild type and Y236F variant) with the guaiacol substrate were conducted using 40 μL of enzyme at 0.5 mM (initial concentration) and adding 2 μL of substrate at 1 M (initial concentration) to produce a final excess of 100-fold. Smaller excesses were also tested. The incubation of CcP with guaiacol was conducted at room temperature for 20 min and prior to the reaction with a 2-fold excess of hydrogen peroxide (10 s mixing time on ice). The EPR spectra of ferric CcP samples were recorded before and after incubation with guaiacol. No spectral changes were observed upon binding of guaiacol (and using a 100-fold excess).
Results and Discussion
The previous reports from different groups with respect to the identification of the site(s) of the Tyr radical(s) in CcP by characterizing single Tyr mutations clearly demonstrated the complexity of the case. Such studies naturally led to the general consensus that once a putative site for the formation of a Tyr radical is suppressed (by site-directed mutagenesis) the radical could be formed on other Tyr(s), with the underlying interpretation that the formation of Tyr radicals in CcP may be a rather random event, with no specific electron transfer pathway and/or catalytic function involved. Later, a more inclusive approach using multiple-site Tyr and Trp mutations on CcP was conducted by one of our laboratories.28 Such CcP variants were designed to incorporate, in an additive fashion, some of the Tyr sites proposed from the previous reports of single-site mutations, as well as the two Trp residues of the proximal and distal heme sides. The kinetic characterization of such CcP multiple mutants by stopped-flow UV–vis spectroscopy showed that the sequential removal of Tyr residues resulted in an increased stability of the otherwise very short-lived Fe(IV)=O Por•+ species (the so-called Compound I intermediate in peroxidases), making possible its detection in the millisecond time range and through its distinct electronic absorption spectrum.28 The subsequent changes observed in the heme absorption spectrum of the reacted CcP were consistent with the spontaneous decay of the Fe(IV)=O Por•+ intermediate to a ferryl species (so-called Compound II). However, as we have shown previously, it is not possible to discriminate between the absorption spectra of an Fe(IV)=O species and an [Fe(IV)=O Tyr•] intermediate in heme enzymes.30−32 Therefore, in this work, we have applied multifrequency EPR spectroscopy to the same set of CcP multiple (Tyr and Trp) variants, as the starting point for the specific characterization of the Tyr• sites formed in CcP and their putative role in substrate oxidation, and in the context of the recently reported crystal structure of CcP with bound guaiacol.
EPR Spectroscopic Characterization of the Tyr Radicals Formed in Multiple Tyr/Trp Mutants in CcP
Figure 1 shows the 285 GHz (panel A) and 9 GHz (panel B) EPR spectra of the multiple-Tyr/Trp variants upon reaction with a 2-fold excess of hydrogen peroxide. These variants include substitutions of the Trp residues that are close to the heme, on the proximal and distal sides (Trp191 and Trp51), and also incorporate, in an additive fashion, putative Tyr• sites.28 The EPR spectra shown in panels A and B of Figure 1 correspond to those of W191F (top, black trace), W191F/W51F (middle, magenta trace) and W191F/W51F/Y187F/Y229F/Y236F (bottom, purple trace). The EPR spectra of two other CcP multiple-site variants, W191F/W51F/Y187F and W191F/W51F/Y187F/Y229F/Y236F/Y36F/Y39F/Y42F, were identical to those of W191F/W51F and W191F/W51F/Y187F/Y229F/Y236F, respectively (Table 1).
Figure 1.
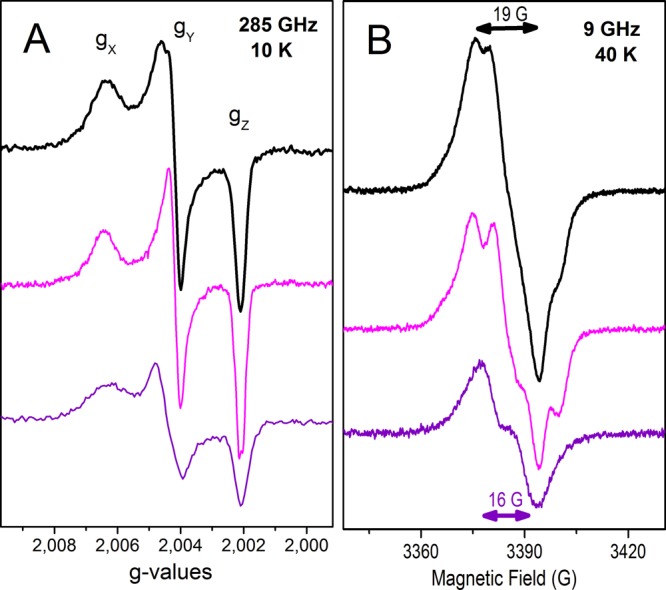
(A) 285 GHz and (B) 9 GHz EPR spectra of Tyr radicals formed in the W191F (top, black), W191F/W51F (middle, magenta), and W191F/W51F/Y187F/Y229F/Y236F (bottom, purple) variants of cytochrome c peroxidase upon reaction with a 2-fold excess of hydrogen peroxide, using a mixing time on ice of 10 s. (A) The 285 GHz spectra (shown in g scale) were recorded at 10 K, using a frequency modulation of 30 kHz; a field modulation of 5 G was used for the top and middle spectra, while a field modulation of 10 G was used for the bottom spectra because of the much lower radical yield. The top and middle spectra result from averages of four scans, while 10 scans on average were used for the bottom spectrum. (B) The 9 GHz spectra (top and middle) were recorded at 40 K, with a modulation amplitude of 1 G, a microwave power of 0.08 mW, and a modulation frequency of 100 kHz. For the bottom spectrum, a modulation amplitude of 3 G and a microwave power of 0.2 mW were required because of the much lower radical yield.
Table 1. Assignments of the Sites for the Two Primary Tyr• Intermediates in Cytochrome c Peroxidase (CcP) and the Reaction with the Guaiacol Substrate, Based on the EPR Characterization of the Designed CcP Variants Shown in Panels A and B of Figures 1 and 4.
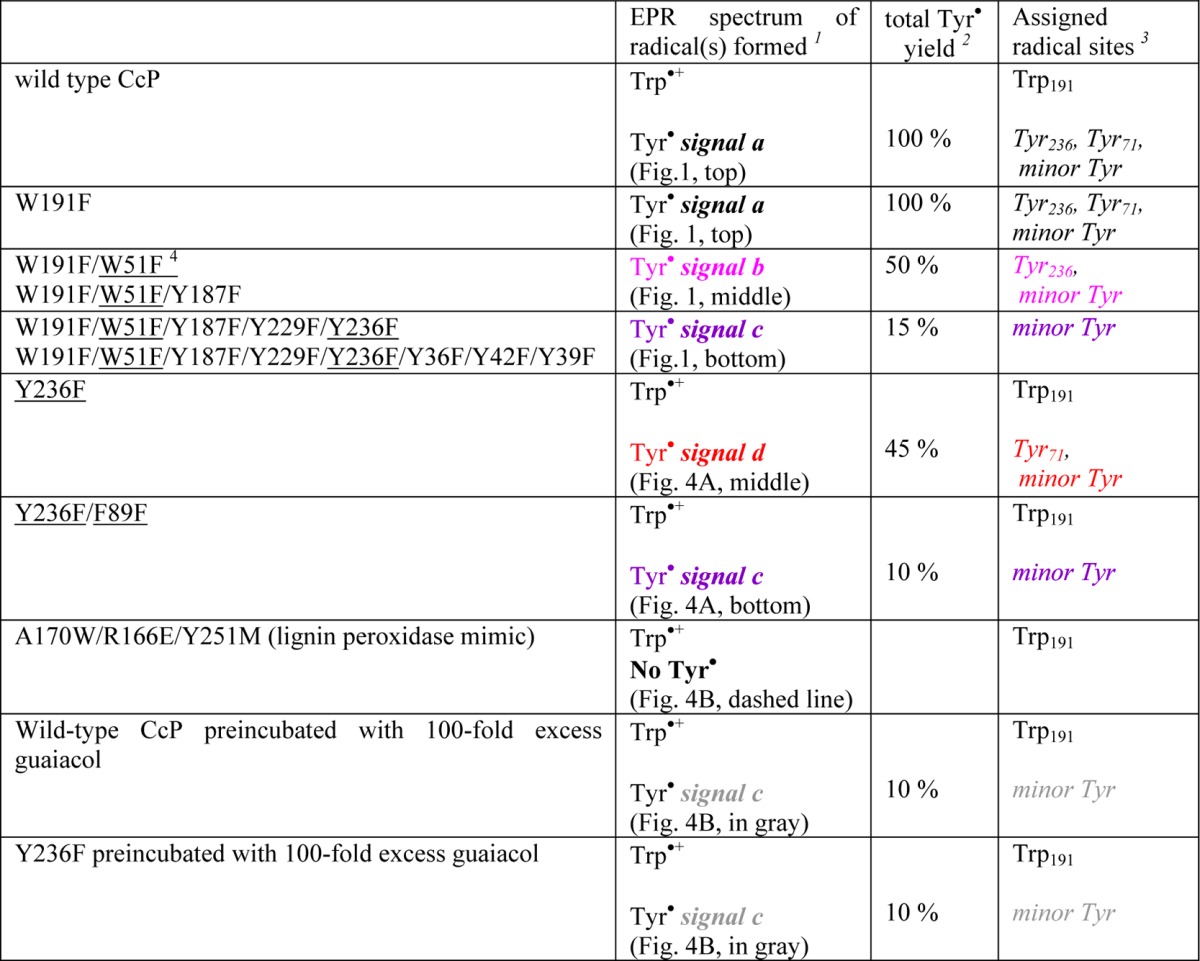
The colors highlight the four different Tyr radical signals that correspond to those in Figures 1 and 4.
The percentage of Tyr radical(s) yield for the variants was estimated by comparison of the signal intensity with that of the wild-type CcP with H2O2 (considered the 100% yield).
Tyr sites that contribute to the Tyr• EPR signal in each sample.
The underlined residues are those inducing EPR spectral changes, consistent with suppression of the primary Tyr radical sites. The W51F variant indirectly impeded the formation of Tyr71• by the disruption of the long-range electron transfer pathway, while the F89L variant destabilized radical formation on Tyr71 by inducing steric effects on its microenvironment (see the text).
The advantageous resolution of the EPR spectra of organic radicals when they are recorded at high magnetic fields (285 GHz and 10 T) showed that the intrinsic g values and g anisotropy, defined as |gx – gz|, of all five CcP variants (Figure 1A) were those expected for tyrosyl radicals.33,34 The HF-EPR spectra of the W191F variant (Figure 1A, black trace) could be best simulated with g values of 2.0066, 2.0041, and 2.0020 for gx, gy, and gz, respectively, including a distribution in gx values with a Gaussian width of 0.0007, to account for the broad low-field edge of the spectrum. The Tyr• spectra of the W191F/W51F and W191F/W51F/Y187F variants (Figure 1A, magenta trace) showed small, yet significant, differences compared to those of the W191F sample (Figure 1A, black trace). In particular, the gx component of the HF-EPR spectrum was narrower, thus requiring a smaller distribution of gx values (Gaussian width of 0.0005) in the spectral simulations. Also, proton hyperfine couplings were well-resolved on the high-field edge of the spectrum (gz component) of the W191F/W51F and W191F/W51F/Y187F variants (Figure 1A, magenta trace; better shown in Figure S2 of the Supporting Information).
The Tyr• spectra of the W191F/W51F/Y187F/Y229F/Y236F and W191F/W51F/Y187F/Y229F/Y236F/Y36F/Y39F/Y42F variants (Figure 1A, purple trace) showed detectable differences on the gy component of the HF-EPR spectra (better shown in Figure S1 of the Supporting Information).
Consistently, the 9 GHz EPR spectra of the same CcP samples (Figure 1B) showed small, yet significant, differences in the overall shapes related to their proton hyperfine couplings, which dominate the EPR spectra of the tyrosyl radicals at this lower frequency. In addition, the W191F/W51F/Y187F/Y229F/Y236F and W191F/W51F/Y187F/Y229F/Y236F/Y36F/Y39F/Y42F variants showed a narrower Tyr radical signal [peak to trough of 16 G (Figure 1B, purple trace)] compared to the signals of the others [peak to trough of 19 G (Figure 1B, black and magenta traces)]. It is of note that the Tyr• yield of the W191F/W51F sample was 50% of that of the W191F CcP sample, while that of the W191F/W51F/Y187F/Y229F/Y236F variant was much lower (15% of that of the W191F CcP sample). The radical yield could be directly estimated from the intensities of the EPR signals because the EPR samples had the same final volume, were prepared using the same conditions (described in Experimental Procedures), and were measured using the same instrument settings. As mentioned in the legend of Figure 1, because of the much lower yield of the Tyr• in the case of the W191F/W51F/Y187F/Y229F/Y236F and W191F/W51F/Y187F/Y229F/Y236F/Y36F/Y39F/Y42F variants, the spectra shown in Figure 1 (purple traces) were recorded using a larger number of scans (at 285 GHz) or a higher modulation amplitude (at 9 GHz) compared to those of W191F, W191F/W51F, and W191F/W51F/Y187F variants.
Taken together, the differences in yields, g values, and proton hyperfine couplings detected in the EPR spectra of the multiple-Tyr/Trp variants of CcP recorded at both 285 and 9 GHz are consistent with two predominant, and chemically different, Tyr radicals contributing to the wild-type Tyr• EPR spectrum of CcP with a minor contribution of a third species, as summarized in Table 1.
Assignments of the Tyr Radical Sites
To rationalize our findings, it was crucial to take into account the fact that the Tyr• EPR spectra of the multiple-substitution variants were identical in a two by two fashion and for both frequencies (see Table 1). Specifically, the W191F variant showed the same Tyr• spectrum as wild-type CcP, thus indicative of no effect of the Trp191 mutation [heme proximal side (see Figure 2)] on the formation of the Tyr radicals, as we have previously shown for the CcP-MKT protein.16 However, a different Tyr• spectrum with half-signal intensity (Figure 1, magenta trace) was observed when substituting Trp51 on the heme distal side in addition to Trp191 (W191F/W51F variant). Because the HF-EPR spectra are inconsistent with a Trp radical,23,33 this result clearly indicates that substitution of Trp51 (see Figure 2) impeded the formation of one of the two primary Tyr radicals in CcP.
Figure 2.
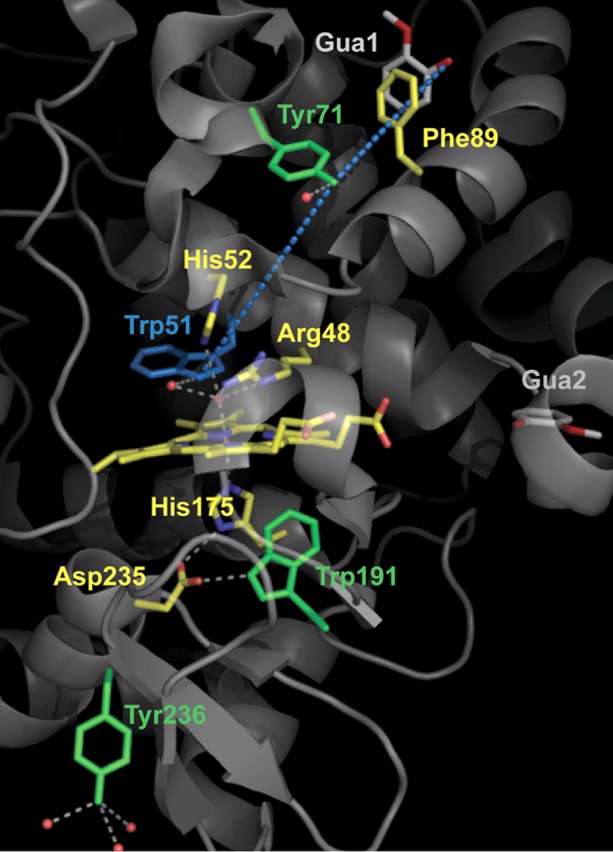
Heme site in cytochrome c peroxidase (CcP), including the catalytically relevant residues of the proximal and distal sides (yellow), and the two binding sites for guaiacol (labeled GUA1 and GUA2) recently reported by Raven and co-workers.25 Tyr71 and Tyr236, the Tyr radical sites identified in this work, as well as the well-characterized Trp191 radical site for the [Fe(IV)=O Trp191•+] intermediate are all colored green. The proposed electron transfer pathway between Tyr71 (the oxidizing species for binding of guaiacol at GUA1) and the heme, including Trp51 and based on the multifrequency EPR study in this work, is shown as blue dotted lines. This figure was made using the published structure of the CcP–guaiacol complex (Protein Data Bank entry 4A6Z).
We have previously reported a comparable situation in the case of BpKatG,23 the bifunctional peroxidase from B. pseudomallei (see Figure 3). Indeed, Trp111 (equivalent to Trp51 in CcP) on the heme distal side was shown to be part of the extended H-bonding network (white dotted trace) that plays a crucial role in the electron transfer pathways (blue dotted traces) to the two remote protein-based radical sites, Trp139 and Trp153.23 Specifically, the EPR characterization of the W111F variant in BpKatG showed that the substitution of Trp111 with Phe hindered the formation of both Trp139• and Trp153•, the experimental evidence being the disappearance of the Trp• signals from the EPR spectrum of the W111F variant (see Figure 5 of ref (23)). It is of note that previous reports of CcP mutants showed cross-links between Trp51 and the heme, in the case of the W191F variant,35 and between Trp51 and Y52, in the case of the H52Y variant,36 indicating that Trp51 is prone to forming a transient radical species as in the case of Trp111 in BpKatG.23
Figure 3.
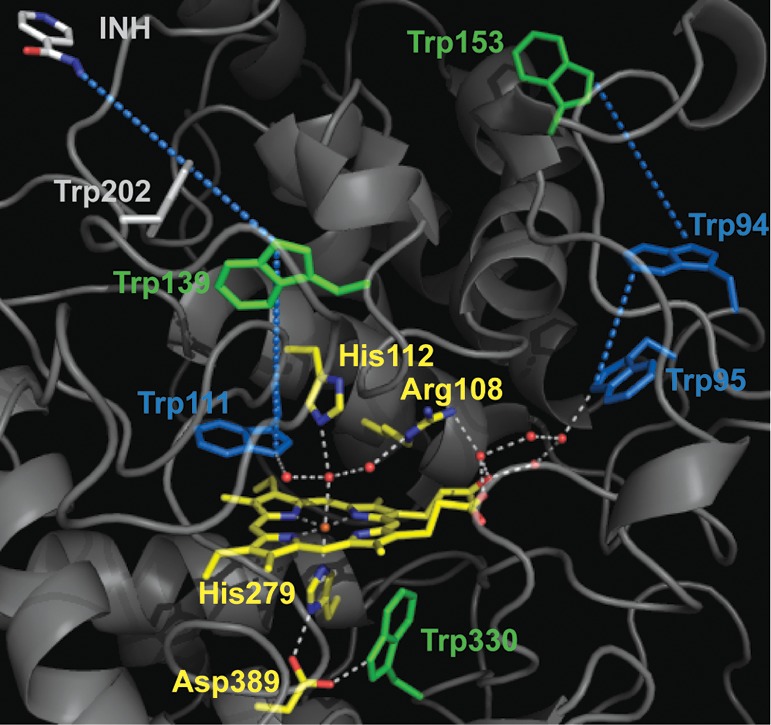
Crystallographic structure of the N-terminal domain of B. pseudomallei catalase-peroxidase (BpKatG) with bound isoniazid (INH), showing the environment of the heme cofactor.24 Trp330, Trp139, and Trp153 (green) are the radical sites identified in a previous multifrequency EPR spectroscopic characterization.23 The electron transfer pathways (blue dotted lines), including the extended H-bonding network of the heme distal side (white dotted lines) and Trp95, Trp94, and Trp111 (all colored blue), are also shown. This figure was made using the published structure of BpKatG cocrystallized with INH (Protein Data Bank entry 3N3N).
Comparison of the W191F/W51F and W191F/W51F/Y187F variants in CcP allowed us to rule out Tyr187 as a radical site, because both variants showed the very same EPR spectrum (g values, distribution in gx values, and proton hyperfine couplings) and radical yields (see Table 1). In contrast, the distinct Tyr• signal of the W191F/W51F variant clearly disappeared when two other Tyr mutations (Tyr229 and Tyr236) were added (Figure 1A,B, magenta traces), thus revealing a different, and much lower in yield, Tyr• spectrum for the W191F/W51F/W187F/Y229F/Y236F variant (Figure 1, purple traces). This result indicates that substitution of Tyr236 or Tyr229 specifically suppressed the other primary Tyr• site in CcP, leaving just the minor species (also Tyr radical, <15% contribution to the wild-type spectrum).
In conclusion, the EPR spectroscopic characterization of the multiple-Tyr/Trp variants in CcP showed that there are two primary Tyr radicals accounting for 85% of the EPR signal detected in the wild-type sample, forming independently of each other and also independently of the Trp191• species. The EPR results also indicated that one of the Tyr• sites should be either Tyr236 or Tyr229 (both located on the heme proximal-side region) and that the other Tyr• site should be on the heme distal-side region of the protein (see Figure 2), because Trp51 was required for its formation.
Identification of the Two Tyr• Sites in CcP
On the basis of the conclusions from the EPR spectroscopic characterization of the multiple-Tyr/Trp variants (summarized in Table 1), we then designed and constructed simplified CcP variants to pin down the two primary Tyr• sites. The Tyr236 substitution was then expected to remove the radical site on the heme proximal-side region.a As shown in Figure 4A, the Tyr• signal of the Y236F variant (red trace) showed measurable differences in the overall shape related to the proton hyperfine couplings of the radical, and a clear lower yield (45%) when compared to the wild-type CcP Tyr• signal (black trace), consistent with the lack of a contribution from the Tyr236• species due to the mutation.
Figure 4.

(A) 9 GHz EPR spectra of Tyr radicals formed in wild-type CcP (black), Y236F (red), and Y236F/F89L (purple) variants, upon reaction with a 2-fold molar excess of hydrogen peroxide. (B) Reaction of wild-type CcP and the Y236F variant with guaiacol monitored by the 9 GHz EPR spectrum of their Tyr radicals. The ferric samples of wild-type CcP and the Y236F variant were incubated with a 100-fold molar excess of guaiacol, prior to being mixed with a 2-fold molar excess of hydrogen peroxide. The resulting spectrum in both cases shows a narrower radical signal (solid line, gray) representing 10% of the Tyr radical signal of wild-type CcP (solid line, black), as estimated from their EPR signal intensities. It is of note that because of the very low radical yield upon reaction in the presence of guaiacol, the spectrum (solid line, gray) was recorded using a modulation amplitude of 3 G and a microwave power of 0.2 mW instead of the modulation amplitude of 1 G and the microwave power of 0.08 mW used for the control (solid line, black). The EPR spectra of the A170W/R166E/Y251M variant of CcP (dashed line, black), which does not form any Tyr radical intermediate upon reaction with hydrogen peroxide (see text), show the g ≈ 2 background signal arising from the gz component of the [Fe(IV)=O Trp191•+] species, because this component is not affected by the exchange coupling interaction.16 The experimental conditions for recording the 9 GHz spectra were the same as those in Figure 1B, except for the temperature (40 K).
To narrow the list of possible candidates for the other Tyr radical site, remote from the heme and located in the distal-side region as indicated by the effect of the Trp51 substitution, we took into consideration the existence of the substrate binding site remote from the heme recently reported by Raven and co-workers.25 Specifically, the crystal structure of CcP with bound guaiacol (named GUA1 and GUA2 in Figure 2) revealed two binding sites, neither of which was the typical binding site for aromatic substrates in peroxidases (namely, close to the δ-heme edge) or the surface binding site for cyt c in CcP. One of the guaiacol molecules (named GUA1 in Figure 2) is found to bind more than 20 Å from the heme. The presence of a potentially redox-active residue, Tyr71, with a distance of 10.2 Å between the phenolic oxygen of Tyr71 and guaiacol (see Figure 2 and Figure S2 of the Supporting Information), makes the overall configuration strikingly comparable to that of BpKatG-INH, in which the three-dimensional structure of the enzyme cocrystallized with isoniazid24 revealed an INH binding site more than 20 Å from the heme (see Figure 3), but close enough for a one-step electron transfer to the protein-based site of the [Fe(IV)=O Trp139•] intermediate.23 Moreover, the presence of a structural water within H-bonding distance of the phenolic oxygen of Tyr71 (see Figure S2 of the Supporting Information) is consistent with the electropositive microenvironment of the Tyr radical shown by the HF-EPR spectrum, thus making Tyr71 a good candidate for the other (primary) Tyr• site in CcP.
A different strategy was chosen to probe the radical on Tyr71. We designed a mutation that would destabilize the radical formation without actual substitution of the Tyr site, the advantage being to avoid changes on the electron transfer pathways related to the radical formation that could result in the formation of a new radical. We have previously shown that changes in the microenvironment of the protein-based radical sites in KatGs,37 in particular disruption of H-bonds, destabilize radical formation.38,39 In CcP, a structural water molecule is within H-bonding distance of Tyr71 (see Figure S2 of the Supporting Information). Accordingly, the substitution of Phe89 was expected to induce subtle steric effects on the microenvironment of Tyr71 and the water (Figure S2 of the Supporting Information), which in turn would compromise the formation of the radical. In addition, and to have an unambiguous EPR result, we combined the substitutions on Phe89 and Tyr236 (see above). Then, the Y236F/F89L double substitution was expected to suppress both primary Tyr• sites in CcP (Tyr236• and Tyr71•) and consequently their contribution to the EPR spectrum. This was indeed the case, with the Y236F/F89L variant (Figure 4A, purple trace) showing a very low yield (10%) of a narrower radical spectrum and a peak to trough of 16 G, as in the case of the multiple-site variant W191F/W51F/Y187F/Y229F/Y236F (Figure 1B, purple trace).
It is of note that the contribution of the minor Tyr radical (10–15%) was always present despite the increasing number of Tyr mutations in CcP, indicating that the electron transfer pathway for its formation is uncorrelated to the two primary sites, Tyr71 and Tyr236. Only in the case of the A170W/R166E/Y251M variant was the minority Tyr• signal not detected (Figure 4B, black dashed trace). This triple mutant served to engineer a new Trp radical site on the enzyme surface, thus mimicking the lignin peroxidase case.27 The inhibition of the formation of the naturally occurring Tyr radicals in CcP induced by the A170W/R166E/Y251M variant can be rationalized by the creation of a new and preferential electron flow to the engineered Trp radical site, because the single mutation of Tyr251 showed that this amino acid residue does not contribute to the Tyr• EPR signal. The substitutions of Tyr251 and Arg166 were necessary to make sufficient space for the insertion of Trp at position 170 in CcP, as well as reproducing the microenvironment of the catalytically active surface Trp in lignin peroxidase.27 The EPR spectrum of the A170W/R166E/Y251M variant upon reaction with hydrogen peroxide showed the formation of the exchange-coupled Trp191 radical as in wild-type CcP, and no other radical signal was detected (Figure 4B, black dashed trace), indicating that the putative Trp170• radical was most probably too short-lived to be trapped, as in the case of lignin peroxidase.27
Reaction of CcP with Guaiacol Monitored by EPR Spectroscopy
CcP is known to utilize the typical heme-edge peroxidase mechanism to oxidize small (aromatic) substrates binding at the δ-heme edge, by means of the Fe(IV)=O Por•+ intermediate (reviewed in ref (11)). In addition, the subsequent [Fe(IV)=O Trp191•+] intermediate serves for the oxidation of cyt c, a substrate binding to the enzyme surface (recently reviewed in ref (10)). Our findings show that the hitherto elusive sites for the formation of the Tyr• intermediates in CcP are Tyr71 and Tyr236, and given the recent report of guaiacol having a binding site close to Tyr71 (Figure 2), we investigated its putative role in guaiacol oxidation.
As we have previously shown, EPR spectroscopy is the technique of choice for assessing and selectively monitoring the reactivity with substrates of the [Fe(IV)=O Trp•] and/or [Fe(IV)=O Tyr•] species, formed as alternative intermediates to the Fe(IV)=O Por•+ intermediate in certain peroxidases.22,26,27,32 As in the previous cases, CcP was incubated with guaiacol (100-fold excess) at room temperature and prior to the reaction with hydrogen peroxide (2-fold molar excess), to prevent competition between the fast formation of the radical intermediates and the binding of guaiacol. The 9 GHz EPR spectra of CcP, recorded at 4 K and upon reaction with a 2-fold excess of H2O2, showed that the yield of the Trp191 radical was the same in the absence and presence of guaiacol (see Figure S3A of the Supporting Information), clearly indicating that the [Fe(IV)=O Trp191•+] intermediate was neither involved nor affected by the reaction with guaiacol. In contrast, the disappearance of the major Tyr radical signal in the 40 K EPR spectrum of the wild-type CcP sample preincubated with guaiacol (Figure 4B, gray trace) indicated that Tyr71 and/or Tyr236 was either involved in or affected by the reaction with guaiacol. Then, the EPR characterization of the reaction, with a 2-fold excess of H2O2, of the Y236F variant preincubated with a 100-fold excess of guaiacol allowed us to rule out Tyr236• as the reactive species. The resulting EPR spectrum was also that of the narrower minority species, as in the case of wild-type CcP (Figure 4B, gray trace). The location of Tyr71, with an OTyr71–Ogua1 distance of 10.2 Å (see Figure S2 of the Supporting Information), is quite advantageous for a direct oxidation of guaiacol at the site named GUA1, while Tyr236 is 41 Å distant. In addition, the reported steady-state kinetic data showed that the kcat values for guaiacol oxidation were essentially the same for wild-type CcP (4.1 s–1) and the M119W variant (5.9 s–1), in which guaiacol binds only at a site close to Tyr71 (GUA1), as shown by the crystal structure of the mutant CcP.25 The corresponding reported Km values for wild-type CcP and the M119W variant are 53 and 26 mM, respectively.25 Taken together, this is strong evidence of Tyr71• being the reactive species with the guaicol that binds at GUA1, the site more remote from the heme (see Figure 2). It is worth mentioning that in the report showing the crystal structure of guaiacol bound to CcP,25 Raven and co-workers concluded that the catalytic binding site for guaiacol is the δ-heme edge, despite the fact that it was not observed at such a site, the rationale being a crystallographic artifact and/or weak binding. However, our EPR characterization of the reaction in solution of the CcP high-valent intermediates with guaiacol rules out both the [Fe(IV)=O Trp191•+] intermediate and the preceding [Fe(IV)=O Por•+] intermediate being the reactive species with guaiacol. Consistently, the reported kcat for guaiacol oxidation by the W191F variant (14 s–1) is ∼3 times higher than that of wild-type CcP (4.1 s–1), with KM being essentially the same.40 This shows that suppressing the preferred electron transfer pathway between Trp191 and the heme by the Trp191 substitution results in an enhancement of the alternate pathway to Tyr71, the latter then becoming more efficient in the oxidation of guaiacol.
Conclusions
The two primary sites for Tyr radical formation in CcP, Tyr71 and Tyr236, were identified using multiple-site Trp/Tyr variants of CcP and multifrequency EPR spectroscopy. Our identification of the surface-exposed Tyr236, with the phenolic oxygen pointing outward, as one of the two radical sites in CcP agrees well with previous reports that were based on MNP labeling and protein cross-linking18 and on its covalent modification upon reaction of W191G CcP with 2-aminotriazole.19 The unprecedented radical site identified in this work as Tyr71 is close to the surface, but not necessarily accessible to spin-labels or cross-linking reactions. Tyr71 is located in an advantageous position to mediate the oxidation of substrates binding at a site, rather distant from the heme (named GUA1 in Figure 2), which has been recently reported in the CcP crystal structures with bound guaiacol and phenol molecules.25 The EPR characterization of the multiple-site Trp/Tyr variants also showed that Trp51, located on the heme distal side, is involved in the electron transfer pathway between Tyr71 and the heme. The overall configuration of the substrate binding site (GUA1) and the associated radical intermediate on Tyr71 (see Figure 2) is thus reminiscent of the situation in BpKatG (see Figure 3). In the latter, the isoniazid binding site is also remote from the heme but close to Trp139,24 and the heme distal-side Trp (Trp111) is essential for the formation of the [Fe(IV) Trp139•] intermediate.23
Furthermore, our finding that Tyr71• is the oxidizing intermediate for guaiacol, as shown by monitoring the reactivity of CcP with guaiacol using EPR spectroscopy, indicates different roles for each Tyr radical. Accordingly, Tyr71 is a true reactive intermediate for substrate oxidation, while the surface-exposed Tyr236 is more likely related to oxidative stress signaling, as previously proposed by English and co-workers.18 Despite the fact that yeast does not naturally produce phenolic compounds as secondary metabolites, the central metabolism provides the necessary amino acid precursors for an introduced phenolic biosynthetic pathway, obtained in engineered yeast strains. Accordingly, phenol-producing Saccharomyces and non-Saccharomyces yeast (so-called phenolic yeast) have been characterized as producing volatile phenols,41 which are released during yeast autolysis and impart specific flavors to wine and beer during the fermentation process.42 It is possible then that the [Fe(IV)=O Tyr71•] intermediate in CcP could play a role equivalent to that of the [Fe(IV)=O Trp171•] intermediate of the lignin peroxidase from white-rot fungi, in which the secondary metabolite, veratryl alcohol, is oxidized by the peroxidase.27 Other endogenous compounds, such as indole-3-acetic acid and flavin-type molecules, that have been extracted from yeast43 could be also metabolized by the Tyr71-based radical intermediate of CcP.
Finally, our findings reinforce the view of CcP being the monofunctional peroxidase that most closely resembles their ancestor enzymes KatGs,23 in terms of the more elaborate strategy for the peroxidase reaction, and includes stabilization of the heme proximal-side Trp• as well as well-defined electron transfer pathways for the oxidation of substrates remote from the heme. The strategy used for identifying the elusive Tyr radical sites in CcP, in particular the design of mutations that destabilize formation of the Tyr• radical without actual substitution of the Tyr site, may be generalized to other heme enzymes containing a large number of Tyr and Trp residues and for which Tyr (or Trp) radicals have been proposed to be involved in their peroxidase or peroxidase-like reaction.
Glossary
Abbreviations
- CcP
cytochrome c peroxidase
- KatG
catalase-peroxidase
- BpKatG
B. pseudomallei catalase-peroxidase
- MtKatG
Mycobacterium tuberculosis catalase-peroxidase
- LiP
lignin peroxidase
- cyt c
cytochrome c
- EPR
Electron Paramagnetic Resonance
- HF-EPR
high-field Electron Paramagnetic Resonance
- ENDOR
electron nuclear double resonance
- MNDO
modified neglect of diatomic overlap
- MNP labeling
labeling with 2-methyl-2-nitrosopropane
- INH
isoniazid (isonicotinic acid hydrazide)
- guaiacol
2-methoxyphenol
- ABTS
2,2′-azino-bis[3-ethylbenzothiazoline sulfonate(6)] diammonium salt.
Supporting Information Available
285 GHz EPR spectra of Tyr radicals formed in the multiple-Trp/Tyr CcP variants (Figure S1), the microenvironment of Tyr71 showing the nearby binding site of guaiacol in CcP (Figure S2), and the 9 GHz EPR spectra, recorded at 4 and 40 K, of the radicals formed upon reaction of wild-type CcP and the Y236F variant with hydrogen peroxide in the presence and absence of guaiacol (Figure S3). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ T.D.P.: Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, P.O. Box B, Frederick, MD 21702.
Author Contributions
K.D.M. and T.D.P. contributed equally to this work.
This work was supported by the French CNRS (UMR 8221), CEA-Saclay, and the CNRS Program PICS-Canada (PICS-05865) (to A.I.), the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery Grant 9600) and the Canada Research Chair Program (to P.C.L.), and the National Institutes of Health (Grant GM06221 to Y.L.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Footnotes
Tyr236 was proposed to be the radical site based on the detection of its covalent modification in the reaction of W191G CcP with 2-aminotriazole.19 Later, Tyr236 was ruled out under the consideration that there was no Asp or Glu H-bond donor or positively charged residue to provide with the electropositive microenvironment predicted by the gx value in the Tyr• HF-EPR spectrum,16 yet the presence of two structural waters within H-bonding distance of the phenolic oxygen of Tyr236 (Figure 2) could account for the low gx value of the Tyr• spectrum (Figure 1A, magenta trace), as shown by the semiempirical MNDO calculations on the variation of the gx values of a p-methylphenoxy radical as a function of the H-bonding distance to two water molecules (Figure 6 of ref (44)) and experimentally shown for the Tyr• radical in ribonucleotide reductase.45
Supplementary Material
References
- Stubbe J.; van der Donk W. (1998) Protein radicals in enzyme catalysis. Chem. Rev. 98, 705–762. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Ener M. E.; Vlček A. Jr.; Winkler J. R.; Gray H. B. (2012) Electron hopping through proteins. Coord. Chem. Rev. 256, 2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunford B. H. (1999) in Heme Peroxidases, Chapters 9 and 10, Wiley-VCH, New York. [Google Scholar]
- Poulos T. L. (2010) Thirty years of heme peroxidase structural biology. Arch. Biochem. Biophys. 500, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos T. L. (2014) Heme enzyme structure and function. Chem. Rev. 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro J. M.; Fishel L. A.; Hazzard J. T.; Meyer T. E.; Tollin G.; Cusanovich M. A.; Kraut J. (1988) Tryptophan-191→Phenylalanine, a proximal-side mutation in yeast cytochrome c peroxidase that strongly affects the kinetics of ferrocytochrome c oxidation. Biochemistry 27, 6243–6256. [DOI] [PubMed] [Google Scholar]
- Erman J. E.; Vitello L. B.; Mauro J. M.; Kraut J. (1989) Detection of an oxyferryl porphyrin π-cation-radical intermediate in the reaction between hydrogen peroxide and a mutant yeast cytochrome c peroxidase. Evidence for tryptophan-191 involvement in the radical site of Compound I. Biochemistry 28, 7992–7995. [DOI] [PubMed] [Google Scholar]
- Sivaraja M.; Goodin D. B.; Smith M.; Hoffman B. (1989) Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase Compound ES. Science 245, 738–740. [DOI] [PubMed] [Google Scholar]
- Erman J. E.; Vitello L. B. (2002) Yeast cytochrome c peroxidase: Mechanistic studies via protein engineering. Biochim. Biophys. Acta 1597, 193–220. [DOI] [PubMed] [Google Scholar]
- Volkov A. N.; Nicholls P.; Worrall J. A. R. (2011) The complex of cytochrome c and cytochrome c peroxidase: The end of the road?. Biochim. Biophys. Acta 1807, 1482–1503. [DOI] [PubMed] [Google Scholar]
- Gumiero A.; Murphy E. J.; Metcalfe C. L.; Moody P. C. E.; Raven E. L. (2010) An analysis of substrate binding interactions in the heme peroxidase enzymes: A structural perspective. Arch. Biochem. Biophys. 500, 13–20. [DOI] [PubMed] [Google Scholar]
- Putnam A.-M. A. H.; Lee Y.-T.; Goodin D. B. (2009) Replacement of an Electron Transfer Pathway in Cytochrome c Peroxidase with a Surrogate Peptide. Biochemistry 48, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel L. A.; Farnum M. F.; Mauro J. M.; Miller M. A.; Kraut J.; Liu Y.; Tan X. L.; Scholes C. P. (1991) Compound I radical in site-directed mutants of cytochrome c peroxidase as probed by electron paramagnetic resonance and electron-nuclear double resonance. Biochemistry 30, 1986–1996. [DOI] [PubMed] [Google Scholar]
- Housseman A. L.; Doan P.; Goodin D. B.; Hoffman B. M. (1993) Comprehensive explanation of the anomalous EPR spectra of wild-type and mutant cytochrome c peroxidase Compound ES. Biochemistry 32, 4430–4443. [DOI] [PubMed] [Google Scholar]
- Huyett J. E.; Doan P. E.; Gurbiel R.; Houseman A. L. P.; Sivaraja M.; Goodin D. B.; Hoffman B. M. (1995) Compound ES of cytochrome c peroxidase contains a Trp π-cation radical: Characterization by CW and Pulsed Q-band ENDOR. J. Am. Chem. Soc. 117, 9033–9041. [Google Scholar]
- Ivancich A.; Dorlet P.; Goodin D. B.; Un S. (2001) Multifrequency high-field EPR study of the tryptophanyl and tyrosyl radical intermediates in wild-type and the W191G mutant of cytochrome c peroxidase. J. Am. Chem. Soc. 123, 5050–5058. [DOI] [PubMed] [Google Scholar]
- Hori H.; Yonetani T. (1985) Powder and single-crystal electron paramagnetic resonance studies of yeast cytochrome c peroxidase and its peroxide compound, Compound ES. J. Biol. Chem. 260, 349–355. [PubMed] [Google Scholar]
- Tsaprailis G.; English A. M. (2003) Different pathways of radical translocation in yeast cytochrome c peroxidase and its W191F mutant on reaction with H2O2 suggest an antioxidant role. JBIC, J. Biol. Inorg. Chem. 8, 248–255. [DOI] [PubMed] [Google Scholar]
- Musah R. A.; Goodin D. B. (1997) Introduction of novel substrate oxidation into cytochrome c peroxidase by cavity complementation: Oxidation of 2-aminothiazole and covalent modification of the enzyme. Biochemistry 36, 11665–11674. [DOI] [PubMed] [Google Scholar]
- Zhang H.; He S.; Mauk A. G. (2002) Radical formation at Tyr39 and Tyr153 following reaction of yeast cytochrome c peroxidase with hydrogen peroxide. Biochemistry 41, 13507–13513. [DOI] [PubMed] [Google Scholar]
- Wright P. J.; English A. M. (2003) Scavenging with TEMPO• to identify peptide- and protein-based radicals by mass spectrometry: Advantages of spin scavenging over spin trapping. J. Am. Chem. Soc. 125, 8655–8665. [DOI] [PubMed] [Google Scholar]
- Singh R.; Switala J.; Loewen P. C.; Ivancich A. (2007) Two [Fe(IV)=O Trp•] intermediates in M. tuberculosis catalase-peroxidase discriminated by multifrequency (9–285 GHz) EPR spectroscopy: Reactivity toward isoniazid. J. Am. Chem. Soc. 129, 15954–15963. [DOI] [PubMed] [Google Scholar]
- Colin J.; Wiseman B.; Switala J.; Loewen P. C.; Ivancich A. (2009) Distinct role of specific tryptophans in facilitating electron transfer or as [Fe(IV)=O Trp•] intermediates in the peroxidase reaction of Burkholderia pseudomallei catalase-peroxidase: A multifrequency EPR spectroscopy investigation. J. Am. Chem. Soc. 131, 8557–8563. [DOI] [PubMed] [Google Scholar]
- Wiseman B.; Carpena X.; Feliz M.; Donald L. J.; Pons M.; Fita I.; Loewen P. C. (2010) Isonicotinic acid hydrazide conversion to isonicotinyl-NAD by catalase-peroxidases. J. Biol. Chem. 285, 26662–26673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E. J.; Metcalfe C. L.; Nnamchi C.; Moody P. C. E.; Raven E. L. (2012) Crystal structure of guaiacol and phenol bound to a heme peroxidase. FEBS J. 279, 1632–1639. [DOI] [PubMed] [Google Scholar]
- Fielding A. J.; Singh R.; Boscolo B.; Ghibaudi E. M.; Ivancich A. (2008) Intramolecular electron transfer versus substrate oxidation in lactoperoxidase: Investigation of radical intermediates by stopped-flow absorption spectrophotometry and (9–285 GHz) electron paramagnetic resonance spectroscopy. Biochemistry 47, 9781–9792. [DOI] [PubMed] [Google Scholar]
- Smith A. T.; Doyle W. A.; Dorlet P.; Ivancich A. (2009) Spectroscopic evidence for an engineered, catalytically-active Trp radical that creates the unique reactivity of lignin peroxidase. Proc. Natl. Acad. Sci. U.S.A. 106, 16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister T. D.; Gengenbach A. J.; Syn S.; Lu Y. (2001) The role of redox-active amino acids on Compound I stability, substrate oxidation, and protein cross-linking in yeast cytochrome c peroxidase. Biochemistry 40, 14942–14951. [DOI] [PubMed] [Google Scholar]
- Un S.; Dorlet P.; Rutherford A. W. (2001) A high-field EPR tour of radicals in photosystems I and II. Appl. Magn. Reson. 21, 341–361. [Google Scholar]
- Ivancich A.; Jouve H. M.; Sartor B.; Gaillard J. (1997) EPR investigation of Compound I in Proteus mirabilis and bovine liver catalases: Formation of porphyrin and tyrosyl radical intermediates. Biochemistry 36, 9356–9364. [DOI] [PubMed] [Google Scholar]
- Jakopitsch C.; Ivancich A.; Schmuckenschlager F.; Wanasinghe A.; Poltl G.; Furtmuller P. G.; Ruker F.; Obinger C. (2004) Influence of the unusual covalent adduct on the kinetics and formation of radical intermediates in Synechocystis catalase peroxidase: A stopped-flow and EPR characterization of the MET275, TYR249, and ARG439 variants. J. Biol. Chem. 279, 46082–46095. [DOI] [PubMed] [Google Scholar]
- Singh R.; Berry R. E.; Yang F.; Zhang H.; Walker F. A.; Ivancich A. (2010) Unprecedented peroxidase-like activity of Rhodnius prolixus nitrophorin 2: Identification of the [FeIV=O Por•]+ and [FeIV=O Por](Tyr38•) intermediates and their role(s) in substrate oxidation. Biochemistry 49, 8857–8872. [DOI] [PubMed] [Google Scholar]
- Un S. (2005) The g-values and hyperfine coupling of amino acid radicals in proteins: Comparison of experimental measurements with ab initio calculations. Magn. Reson. Chem. 43, S229–S236. [DOI] [PubMed] [Google Scholar]
- Un S.; Atta M.; Fontecave M.; Rutherford A. W. (1995) g-values as a probe of the local protein environment: High-field EPR of tyrosyl radicals in ribonucleotide reductase and photosystem-II. J. Am. Chem. Soc. 117, 10713–10719. [Google Scholar]
- Pipirou Z.; Guallar V.; Basran J.; Metcalfe C. L.; Murphy E. J.; Bottrill A. R.; Mistry S. C.; Raven E. L. (2009) Peroxide-dependent formation of a covalent link between Trp51 and the heme in cytochrome c peroxidase. Biochemistry 48, 3593–3599. [DOI] [PubMed] [Google Scholar]
- Bhaskar B.; Immoos C. E.; Shimizu H.; Sulc F.; Farmer P. J.; Poulos T. L. (2003) A novel heme and peroxide-dependent tryptophan-tyrosine cross-link in a mutant of cytochrome c peroxidase. J. Mol. Biol. 328, 157–166. [DOI] [PubMed] [Google Scholar]
- Un S.; Gerez C.; Elleingand E.; Fontecave M. (2001) Sensitivity of tyrosyl radical g-values to changes in protein structure: A high-field EPR study of mutants of ribonucleotide reductase. J. Am. Chem. Soc. 123, 3048–3054. [DOI] [PubMed] [Google Scholar]
- Ivancich A.; Jakopitsch C.; Auer M.; Un S.; Obinger C. (2003) Protein-based radicals in the catalase-peroxidase of Synechocystis PCC6803: A multifrequency EPR investigation of wild-type and variants on the environment of the heme active site. J. Am. Chem. Soc. 125, 14093–14102. [DOI] [PubMed] [Google Scholar]
- Jakopitsch C.; Obinger C.; Un S.; Ivancich A. (2006) Identification of Trp106 as the tryptophanyl radical intermediate in Synechocystis PCC6803 catalase-peroxidase by multifrequency electron paramagnetic resonance spectroscopy. J. Inorg. Biochem. 100, 1091–1099. [DOI] [PubMed] [Google Scholar]
- Murphy E. J.; Metcalfe C. L.; Basran J.; Moody P. C. E.; Raven E. L. (2008) Engineering the substrate specificity and reactivity of a heme protein: Creation of an ascorbate binding site in cytochrome c peroxidase. Biochemistry 47, 13933–13941. [DOI] [PubMed] [Google Scholar]
- Thornton R. J.; Bunker A. (1992) Characterization of wine yeasts for genetically modifiable properties. J. Inst. Brew. 95, 181–184. [Google Scholar]
- Shinohara T.; Kubodera S.; Yanagida F. (2000) Distribution of phenolic yeast and production of phenolic off-flavors in wine fermentation. J. Biosci. Bioeng. 90, 90–97. [DOI] [PubMed] [Google Scholar]
- Bau Y.-S. (1981) Indole compounds in Saccharomyces cervisiae and Aspergilus niger. Bot. Bull. Acad. Sin. 22, 13–130. [Google Scholar]
- Ivancich A.; Mattioli T. A.; Un S. (1999) Effect of Protein Microenvironment on Tyrosyl Radicals. A High-Field (285 GHz) EPR, Resonance Raman, and Hybrid Density Functional Study. J. Am. Chem. Soc. 121, 5743–5753. [Google Scholar]
- van Dam P. J.; Willems J. P.; Schmidt P. P.; Po1tsch S.; Barra A. L.; Hagen W. R.; Hoffman B. M.; Andersson K. K.; Graslund A. (1998) High-Frequency EPR and Pulsed Q-Band ENDOR Studies on the Origin of the Hydrogen Bond in Tyrosyl Radicals of Ribonucleotide Reductase R2 Proteins from Mouse and Herpes Simplex Virus Type 1. J. Am. Chem. Soc. 120, 5080–5085. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.