

Abstract
A highly diverse set of protein kinases functions as early responders in the mitogen- and stress-activated protein kinase (MAPK/SAPK) signaling pathways. For instance, humans possess 14 MAPK kinase kinases (MAP3Ks) that activate Jun kinase (JNK) signaling downstream. A major challenge is to decipher the selective and redundant functions of these upstream MAP3Ks. Taking advantage of the relative simplicity of Drosophila melanogaster as a model system, we assessed MAP3K signaling specificity in several JNK-dependent processes during development and stress response. Our approach was to generate molecular chimeras between two MAP3K family members, the mixed lineage kinase, Slpr, and the TGF-β activated kinase, Tak1, which share 32% amino acid identity across the kinase domain but otherwise differ in sequence and domain structure, and then test the contributions of various domains for protein localization, complementation of mutants, and activation of signaling. We found that overexpression of the wild-type kinases stimulated JNK signaling in alternate contexts, so cells were capable of responding to both MAP3Ks, but with distinct outcomes. Relative to wild-type, the catalytic domain swaps compensated weakly or not at all, despite having a shared substrate, the JNK kinase Hep. Tak1 C-terminal domain-containing constructs were inhibitory in Tak1 signaling contexts, including tumor necrosis factor-dependent cell death and innate immune signaling; however, depressing antimicrobial gene expression did not necessarily cause phenotypic susceptibility to infection. These same constructs were neutral in the context of Slpr-dependent developmental signaling, reflecting differential subcellular protein localization and by inference, point of activation. Altogether, our findings suggest that the selective deployment of a particular MAP3K can be attributed in part to its inherent sequence differences, cellular localization, and binding partner availability.
Keywords: Drosophila, signal transduction, specificity, Jun kinase, dorsal closure, innate immunity, cell death, complex genetics, tolerance, complex immunity, infection, resistance
PROTEIN kinases are common transducers of information within cells. Indeed, reversible phosphorylation of substrates, by the opposing activities of kinases and phosphatases, is a major currency in cells forming the basis for information relay in many signaling pathways, ultimately transforming cell behavior in response to a changing environment. Unregulated kinase activity, however, has been implicated in numerous diseases of medical concern, notably cancer. One family in particular, the mitogen-activated protein kinases (MAPKs), composed of ERK, p38, and JNK enzymes, are central to a vast array of cellular and pathological processes (Chang and Karin 2001; Johnson and Nakamura 2007; Wagner and Nebreda 2009; Keshet and Seger 2010; Sabapathy 2012). Converging on the activation of MAPKs are typically two additional levels of kinases within a hierarchical three-tiered core, namely the MAPK kinases or MAP2Ks, and their activators, the MAPK kinase kinases, or MAP3Ks. While MAPK enzymes have been extensively studied at biochemical, structural, and physiological levels, the MAP3Ks are less well understood, more diverse, and greater in number. For example, in mammals there exist at least 20 different MAP3K family members, 14 of which impinge downstream upon three JNK stress-activated protein kinases (SAPKs) (Cuevas et al. 2007; Johnson and Nakamura 2007; Craig et al. 2008). From an evolutionary standpoint, the diversity of MAP3Ks may allow cells to respond to a greater breadth of stimuli or with greater sensitivity to discrete signals. Emerging evidence suggests that MAP3Ks can work selectively or cooperatively downstream of different signals to tune a MAPK network response (Chen et al. 2002; Cronan et al. 2012). The selective function of MAP3Ks can presumably be harnessed to provide specific alternative therapeutic targets for MAPK pathway-associated disease intervention. On the other hand, if MAP3Ks act cooperatively to fine tune a response, then targeting individual members could result in minimal efficacy. Thus, elucidation of the context-dependent functions and mechanisms of signaling specificity among MAP3K proteins is the focus of current research.
Context-dependent influences, like environmental, cellular, developmental, or spatial influences, are pervasive in tuning signaling networks. As such, a major challenge is to understand the molecular mechanisms by which context imparts distinct properties to a system. Recent work has provided some mechanistic insight. For example, within a single cell, related kinases might avoid inappropriate crosstalk by deploying nonoverlapping substrates or by compartmentalization of their function in cellular space or time (Alexander et al. 2011). Considering the conserved three-tier kinase organization within the MAPK pathways, the core pathway may incorporate distinct upstream transducers, as is the case with the diversity of MAP3K proteins, to shift the outcome of signaling in response to distinct stimuli. Two general approaches to the challenge of identifying context-dependent influences on signaling have been applied: first, to alter the context of a constant set of components, for example, by adding a stimulatory ligand, and second, to change a system component while keeping the context constant. The latter experiment can be useful to test redundancy and specificity among related proteins. If one component is swapped for another within the same context and a different outcome is observed, there must be intrinsic differences in the components. To determine how individual MAP3Ks confer specificity in their responses in vivo, we have focused on two members of the tyrosine kinase-like (TKL) group (Manning et al. 2002) in the Drosophila model system, mixed lineage kinase (MLK) encoded by the slpr gene and transforming growth factor-β activated kinase (Tak1).
Among the MAP3Ks that stimulate JNK activation, the mixed lineage kinase group consisting of the MLKs, the dual leucine zipper kinases (DLKs), and zipper sterile alpha kinase (ZAK), is the largest, related by sequence homology within the kinase domain and the presence of leucine zipper (LZ) dimerization motifs (Gallo and Johnson 2002). MLK family members mediate MAPK-dependent responses to cytokines, ceramide, fatty acids, and other stresses (Sathyanarayana et al. 2002; Jaeschke and Davis 2007; Korchnak et al. 2009; Kant et al. 2011). Consequently, they are implicated in metabolic and neurodegenerative diseases, epithelial migration and healing, and tumor growth and metastasis, reflecting their broad tissue distribution in epithelia and the nervous system (Silva et al. 2005; Jaeschke and Davis 2007; Chen et al. 2010; Velho et al. 2010; Cronan et al. 2012; Stark et al. 2012; Zhan et al. 2012). Their roles in development have been more difficult to ascertain, as single and double gene knockouts in mice are viable (Brancho et al. 2005; Bisson et al. 2008). MLK proteins are distinguished by an N-terminal SH3 domain, followed by the kinase, LZ, and CRIB domains mediating catalysis, dimerization, and Rac or Cdc42 GTPase binding, respectively (Gallo and Johnson 2002). These functional domains are followed by a long C-terminal region lacking notable domains but enriched in phosphorylation motifs thought to modulate protein function and/or localization (Vacratsis et al. 2002). Multistep activation of MLKs by upstream signals involves GTPase binding, relief of autoinhibition, dimerization, and phosphorylation by MAP4K proteins (Bock et al. 2000; Vacratsis and Gallo 2000; Zhang and Gallo 2001; Du et al. 2005; Garlena et al. 2010; Kant et al. 2011).
More distantly related and lacking overt LZ motifs, Tak1 is a pivotal activator of NF-κB and MAPK signaling in inflammatory, immune, and stress responses (Cuevas et al. 2007, 2008; Sakurai 2012). Tak1 also participates in noncanonical (Smad independent) TGF-β signaling, reflecting its moniker (Yamaguchi et al. 1995). Conditional and complete Tak1 knockouts in mice provide evidence for essential roles in embryonic development and differentiation of immune cells, skin, and vasculature (Shim et al. 2005; Jadrich et al. 2006; Omori et al. 2006). Tak1 signals as part of a protein complex with the partners Tab1 and Tab2/3, which interact with the N-terminal kinase domain and C-terminal regulatory domain of Tak1, respectively (Shibuya et al. 1996; Takaesu et al. 2000; Besse et al. 2007). Growing evidence suggests that an important component of Tak1 activation involves the binding of K63-linked polyubiquitin chains by Tab2/3, leading to Tak1 autophosphorylation and kinase activity (Wang et al. 2001; Kanayama et al. 2004; Xia et al. 2009).
Our previous work has focused on MAP3K family members in Drosophila, which is intermediate in complexity between single cell and vertebrate systems with respect to genetic redundancy and cellular diversity. In flies, there are eight recognizable homologs to the 14 mammalian proteins implicated in stimulating JNK activity. Of these, Mekk1, Pk92B/Ask1, Tak1, Slpr/MLK, and Wnd/DLK have definitive roles in JNK signaling (Igaki et al. 2002; Kuranaga et al. 2002; Stronach and Perrimon 2002; Collins et al. 2006; Ryabinina et al. 2006; Kang et al. 2012). Genetic and cell culture experiments have demonstrated both unique and overlapping functions for some of them, but the intrinsic properties of the individual family members that confer particular responses to distinct signals are still poorly characterized. Here, we address this question using chimeric constructs. Protein chimeras have been used widely, in cellular and in vitro assays, to discern the specific contributions of related domains in various types of proteins (e.g., (Walker et al. 1995; Sanchez-Hernandez et al. 2012; Anisimov et al. 2013). Given that there are processes uniquely dependent on Slpr, such as embryonic epidermal dorsal closure, and on Tak1, such as innate immune response, the separation of functions provides a platform upon which to study the specific contributions to signaling for the two different proteins (Mihaly et al. 2001; Silverman et al. 2003; Polaski et al. 2006). Moreover, since Slpr and Tak1 share at least one common substrate, Hep, a MAP2K related to mammalian MKK7 (Holland et al. 1997; Sathyanarayana et al. 2003), we sought to test directly if the catalytic kinase domain is functionally equivalent and if integration into an alternate context, by sequences outside the kinase domain, is sufficient to alter signaling specificity.
Materials and Methods
Transgenic constructs
All UAS constructs generated for this study were made in the pUASp vector (Rorth 1998) and transgenic lines were established after injection of DNA by Genetic Services (Sudbury, MA). All transgenic proteins generated herein were tagged at the C terminus with two copies of the HA epitope tag. Using site-directed mutagenesis by PCR overlap extension (Ho et al. 1989), the following chimeric constructs were created: the Slpr-Tak kinase swap, STK, consists of Slpr aa 1–128, Tak1 aa 19–271, and Slpr aa 383–1148, in that order. The Slpr-Tak C-terminus swaps, STCt and SAAATCt, consist of Slpr aa 1–516, with either a wild-type kinase domain or with activation loop alanine mutations, respectively (Garlena et al. 2010), followed directly by Tak1 aa 272–678. The alternate Tak-Slpr kinase swap chimeras, TSK and TSAAA, consist of Tak1 aa 1–18, Slpr wild-type or triple alanine mutant kinase domain aa 128–385, followed directly by Tak1 aa 272–678. Finally, we also generated the Tak1 C terminus alone, TCt encoding aa 272–678, with the 5′ UTR and starting methionine codon from the wild-type Tak1 transcript upstream. All constructs were verified by DNA sequencing.
Fly strains
Stocks were maintained at 22° on cornmeal–molasses–agar medium. Crosses were raised at 25° in 50 ± 10% relative humidity unless noted otherwise. w1118 was used as a control. For mutants and transgenics, Bloomington (BL) Stock Center numbers are given if appropriate: UAS-Slpr, UAS-SlprAAA, and UAS-SKLC (Garlena et al. 2010), slprBS06 (Polaski et al. 2006), Tak12 BL# 26272 (Vidal et al. 2001), UAS-Tak1 and UAS-Tak1K46R (Mihaly et al. 2001), egrGS9830 (UAS-eiger) (Igaki et al. 2002), pucE69 (puc-lacZ) (Ring and Martinez Arias 1993), and UAS-srcEGFP BL# 5432. For constructs under the control of UAS sequences, expression was regulated by the Gal4 transcription factor (Brand and Perrimon 1993). arm-Gal4 BL# 1560 (Sanson et al. 1996) and da-Gal4 BL# 5460 (Wodarz et al. 1995) were used for ubiquitous expression; pnr-Gal4 BL# 3039 (Calleja et al. 1996) was used for expression in the dorsal ectoderm of the embryo, though it directs expression in other cells and tissues throughout development; Yp1-Gal4 (yolk-Gal4) (Vidal et al. 2001) was used for expression in the adult female fat body starting around day 2 after eclosion; r4-Gal4 BL# 33832 (Lee and Park 2004) drives expression in the larval and adult fat body of both sexes; and GMR-Gal4 BL# 1104 was used for expression in the developing eye tissue (Freeman 1996). The genetic rescue experiment with the gtX11 slpr921 double mutant chromosome has been described previously (Stronach and Perrimon 2002).
Tissue immunofluorescence, X-gal staining, and immunoblot
Embryos were collected overnight on grapejuice plates, dechorionated, washed, and then fixed at room temperature for 20 min with equal volumes of 4% formaldehyde in PEM buffer (100 mM Pipes, 2 mM EGTA, 1 mM MgSO4) and heptane. After devitellinization in methanol, subsequent washes and processing were done in PBS plus 0.1% Triton X-100. For immunofluorescent staining of fat body, larvae were coarsely dissected and fixed in PBS plus 4% formaldehyde overnight at 4°. Subsequent washes and incubations were in PBS plus 0.1% Tween-20. The following antibodies and dilutions were used: mouse α-HA (16B12, Covance) at 1:500–1:1000, rabbit α-β-galactosidase preadsorbed at 1:1000 (Cappel), mouse α-fasciclin 3 (7G10, Developmental Studies Hybridoma Bank) at 1:50, and rat α-Tak1 peptide antibody at 1:250 (custom antibody services, GenScript). The immunogenic peptide sequence was 440-SSTNAKSDGRERLT-453. Secondary antibodies were FITC- or TxRed-conjugates from Jackson ImmunoResearch Laboratories, used at 1:200 or were Alexa Fluor conjugates from Invitrogen/Molecular Probes used at 1:500–1:750. For detection of the puc-lacZ reporter in adult fat body, 3- to 4-day-old mated females were collected and their abdomens were cut off in cold PBS with fine tissue scissors. Then while grasping the terminalia with a forceps, an incision was made through the cuticle at the dorsal midline with scissors. The tissue was fixed and then stained with X-Gal reagent overnight at 25° according to a published protocol (Romeo and Lemaitre 2008). The stained abdominal tissue was washed, filleted open, and mounted in 70% glycerol in PBS. Protein lysates for Western immunoblots were made by homogenizing, in 150 μl RIPA buffer, four wandering third instar larvae, programmed to express transgenic proteins with the r4-Gal4 driver. An equal volume of lysate was separated by SDS–PAGE and blots were probed with mouse α-HA (16B12, Covance) diluted 1:1000 or mouse α-GFP (GF28R, Pierce) at 1:1500. Expression was quantified by chemiluminescent imaging using the analysis tools provided with the ProteinSimple FluorChem E system software.
Image capture and processing
Images of adult flies were obtained with NIS-Elements software using a Nikon DS-Fi1 digital camera mounted on a Nikon SMZ1500 stereomicroscope. Fluorescent images of stained embryos and larval tissues were obtained by laser-scanning confocal microscopy using an Olympus FV1000 Fluoview system on an IX81 compound inverted microscope and assembled in Adobe Photoshop. For quantification of puc-lacZ induction in the embryo as a measure of JNK signaling intensity, β-galactosidase-positive nuclei from five consecutive segments along the leading edge were marked using the COUNT tool in Adobe Photoshop. The data from four to eight embryos were averaged. puc-lacZ intensity in the adult fat body was obtained by selecting a 100 × 100 pixel region of interest along the central ventral section of the image in the red channel only and measuring “integrated density” in Adobe Photoshop. Values from 5–22 specimens were averaged. Graphing and statistical analysis was performed with GraphPad Prism.
Innate immune assays
Crosses between Tak12; da-Gal4 females and w1118/Y; UAS-transgene males were reared at 22°. Newly eclosed adults were aged 2–4 days at 25°. For infection, adults were pricked once below the wing with a needle dipped in a loose pellet of overnight Escherichia coli DH5α cell culture. Flies were then maintained at 29° and monitored daily for viability. Data from multiple trials with two independent insertion lines were combined, plotted as survival curves, and analyzed using the log-rank test (Mantel–Cox) in GraphPad Prism. A control cross between da-Gal4 and UAS-GFP confirmed that the Gal4 line directs expression ubiquitously throughout development and we note in particular that GFP is expressed highly in newly eclosed adults. Adults with the genotypes da-Gal4 > UAS-Tak1WT or da-Gal4 > UAS-SlprWT were not recovered in sufficient quantity to test.
cDNA synthesis and quantitative real-time PCR
Crosses were raised at 25° and 2- to 4-day-old adult mated females (Yp1-Gal4 > UAS-transgene) were collected, at which time, half of them were infected as described above. After 6 hr at 29°, 7–10 flies were homogenized in 300 μl of TRIzol (Invitrogen). RNA was extracted according to the manufacturer’s recommendations and suspended in 20–25 μl of water. First strand cDNA was synthesized by transcribing 2 μg of RNA template using the Maxima Reverse Transcriptase kit (Thermo Scientific) and random primers. In an Applied Biosystems 7900HT thermal cycler, transcript amplification was monitored with Sybr green dye (Thermo Scientific) using ∼100 ng input cDNA. The following primer pairs were used: RpL32 (forward) 5′-ACCGCAGTACCCACTCAATC-3′ and (reverse) 5′-CAATCTCCTTGCGCTTCTTG-3′, Diptericin (forward) 5′-ACCGCAGTACCCACTCAATC-3′ and (reverse) 5′-ACTTTCCAGCTCGGTTCTGA-3′. Four biological replicates (consisting of two independent transgenic lines per construct) were collected for each genotype except Tak1K46R, which had three replicates. Relative gene expression, compared to a no transgene control, was calculated by normalizing to RpL32 expression levels according to the comparative Ct method (Schmittgen and Livak 2008). In five instances out of 86 data points total (11 genotypes, three or four trials, and two probes), a trial was excluded as an outlier if values exceeded the mean of the remaining values by a factor of five.
Results
Design and construction of MAP3K chimeras
If the primary functions of a kinase catalytic domain are to recognize, bind, and phosphorylate substrate, then two kinase domains that recognize and phosphorylate the same substrate are predicted to be interchangeable. To test this assertion, we engineered Slpr and Tak1 proteins with kinase domain swaps. For example, we generated a full-length Slpr construct with the kinase domain from Tak1 swapped in to replace the endogenous Slpr kinase domain and vice versa, creating STK and TSK, respectively (Figure 1). Given that one of the assays used to monitor a requirement for Tak1 is based on dominant interference of endogenous activity, we also generated a kinase domain swap in Tak1, TSAAA, using a Slpr kinase domain mutated in the activation loop to prevent activating phosphorylation. Our previous work demonstrated that this combination of alanine mutations disrupts phosphorylation and renders Slpr nonfunctional due to its inability to activate downstream JNK signaling (Garlena et al. 2010).
Figure 1.

Slpr and Tak1 domain organization and derived mutant or chimeric constructs. Black lines represent Slpr sequences and red lines indicate Tak1 sequences. The number of amino acids encoded by each construct, minus the epitope tag is given. Slpr encodes four recognizable domains: Src-homology 3 (SH3), kinase, leucine zipper (LZ), and Cdc42/Rac interactive binding motif (CRIB), clustered in the N-terminal half of the protein. Tak1 encodes a protein with an N-terminal kinase domain and a conserved C-terminal domain (CTD) as shown. Specific amino acid point mutations are indicated with an “X.”
The ability of Slpr to localize to the cell cortex in embryonic epithelium is attributed to the C-terminal half of the protein, and though this activity was nonessential in mutant rescue experiments, it contributed to maximal Slpr function (Garlena et al. 2010). The C terminus of the Tak1 protein harbors a putative regulatory domain identifiable by an island of sequence conservation among homologs (Takatsu et al. 2000; Mihaly et al. 2001). This region may contribute to Tak1 localization or protein interactions with signaling partners, as suggested by cell culture and biochemical assays (Takaesu et al. 2000; Zhou et al. 2005; Besse et al. 2007; Guntermann and Foley 2011). Based on this evidence, we reasoned that sequences encompassing this domain might direct Tak1 to specific signaling complexes for which Slpr is excluded, as a specificity-determining mechanism. To test this idea, we replaced amino acids C terminal to the CRIB domain of Slpr with Tak sequences beginning immediately after the kinase domain (Figure 1), both in the context of a wild-type (STCt) and a nonphosphorylatable Slpr kinase domain (SAAATCt). This part of Tak1, lacking the kinase domain, was also expressed on its own (TCt). Using these transgenic reagents, we tested protein localization, function, and specificity in both Slpr-dependent and Tak1-dependent processes during Drosophila development, cell death, and immunity.
Differential localization of chimeric proteins in two tissue contexts is attributable to C-terminal sequences
All transgenic proteins generated in this study were detectable by indirect immunofluorescence with antiserum directed against the C-terminal HA tag and were therefore expressed as full-length proteins. Wild-type Slpr, SlprAAA, and STK displayed strong enrichment at the cell cortex in embryonic epithelia (Figure 2, A and B and Garlena et al. 2010). All of these constructs have the normal Slpr sequence C terminal to the CRIB domain. In contrast, STCt and SAAATCt, which contain the Tak1 C-terminal domain swap, instead localized predominantly in the cytoplasm and showed minimal if any enrichment at the cortex (Figure 2, C and D). This distribution was reminiscent of a previously characterized construct, SKLC (Garlena et al. 2010), which is truncated directly after the CRIB domain of Slpr, suggesting that the Tak1 C-terminal replacement had a minimal effect on localization beyond the loss of the Slpr C terminus. Nevertheless, to determine if the cytoplasmic localization of the chimeras reflected that of the Tak1 C terminus, we assessed the distribution of this portion of Tak1 in isolation. Indeed, the TCt protein had a similar distribution predominantly in the cytoplasm, but in addition appeared to localize partially in the nucleus, though it was not enriched there (Figure 2G). Together, these results align with our previous studies demonstrating that the C-terminal half of the Slpr protein directs its enrichment at the plasma membrane (Garlena et al. 2010).
Figure 2.
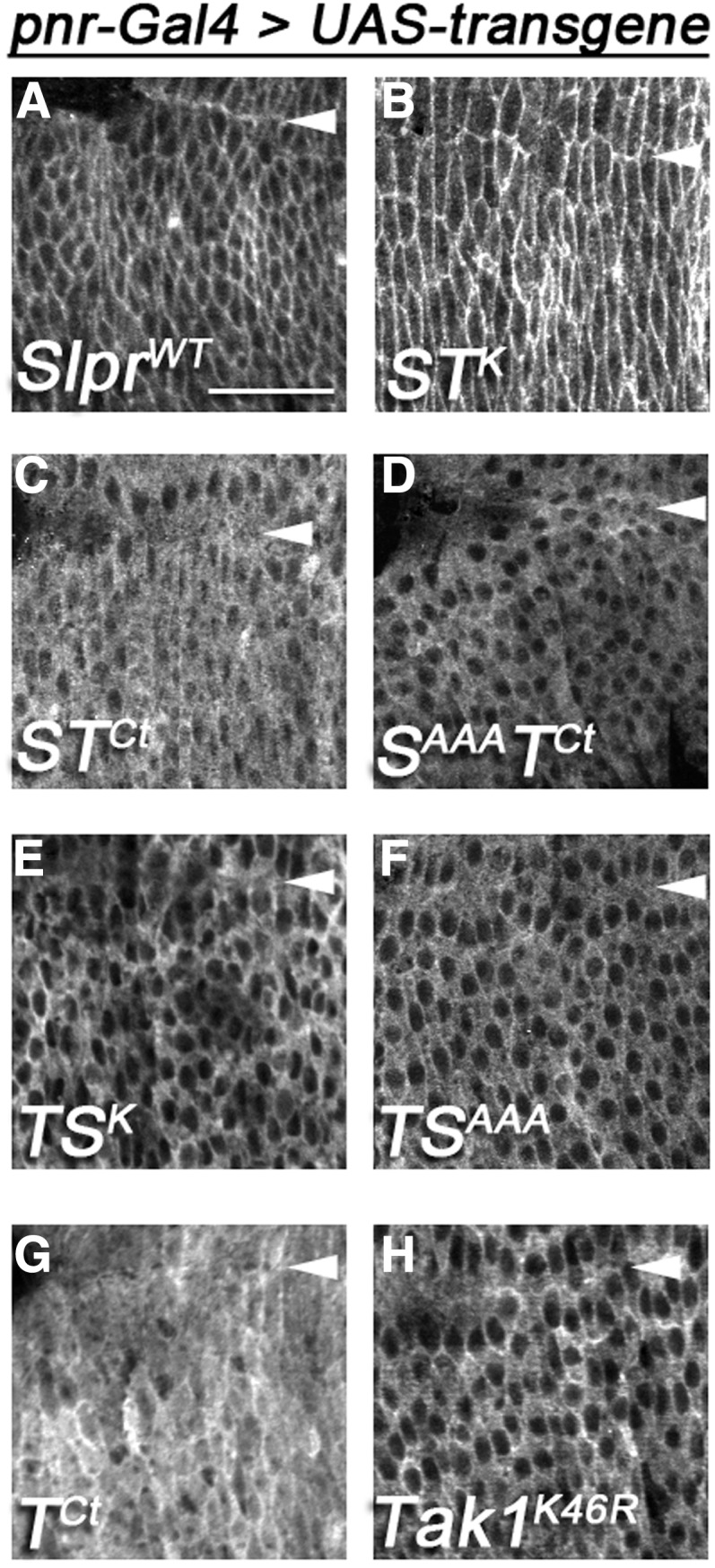
Differential localization of transgenic proteins in embryonic dorsal epidermis maps to the C terminus. (A–G) Anti-HA and (H) anti-Tak1 immunostaining. The indicated constructs were expressed in the embryo with the pnr-Gal4 driver. Images are single confocal slices ∼2 μm below the apical surface of the epidermis. Views are dorsolateral, surrounding the posterior canthus of the zippering epidermis during dorsal closure in stage 15 embryos. Arrowheads indicate the dorsal midline. Bar, 20 μm.
Since the C-terminal portion of Tak1 was detected in the cytoplasm and nucleus, we next determined whether this distribution reflected that of the full-length Tak1 protein and Tak/Slpr chimeras. To that end, immunofluorescence was performed using either the anti-HA antiserum to detect the chimeras or an anti-Tak1 antibody to detect the untagged Tak1K46R transgenic protein, a kinase-dead form of Tak1 (Mihaly et al. 2001). In the embryonic epidermis, overexpressed Tak1K46R localized in the cytoplasm, absent from nuclei. In addition, we observed some association with the cell cortex, as evidenced by a prominent signal at cell boundaries upon completion of dorsal closure (Figure 2H). We did not attempt to localize overexpressed wild-type Tak1 due to its strong proapoptotic effects and disruption of epithelial integrity. Also, we note here that under conditions suitable for detection of the transgenic Tak1 protein, appreciable levels of endogenous Tak1 were not observed, though maternal, and later, ubiquitious expression is reported in FlyBase (Drysdale and FlyBase 2008; Graveley et al. 2011). Finally, the distributions of the chimeric transgenes replacing the kinase domain of Tak1 with that of Slpr appeared identical to that of Tak1K46R, with prominent cytoplasmic staining and occasional cortical localization (Figure 2, E and F). Taken together these localization data suggest that the determinants of subcellular location likely reside outside the kinase domains.
While the embryonic epidermis requires endogenous Slpr function for morphogenesis, the fat body is an important organ for antimicrobial defense during innate immunity (Hultmark 1993), a process mediated by Tak1 in response to Gram-negative bacterial infection (Vidal et al. 2001). With this in mind, we also investigated protein localization in the larval fat body (Figure 3) using the r4-Gal4 driver (Lee and Park 2004) and UAS-srcEGFP, encoding a membrane-associated form of GFP, as a means to compare how tissue context influences protein distribution. Although fat body cells are adherent to one another forming an irregular-shaped organ, their composition and morphology are distinct from typical columnar epidermal epithelia. Despite these differences, the subcellular distributions of the chimeric proteins in the larval fat body mimicked what we observed in the embryonic epidermis (Figure 2 and Figure 3). Proteins with the Slpr C terminus (SlprWT, SlprAAA, and STK) were strongly associated with the plasma membrane and relatively depleted from the cytoplasm (Figure 3, B, C, and F). In contrast, the proteins containing the Tak C-terminus (STCt, SAAATCt, TCt, TSK, and TSAAA) were distributed more uniformly throughout the cell, though membrane staining was still prominent in some instances (Figure 3, D, E, and G–I). A difference in the relative levels of transgenic proteins was evident by immunofluorescence detection (Figure 3, I and Ii; see legend for details). Consistent with these results, Western immunoblot analysis revealed that mutants or chimeras with the Slpr backbone were expressed at relatively low levels compared to those in the Tak1 backbone such that the Tak1Ct-bearing proteins accumulated to a greater extent (Figure 3J). All the transgenic proteins were overexpressed relative to their endogenous counterparts based on both immunofluorescence and RT-PCR analysis of transcripts (Supporting Information, Figure S2). Altogether, from these localization studies, we conclude that the cellular distribution of Slpr and Tak1 is distinct and primarily determined by the protein sequences, not the tissue contexts tested here.
Figure 3.

Differential localization and expression of transgenic proteins in the larval fat body. (A) GFP fluorescence and (B–Ii) anti-HA immunostaining. The indicated constructs were expressed in larvae with the r4-Gal4 driver. Images are single confocal sections. (B–F, Ii) Fluorescence intensity is comparable among panels. (G–I) Images were captured at half laser power compared to panels B–F to reflect differences in expression levels or protein stability. The inset panel (Ii) shows fluorescence intensity captured with the same settings used for panels B–F. Bar, 50 μm. (J) Transgenic protein expression levels in larval lysates were determined relative to GFP. Coomassie-stained membrane shows similar loading of whole larval lysates expressing the indicated transgenes and GFP under the control of the r4-Gal4 driver. Western immunoblots (IB) with the respective antibodies reveal levels of protein expression, graphed below as the ratio of HA:GFP, averaged over three replicates and normalized to the transgene with the highest expression ratio. Bars are the means ± SEM. Molecular weight markers in kilodaltons are indicated.
Rescue of Slpr-dependent dorsal closure and mutant lethality demonstrates kinase specificity
Among all of the Drosophila MAP3K proteins, the function of Slpr is selectively required in the activation of JNK signaling to orchestrate morphogenesis of epithelial tissues during embryonic development and adult metamorphosis. This is borne out by genetic analysis of slpr mutants. Zygotic lethal alleles of slpr cause a failure of dorsal closure, leaving the embryonic epidermis unclosed, resulting in embryonic death (Stronach and Perrimon 2002; Polaski et al. 2006). Animals mutant for another allele, slprBS06, transition through embryogenesis but emerge as adults with reduced Mendelian frequency of ∼5–10% of normal (Polaski et al. 2006). The mutant adults that do eclose variably display defects in morphogenesis of the adult thorax, genitalia, and maxillary palps, as well as reduced longevity (Polaski et al. 2006; Gonda et al. 2012). Using slpr alleles of different severity, it was possible to test for the ability of the ubiquitously expressed transgenes to rescue Slpr function acutely during embryonic dorsal closure or throughout development, restoring survival to adulthood. For example, only three transgenes improved survival over the course of development relative to no transgene expression (Figure 4A). These were SlprWT as expected, SKLC, as shown previously (Garlena et al. 2010), and STCt. Expression of all the other transgenes depressed the frequency of slprBS06 adult recovery to a greater extent than without transgene expression, effectively acting as dominant negative proteins.
Figure 4.

Rescue of slpr mutant viability or dorsal closure demonstrates kinase specificity. (A) Floating bar plot showing the degree of rescue provided by expression of the indicated transgenes (x-axis), as a ratio of slprBS06 mutant to sibling FM7c male flies (y-axis). Bars span minimum to maximum values and horizontal lines indicate the mean ratio for three to six independent trials except SlprAAA and SAAATCt, which were each two trials, testing a minimum of two different transgenic insertions per genotype. In the absence of a UAS construct (no Tg), the eclosion ratio is ∼0.05. The total number (N) of males counted is shown below each bar. Expression of HA-tagged SlprWT provides a significant degree of rescue (P < 0.001) using one-way ANOVA with Bonferroni’s multiple comparisons test vs. the control. (B) Bar graph of the phenotype of gt slpr mutant cuticles recovered among progeny of the indicated cross. In the absence of transgene expression, a majority of severe (dorsal and anterior head open) and some moderate (dorsal hole but head in) dorsal open (DO) cuticles are observed. Rescue of dorsal closure by transgene expression (x-axis) decreases the percentages of severe and moderate cuticle phenotypes while increasing the proportions of cuticles with mild (small holes, scabs, head defects) or no defects (∼WT, resembling wild type). The total number (N) of cuticles counted for each genotype is shown above the bars.
A requirement to rescue slprBS06 mutants to adulthood is a stringent criterion for function and only the wild-type Slpr transgene provided significant rescuing function. Thus, to measure functional properties of the expressed transgenes over a shorter developmental time period, we asked whether each protein was capable of rescuing the dorsal closure phenotype of the embryonic lethal slpr921 allele (Figure 4B). Mirroring the previous rescue experiment, we found that SlprWT, SKLC, and STCt provided substantial rescuing function compared to no transgene expression, reducing the percentage of embryos with a severe dorsal open (DO) phenotype (solid), while increasing the recovery of embryos with no dorsal closure defects or only head defects (open). Only one additional construct, STK, showed an improvement in phenotype upon expression, though to a lesser extent than those mentioned. Thus, the N-terminal half of Slpr, namely the SKLC domains, provided nearly full functional rescue of embryogenesis and some rescue to adulthood, implying that the C terminus is nonessential for function under conditions of high level expression. The presence of the Tak C terminus attached to Slpr SKLC was essentially neutral in both assays acting similarly to SKLC alone. Interestingly, while the Slpr/Tak kinase swap, STK, provided some function during embryogenesis compared to the control, it did not suffice to functionally compensate for all Slpr functions throughout development (compare A and B in Figure 4). Importantly, the ability to rescue developmental defects in the short or long term was independent of transgene expression level.
Localized and specific kinase sequences are key to optimal JNK signaling during dorsal closure
To delve into the basis for the rescue data, we assessed the effect of transgene expression on the expression of puc-lacZ, a molecular reporter for JNK pathway activity used extensively in Drosophila. puc-lacZ is an enhancer trap allele of the puckered gene encoding JNK phosphatase, a negative feedback regulator (Martin-Blanco et al. 1998). As benchmarks for comparison, puc-lacZ induction was assessed in embryos expressing wild-type or dominant negative slpr constructs in the dorsal epidermis using pnr-Gal4 as the driver. As shown in Figure 5, B–Bii and quantified, SlprWT induced a twofold increase in the number of cells expressing puc-lacZ away from the leading edge of the dorsal epidermis at mid and late stages of dorsal closure compared with control embryos that express puc-lacZ in one row of dorsalmost cells flanking the central amnioserosa tissue (Figure 5, A–Aii). In contrast, SlprAAA inhibited JNK-dependent puc-lacZ expression completely (Figure 5, C–Cii). Deleting the C-terminal half of Slpr (SKLC construct) or replacing it with that of Tak1 (STCt construct) resulted in similar rescuing ability but a minimal effect on puc-lacZ expression (Figure 5, E–Eii and Garlena et al. 2010). Notably, if the kinase catalytic domain of Slpr was mutant, however, the presence of the Tak1 C terminus made the SAAATCt protein a less effective inhibitor of puc-lacZ induction than full-length SlprAAA (compare Fii and Cii in Figure 5), presumably due to mislocalization in the cytosol. Expression of Slpr with the Tak1 kinase domain (STK) induced mild ectopic puc-lacZ expression beyond the dorsalmost cells, demonstrating catalytic competency, though not to the extent of SlprWT, consistent with the embryonic rescue data (Figure 5, D–Dii). Expression of the Tak1 derivative constructs, including the C terminus alone (TCt), kinase dead (Tak1K46R), and the kinase swaps (TSK and TSAAA), were also nearly neutral in this assay, neither inducing nor inhibiting puc-lacZ relative to controls (Figure 5, G–Jii), though they were highly expressed. These data attest to the specificity of Slpr function in the embryonic epidermis and suggest that the Tak1 kinase domain cannot compensate for that of Slpr, nor can the nonkinase domains of Tak1 engage the protein in productive signaling complexes in those cells under conditions where they are normally responsive to Slpr.
Figure 5.

Specificity of Slpr vs. Tak1 signaling in activation of JNK target gene expression during dorsal closure. Early and late progression of dorsal closure (stage 13–14, left; stage 15, right) is shown in merged panels (A–J) and in individual channels, with immunostaining for either Fas3 (Ai–Ji) or β-gal to detect puc-lacZ enhancer trap expression (Aii–Jii). Transgenes indicated in the lower left of each panel (A–J) are expressed in the dorsal ectoderm and amnioserosa under the control of pnr-Gal4. Embryos are shown dorsally with anterior to the left. Bar, 20 μm. Quantification of puc-lacZ in stage 15 embryos as a proxy for JNK pathway activity is given in the rightmost panels as the mean number of β-gal positive nuclei per five hemisegments ± SD based on 4–8 embryos. Significant differences compared to the no Tg control (Aii) are indicated based on one-way ANOVA using Bonferroni’s multiple comparisons test vs. the control. ***P < 0.005, **P < 0.01, *P < 0.05.
Eiger/tumor necrosis factor-induced cell death engages the Tak1 C terminus
A well-defined role for Tak1 is to mediate cellular responses to tumor necrosis factor (TNF) signaling. In flies, Tak1 and its partner Tab2 mediate JNK activation in response to ectopic expression of Eiger, the sole ortholog of mammalian TNF (Igaki et al. 2002; Geuking et al. 2005). This results in cell death of the developing eye tissue, such that the adult eye is severely reduced in size (Figure 6A). Loss of Tak1 signaling by mutation, RNA interference, or expression of dominant negative constructs, suffices to block Eiger-induced cell death (Igaki et al. 2002; Moreno et al. 2002), restoring adult eye tissue (Figure 6B); and this effect is specific to Tak1 in comparison with Slpr (Polaski et al. 2006). Thus, we turned to this assay to define domains that are essential for Eiger signaling in this context. Upon crossing the experimental transgenic lines to a GMR-Gal4, UAS-eiger tester stock, in which high levels of eiger expression are induced in the developing larval eye imaginal discs (Igaki et al. 2002), we observed a striking pattern of results. Expression of the C-terminal region of Tak1 alone (Figure 6C) or in combination with any other sequences (Figure 6, E, F, H, and I) showed strong inhibition of cell death, whether the linked kinase domain was wild type or not. For example, even the Tak/Slpr kinase swap (TSK), wherein the Slpr kinase domain is wild type, blocked the cell death phenotype. In contrast, Slpr constructs characterized as dominant negative or the Slpr/Tak kinase swap (STK) failed to interfere with Eiger signaling (Figure 6, D and G). Moreover, expression of these constructs in the absence of Eiger did not phenocopy Eiger overexpression (not shown). In fact, none of the forms of Slpr we have expressed in flies are sufficient to dominantly suppress Eiger-induced cell death. Thus, we conclude that the region responsible for integration of Tak1 into the Eiger/TNF signaling network resides downstream of the kinase domain, in the C-terminal region. Given that Tab2 binds to the C terminus of Tak1 and that Tab2 is required for Eiger-JNK signaling (Takaesu et al. 2000; Geuking et al. 2005; Zhuang et al. 2006), we speculate that excess transgenic protein may sequester Tab2 or other binding partners in unproductive complexes.
Figure 6.

The C-terminal region of Tak1 is sufficient to inhibit ectopic eiger-induced cell death. (A–I) Images of adult eyes from individuals expressing eiger under the control of GMR-Gal4 without (A) or with (B–I) coexpression of transgenic slpr, Tak1, or other indicated constructs. Expression of constructs lacking Tak1 C-terminal sequences fail to suppress cell death (D and G). Expression of transgenes encoding the Tak1 C terminus alone (C) or in combination with other Tak1 or slpr sequences (B, E, F, H, and I), regardless of kinase activity, strongly suppress eiger signaling.
Probing Tak1-dependent innate immune response
Tak1 mutants are viable as adults but susceptible to Gram-negative bacterial infection (Vidal et al. 2001). This observation along with numerous other studies have defined the so-called immune deficiency (Imd) pathway (Lemaitre et al. 1995), in which Tak1 plays a central role in the induction of antimicrobial and stress defenses through the activation of Relish (Rel)/NFκB- and JNK-dependent transcriptional programs (Georgel et al. 2001; Vidal et al. 2001; Silverman et al. 2003; Aggarwal and Silverman 2008). To test the specificity of MAP3K signaling in this process, both infection susceptibility and target gene expression were monitored in adults expressing the various transgenic proteins. First, we generated a stock of the Tak12 allele, encoding an early stop codon (Vidal et al. 2001), in combination with a ubiquitous driver, da-Gal4. It was then possible to cross females from this stock to the UAS transgenic lines. From this cross, male progeny hemizygous mutant for Tak12 were assessed for rescue of the immune deficiency upon challenge with E. coli. In parallel, female progeny heterozygous for Tak12 were also challenged to test whether expression of any transgenic constructs dominantly enhanced the heterozygous loss of Tak1 signaling. Results of these experiments are given in Figure 7. In our hands, more than half of the Tak1 mutant males died over the course of a week after challenge (Figure 7A). Though we were unable to complement the susceptibility by expressing wild-type Tak1 due to early embryonic lethality, none of the transgenic proteins were sufficient to rescue the mutant susceptibility, including TSK. Among the experiments with females (Figure 7B), the heterozygotes were normal, demonstrating that Tak1 is not haploinsufficient, but the homozygous individuals were susceptible as expected. Intriguingly, expression of only two transgenic constructs showed any significant perturbation of the immune response in the heterozygous background. One was Tak1K46R, a dominant negative form of Tak1. Although this result was anticipated (Vidal et al. 2001), its expression did not fully recapitulate the homozygous mutant phenotype. The other transgene that depressed the immune response in females similar to the dominant negative construct was SAAATCt. Given that the mutant kinase domain of Slpr in the context of the full-length Slpr protein (SlprAAA) did not show an effect, this result seems to point to the juxtaposition of the mutant kinase with the Tak1 C terminus, which defined a different spatial context for the chimera according to the localization results (Figure 2 and Figure 3). However, TSAAA expression also had no effect. The only sequence difference between the constructs, SAAATCt and TSAAA, is the N-terminal nonkinase domains of Slpr, including the SH3, LZ, and CRIB domains, which in combination with an inactive kinase domain, might disrupt some important step in the activation of the pathway by the remaining endogenous Tak1 protein. We also note that expression of the Tak1 C terminus alone with da-Gal4 or a fat body-specific Gal4 driver, r4-Gal4, did not inhibit the immune response, contrasting with the context of Eiger-dependent cell death.
Figure 7.

Tak1-dependent antibacterial defense in the absence or presence of ectopic chimera protein expression. (A) Survival curves of Tak12 mutant males after infection with E. coli, without or with expression of indicated transgenes under the control of da-Gal4. Mutant males are susceptible to infection (red) and expression of the transgenic proteins did not significantly rescue the susceptibility. The total number (N) of adult flies tested is shown. (B) Survival curves of females homozygous for Tak12 or heterozygous mutant plus expression of chimeric proteins with the ubiquitous da-Gal4 driver and infected with E. coli. In the absence of transgene expression, homozygous Tak12 females are significantly more susceptible to infection (red) than the heterozygous females (gray), which are not. Expression of dominant-negative Tak1K46R (light blue) or SAAATCt (purple) transgenes renders the heterozygous Tak12 females modestly, but significantly, more sensitive than without exogenous protein. The total number (N) of adult flies tested is shown. ***P < 0.0001 according to the log-rank (Mantel–Cox) test.
A second approach to assess the effects of Slpr and Tak1 in the immune signaling pathways involved monitoring induction of Rel and JNK pathway target genes. It has been demonstrated that ectopic expression of Tak1 or an upstream activator, imd, can dominantly induce antimicrobial peptide (AMP) expression even in the absence of challenge (Georgel et al. 2001; Vidal et al. 2001), though expression levels are below that induced by bacterial infection. Based on this evidence, we assessed induction of a Rel target AMP encoded by Diptericin (Dpt), using quantitative real-time PCR upon expression of the wild-type or chimeric constructs in the adult fat body with Yp1-Gal4 as a driver (Figure 8 and Figure S1). We observed significant induction of basal Dpt levels upon expression of wild-type Tak1, with an average eightfold increase compared to no transgene (Figure 8, A and B). In contrast, expression of the other transgenes failed to induce ectopic Dpt expression under basal conditions (Figure 8B). To determine instead whether the transgenic proteins specifically potentiated or interfered with Tak1-dependent signaling under induced conditions, the experiment was also performed after immune challenge with E. coli. Pairwise comparisons of the individual transgenic lines first revealed that only Tak1WT and the no transgene control samples significantly activated Dpt expression upon challenge (Figure 8A). Among the challenged samples, kinase-dead Tak1 significantly inhibited Dpt upregulation as expected, along with the other Tak1 C-terminal domain-bearing transgenics (STCt, SAAATCt, TSK, TSAAA, and TCt) (Figure 8A) similar to their effects on Eiger signaling. Although Dpt induction was also reduced by expression of SlprWT and STK relative to no transgene expression, the differences were not significant, suggesting that they were neutral in the context of activated Tak1 signaling. Intriguingly, expression of dominant negative Slpr also significantly attenuated Dpt induction. These results could be interpreted to support the contention that JNK signaling is required for optimal AMP expression (Kallio et al. 2005; Delaney et al. 2006). Finally, though induced Dpt expression was dampened in flies expressing many of these transgenes, there was not a strict correlation with overall susceptibility to immune challenge as shown in Figure 7 or with relative expression levels of the constructs (Figure 3 and Figure S2), thus the full response to expression of the chimeras undoubtedly involves regulation of additional genes or pathways.
Figure 8.

The C-terminal region of Tak1 is sufficient to inhibit induction of Rel target gene, Diptericin, in adult females challenged with E. coli. (A) Quantitative real-time PCR results of relative Diptericin (Dpt) antimicrobial gene expression in females expressing the indicated transgenes relative to the Yp1-Gal4 driver-alone control (no Tg) in the absence and presence of bacterial challenge. Values were normalized against RpL32 expression to control for variation in input cDNA and shown as the means ± SEM for three to four independent biological replicates. Statistical comparisons were first performed on each pair (control vs. +Ec) using one-way ANOVA with Bonferroni’s multiple comparisons test. Asterisks indicate significant differences (****P < 0.001) in Dpt induction upon challenge. One-way ANOVA with Bonferroni’s post-test was also used to compare only the values of E. coli challenged groups vs. the control (no Tg +Ec) indicating significant depression of Dpt induction (##P < 0.01, #P < 0.05). (B) Bar graph displaying mean Dpt expression ± SEM values taken from graph in A to compare relative Dpt expression levels in the indicated groups under basal (unchallenged) conditions only. ANOVA analysis comparing all groups to the no Tg control highlights significant induction by Tak1WT only (****P < 0.001).
With respect to the JNK signaling axis, rather than measuring small and transient changes in puckered transcript expression at the population level with real-time PCR, we chose to monitor induction of the puc-lacZ reporter construct in individual females, again using Yp1-Gal4 as a tissue-specific driver (Figure S1). Unlike Dpt, however, pairwise comparisons of individual lines revealed no significant stimulation of JNK activity after bacterial challenge, including those flies expressing no transgene (Figure 9, A and Ai). Regardless of infection, though, we observed that the wild-type forms of Tak1 and Slpr induced robust JNK reporter expression in the fat body (Figure 9, A and B), whereas Tak1K46R-expressing flies resembled those with no transgene in having the lowest puc-lacZ expression. The other trasngenes spurred intermediate reporter expression. Notably, SlprWT was the only transgene to activate puc-lacZ in the oenocytes, an early component of the Yp1-Gal4 expression pattern, as well as fat body (Figure 9B and Figure S1). Also, flies with ectopic Tak1 expression were noticeably unhealthy and showed altered organization and loss of fat body tissue over the course of a few days (Figures 9Bi and Figure S3) consistent with other observations on the detrimental consequences of wild-type Tak1 overexpression. Thus, for this experiment, the chimeras with domain swaps were determined to be nonequivalent to the parental wild-type forms in their ability to ectopically activate JNK signaling, whereas dominant negative Tak1 was the most effective inhibitor of puc-lacZ expression.
Figure 9.

JNK-dependent puc-lacZ induction by Slpr and Tak1 in adult female fat body. (A) X-gal staining of adult female abdominal fillets showing induction of puc-lacZ as indicated by the blue product upon expression of various transgenes compared to a Gal4-only control (no Tg) in the absence (left column) or presence (right column) of E. coli infection. Cells of the dorsal vessel have endogenous galactosidase activity. (Ai) Quantification of β-gal staining intensity in arbitrary units is shown as a floating bar graph representing minimum to maximum values for 5–22 individuals with a vertical line at the mean. Data from two independent transgenes were combined. Transgene identities are aligned with the corresponding stained images from A. All pairwise comparisons of puc-lacZ induction, with and without E. coli challenge, are not significantly different; however, all the individual means compared to the control (without infection) are significantly different except Tak1K46R. Analysis by ANOVA with Bonferroni post-test (P < 0.05). (B and Bi) Magnified images of X-gal staining across one abdominal segment in the fat body (fb) and oenocytes (oe) in response to expression of wild-type Slpr (B) or Tak1 (Bi) using the Yp1-Gal4 driver. Tak1 expression results in disorganization and progressive loss of fat body tissue. Bar, 100 μm.
Discussion
Biological responses to developmental, immune, and cell death signals, are mediated in part by the activation of JNK signaling through numerous upstream MAP3K and MAP2K transducers. Genetic analyses in model organisms and biochemical studies in cultured cells have revealed that different JNK-dependent responses require selective use of various MAP3K proteins (Chen et al. 2002; Stronach 2005; Cuevas et al. 2007; Craig et al. 2008; Cronan et al. 2012). Understanding the factors that determine selective or combinatorial action of upstream transducers is important for the prospect of therapeutic intervention in diseases of unregulated JNK signaling (Manning and Davis 2003). Sequences that contribute to selective functions in vivo were investigated here using molecular chimeras of the Drosophila MAP3K family members, Slpr, a MLK homolog, and Tak1. Three different contexts were examined including embryonic dorsal closure morphogenesis, Eiger/TNF-dependent cell death during eye development, and systemic innate immunity in adults, asking what protein domains are required by Slpr and Tak1 to inhibit endogenous JNK signaling or to induce ectopic signaling.
Kinase domain specificity
It has been established that Tak1 and Slpr/MLK both transduce signals directly to Hep/MKK7 protein kinase as an intermediate to JNK activation (Sathyanarayana et al. 2003; Geuking et al. 2009), but that Tak1 can phosphorylate other substrates as well to activate the Rel/NF-κB pathway (Silverman et al. 2003). Given the different contexts where both MAP3Ks are expressed, we investigated what controls the use of one transducer over the other and whether the kinase activity of one MAP3K would suffice for the other. Our findings indicate that the kinase domains of Slpr and Tak1 do not functionally compensate for one another, even when introduced into the alternate signaling context by way of additional nonkinase domains. STK was feeble in rescuing the embryonic function of slpr mutants and detrimental over the course of development (Figure 4). Yet, the localization of the transgenic protein was indistinguishable from wild-type Slpr in two tissue contexts (Figure 2 and Figure 3) and overexpression resulted in ectopic induction of puc-lacZ in the embryo, an indication that catalytic activity was intact, though perhaps not maximal (Figure 5). Similarly, TSK did not support Tak1-mediated immune or cell death responses (Figure 6 and Figure 7), nor did it induce robust Tak1-dependent transcriptional targets (Figure 8 and Figure 9). The catalytic activity of TSK is unknown; however, the protein was expressed highly and localized comparably with Tak1K46R protein in the cytosol (Figure 1, Figure 2, and Figure 3). These data suggest that precise exchange of the kinase domains between Tak1 and Slpr does not reconstitute functional signal transducers contrasting with studies of protein kinase C catalytic domain swaps, which reconstituted functional enzymes with altered specificity (Walker et al. 1995). In that case, the degree of conservation was much higher, whereas the kinase domains of MLK and Tak1 are only 32% identical. We suggest that the mechanics of catalytic activation may have been uncoupled from the kinase domains in our swaps. To elaborate, ubiquitylation is required at multiple steps during Tak1-dependent innate immune signaling to regulate protein activation and degradation (Park et al. 2004; Tsuda et al. 2005; Zhou et al. 2005). It has also been shown that Tak1 catalysis can be reconstituted in vitro by unanchored K63-polyubiquitin chains bound to Tab2/3 (Kanayama et al. 2004; Xia et al. 2009). Though the precise details of this mechanism are still unclear, the Tab2–ubiquitin complexes may be ineffective toward the activation of the Slpr kinase domain even in the context of the remaining Tak1 sequences. The kinase domains are also sites of interaction with unique protein partners likely to contribute to specific responses. For instance, mammalian Tak1 signaling is regulated by Tab1, a pseudophosphatase, via interaction with the kinase domain (Shibuya et al. 1996; Sakurai et al. 2000; Conner et al. 2006). MLKs on the other hand, have the potential to bind numerous regulators at the kinase domain including Rho GTPase (Neisch et al. 2010), a RhoGEF (Swenson-Fields et al. 2008), Pak kinase (Poitras et al. 2003), and an Hsp90/p50 co-complex (Zhang et al. 2004). Thus, the differential kinase functions observed in our studies could be attributable to nonoverlapping cohorts of binding partners, modifications, activation mechanisms, and possibly spatial context within the cell.
Contributions of nonkinase domains
In regard to subcellular spatial localization as a possible contributor to signaling specificity, the C-terminal half of the Slpr protein facilitates cortical subcellular localization in both epithelia and fat body tissue (Figure 2 and Figure 3). Comparing SlprWT to SKLC or STCt under conditions of overexpression, the C-terminal region was not absolutely essential for viability, but clearly bolstered Slpr function, including activation of puc-lacZ in the embryo and the adult (Figure 4, Figure 5, and Figure 9). Swapping the Slpr C terminus for that of Tak1 did not alter Slpr specificity in dorsal closure or immunity. Instead, STCt supported a moderate degree of signaling, as evidenced by the slpr rescue experiments, and SAAATCt showed limited interference with endogenous JNK signaling during dorsal closure (Figure 4 and Figure 5), indicating residual functional interactions with the SH3, kinase, LZ, and CRIB domains of Slpr. In the context of innate immune signaling, addition of the Tak1 C terminus to Slpr SKLC to make STCt also failed to impart the ability to respond systemically or transcriptionally (Figure 7 and Figure 8). Altogether, with respect to Slpr-dependent JNK activation, we argue that localization at the cortex of the cell, mediated by sequences in the C-terminal half of the Slpr protein, coupled with the presence of the SH3, LZ, and CRIB domains, which allow interactions with upstream activators (Garlena et al. 2010), are required for optimal signaling and target gene expression during dorsal closure. Since Tak1 lacks these interaction domains and localization at the membrane, endogenous Tak1 and the Tak1-based chimeric transgenes are unproductive in engaging JNK signaling during dorsal closure. This is not likely to reflect the absence of appropriate signaling partners, however. Given that overexpression of wild-type Tak1 robustly induces JNK-dependent cell death in the epidermis similar to its effect in larval imaginal discs (Takatsu et al. 2000; Mihaly et al. 2001), the machinery for productive Tak1-dependent JNK signaling is presumably present, but latent.
Just as the C terminus of Slpr is important for maximal Slpr function, the Tak1 C-terminal region was key to participation in Eiger-dependent cell death. The small eye phenotype resulting from ectopic Eiger expression was strongly suppressed by coexpression with any construct that contained the C-terminal portion of Tak1, suggesting that interactions within this region are rate limiting for Eiger signaling. One explanation for these results is sequestration of Tab2, whose levels are critical for appropriate signal transduction from Eiger (Geuking et al. 2005). In line with these results, cytokine-stimulated Tak1 signaling in cultured human and mouse cells is also dependent on functional interactions with Tab2/3, which map to residues in the C terminus of Tak1 (Besse et al. 2007). Our additional findings that no individual Slpr mutant or deletion constructs were sufficient to dominantly block Eiger signaling (Figure 6 and Polaski et al. 2006) are also consistent; these constructs lacked docking sites for Tak1 C-terminal binding partners, trumping residual interactions with the substrate Hep kinase. Another factor possibly contributing to the unsuccessful phenotypic suppression of Eiger by transgenic Slpr proteins is the MAP2K, Mkk4, which is required in a nonredundant manner with Hep/Mkk7 downstream of Tak1 (Geuking et al. 2009). Mkk4 mutants are viable, however, suggesting a lack of functional requirements in Slpr-dependent developmental signaling contexts. Thus, the genetic requirements and binding interactions of Mkk4 and Tab2 with Tak1 in JNK activation would provide a feasible explanation for the context-dependent selective signaling of Tak1, rather than Slpr, downstream of Eiger/TNF. Lastly, recent studies implicate Eiger-dependent JNK signaling associated with endocytic compartments (Igaki et al. 2009), which may also facilitate specificity through spatial separation of transducers. Taken together, these data indicate that the C-terminal regions of Slpr and Tak1 contribute to localization and selective integration into the appropriate signaling pathways in a context-dependent manner.
Intriguingly, in the context of the innate immune response, which requires Tak1-dependent activation of JNK and Rel signaling in combination with Tab2 (Kleino et al. 2005; Zhuang et al. 2006), expression of the Tak1 C-terminal region on its own did not impair an effective immune response against E. coli infection, even in a heterozygous Tak1 mutant background (Figure 7). Yet, phenotypic susceptibility was observed with expression of Tak1K46R and SAAATCt. To get a handle on the extent to which the phenotypes reflected effects on AMP expression, we evaluated basal and induced Diptericin levels in flies expressing the various transgenes. Basal immune signaling is actively repressed, but overexpression of Tak1 is sufficient for Rel-dependent AMP induction in vivo in the absence of bacterial challenge (Vidal et al. 2001; Leulier et al. 2002). Our findings also demonstrate that Tak1 can induce constitutive Dpt expression above basal levels as expected, but the other chimeras and SlprWT had no effect (Figure 8). The latter observation is consistent with the absence of immunity phenotypes of slpr mutants (not shown), the resistance of adults expressing dominant negative SlprAAA to E.coli infection (Figure 7), and previous reports that expression of activated Hep failed to induce ectopic dpt expression without bacterial challenge (Delaney et al. 2006). Thus, in the context of the Rel signaling branch, Tak1 is highly specific vs. Slpr. Upon infection, Dpt expression levels increased a 100-fold or more in several hours. Under these conditions, SlprWT and STK had a minor insignificant effect, but SlprAAA blocked full induction. Tak1Ct-bearing proteins inhibited induction of Dpt at least as well as Tak1K46R, whose expression was actually far greater based on RT-PCR amplification with Tak1 gene-specific primers (Figure 8 and Figure S2). Thus, there was a partial disconnect between Dpt regulation and infection susceptibility vis-à-vis expression of the TCt and SlprAAA constructs, the latter of which might be due to its influence on JNK signaling, resulting in submaximal AMP induction upon infection as noted by others (Kallio et al. 2005; Delaney et al. 2006). Given that innate immune signaling is highly complex and regulated at many levels to prevent unnecessary activation or prolonged response (Schneider 2007), it is perhaps not surprising that the effects on Dpt induction did not fully account for the overall systemic response.
With respect to the JNK signaling arm, puc is known to be upregulated transiently and at relatively low levels in the event of infection (Boutros et al. 2002; Park et al. 2004; Guntermann and Foley 2011). Here, both Tak1 and Slpr induced puc-lacZ levels significantly in the fat body regardless of infection (Figure 9), indicating that these cells have the capability to activate JNK signaling in response to more than one MAP3K. However, the effects of Tak1 were much more severe, presumably attributable to activation of other factors like Rel. No other construct induced a response similar to their parental constructs consistent with results on basal Dpt induction.
In summary, Tak1 is dispensable in the Slpr-dependent process of dorsal closure; it does not induce or inhibit morphogenetic JNK signaling. Similarly, Slpr is dispensable for Eiger/TNF-induced cell death and innate immune response mediated by Tak1. In exploring the protein contributions to this context-dependent specificity, our findings substantiate the following conclusions. First, the kinase catalytic domains are distinct in the chimeras, inferring that they contribute to inherent specificity of the proteins and pathways in which they function. Second, the C-terminal regions direct integration of the proteins into proper signaling contexts spatially and through interactions with relevant activators. Third, the properties afforded by certain domains, e.g., the C-terminal region of Tak1, are also subject to context-specific influences such that interactions that are rate limiting in one signaling context may not be in another.
Acknowledgments
We are grateful to A. Green, Z. Sailor, T. Zion, L. O’Neill, J. Wlodarczyk, and B. Fritchmann for their technical contributions and fly stock maintenance during the course of this work. We also appreciate the generosity of the fly community including L. Kockel, M. Miura, N. Silverman, E. Spana, and the Bloomington Stock Center for stocks used in this study. Fas3 antibody was acquired from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biology. This work was funded by the National Institutes of Health (HD045836).
Footnotes
Communicating editor: L. Cooley
Literature Cited
- Aggarwal K., Silverman N., 2008. Positive and negative regulation of the Drosophila immune response. BMB Rep 41: 267–277 [DOI] [PubMed] [Google Scholar]
- Alexander J., Lim D., Joughin B. A., Hegemann B., Hutchins J. R., et al. , 2011. Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci. Signal. 4: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimov A., Leppanen V. M., Tvorogov D., Zarkada G., Jeltsch M., et al. , 2013. The basis for the distinct biological activities of vascular endothelial growth factor receptor-1 ligands. Sci. Signal. 6: ra52. [DOI] [PubMed] [Google Scholar]
- Besse A., Lamothe B., Campos A. D., Webster W. K., Maddineni U., et al. , 2007. TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J. Biol. Chem. 282: 3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson N., Tremblay M., Robinson F., Kaplan D. R., Trusko S. P., et al. , 2008. Mice lacking both mixed-lineage kinase genes Mlk1 and Mlk2 retain a wild type phenotype. Cell Cycle 7: 909–916 [DOI] [PubMed] [Google Scholar]
- Bock B. C., Vacratsis P. O., Qamirani E., Gallo K. A., 2000. Cdc42-induced activation of the mixed-lineage kinase SPRK in vivo. Requirement of the Cdc42/Rac interactive binding motif and changes in phosphorylation. J. Biol. Chem. 275: 14231–14241 [DOI] [PubMed] [Google Scholar]
- Boutros M., Agaisse H., Perrimon N., 2002. Sequential activation of signaling pathways during innate immune responses in Drosophila. Dev. Cell 3: 711–722 [DOI] [PubMed] [Google Scholar]
- Brancho D., Ventura J. J., Jaeschke A., Doran B., Flavell R. A., et al. , 2005. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol. Cell. Biol. 25: 3670–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N., 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415 [DOI] [PubMed] [Google Scholar]
- Calleja M., Moreno E., Pelaz S., Morata G., 1996. Visualization of gene expression in living adult Drosophila. Science 274: 252–255 [DOI] [PubMed] [Google Scholar]
- Chang L., Karin M., 2001. Mammalian MAP kinase signalling cascades. Nature 410: 37–40 [DOI] [PubMed] [Google Scholar]
- Chen J., Miller E. M., Gallo K. A., 2010. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene 29: 4399–4411 [DOI] [PubMed] [Google Scholar]
- Chen W., White M. A., Cobb M. H., 2002. Stimulus-specific requirements for MAP3 kinases in activating the JNK pathway. J. Biol. Chem. 277: 49105–49110 [DOI] [PubMed] [Google Scholar]
- Collins C. A., Wairkar Y. P., Johnson S. L., Diantonio A., 2006. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51: 57–69 [DOI] [PubMed] [Google Scholar]
- Conner S. H., Kular G., Peggie M., Shepherd S., Schuttelkopf A. W., et al. , 2006. TAK1-binding protein 1 is a pseudophosphatase. Biochem. J. 399: 427–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig E. A., Stevens M. V., Vaillancourt R. R., Camenisch T. D., 2008. MAP3Ks as central regulators of cell fate during development. Dev. Dyn. 237: 3102–3114 [DOI] [PubMed] [Google Scholar]
- Cronan M. R., Nakamura K., Johnson N. L., Granger D. A., Cuevas B. D., et al. , 2012. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene 31: 3889–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas B. D., Abell A. N., Johnson G. L., 2007. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene 26: 3159–3171 [DOI] [PubMed] [Google Scholar]
- Delaney J. R., Stoven S., Uvell H., Anderson K. V., Engstrom Y., et al. , 2006. Cooperative control of Drosophila immune responses by the JNK and NF-kappaB signaling pathways. EMBO J. 25: 3068–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drysdale R., Flybase C., 2008. FlyBase: a database for the Drosophila research community. Methods Mol. Biol. 420: 45–59 [DOI] [PubMed] [Google Scholar]
- Du Y., Bock B. C., Schachter K. A., Chao M., Gallo K. A., 2005. Cdc42 induces activation loop phosphorylation and membrane targeting of mixed lineage kinase 3. J. Biol. Chem. 280: 42984–42993 [DOI] [PubMed] [Google Scholar]
- Freeman M., 1996. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell 87: 651–660 [DOI] [PubMed] [Google Scholar]
- Gallo K. A., Johnson G. L., 2002. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 3: 663–672 [DOI] [PubMed] [Google Scholar]
- Garlena R. A., Gonda R. L., Green A. B., Pileggi R. M., Stronach B., 2010. Regulation of mixed-lineage kinase activation in JNK-dependent morphogenesis. J. Cell Sci. 123: 3177–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgel P., Naitza S., Kappler C., Ferrandon D., Zachary D., et al. , 2001. Drosophila immune deficiency (IMD) is a death domain protein that activates antibacterial defense and can promote apoptosis. Dev. Cell 1: 503–514 [DOI] [PubMed] [Google Scholar]
- Geuking P., Narasimamurthy R., Basler K., 2005. A genetic screen targeting the tumor necrosis factor/Eiger signaling pathway: identification of Drosophila TAB2 as a functionally conserved component. Genetics 171: 1683–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuking P., Narasimamurthy R., Lemaitre B., Basler K., Leulier F., 2009. A non-redundant role for Drosophila Mkk4 and hemipterous/Mkk7 in TAK1-mediated activation of JNK. PLoS ONE 4: e7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda R. L., Garlena R. A., Stronach B., 2012. Drosophila heat shock response requires the JNK pathway and phosphorylation of mixed lineage kinase at a conserved serine-proline motif. PLoS ONE 7: e42369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley B. R., Brooks A. N., Carlson J. W., Duff M. O., Landolin J. M., et al. , 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntermann S., Foley E., 2011. The protein Dredd is an essential component of the c-Jun N-terminal kinase pathway in the Drosophila immune response. J. Biol. Chem. 286: 30284–30294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R., 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59 [DOI] [PubMed] [Google Scholar]
- Holland P. M., Suzanne M., Campbell J. S., Noselli S., Cooper J. A., 1997. MKK7 is a stress-activated mitogen-activated protein kinase kinase functionally related to hemipterous. J. Biol. Chem. 272: 24994–24998 [DOI] [PubMed] [Google Scholar]
- Hultmark D., 1993. Immune reactions in Drosophila and other insects: a model for innate immunity. Trends Genet. 9: 178–183 [DOI] [PubMed] [Google Scholar]
- Igaki T., Kanda H., Yamamoto-Goto Y., Kanuka H., Kuranaga E., et al. , 2002. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21: 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T., Pastor-Pareja J. C., Aonuma H., Miura M., Xu T., 2009. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell 16: 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadrich J. L., O’Connor M. B., Coucouvanis E., 2006. The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development 133: 1529–1541 [DOI] [PubMed] [Google Scholar]
- Jaeschke A., Davis R. J., 2007. Metabolic stress signaling mediated by mixed-lineage kinases. Mol. Cell 27: 498–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson G. L., Nakamura K., 2007. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim. Biophys. Acta 1773: 1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio J., Leinonen A., Ulvila J., Valanne S., Ezekowitz R. A., et al. , 2005. Functional analysis of immune response genes in Drosophila identifies JNK pathway as a regulator of antimicrobial peptide gene expression in S2 cells. Microbes Infect. 7: 811–819 [DOI] [PubMed] [Google Scholar]
- Kanayama A., Seth R. B., Sun L., Ea C. K., Hong M., et al. , 2004. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol. Cell 15: 535–548 [DOI] [PubMed] [Google Scholar]
- Kang M. J., Chung J., Ryoo H. D., 2012. CDK5 and MEKK1 mediate pro-apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat. Cell Biol. 14: 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant S., Swat W., Zhang S., Zhang Z. Y., Neel B. G., et al. , 2011. TNF-stimulated MAP kinase activation mediated by a Rho family GTPase signaling pathway. Genes Dev. 25: 2069–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet Y., Seger R., 2010. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 661: 3–38 [DOI] [PubMed] [Google Scholar]
- Kleino A., Valanne S., Ulvila J., Kallio J., Myllymaki H., et al. , 2005. Inhibitor of apoptosis 2 and TAK1-binding protein are components of the Drosophila Imd pathway. EMBO J. 24: 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korchnak A. C., Zhan Y., Aguilar M. T., Chadee D. N., 2009. Cytokine-induced activation of Mixed Lineage Kinase 3 requires TRAF2 and TRAF6. Cell Signal. 21: 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuranaga E., Kanuka H., Igaki T., Sawamoto K., Ichijo H., et al. , 2002. Reaper-mediated inhibition of DIAP1-induced DTRAF1 degradation results in activation of JNK in Drosophila. Nat. Cell Biol. 4: 705–710 [DOI] [PubMed] [Google Scholar]
- Lee G., Park J. H., 2004. Hemolymph sugar homeostasis and starvation-induced hyperactivity affected by genetic manipulations of the adipokinetic hormone-encoding gene in Drosophila melanogaster. Genetics 167: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B., Kromer-Metzger E., Michaut L., Nicolas E., Meister M., et al. , 1995. A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proc. Natl. Acad. Sci. USA 92: 9465–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulier F., Vidal S., Saigo K., Ueda R., Lemaitre B., 2002. Inducible expression of double-stranded RNA reveals a role for dFADD in the regulation of the antibacterial response in Drosophila adults. Curr. Biol. 12: 996–1000 [DOI] [PubMed] [Google Scholar]
- Manning A. M., Davis R. J., 2003. Targeting JNK for therapeutic benefit: from junk to gold? Nat. Rev. Drug Discov. 2: 554–565 [DOI] [PubMed] [Google Scholar]
- Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S., 2002. The protein kinase complement of the human genome. Science 298: 1912–1934 [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E., Gampel A., Ring J., Virdee K., Kirov N., et al. , 1998. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12: 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaly J., Kockel L., Gaengel K., Weber U., Bohmann D., et al. , 2001. The role of the Drosophila TAK homologue dTAK during development. Mech. Dev. 102: 67–79 [DOI] [PubMed] [Google Scholar]
- Moreno E., Yan M., Basler K., 2002. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12: 1263–1268 [DOI] [PubMed] [Google Scholar]
- Neisch A. L., Speck O., Stronach B., Fehon R. G., 2010. Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J. Cell Biol. 189: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori E., Matsumoto K., Sanjo H., Sato S., Akira S., et al. , 2006. TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. J. Biol. Chem. 281: 19610–19617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. M., Brady H., Ruocco M. G., Sun H., Williams D., et al. , 2004. Targeting of TAK1 by the NF-kappa B protein Relish regulates the JNK-mediated immune response in Drosophila. Genes Dev. 18: 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitras L., Jean S., Islam N., Moss T., 2003. PAK interacts with NCK and MLK2 to regulate the activation of jun N-terminal kinase. FEBS Lett. 543: 129–135 [DOI] [PubMed] [Google Scholar]
- Polaski S., Whitney L., Barker B. W., Stronach B., 2006. Genetic analysis of slipper/mixed lineage kinase reveals requirements in multiple Jun-N-terminal kinase-dependent morphogenetic events during Drosophila development. Genetics 174: 719–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring J. M., Martinez Arias A., 1993. puckered, a gene involved in position-specific cell differentiation in the dorsal epidermis of the Drosophila larva. Dev. Suppl. 251–259 [PubMed] [Google Scholar]
- Romeo Y., Lemaitre B., 2008. Drosophila immunity: methods for monitoring the activity of Toll and Imd signaling pathways. Methods Mol. Biol. 415: 379–394 [DOI] [PubMed] [Google Scholar]
- Rorth P., 1998. Gal4 in the Drosophila female germline. Mech. Dev. 78: 113–118 [DOI] [PubMed] [Google Scholar]
- Ryabinina O. P., Subbian E., Iordanov M. S., 2006. D-MEKK1, the Drosophila orthologue of mammalian MEKK4/MTK1, and Hemipterous/D-MKK7 mediate the activation of D-JNK by cadmium and arsenite in Schneider cells. BMC Cell Biol. 7: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K., 2012. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Transl. Sci. 106: 145–169 [DOI] [PubMed] [Google Scholar]
- Sakurai H., 2012. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 33: 522–530 [DOI] [PubMed] [Google Scholar]
- Sakurai H., Miyoshi H., Mizukami J., Sugita T., 2000. Phosphorylation-dependent activation of TAK1 mitogen-activated protein kinase kinase kinase by TAB1. FEBS Lett. 474: 141–145 [DOI] [PubMed] [Google Scholar]
- Sanchez-Hernandez D., Sierra J., Ortigao-Farias J. R., Guerrero I., 2012. The WIF domain of the human and Drosophila Wif-1 secreted factors confers specificity for Wnt or Hedgehog. Development 139: 3849–3858 [DOI] [PubMed] [Google Scholar]
- Sanson B., White P., Vincent J. P., 1996. Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 383: 627–630 [DOI] [PubMed] [Google Scholar]
- Sathyanarayana P., Barthwal M. K., Kundu C. N., Lane M. E., Bergmann A., et al. , 2002. Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Mol. Cell 10: 1527–1533 [DOI] [PubMed] [Google Scholar]
- Sathyanarayana P., Barthwal M. K., Lane M. E., Acevedo S. F., Skoulakis E. M., et al. , 2003. Drosophila mixed lineage kinase/slipper, a missing biochemical link in Drosophila JNK signaling. Biochim. Biophys. Acta 1640: 77–84 [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D., Livak K. J., 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Schneider D. S., 2007. How and why does a fly turn its immune system off? PLoS Biol. 5: e247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya H., Yamaguchi K., Shirakabe K., Tonegawa A., Gotoh Y., et al. , 1996. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 272: 1179–1182 [DOI] [PubMed] [Google Scholar]
- Shim J. H., Xiao C., Paschal A. E., Bailey S. T., Rao P., et al. , 2005. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19: 2668–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R. M., Kuan C. Y., Rakic P., Burke R. E., 2005. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson’s disease. Mov. Disord. 20: 653–664 [DOI] [PubMed] [Google Scholar]
- Silverman N., Zhou R., Erlich R. L., Hunter M., Bernstein E., et al. , 2003. Immune activation of NF-kappaB and JNK requires Drosophila TAK1. J. Biol. Chem. 278: 48928–48934 [DOI] [PubMed] [Google Scholar]
- Stark M. S., Woods S. L., Gartside M. G., Bonazzi V. F., Dutton-Regester K., et al. , 2012. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat. Genet. 44: 165–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronach B., 2005. Dissecting JNK signaling, one KKKinase at a time. Dev. Dyn. 232: 575–584 [DOI] [PubMed] [Google Scholar]
- Stronach B., Perrimon N., 2002. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 16: 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson-Fields K. I., Sandquist J. C., Rossol-Allison J., Blat I. C., Wennerberg K., et al. , 2008. MLK3 limits activated Galphaq signaling to Rho by binding to p63RhoGEF. Mol. Cell 32: 43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaesu G., Kishida S., Hiyama A., Yamaguchi K., Shibuya H., et al. , 2000. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell 5: 649–658 [DOI] [PubMed] [Google Scholar]
- Takatsu Y., Nakamura M., Stapleton M., Danos M. C., Matsumoto K., et al. , 2000. TAK1 participates in c-Jun N-terminal kinase signaling during Drosophila development. Mol. Cell. Biol. 20: 3015–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M., Langmann C., Harden N., Aigaki T., 2005. The RING-finger scaffold protein plenty of SH3s targets TAK1 to control immunity signalling in Drosophila. EMBO Rep. 6: 1082–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacratsis P. O., Gallo K. A., 2000. Zipper-mediated oligomerization of the mixed lineage kinase SPRK/MLK-3 is not required for its activation by the GTPase cdc 42 but is necessary for its activation of the JNK pathway. Monomeric SPRK L410P does not catalyze the activating phosphorylation of Thr258 of murine mitogen-activated protein kinase kinase 4. J. Biol. Chem. 275: 27893–27900 [DOI] [PubMed] [Google Scholar]
- Vacratsis P. O., Phinney B. S., Gage D. A., Gallo K. A., 2002. Identification of in vivo phosphorylation sites of MLK3 by mass spectrometry and phosphopeptide mapping. Biochemistry 41: 5613–5624 [DOI] [PubMed] [Google Scholar]
- Velho S., Oliveira C., Paredes J., Sousa S., Leite M., et al. , 2010. Mixed lineage kinase 3 gene mutations in mismatch repair deficient gastrointestinal tumours. Hum. Mol. Genet. 19: 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S., Khush R. S., Leulier F., Tzou P., Nakamura M., et al. , 2001. Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-kappaB-dependent innate immune responses. Genes Dev. 15: 1900–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E. F., Nebreda A. R., 2009. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9: 537–549 [DOI] [PubMed] [Google Scholar]
- Walker S. D., Murray N. R., Burns D. J., Fields A. P., 1995. Protein kinase C chimeras: catalytic domains of alpha and beta II protein kinase C contain determinants for isotype-specific function. Proc. Natl. Acad. Sci. USA 92: 9156–9160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., et al. , 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412: 346–351 [DOI] [PubMed] [Google Scholar]
- Wodarz A., Hinz U., Engelbert M., Knust E., 1995. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell 82: 67–76 [DOI] [PubMed] [Google Scholar]
- Xia Z. P., Sun L., Chen X., Pineda G., Jiang X., et al. , 2009. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461: 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., et al. , 1995. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270: 2008–2011 [DOI] [PubMed] [Google Scholar]
- Zhan Y., Abi Saab W. F., Modi N., Stewart A. M., Liu J., et al. , 2012. Mixed lineage kinase 3 is required for matrix metalloproteinase expression and invasion in ovarian cancer cells. Exp. Cell Res. 318: 1641–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Gallo K. A., 2001. Autoinhibition of mixed lineage kinase 3 through its Src homology 3 domain. J. Biol. Chem. 276: 45598–45603 [DOI] [PubMed] [Google Scholar]
- Zhang H., Wu W., Du Y., Santos S. J., Conrad S. E., et al. , 2004. Hsp90/p50cdc37 is required for mixed-lineage kinase (MLK) 3 signaling. J. Biol. Chem. 279: 19457–19463 [DOI] [PubMed] [Google Scholar]
- Zhou R., Silverman N., Hong M., Liao D. S., Chung Y., et al. , 2005. The role of ubiquitination in Drosophila innate immunity. J. Biol. Chem. 280: 34048–34055 [DOI] [PubMed] [Google Scholar]
- Zhuang Z. H., Sun L., Kong L., Hu J. H., Yu M. C., et al. , 2006. Drosophila TAB2 is required for the immune activation of JNK and NF-kappaB. Cell. Signal. 18: 964–970 [DOI] [PubMed] [Google Scholar]