

Abstract
Vegetative incompatibility (vic), a form of nonself allorecognition, operates widely in filamentous fungi and restricts transmission of virulence-attenuating hypoviruses in the chestnut blight fungus Cryphonectria parasitica. We report here the use of a polymorphism-based comparative genomics approach to complete the molecular identification of the genetically defined C. parasitica vic loci with the identification of vic1 and vic3. The vic1 locus in the C. parasitica reference strain EP155 consists of a polymorphic HET-domain-containing 771-aa ORF designated vic1a-2, which shares 91% identity with the corresponding vic1a-1 allele, and a small (172 aa) idiomorphic DUF1909-domain-containing ORF designated vic1b-2 that is absent at the vic1-1 locus. Gene disruption of either vic1a-2 or vic1b-2 in strain EP155 eliminated restrictions on virus transmission when paired with a vic1 heteroallelic strain; however, only disruption of vic1a-2 abolished the incompatible programmed cell death (PCD) reaction. The vic3 locus of strain EP155 contains two polymorphic ORFs of 599 aa (vic3a-1) and 102 aa (vic3b-1) that shared 46 and 85% aa identity with the corresponding vic3a-2 and vic3b-2 alleles, respectively. Disruption of either vic3a-1 or vic3b-1 resulted in increased virus transmission. However, elimination of PCD required disruption of both vic3a and vic3b. Additional allelic heterogeneity included a sequence inversion and a 8.5-kb insertion containing a LTR retrotransposon sequence and an adjacent HET-domain gene at the vic1 locus and a 7.7-kb sequence deletion associated with a nonfunctional, pseudo vic locus. Combined gene disruption studies formally confirmed restriction of mycovirus transmission by five C. parasitica vic loci and suggested dedicated roles in allorecognition. The relevance of these results to the acquisition and maintenance of vic genes and the potential for manipulation of vic alleles for enhanced mycovirus transmission are discussed.
Keywords: Cryphonectria parasitica, hypovirulence, mycovirus transmission, nonself recognition, vegetative incompatibility
ALLORECOGNITION genetic systems, which provide the ability to distinguish self from nonself, play important functional roles in microbial and multicellular organisms. These systems range from restriction endonucleases in bacteria (Meselson and Yuan 1968) to somatic histocompatibility in protocordates (De Tomaso et al. 2005), self-infertility in plants (Nasrallah 2005), and innate immunity in vertebrates (Medzhitov and Janeway 2002) (reviewed by Aanen et al. 2008; Nydam and De Tomaso 2011; Rosengarten and Nicotra 2011). Allorecognition operates widely in filamentous fungi in both the sexual and the vegetative growth phases (Saupe 2000). The role of the mating-type locus in controlling sexual recognition and promoting outbreeding in yeast and filamentous fungi is well understood (reviewed by Coppin et al. 1997). It is also known that somatic or vegetative fusion of fungal cells (termed “anastomosis”) occurs at a high frequency within and between individuals promoting network formation (Rayner 1996) and facilitating foraging, the pooling of resources (Rayner 1996), and the introduction of genetic variation via the parasexual cycle (Pontecorvo 1956). However, most fusion events between genetically distinct individuals of the same species result in an incompatible reaction that triggers localized cell death (reviewed by Glass et al. 2000; Saupe 2000). This form of conspecific nonself recognition is know as heterokaryon incompatibility or, as used here, vegetative incompatibility, and the corresponding genetic determinants are known as het or vic loci (Smith and Lafontaine 2013).
While vegetative incompatibility is widespread in filamentous fungi (reviewed in Smith and Lafontaine 2013), the origins and biological functions of these fungal nonself recognition systems remain enigmatic. Two nonexclusive hypotheses have been forwarded (Saupe 2000). The accidental hypothesis proposes that vegetative incompatibility does not have a function but is the result of some evolutionary coincidence that resulted in the formation of polymorphic alleles that are detrimental when combined, e.g., as a result of anastomosis. A prediction of this hypothesis is that the vic genes would not be dedicated to allorecognition but would have biological functions in addition to nonself recognition. The allorecognition hypothesis proposes that vegetative incompatibility functions primarily to reduce transmission of deleterious genetic elements such as mycoviruses (Caten 1972), transposable elements (Kinsey 1990), or parasitic nuclei (Debets and Griffiths 1998). A prediction of this hypothesis is that vic genes are dedicated to nonself recognition and that dual functions would be rare.
The genetic and molecular characterization of vic genes has been limited to three ascomycetes, the model fungi Neurospora crassa and Podospora anserina and, more recently, the plant pathogenic fungus Cryphonectria parasitica, the chestnut blight fungus. Genes have been characterized for 3 of the 11 genetically defined N. crassa incompatibility loci and 3 of the 9 P. anserina incompatibility loci (reviewed by Smith and Lafontaine 2013). The characterized vic genes exhibited high levels of allelic polymorphism as a result of diversifying selection (Hall et al. 2010; Bastiaans et al. 2014), and several have been shown to have dual functions; e.g., N. crassa mat-A1 and mat-a1 genes are involved in transcriptional regulation of mating type (Glass et al. 1990; Staben and Yanofsky 1990), N. crassa un-24 encodes the large subunit of ribonucleotide reductase (Lafontaine and Smith 2012), and P. anserina het-C encodes a glycolipid transferase protein (Saupe et al. 1994). While diversifying selection of vic genes supports the allorecognition hypothesis and dual function of vic genes supports the accidental hypothesis, the analyses are limited to a subset of the genetically defined vic loci for the two fungi.
Unlike N. crassa and P. anserina, C. parasitica is known to be subjected to widespread infections in nature by a family of viruses, the Hypoviridae, that can reduce ecological fitness, e.g., loss of female fertility, reduced asexual reproduction, and reduced virulence on the plant host (reviewed by Nuss 2005; Dawe and Nuss 2013). Hypovirus-mediated attenuation of C. parasitica virulence, termed “transmissible hypovirulence,” naturally moderated the chestnut blight epidemic in Europe and provided the basis for applied biological control (Anagnostakis, 1982a, 1983, 1988). Because mycoviruses generally replicate without an extracellular phase, they are transmitted primarily by fungal anastomosis (Ghabrial and Suzuki 2009). The incompatible reaction and accompanying localized cell death triggered by the vic system is predicted to constrain cytoplasmic exchange, thus restricting virus transmission.
Genetic studies have defined six C. parasitica vic loci with only two alleles at each locus (Anagnostakis 1982b; Huber 1996; Cortesi and Milgroom 1998). Subsequent studies have confirmed that allelic differences at any of the individual vic loci, except vic4, restrict hypovirus transmission with a magnitude related to the rate at which programmed cell death (PCD) occurs after hyphal fusion (Liu and Milgroom 1996; Cortesi et al. 2001; Biella et al. 2002). However, it must be noted that, since the heteroallelic strains were not isogenic, it has not been possible to rule out the possibility that non-vic genetic determinants contribute to restriction of virus transmission. In the strictest sense, the allorecognition hypothesis would predict that the C. parasitica vic genes should be primarily, if not exclusively, dedicated to nonself recognition and that their disruption would allow unrestricted virus transmission without significant negative effects on other biological functions.
Difficulties in identifying vic genes have hampered progress in understanding the molecular mechanisms of fungal vegetative incompatibility. Recent efforts to identify fungal vic genes based on sequence homology with N. crassa and P. anserina vic genes have not been fruitful (Fournier et al. 2003; Kerenyi et al. 2006; Pal et al. 2007; Van Diepeningen et al. 2008; Iotti et al. 2012; Van Der Nest et al. 2014). Using a strategy based on polymorphism-guided comparative genomics and on information about vic genetic markers, we recently identified seven incompatibility genes associated with four of the six C. parasitica vic loci (Choi et al. 2012). We now report an extension of this approach to identify the remaining vic loci, vic1 and vic3. In addition to polymorphisms in candidate vic genes, allelic heterogeneity includes a sequence inversion and a sequence insertion associated with vic1 as well as a sequence deletion at a nonfunctional pseudo vic locus. Functional analysis revealed evidence for both polymorphic and idiomorphic allelic components as well as allelic and nonallelic interactions. These studies formally demonstrate that five of the six C. parasitica vic loci contribute to restriction of virus transmission; as noted above, vic4 was previously shown not to restrict virus transmission. Disruption of 11 vic genes associated with the same five vic loci resulted in no detectable phenotypic changes in addition to altered incompatibility reactions or virus transmission, which is consistent with a dedicated allorecognition function of the vic loci. The molecular identification of the full complement of genetically defined C. parasitica loci provides new prospects for manipulation of the vic alleles to enhance hypovirus transmission and biological control.
Materials and Methods
Fungal strains and growth conditions
C. parasitica strains used in this study included wild-type and genome reference strain EP155 (ATCC 38755) and vic-genotyped tester strains (Cortesi and Milgroom 1998) EU31 (ATCC MYA-1074), EU40 (ATCC MYA-1083), EU55 (ATCC MYA-1098), and EU60 (ATCC MYA-1103). C. parasitica strain DK80, a mutant of EP155 containing a disruption of the nonhomologous end-joining DNA repair pathway ku80 gene homolog to promote homologous recombination (Lan et al. 2008), was used for most gene disruption analyses. C. parasitica field isolates collected from Japan (Liu and Milgroom 2007) and Maryland (Milgroom and Cortesi 1999) were kindly provided by Michael Milgroom (Cornell University). All strains were cultured on potato dextrose agar (PDA) at room temperature on the bench top, unless indicated otherwise.
Genome sequencing and analysis
Genome sequence data and gene model predictions for C. parasitica strain EP155 are accessible through the Joint Genome Institute of the U.S. Department of Energy (http://genome.jgi.doe.gov/Crypa2/Crypa2.home.html). Genome sequencing of C. parasitica vic-genotyped tester strains EU31, EU40, and EU60 was performed at the University of Maryland DNA sequencing facility according to Illumina HiSeq protocols for generating 100-base paired-end reads. Additionally, strain EU55 DNA was taken through two rounds of sequencing according to the Roche 454 GS FLX Titanium protocols for library preparation, emulsion PCR reaction, and sequencing (Margulies et al. 2005). Sequence reads were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive under study accession no. SRP040304. Genomic DNA used for sequencing was prepared after the method of Borges et al. (1990). Sequence reads were mapped by similarity to the EP155 reference genome scaffold assemblies with CLC Genomics Workbench 6.0 (CLCBio, Cambridge, MA). Sequence assembly contigs containing candidate vic alleles were analyzed with FGENESH (hosted by Softberry at http://linux1.sofberry.com) for gene and protein predictions and with NCBI’s conserved domain searches (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) to identify conserved protein domains in FGENESH-predicted ORFs. The sequences of assembled contigs generated from raw sequence reads for the vic1 allele of the nonreference strain, e.g., the vic1-1 allele in strain EU31, was confirmed by DNA sequencing of overlapping PCR amplicons generated from genomic DNA of the corresponding resequenced strains. Protein sequence alignments were performed with MUltiple Sequence Comparison by Log Expectation (MUSCLE at http://www.ebi.ac.uk/Tools/msa/muscle) and alignments visualized in JalView (http://www.jalview.org). The vic1 alleles were further analyzed by BLAST alignment of the corresponding nucleotide sequences in WebACT (http://www.webact.org/WebACT/home) for visualization of the relative genetic organization at each allele with the Artemis Comparison Tool (ACT) (Sanger Institute, Cambridge, UK).
GenBank accession numbers for the candidate vic genes are as follows: vic1a-1, HG799041; vic1a-2, HG799042; vic1b-2, HG799043; vic1c-1, HG799048; vic1d-1, HG799049; vic3a-1, HG799044; vic3a-2, HG799045; vic3b-1, HG799046; vic3b-2, HG799047; pseudo vic HET-domain gene, HG799050; and pseudo vic GTPase-domain gene, HG799051.
Disruption of candidate vic genes
Disruption of candidate vic genes was performed by homologous recombination in C. parasitica strain DK80 using PCR-generated disruption fragments based on the strategy of Kuwayama et al. (2002) with modifications as described by Zhang et al. (2013). Disruption of both vic candidate genes vic3a-2 and vic3b-2 was also accomplished in strain EU60. DNA-mediated transformation was performed according to the method of Churchill et al. (1990), followed by selection of putative transformants grown in the presence of 40 μg/ml hygromycin B or 25 μg/ml G418 for neomycin-resistance selection. Gene disruption was confirmed by PCR analysis of the putative transformants, followed by selection of uninuclear single conidial isolates on antibiotic-containing PDA to eliminate heterokaryons and by a final PCR confirmation. Conditions used for PCR were as described in Zhang et al. (2013) on fungal DNA extracted according to Spiering et al. (2008).
Analysis of fungal phenotype, mycelial incompatibility, and virus transmission
Colony morphology, asexual conidia production, and fungal virulence were assessed according to standard procedures (Hillman et al. 1990). Mycelial incompatibility and virus transmission assays were performed as described in Choi et al. (2012). Hypovirus CHV-1/EP713 infection was established in the different vic-genotyped tester strains, strain DK80, and vic1 and vic3 disruption strains by transfecting fungal spheroplasts with CHV-1/EP713 transcripts generated in vitro from a full-length viral complementary DNA as described by Chen et al. (1994).
Results
Polymorphism-based identification of the C. parasitica vic1 and vic3 loci
The polymorphism-based comparative genomics approach used for molecular identification of vic2, vic4, vic6, and vic7 (Choi et al. 2012) was extended here to identify the vic1 and vic3 loci. Genotyped vic tester strains (Cortesi and Milgroom 1998) that differed from the reference strain EP155 at vic1 and/or vic3 were resequenced by high-throughput sequencing protocols. The convention for vic genotypes specifies which allele, designated 1 or 2, is present at the individual vic genetic loci; e.g., the EP155 vic genotype is vic1-2, vic2-2, vic3-1, vic4-1, vic6-2, and vic7-2 (abbreviated to 2211-22). Raw sequence reads were mapped to the EP155 reference genome assembly by sequence similarity, and regions of significant hypervariability, appearing as gaps in the sequence read-density plots, were identified. Molecular markers linked to the vic1 locus were used as guides to locate vic1 candidate polymorphic regions. The absence of vic3-specific molecular linkage markers required the use of manual inspection to identify polymorphic vic3 candidates. The resequenced strains included EU-31 (1211-22), which differs from the reference strain EP155 (2211-22) at the vic1 locus; EU-60 (2221-22), which differs from EP155 at the vic3 locus; EU-55 (1221-22), which differs from EP155 at both the vic1 and vic3 loci; and EU-40 (1122-11), which differs from EP155 at all six genetically identified vic loci. Illumina HiSequation 1000 protocols for generating 100-base paired-end reads were used for strains EU-31, EU-40, and EU-60. Strain EU-55 DNA was sequenced using Roche 454 GS FLX Titanium protocols. More than 15 million reads for each of the EU strains were mapped to the EP155 reference sequence using CLC Genomics Workbench.
Identification of the candidate vic1 locus
Inspection of the EU-31 (1211-22; vic1, allele 1) reads mapped to EP155 (2211-22; vic1, allele 2) Scaffold 5 in a region that contained vic1 linkage markers (Supporting Information, Figure S1) resulted in the identification of a gap in the read-density plot (Scaffold 5: 921500–926500), suggesting polymorphism located ∼6 kb from the linkage marker E19 0825 (Figure 1). A similar gap was observed in the density plots for EU55 and EU40, which also contained the vic1-1 allele, but not for EU60, which contained the vic1-2 allele also present in reference strain EP155. This polymorphic region in strain EP155 consisted of an ORF encoding a 771-amino-acid-long protein containing an heterokaryon or HET domain (Pfam:PF06985) (Smith et al. 2000), conserved in genes found in the three characterized vic loci in N. crassa and two of the three characterized vic loci in P. anserina (reviewed in Smith and Lafontaine 2013) and an adjacent ORF that encodes a 172-amino-acid-long hypothetical protein containing a DUF1909 (conserved domain of unknown function) domain. The ORF encoding the HET-domain protein in strains EP155 and EU60 was designated vic1a-allele 2 (vic1a-2), and the corresponding ORF in strains EU31, EU40, and EU55 was designated vic1a-allele 1 (vic1a-1). The corresponding vic1a-1 and vic1a-2 HET proteins showed 91% identity at the amino acid level (Figure S2). This level of sequence heterogeneity was lower than that observed for the other C. parasitica vic genes [e.g., the HET-domain-containing vic6 alleles show 53% overall amino acid identity (Choi et al. 2012)]. The ORF encoding the DUF1909-domain protein in strain EP155 was designated vic1b-2. Interestingly, vic1b-2 in strain EP155 is idiomorphic rather than polymorphic. That is, the vic1b-2 sequence is absent in the corresponding vic1-1 region.
Figure 1.

The candidate vic1 locus. Density plots of sequence reads that match by sequence similarity to a corresponding portion of Scaffold 5 of the EP155 reference genome assembly (coordinates indicated at the top) near a vic1 linkage marker (E190825) are shown for three strains that differ from EP155 (2211-22) at the vic1 locus (allele 1), EU55 (1221-22), EU31 (1211-22), and EU40 (1122-11) and for strain EU60 (2221-22) that is the same as EP155 at the vic1 locus (allele 2). Strain EU55 was sequenced using Roche 454 GS FLX Titanium protocols, and the other strains were sequenced with Illumina HiSeq protocols. Forward reads are indicated in green, reverse reads are indicated in red, paired reads are indicated in blue. The region containing the highly polymorphic sequences is indicated by the absence of matched reads (gap). The ORFs located within a ∼14-kb region containing the gap are shown below the sequence read-density plots. A polymorphic gene encoding a HET-domain-containing protein (91% amino acid identity between alleles 1 and 2) was designated as vic1a-1 for allele 1 present in EU31, EU40, and EU55 and as vic1a-2 for allele 2 present in reference strain EP155 and strain EU60. The gene encoding the DUF1909-domain-containing protein present in allele 2, but absent in allele 1 (i.e., idiomorphic), was designated vic1b-2. The position of the vic1 linkage marker E190825 relative to the vic1 gene candidates is also indicated. The C. parasitica strain EP155 reference genome assembly is available at http://genome.jgi.doe.gov/Crypa2/Crypa2.home.html. The Joint Genome Institute (JGI) protein identity (ID) numbers for vic1a-2 and vic1b-2 are 330677 and 356517, respectively.
Further analysis and confirmation of the nucleotide sequence assembly contigs spanning the entire vic1-1 allele by sequencing of overlapping PCR amplicons generated from EU31 and EU55 genomic DNAs revealed evidence of a sequence inversion and a sequence insertion in vic1-1 relative to the vic1-2 sequence. A conserved ORF encoding a clavaminate synthase-like protein (Figure S3) was found to be in opposite orientation in vic1-1 and vic1-2 relative to the orientation of the predicted HET-domain genes. Interestingly, an insertion of ∼8.5 kb that contained two ORFs encoding a 1619-aa-long TY1/copia-like LTR retrotransposon (designated vic1c-1) and an adjacent 689-aa-long HET-domain protein (designated vic1d-1) was found next to the inverted clavaminate synthase-like gene in vic1-1. The relative genetic organization of the two alleles is shown in Figure 2, visualized as a BLAST alignment of the vic1-1 and vic1-2 nucleotide sequences in the Artemis Comparison Tool (available at WebACT at http://www.webact.org/WebACT/home) with a depiction of the vic1-1 sequence insert drawn to approximate scale.
Figure 2.

Comparison of the gene order at the vic1-1 and vic1-2 alleles. The sequences at the two alleles were BLAST-aligned in WebACT (http://www.webact.org/WebACT/home) for visualization of the genetic organization at each allele relative to that at the other with the Artemis Comparison Tool. High-scoring alignments are visualized in red (forward matches) and blue (reverse matches); the cutoff for an alignment was set at an ID score of ≥16. The vic1 locus contains a region that has undergone a sequence inversion affecting the orientation of a putative clavaminate synthatase-like gene relative to that of the predicted HET gene. Allele 1 is indicated below the alignment drawn to approximate scale. Note that allele 1 also contains a ∼8.5-kb insertion that is absent in allele 2. The allele-1-specific sequence contained two ORFs encoding a 689-aa HET-domain-containing protein (designated vic1d-1) and a 1619-aa TY1/copia-like LTR retrotransposon (designated vic1c-1) as indicated below the allele sequence alignment.
Disruption and functional analysis of the candidate vic1-2 genes
Where colonies of two vic incompatible C. parasitica strains come into contact, a zone of demarcation (called a “barrage”) forms, which is composed of lysed and dead cells (Powell 1995). Barrage formation is considered a form of mycelial incompatibility (Smith et al. 2006), and the incompatibility reaction is described as having the hallmarks of PCD (Biella et al. 2002 and reviewed in Smith and Lafontaine 2013).
The candidate genes at allele 2 of the vic1 locus, vic1a-2 and vic1b-2, were independently disrupted in the DK80 nonhomologous end-joining mutant strain of EP155 as described in Figure S4. While strains DK80 and EU-31 underwent an incompatible reaction, clearly forming a barrage when paired, the Δvic1a-2 disruption mutant was compatible with EU-31 (Figure 3A). Thus, disruption of the vic1a allele in just one of the paired vic1 heteroallelic strains resulted in loss of barrage formation.
Figure 3.

Mycelial incompatibility assay for strains disrupted in candidate vic alleles vic1a and vic1b in strain DK80 (2211-22) (barrage lines indicated by red arrows). (A) Barrage formation resulting from an incompatible reaction occurred when strain DK80 (2211-22) was paired with strain EU31 (1211-22), but was abolished when DK80 vic1a-2-disrupted strain Δvic1a-2 (2211-22) was paired with strain EU31 (1211-22). (B) Barrage formation was retained, but was less pronounced, when DK80 vic1b-2-disrupted strain Δvic1b-2 (2211-22) was paired with tester strain EU31 (1211-22).
The influence of heteroallelism at the vic1 locus on virus transmission is highly asymmetric (Cortesi et al. 2001). No resistance to virus transmission is observed if the recipient strain contains allele 1, while transmission is only ∼10% if the recipient strain contains allele 2. As indicated in Table 1, deletion of the vic1a-2 allele resulted in an increase in virus transmission from ∼10 to 100% in pairings in which the mutant strain served as the virus recipient and strain EU-31 served as the virus donor. The combined loss of barrage formation and increased virus transmission observed for the Δvic1a-2 mutant strain, coupled with the concordance between the allele nucleotide sequence specificity and vic genotypes for EP155, EU55, EU31, EU40, and EU60 (Figure 1), provide strong evidence that the vic1a-2 gene is a functional component of the vic1-locus.
Table 1. Hypovirus transmission frequency for disruption mutants of candidate vic1 alleles.
| Donor | Recipient | Transmission | Note |
|---|---|---|---|
| DK80 (2211-22) | EP155 (2211-22) | 20/20 | |
| EP155 (2211-22) | DK80 (2211-22) | 20/20 | |
| DK80 (2211-22) | EU31 (1211-22) | 20/20 | PDA |
| DK80 Δvic1a-2 (2211-22) | EU31 (1211-22) | 20/20 | Medium |
| EU31 (1211-22) | DK80 (2211-22) | 2/20 | |
| EU31 (1211-22) | DK80 Δvic1a-2 (2211-22) | 20/20 | |
| DK80 (2211-22) | EP155 (2211-22) | 20/20 | |
| EP155 (2211-22) | DK80 (2211-22) | 20/20 | |
| DK80 (2211-22) | EU31 (1211-22) | 20/20 | GBR |
| DK80 Δvic1b-2 (2211-22) | EU31 (1211-22) | 20/20 | Medium |
| EU31 (1211-22) | DK80 (2211-22) | 6/20 | |
| EU31 (1211-22) | DK80 Δvic1b-2 (2211-22) | 20/20 |
GBR: PDA supplemented with 7 g/liter malt extract, 2 g/liter yeast extract, 0.8 g/liter tannic acid, 50 mg/ liter Bromocresol Green, 20 g/liter additional agar, and 12 drops/liter of Tween 20) (modified from Powell 1995).
Disruption of the vic1b-2 allele (Figure S4) resulted in reduced growth and altered morphology (Figure S5). This altered phenotype complicated the interpretation of barrage formation and virus transmission experiments. However, the growth and morphology alterations were ameliorated when the mutants were grown on enriched medium (Figure S5). Under these conditions, when paired with strain EU31, the Δvic1b-2 mutant strain showed reduced, but not loss of, barrage formation (Figure 3B) and increased virus transmission from 20–30% to 100% when serving as the recipient (Table 1). These combined results demonstrate that the vic1b-2 ORF also contributes to the vic-1-associated incompatible reaction.
Interestingly, the Δvic1b-2 slow-growth phenotype was more severe when the fungus was grown in the dark, while the growth difference between the Δvic1b-2 and DK80 strains was less pronounced when both strains were infected with hypovirus CHV-1/EP713 (Figure S5). Although the Δvic1b-2 mutant exhibited a reduced growth rate, asexual sporulation per unit of mycelial mass was several fold higher than that of the parental strain under standard culture conditions (data not shown). Consistent with the slower growth in culture, the Δvic1b-2 mutant strain formed cankers on dormant chestnut stems that were ∼35% smaller than those formed by strain DK80, i.e., demonstrated a moderate level of reduced virulence (data not shown). These phenotypic changes were observed for multiple independent vic1b-2-disruption mutants. Extended sequence analysis of two of these independent disruption mutants showed no unintended sequence modifications extending upstream into vic1a-2 and downstream through the entire clavaminate synthase gene (results not shown). Note that vic1b-2 is the only C. parasitica candidate vic gene for which a phenotypic change has been observed upon disruption.
Identification of the candidate vic3 locus
Since the parental strains used to generate the C. parasitica linkage map (Kubisiak and Milgroom 2006) were homoallelic at vic3, no molecular linkage marker sequences were available to serve as guides to the location of the vic3 locus. Consequently, the sequence read-density plots were examined manually for gaps that occurred when reads from vic3-2 allelic strains EU55 (1221-22), EU40 (1122-11), and EU60 (2221-22) were mapped to the vic3-1-containing EP155 (2211-22) genome sequence, but that were absent in the plot for strain EU31 (1211-22), which also contains the vic3-1 allele.
A 7-kb-long span containing two such gaps was identified on Scaffold 5 ∼2 Mb away from the vic1 locus at map position 2,993,000–3,000,000 (Figure 4). Gap 1 was 2.4 kb in size and included the coding sequence for a hypothetical protein consisting of 599 and 614 amino acids in vic3-1 and vic3-2, respectively, designated vic3a-1 and vic3a-2. With only 46% identity between vic3a-1 and vic3a-2, this protein displayed significant allelic sequence divergence (Figure S6). Gap 2 consisted of a short ∼200-bp-long region located 3.4 kb away from gap 1 and contained the 3′-terminal end of an ORF for a short glycine–glutamine-rich protein of 102 and 108 amino acids in vic3-1 and vic3-2, respectively, designated vic3b-1 and vic3b-2. The predicted vic3b proteins had significant (e-value <1e-05) similarity to glutamate-receptor-associated, Life-guard-1-like proteins. The predicted vic3b-2 protein also contained a 2Fe-2S ferredoxin-type iron-sulfur-binding region signature (PS00197), absent in the vic3b-1 protein, and the two allelic proteins displayed moderately high similarity (85% identity) (Figure S7). The two polymorphic ORFs were separated by a highly conserved ORF encoding an actin-binding-like protein (Figure 4).
Figure 4.

The candidate vic3 locus. Read-density plots of sequence reads mapped by homology to a ∼7.5-kb portion of Scaffold 5 of the EP155 reference genome assembly identified by manual inspection to contain a region of sequence polymorphism (gaps) with a pattern consistent with that for allele distributions at the vic3 locus. Density plots are shown for strains EU55 (1221-22), EU40 (1122-11), and EU60 (2221-22) that differ at the vic3 genetic locus (i.e., contain allele 2) from reference strain EP155 (2211-22) and strain EU31 (1211-22) that, like strain EP155, contains allele 1 at the vic3 locus (Cortesi and Milgroom 1998). The ORFs located within this region are shown for the two alleles below the sequence read-density plots. Polymorphic (46% amino acid identity) ORFs encoding a hypothetical protein were designated vic3a-1 (599 aa) for allele 1 in strains EP155 and EU31 and vic3a-2 (614 aa) for allele 2 present in strains EU55, EU40, and EU60, while two small polymorphic (85% amino acid identity) ORFs encoding a Life-guard-1-like protein were designated vic3b-1 (102 aa) and vic3b-2 (108 aa). These two polymorphic genes were separated by a highly conserved ORF encoding an actin-binding-like protein. Sequencing protocols and color coding of reads are described in Figure 1. The JGI protein ID numbers for vic3a-1 and vic3b-1 are 331201 and 340400, respectively. The protein ID number for the predicted actin-binding-like protein is 331200.
Disruption and functional analysis of the candidate vic3 genes
Similar to the strategy used for disruption of vic1a-2 and vic1b-2, the vic3 candidate genes vic3a-1 and vic3b-1 in strain DK80 were independently disrupted as described in Figure S8A. Unlike the observation for the vic1a-2 disruption mutant, the individual Δvic3a-1 and Δvic3b-1 mutant strains retained the ability to form a barrage when paired with the vic3 heteroallelic strain EU60 (2221-22) (Figure 5, A and B). Consequently, deletion of both vic3a and vic3b ORFs was undertaken. A double vic3a-1/vic3b-1 mutant in strain DK80 (2211-22) was generated by disrupting vic3b-1 in the existing vic3a-1 mutant, leaving the actin-binding-like gene intact (Figure S8A). A vic3-2 double mutant was also generated by disrupting vic3a-2, vic3b-2, and the intervening actin-binding-like gene in strain EU60 (2221-22), as described in Figure S8B. Barrage formation was eliminated when the DK80 Δvic3a-1/Δvic3b-1 double-mutant strain was paired with EU60 (2221-22) and when the EU60 Δvic3a-2/Δvic3b-2 double mutant was paired with DK80 (2211-22) (Figure 5, C and D). As expected, no barrage formed when either of the double-mutant strains was paired with the heteroallelic mutant strain deleted for vic3a or vic3b (Figure 5, D–F).
Figure 5.

Mycelial incompatibility assay for strains disrupted in candidate vic alleles vic3a and vic3b in the incompatible strains DK80 (2211-22) and EU60 (2221-22). Barrage formation resulting from an incompatible reaction occurred when vic3a mutant DK80 Δvic3a-1 (2211-22) was paired with tester strain EU60 (2221-22) (A) and when vic3b mutant DK80 Δvic3b-1 (2211-22) was paired with EU60 (2221-22) (B) (barrage lines indicated by red arrows). In contrast, barrage formation was abolished when double vic3a-1/vic3b-1 mutant strain DK80 ΔΔvic3-1 (2211-22) was paired with EU60 (2221-22) (C). Barrage formation was also abolished for the reciprocal pairing involving the EU60 double vic3a-2/vic3b-2 mutant strain EU60 ΔΔvic3-2 (2221-22) and DK80 (2211-22) (D). As expected, no barrage formed when the EU60 ΔΔvic3-2 (2221-22) mutant strain was paired with either of the DK80 ΔΔvic3-1 (2211-22) double-mutant strain (F) or the DK80 Δvic3a-1 (2211-22) or DK80 Δvic3b-1 (2211-22) single-mutant strains (D and E, respectively).
The effect of vic3 component genes on hypovirus transmission was somewhat complicated as indicated in Figure 6. As shown in Figure 6, heteroallism at the vic3 locus for DK80 (2211-22) and EU60 (2221-22) resulted in a significant resistance to hypovirus transmission: 10–17% of pairings resulted in virus transmission no matter which strain was the donor or the recipient. Disruption of the vic3a-1 gene in DK80 resulted in 100% virus transmission, but only when the mutant strain served as the recipient. In contrast, disruption of the vic3b-1 gene in DK80 resulted in 100% transmission, but only if the mutant strain served as the donor. Similar to previous observations for the vic6 locus (Choi et al. 2012), these asymmetric increases in virus transmission occurred even though barrage formation was still observed. As expected, disruption of both vic3a and vic3b in DK80 completely removed resistance to virus transmission in both directions when paired with strain EU60. Similarly, virus transmission was 100% in both directions when an EU60 strain containing disruptions of both vic3a-2 and vic3b-2 was paired with strain DK80. These results clearly demonstrate that the vic3a and vic3b genes are functional components of the vic3 locus.
Figure 6.

The effect of vic3 locus mutations on hypovirus transmission. Pairing of vic3 heteroallelic strains DK80 (2211-22) and EU60 (2221-22), indicated at the top, resulted in a significant and symmetrical restriction in virus transmission: only 10–17% of pairings resulted in hypovirus transmission independent of which strain served as virus donor. The arrows indicate direction of virus transmission, with thin arrows indicating a level of restriction shown in parentheses and thick arrows indicating 100% (60/60) transmission. Virus transmission increased to 100% when vic3a-1 was disrupted in strain DK80 (DK80Δvic3a-1) and paired with EU60, if the mutant strain served as the recipient, and increased only slightly (from 10/60 to 15/60) if the mutant served as the donor. Disruption of vic3b-1 in DK80 (DK80Δvic3b-1) also increased virus transmission to 100%, but only when the mutant strain served as the donor, and increased from 6/60 to 22/60 when the mutant served as the recipient. Virus transmission increased to 100% in both directions when both vic3a-1 and vic3b-1 in DK80 were disrupted (DK80ΔΔvic3-1) and paired with EU60. Unexpectedly, virus transmission remained slightly restricted when the DK80 ΔΔvic3-1 and EU60 ΔΔvic3-2 double-mutant strains were paired, but only when the DK80 mutant strain was the donor. An unexpected slight restriction was also observed when the DK80 Δvic3b-1 mutant was paired as donor with the EU60 ΔΔvic3-2 double-mutant strain. No restriction was observed when DK80 or DK80Δvic3a-1 was paired with the EU60 ΔΔvic3-2 double mutant, similar to the observation for pairing of EU60 with the DK80ΔΔvic3-1 double mutant.
Curiously, virus transmission remained slightly restricted between DK80 containing the Δvic3b-1 disruption and EU60 containing the vic3a-2 + vic3b-2 double disruption (45/60) only when the DK80 vic3b-1 mutant was the donor and between the EU60 double and the DK80 double vic3a + vic3b disruption mutants when the DK80 double mutant was the donor (51/60). Additional in-depth studies of vic3 component gene interactions will be required to understand the basis of this residual resistance, e.g., the possible influence of eliminating the actin-binding-like gene in the EU60 vic3 double mutant.
Polymorphism-based identification of a nonfunctional pseudo vic locus containing a HET-domain gene deletion
The manual inspection of the sequence read-density plots conducted during the search for vic3-related polymorphic regions identified an additional polymorphic region that contained genes similar to those found at other vic loci. Located on Scaffold 6, spanning region 599514-606808 (Figure 7), the gap was found for strains EU31, EU55, and EU60, but not for strain EU40. Two ORFs were identified at this position in the EP155 sequence assembly, arbitrarily designated allele 1, that encoded a 1211-aa HET-domain protein and a 407-aa protein with a GTPase domain (Figure 8). Interestingly, there was no correspondence between the pattern of allele specificity at this position and the pattern of allele distributions at the genetically defined vic loci for the resequenced strains. For example, EP155 (2211-22) and EU40 (1122-11) are heteroallelic at all six genetically defined vic loci, but are homoallelic at this locus.
Figure 7.

Polymorphic nonfunctional pseudo vic locus. Density plots of sequence reads mapped by homology to a portion of EP155 (2211-22) sequence assembly Scaffold 6 extending from map coordinates ∼588,000 to 624,000 are shown for strains EU55 (1221-22), EU31 (1211-22), EU40 (1122-11), and EU60 (2221-22). The defined gap observed at 599,514–606,808 for strains EU55, EU31, and EU60 relative to EP155 and EU40 corresponds to allelic differences in genome organization at this locus as shown in Figure 8 for allele 1 (in strains EP155 and EU40) and allele 2 (in strains EU55, EU31, and EU60). There is no concordance between the polymorphic pattern and allele specificity at any of the genetically defined vic loci; e.g., EP155 (2211-22) and EU40 (1122-11) are heteroallelic at all six genetically defined vic loci, but are homoallelic at this locus.
Figure 8.
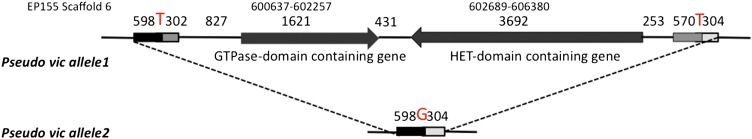
Organization of polymorphic nonfunctional pseudo vic locus alleles. Inspection of the sequence contigs for the two alleles corresponding to the polymorphic region identified on Scaffold 6 spanning positions 599514–606808 in the EP155 reference genome assembly revealed that an ∼7.7-kb portion of the sequence present in EP155 and EU40 (allele 1) was deleted in strains EU55, EU31, and EU60 (allele 2). The allele 1 sequence contained two ORFs not present in allele 2. These encoded a 407-aa protein containing a GTPase domain and a 1211-aa-long HET-domain-containing protein. The two ORFs were flanked by ∼900-bp imperfect (88% identity) direct repeats. The allele 2 sequence lacked the ∼7.7-kb region harboring the GTPase and HET-domain ORFs and contained only one copy of the flanking repeats that consisted of the 5′-proximal two-thirds of the left repeat and the 3′-proximal one-third of the right repeat and a G-C base pair replacing a T-A base pair at the breakpoint as indicated by the red letters. (Figure S9).
Inspection of sequence contigs for the allele present in EU31, EU55, and EU60, designated allele 2, indicated that the polymorphism was due to the absence of the GTPase and HET-domain ORFs and flanking noncoding regions as shown in Figure 8. Note the presence of ∼900-bp direct repeats of 88% identity flanking the polymorphic region in allele 1 and the presence of a single chimeric repeat in the corresponding allele 2. Close inspection showed that the remaining single copy of the repeat sequence present in allele 2 is chimeric with two-thirds of the sequence coming from the left-flanking repeat and one-third from the right-flanking repeat (Figure S9). The chimeric nature of the remaining repeat sequence is consistent with deletion of the region containing the HET-domain gene and the GTPase gene by a homologous recombination event.
Indirect evidence indicated that this locus does not currently function in the vic nonself recognition system. Barrage formation between EU31 (1211-22) and DK80 (2211-22) was eliminated (Figure 3) by disruption of the vic1a-2 allele, and unrestricted virus transmission was observed (Table 1) even though, as shown in Figure 7, the DK80/Δvic1a-2 mutant and EU31 are heteroallelic at the polymorphic Scaffold 6 locus. However, the possibility that another incompatible allelic form of this locus is present in some C. parasitica population cannot be ruled out.
It was of interest to determine whether the HET-domain gene deletion (in allele 2) associated with this nonfunctional locus was more widely distributed in the C. parasitica population. Four primer pairs (Table S1) were designed to test for the presence/absence of the left repeat, GTPase gene, HET-domain gene, and chimeric repeat. As shown in Table 2, allele 2 was found in 4 of 14 field isolates obtained from a North American population in Finzel, Maryland (Milgroom and Cortesi 1999), and in 2 of 4 field isolates sampled from an Asian population from Iwama, Ibaraki Prefecture, Japan (Liu and Milgroom 2007). The presence of only alleles 1 and 2 in both North American and Asian C. parasitica populations indicates that the deletion at this locus was not a recent event.
Table 2. PCR-based differentiation of alleles for pseudo vic locus at C. parasitica genome Scaffold 6: 599,514–606,808.
| Isolate | Location | Pseudo vic allele | Isolate | Location | Pseudo vic allele |
|---|---|---|---|---|---|
| JA01 | Chiyodo, Ibaraki, Japan | 1 | MD-01 | Finzel, MD | 1 |
| JA02 | Chiyodo, Ibaraki, Japan | PCR failure | MD-05 | Finzel, MD | 1 |
| JA03 | Chiyodo, Ibaraki, Japan | 1 | MD-07 | Finzel, MD | 1 |
| JA04 | Chiyodo, Ibaraki, Japan | 1 | MD-09 | Finzel, MD | 1 |
| JA06 | Chiyodo, Ibaraki, Japan | 1 | MD-11 | Finzel, MD | 1 |
| JA11 | Chiyodo, Ibaraki, Japan | 1 | MD-16 | Finzel, MD | 2 |
| JA12 | Chiyodo, Ibaraki, Japan | 1 | MD-18 | Finzel, MD | 1 |
| JA19 | Iwama, Ibaraki, Japan | 1 | MD-21 | Finzel, MD | 1 |
| JA20 | Iwama, Ibaraki, Japan | 2 | MD-25 | Finzel, MD | 1 |
| JA21 | Iwama, Ibaraki, Japan | 1 | MD-28 | Finzel, MD | 1 |
| JA23 | Iwama, Ibaraki, Japan | 2 | MD-30 | Finzel, MD | 1 |
| JA60 | Yasato, Ibaraki, Japan | 1 | MD-32 | Finzel, MD | 2 |
| EU40 (control) | 1 | MD-34 | Finzel, MD | 2 | |
| EU31 (control) | 2 | MD-36 | Finzel, MD | 2 |
The C. parasitica genome Scaffold 6: 599,514–606,808 can be found at http://genome.jgi.doe.gov/Crypa2/Crypa2.home.html. Isolates from Japan and Finzel, Maryland, were used in previous population studies described by Liu and Milgroom (2007) and Milgroom and Cortesi (1999), respectively.
Discussion
Interest in the vic nonself recognition system operating in C. parasitica derives primarily from reports that it restricts transmission of viruses that mediate biological control of chestnut blight (Van Alfen et al. 1975; Anagnostakis 1982a; Macdonald and Fulbright 1991; Nuss 1992; Heiniger and Rigling 1994; Milgroom and Cortesi 2004). A major advancement in understanding the genetic structure of this system came from analyses by Cortesi and Milgroom (1998) that identified six di-allelic loci controlling vic in European C. parasitica populations and provided a collection of 64 vic tester strains with defined genotypes corresponding to all combinations of the vic alleles. The ability to define the vic genotype of C. parasitica field isolates and laboratory strains has enabled more precise examinations of the relationship between vic diversity and virus transmission and the influence of different vic loci on virus transmission, leading to the identification of allele-specific influences on the frequency and symmetry of virus transmission (Huber and Fulbright 1994; Cortesi et al. 2001; Biella et al. 2002). The molecular identification of the vic1 and vic3 loci described here, along with the report by Choi et al. (2012), completes the molecular identification of all six genetically defined C. parasitica vic loci. The identification of the gene compositions of the respective vic loci has allowed gene disruption analysis to definitively demonstrate that vic genes contribute to restriction of mycovirus transmission and to test whether they have biological functions in addition to allorecognition. This information will also enable more precise population genetic studies, create opportunities to determine molecular mechanisms underlying vic-mediated triggering of PCD, and provide the means for manipulating the vic system to enhance the transmission and utility of hypoviruses for biological control.
The gene organization and composition uncovered for the vic1 and vic3 loci extend the overall level of diversity observed for the C. parasitica vic loci. The composition of the vic1-2 allele is unusual in that it contains the combination of a polymorphic HET-domain gene (vic1a-2) and an idiomorphic DUF1909-domain gene (vic1b-2). Polymorphic HET-domain genes were also found at the vic6 and vic7 loci. However, the association of HET-domain proteins with only three of the six genetically defined C. parasitica vic loci contrasts with the observations in N. crassa, where all three of the characterized het (vic equivalent) complexes involve a HET-domain gene, and P. anserina, where HET-domain genes are associated with two of the three characterized het loci (reviewed in Smith and Lafontaine 2013).
While the vic1b-2 gene does not have a corresponding polymorphic ORF present in the vic1-1 allele, remnants of the vic1b-2 sequence are evident in vic1-1 and are associated with the sequence inversion that includes the clavaminate synthase-like gene, as indicated by the alignment highlighted in blue shown in Figure 2. This differs from the previously described vic4 idiomorphs (Choi et al. 2012), where a protein kinase c-like gene present in vic4-1 is replaced with a completely unrelated NACHT-NTP/WD repeat-encoding gene in vic4-2. In this regard, the vic1-1 idiomorph does contain an LTR retrotransposon (vic1c-1) and a HET-domain protein gene (vic1d-1) that are completely absent in vic1-2. vic1 and vic4 therefore join the N. crassa mat-A1/mat-a1/tol locus (Glass et al. 1988) as fungal allorecognition loci involving idiomorphic alleles. The vic1b-2 and vic1d-1 sequences are unique and were not found elsewhere in the C. parasitica reference genome sequence. However, transposon-like sequences, some having similarity to vic1c-1, are common, and their distribution in the genomes of different C. parasitica strains is currently under investigation.
Disruption analysis of vic1-2 genes uncovered additional differences with the other C. parasitica vic loci. Disruption of the HET-domain gene vic1a-2 resulted in elimination of barrage formation and in unrestricted virus transmission when the disrupted strain was paired with one that was heteroallelic at vic1 (Figure 3). This contrasts with the results for vic2-2, vic6-2, and vic7-2 where disruption of their HET-domain genes increased virus transmission but did not eliminate barrage formation (Choi et al. 2012). In the case of vic6, nonallelic interactions between the vic6 gene and the adjacent pix6 gene triggered PCD (Choi et al. 2012). The observation that disruption of the vic1a allele in just one of the paired vic1 heteroallelic strains resulted in loss of barrage formation suggests that an allelic interaction between vic1a-1 and vic1a-2 is sufficient to trigger PCD, although vic1a-1 disruption is required for confirmation.
In contrast to the result of vic1a-2 disruption, vic1b-2 disruption failed to eliminate barrage formation. However, as was observed previously for disruption of vic2-2, vic6-2, and vic7-2 (Choi et al. 2012), disruption of vic1b-2 increased virus transmission without elimination of barrage formation, presumably due to a slower onset of PCD (Table 1). These results suggest that the vic1b-2 product is involved in a nonallelic interaction to trigger PCD. The vic1-1 allele remains to be functionally characterized, and the product of the HET-domain gene vic1d-1 certainly provides a potential candidate for that interaction. It will also be of interest to determine whether disruption of the LTR-retrotransposon gene, vic1c-1, affects nonself recognition or the fungal phenotype.
The vic3 locus contains two polymorphic genes that are unrelated to any previously identified C. parasitica vic genes. The vic3a gene products are highly polymorphic (∼46% identical) and lack recognizable conserved sequence domains. The vic3b gene products are much less polymorphic, but are distinctive in the unusually high level of glycine and glutamine repeats. Since both vic3a and vic3b alleles must be deleted to eliminate barrage formation, it is likely, but not confirmed, that allelic interactions are insufficient to trigger PCD.
As reported for the nonallelic interactions observed for the vic6 locus (Choi et al. 2012), disruption of vic3a-1 and vic3b-1 independently caused an asymmetric change in virus transmission without eliminating barrage formation: the Δvic3a-1 disruption mutant became a good recipient, while the Δvic3b-1 mutant became a good virus donor (Figure 6). Notably, the virus transmission rates of ∼10–17% observed in this study for pairing of the vic3 heteroallelic strains DK80 (2211-22) and EU60 (2221-22) (Figure 6) differ significantly from the rates of 60–80% reported by Cortesi et al. (2001) for other paired vic3 heteroallelic strains. Virus transmission rates were in excellent agreement in these different studies for paired strains that are heteroallelic at each of the other five vic loci. It should be noted that the specific vic genotypes of the vic3-heteroallelic paired strains used in the Cortesi et al. (2001) study did not include the DK80 (2211-22) vs. EU60 (2221-22) genotypes used in this study. Irrespective of these transmission rate differences, the results of the vic3 gene disruption analysis for DK80 and EU60 confirm that vic3a and vic3b alleles contribute to restriction of virus transmission.
Combined, Choi et al. (2012) and this study have identified and characterized 8 polymorphic genes and 5 idiomorphic genes associated with 6 C. parasitica vic loci (Table 3). This compares with 5 polymorphic and 2 idiomorpic genes characterized at 3 incompatibility loci in N. crassa and 5 genes characterized at 3 loci in P. anserina (reviewed in Smith and Lafontaine 2013). Thus, with this recent work on the C. parasitica system, the number of fungal allorecognition-associated genes identified at the molecular level has more than doubled. A total of 11 polymorphic and 1 idiomorphic C. parasitica vic genes have been disrupted. One potential allelic interaction was detected with the loss of barrage formation upon disruption of vic1a-2 (Figure 3). Nonallelic interactions were demonstrated for the vic6 and pix6 alleles (Choi et al. 2012). Nonallelic interactions were inferred for vic2, vic3, and vic7 based on no loss of barrage formation following disruption of one gene at the corresponding loci.
Table 3. Summary of C. parasitica vic-associated genes identified at the molecular level.
| Genea | P/Ib | Allelic/nonallelic | Protein features | Gene disruption increases virus transmissionc |
|---|---|---|---|---|
| vic1a | P | Allelicd | HET domain | Yes |
| vic1b-2 | I | Nonelleice | DUF domain | Yes |
| vic1c-1 | I | NDf | LTR retrotransposon | ND |
| vic1d-1 | I | ND | HET domain | ND |
| vic2 | P | Nonallelice | Patatin-like phospholipase | Yes |
| vic2a | P | ND | Sec9-like | ND |
| vic3a | P | Nonallelice | Hypothetical protein | Yes |
| vic3b | P | Nonallelice | Life guard-1-like | Yes |
| vic4-1 | I | ND | Protein kinase c-like | NAg |
| vic4-2 | I | ND | NACHT + WD40 domains | NA |
| vic6 | P | Nonallelic | HET domain | Yes |
| pix6 | P | Nonallelic | DUF1040 domain | Yes |
| vic7 | P | Nonallelice | HET domain | Yes |
Molecular identification of vic2, vic4, vic6, and vic7 reported in Choi et al. (2012). Molecular identification of vic1 and vic3 reported in this study. Allele specificity is indicated for idiomorphic genes and not polymorphic genes.
P, polymorphic; I, idiomorphic.
Transmission increased when the disrupted strain was paired with a nonmutant strain that is heteroallelic only at the vic locus being analyzed, e.g., Δvic1a-2 (2211-22) vs. EU31 (1211-22).
Potential allelic interaction that requires vic1a-1 disruption for confirmation.
Inferred as nonallelic based on the presence of two polymorphic or idiomorphic genes at locus and on the observation that incompatibility reaction is not eliminated when one gene at locus is disrupted.
ND, not determined.
NA, not applicable. Virus transmission is not restricted by allelic differences at vic4.
Evidence that the C. parasitica vic system restricts mycovirus transmission, while strong, has been only correlative in nature. The results of extensive field studies in Europe and North America are in general agreement that hypovirus transmission and biological control of chestnut blight are more effective when the C. parasitica populations exhibit a low diversity of vic genotypes (Anagnostakis et al. 1986; Heiniger and Rigling 1994; Robin et al. 2000, 2009; Milgroom and Cortesi 2004). Laboratory transmission studies have shown vic loci-dependent variations in virus transmission, including allele-specific asymmetry, apparently resulting from differences in the rate at which PCD occurs in either or both donor and recipient strains after cell fusion (Cortesi et al. 2001; Biella et al. 2002). However, the heteroallelic strains used in those studies were not isogenic, raising the possibility of contributions from non-vic genes. The vic gene disruption studies reported here and in Choi et al. (2012) provide formal confirmation of contributions to restriction of virus transmission for vic1a-2, vic1b-2, vic2-2, vic3a-1, vic3a-2, vic3b-1, vic3b-2, vic6-1, vic6-2, pix6-1, pix6-2, and vic7-2, representing the five vic loci previously implicated.
The vic gene disruption studies also strongly support the allorecognition hypothesis rather than the accidental hypothesis of vic origins. The former hypothesis proposes that vegetative incompatibility systems arose through positive Darwinian selection because they provide some specific advantages (Saupe 2000; Smith and Lafontaine 2013). One often-mentioned advantage is protection against the transmission of deleterious genetic elements (Caten 1972). Because natural C. parasitica populations worldwide are known to be infected by mycoviruses that have a deleterious effect on host functions and reproduction (Anagnostaksis 1982a; Heiniger and Rigling 1994; Milgroom and Cortesi 2004), the molecular identification of the C. parasitica vic genes provides new opportunities, not available with N. crassa and P. anseria, for testing this hypothesis. A major prediction of the allorecognition hypothesis is that the C. parasitica vic genes that have been demonstrated to contribute to restriction of mycovirus transmission as a result of the incompatibility reaction would be unlikely to have other biological functions. As reported here and in Choi et al. (2012), no phenotypic changes, other than reduced rate (increased virus transmission) or loss of PCD, were detected for disruption mutants of 11 polymorphic genes associated with vic1, vic2, vic3, vic6, and vic7, consistent with dedicated roles in allorecognition. The molecular identification of the C. parasitica vic loci also provides an unparalleled opportunity to test predictions of diversifying and balanced selection of vic gene polymorphism in a fungus under pressure of natural infection by a deleterious pathogen.
The one exception where disruption of a vic gene did have additional phenotypic consequences, i.e., disruption of vic1b-2, requires further study. The reduced growth, irregular colony morphology, increased asexual sporulation, and reduced virulence exhibited by the Δvic1b-2 disruption mutant certainly suggests functional roles for the corresponding gene product in cellular processes other than those for nonself recognition. However, while vic1b-2 disruption did increase virus transmission if the mutant strain served as a recipient, disruption of the Het-domain gene at this locus, vic1a-2, in just one of two heteroallelic strains was sufficient to completely abolish PCD and allowed virus transmission in both directions. This raises questions about the role of vic1b in the incompatibility reaction. Does it play an auxiliary role, modifying the activity of vic1a? Does it interact with vic1a? In analogy with the guard hypothesis in plant disease resistance, does the vic1b product serve as a guard of the vic1a product and are the phenotypic consequences of vic1b gene disruption a result of vic1a protein activation in the absence of vic1b? All of these possibilities are testable and will be the subject of future studies.
The polymorphism-based comparative genomics approach used to identify the C. parasitica vic loci (this study and Choi et al. 2012) should have general applicability for identifying fungal allorecognition loci. The availability of a set of genotyped vic tester strains and linkage map markers for five of the six vic loci was certainly useful in their identification. However, the identification of the vic3 locus in the absence of linkage marker guides (Figure 4) suggests that manual inspection of sequence read-density plots for polymorphism-related gaps in alignment of resequenced and reference genome sequences will provide a good list of candidate vic loci for functional characterization. It is noteworthy that >100,000 polymorphisms were found when comparing the EP155 reference genome sequence and the sequence generated by resequencing strain EP146 (Choi et al. 2012). Of these, only 3681 were located in predicted coding regions, and only 2079 of those were predicted to result in nonsynonymous changes in a protein product. Highly polymorphic regions that did not involve repetitive sequence elements, such as the identified vic loci, were rare. Of course, the presence of a HET-domain or WD repeat gene in the region of polymorphism would indicate a prime candidate vic locus.
The identification of the nonfunctional pseudo vic locus on Scaffold 6 (Figure 7 and Figure 8) also indicates that the polymorphism-based comparative genomic approach holds promise for providing insights into the origins and evolution of vic loci. The polymorphism, or indel, in this case involved an apparent deletion of a HET-domain gene and of an adjacent GTPase-domain gene. That the difference is due to a deletion rather than an insertion is suggested by the observation that the two flanking direct imperfect repetitive elements present in allele 1 (Figure 8) are replaced by a single chimeric sequence consisting of 598 bp of the left repeat and 304 bp of the right repeat with a G-C base pair replacing a T-A base pair at the breakpoint (Figure S9). Interestingly, this presumptive deletion event is not recent, since it was found in C. parasitica field isolates collected in Asia, Europe, and North America (Table 2). Repetitive elements have also been identified associated with a sequence inversion event at the het-6 locus of N. crassa (Micali and Smith 2006). Combined with the LTR retrotransposon associated with the sequence inversion and sequence insertion at the vic1 locus (Figure 2), these observations provide additional support for the proposal that mobile genetic elements contributed to the origins and evolution of vic loci (Smith and Lafontaine 2013). It is anticipated that analysis of the corresponding genomic sequences in species closely related to C. parasitica—e.g., C. radicalis, C. nitschkei, C. megaspore (Gryzenhout et al. 2006), and C. naterciae (Braganca et al. 2011)—will provide additional insights into the origins and maintenance of vegetative incompatibility.
The use of hypovirus-infected hypovirulent C. parastica strains to control chestnut blight by treating individual cankers on blighted trees has been well demonstrated (Anagnostakis and Kranz 1987) and is used in orchard settings (Heiniger and Rigling 1994) and to preserve chestnut germplasm used in backcross resistance breeding programs (Jacobs et al. 2013). Application of the virus-infected strain to the margin of a canker can result in viral transmission and spread of the virus in the recipient strain, resulting in conversion to hypovirulence and shutdown of canker expansion. Such treatment is 100% effective if the donor and recipient strains have identical vic genotypes. However, treatment effectiveness decreases as the number heteroallelic vic genes increases. The molecular identification of the six genetically defined vic loci now provides the opportunity to engineer a universal hypovirus donor strain for effective and efficient treatment of individual blight cankers irrespective of the recipient vic genotype. The use of a universal donor strain as a vector for delivery of hypoviruses into a forest ecosystem may also find some enhanced utility for woodland restoration of the American chestnut.
Acknowledgments
This work was supported by National Science Foundation awards DBI-MRI-0821806 (Peter Houde, New Mexico State University, Las Cruces, NM, PI), MCB-1051453 (A.L.D.), and MCB-1051331 (D.L.N.).
Footnotes
Communicating editor: J. Heitman
Literature Cited
- Aanen D. K., Debets A. J. M., de Visser J. A. G. M., Hoekstra R. F., 2008. The social evolution of somatic fusion. Bioessays 30: 1193–1203 [DOI] [PubMed] [Google Scholar]
- Anagnostakis S. L., 1982a Biological control of chestnut blight. Science 215: 466–471 [DOI] [PubMed] [Google Scholar]
- Anagnostakis S. L., 1982b Genetic analysis of Endothia parasitica: linkage map of four single genes and three vegetative compatibility types. Genetics 102: 25–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostakis S. L., 1983. Conversion to curative morphology in Endothia parasitica and its restriction by vegetative compatibility. Mycologia 79: 23–37 [Google Scholar]
- Anagnostakis S. L., 1988. Cryphonectria parasitica, cause of chestnut blight. Adv. Plant Pathol. 6: 123–136 [Google Scholar]
- Anagnostakis S. L., Kranz J., 1987. Population dynamics of Cryphonectria parasitica in a mixed-hardwood forest in Connecticut. Phytopathology 77: 751–754 [Google Scholar]
- Anagnostakis S. L., Hau B., Kranz J., 1986. Diversity of vegetative compatibility groups of Cryphonectria parasitica in Connecticut and Europe. Plant Dis. 70: 536–538 [Google Scholar]
- Bastiaans E., Debets A. J. M., Aanen D. K., van Diepeningen A. D., Saupe S. J., et al. , 2014. Natural variation of heterokaryon incompatibility gene het-c in Podospora anserina reveals diversifying selection. Mol. Biol. Evol. 31: 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biella S., Smith M. L., Aist J. R., Cortesi P., Milgroom M. G., 2002. Programmed cell death correlates with virus transmission in a filamentous fungus. Proc. Biol. Sci. 269: 2269–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges M. J., Azevedo M. O., Bonatelli R., Jr, Felipe M. S. S., Astolfi-Filho S., 1990. A practical method for the preparation of total DNA from filamentous fungi. Fungal Genet. Newsl. 37: 10 [Google Scholar]
- Braganca H., Rigling D., Diogo E., Capelo J., Phillips A., et al. , 2011. Cryphonectria naterciae: a new species in the Cryphonectria-Endothia complex and diagnostic molecular markers based on microsatellite-primed PCR. Fungal Biol. 115: 852–861 [DOI] [PubMed] [Google Scholar]
- Caten C. E., 1972. Vegetative incompatibility and cytoplasmic infections in fungi. J. Gen. Microbiol. 72: 221–229 [DOI] [PubMed] [Google Scholar]
- Chen B., Choi G. H., Nuss D. L., 1994. Attenuation of fungal virulence by synthetic infectious hypovirus transcripts. Science 264: 1762–1764 [DOI] [PubMed] [Google Scholar]
- Choi G. H., Dawe A. L., Churbanov A., Smith M. L., Milgroom M. G., et al. , 2012. Molecular characterization of vegetative incompatibility genes that restrict hypovirus transmission in the chestnut blight fungus Cryphonectria parasitica. Genetics 190: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill A. C. L., Ciufetti L. M., Hansen D. R., Van Etten H. D., Van Alfen N. K., 1990. Transformation of the fungal pathogen Cryphonectria parasitica with a variety of heterologous plasmids. Curr. Genet. 17: 25–31 [Google Scholar]
- Coppin E., Debuchy R., Arnaise S., Picard M., 1997. Mating types and sexual development in filamentous ascomycetes. Microbiol. Mol. Biol. Rev. 61: 411–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesi P., Milgroom M. G., 1998. Genetics of vegetative incompatibility in Cryphonectria parasitica. Appl. Environ. Microbiol. 64: 2988–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesi P., McCulloch C. E., Song H., Lin H., Milgroom M. G., 2001. Genetic control of horizontal virus transmission in the chestnut blight fungus, Cryphonectria parasitica. Genetics 159: 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe A. L., Nuss D. L., 2013. Hypovirus molecular biology: from Koch’s postulates to host self-recognition genes that restrict virus transmission. Adv. Virus Res. 86: 109–147 [DOI] [PubMed] [Google Scholar]
- Debets A. J. M., Griffiths A. J. F., 1998. Polymorphisms in het-genes prevents resource plundering in Neurospora crassa. Mycol. Res. 102: 1343–1349 [Google Scholar]
- De Tomaso A. W., Nyholm S. V., Palmeri K. J., Ishizuka K. J., Ludington W. B., et al. , 2005. Isolation and characterization of a protochordate histocompatibility locus. Nature 438: 454–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier E., Levis C., Fortini D., Leroux P., Giraud T., et al. , 2003. Characterization Bc-hch, the Botrytis cinerea homolog of the Neurospora crassa het-c vegetative incompatibility locus, and its use as a population marker. Mycologia 95: 251–261 [PubMed] [Google Scholar]
- Ghabrial S. A., Suzuki N., 2009. Viruses of plant pathogenic fungi. Annu. Rev. Phytopathol. 47: 353–384 [DOI] [PubMed] [Google Scholar]
- Glass N. L., Vollmer S. J., Staben C., Metzenbereg R. L., Yanofski C., 1988. DNAs of the two mating type alleles of Neurospora crassa are highly dissimilar. Science 241: 570–573 [DOI] [PubMed] [Google Scholar]
- Glass N. L., Grotelueschen J., Metzenberg R. L., 1990. Neurospora crassa A mating-type proteins. Proc. Natl. Acad. Sci. USA 87: 4912–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass N. L., Jacobson D. J., Shiu P. K., 2000. The genetics of hyphal fusion and vegetative incompatibility in filamentous ascomycete fungi. Annu. Rev. Genet. 34: 165–186 [DOI] [PubMed] [Google Scholar]
- Gryzenhout M., Wingfield B., Wingfield M., 2006. New taxonomic concepts for the important forest pathogens Cryphonectria parasitica and related fungi. FEMS Microbiol. Lett. 258: 161–172 [DOI] [PubMed] [Google Scholar]
- Hall, C., J. Welch, D. J. Kowbel, and N. L. Glass, 2010 Evolution and diversity of a fungal self/nonself recognition locus. PLoS ONE 5: el4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiniger U., Rigling D., 1994. Biological control of chestnut blight in Europe. Annu. Rev. Phytopathol. 32: 581–599 [Google Scholar]
- Hillman B. I., Shapira R., Nuss D. L., 1990. Hypovirulence-associated suppression of host functions in Cryphonectria parasitica can be partially relieved by high light intensity. Phytopathology 80: 950–956 [Google Scholar]
- Huber, D. H., 1996 Genetic analysis of vegetative incompatibility polymorphisms and horizontal transmission in the chestnut blight fungus Cryphonectria parasitica Ph.D. Thesis, Michigan State University, East Lansing, MI. [Google Scholar]
- Huber, D. H., and D. W. Fulbright, 1994 Preliminary investigations on the effect of individual genes upon the transmission of dsRNA in Cryphonectria parasitica, pp. 15–19 in Proceedings of the International Chestnut Conference, edited by M. L. Double and W. L. MacDonald. West Virigina University Press, Morgantown, WV. [Google Scholar]
- Iotti M., Rubini A., Tisserant E., Kholer A., Paolocci F., et al. , 2012. Self/nonself recognition in Tuber melanosporum is not mediated by a heterokaryon incompatibility system. Fungal Biol. 116: 261–275 [DOI] [PubMed] [Google Scholar]
- Jacobs D. F., Dalgleish H. J., Nelson C. D., 2013. A conceptual framework for restoration of threatened plants: the effective model of American Chestnut (Castanea dentata) reintroduction. New Phytol. 197: 378–393 [DOI] [PubMed] [Google Scholar]
- Kerenyi Z., Olah B., Jeney A., Hornok L., Leslie J. F., 2006. The homologue of het-c of Neurospora crassa lacks vegetative compatibility function in Fusarium proliferatum. Appl. Environ. Microbiol. 72: 6527–6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey J. A., 1990. Tad, a LINE-like transposable element in Neurospora, can transpose between nuclei in heterokaryons. Genetics 126: 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubisiak T. L., Milgroom M. G., 2006. Markers linked to vegetative incompatibility (vic) genes and a region of high heterogeneity and reduced recombination near the mating type locus (MAT) in Cryphonectria parasitica. Fungal Genet. Biol. 43: 453–463 [DOI] [PubMed] [Google Scholar]
- Kuwayama H., Obara S., Morio T., Katoh M., Urushihara H., et al. , 2002. PCR-mediated generation of a gene disruption construct without the use of DNA ligase and plasmid vectors. Nucleic Acids Res. 15: E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine D. L., Smith M. L., 2012. Diverse interactions mediate asymmetric incompatibility by the het-6 supergene complex in Neurospora crassa. Fungal Genet. Biol. 49: 65–73 [DOI] [PubMed] [Google Scholar]
- Lan X., Yao Z., Zhou Y., Shang J., Lin H., et al. , 2008. Deletion of the cpku80 gene in the chestnut blight fungus, Cryphonectria parasitica, enhances gene disruption efficiency. Curr. Genet. 53: 59–66 [DOI] [PubMed] [Google Scholar]
- Liu Y. C., Milgroom M. G., 1996. Correlation between hypovirus transmission and the number of vegetative incompatibility (vic) genes different among isolates from a natural population of Cryphonectria parasitica. Phytopathology 86: 79–86 [Google Scholar]
- Liu Y. C., Milgroom M. G., 2007. High diversity of vegetative compatibility types in Cryphonectria parasitica in Japan and China. Mycologia 99: 279–284 [DOI] [PubMed] [Google Scholar]
- MacDonald W. L., Fulbright D. W., 1991. Biological control of chestnut blight: use and limitations of transmissible hypovirulence. Plant Dis. 75: 656–661 [Google Scholar]
- Margulies M., Egholm M., Altman W. E., Attiya S., Bader J. S., et al. , 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R., Janeway C. A., Jr, 2002. Decoding the patterns of self and nonself by the innate immune system. Science 296: 298–300 [DOI] [PubMed] [Google Scholar]
- Meselson M., Yuan R., 1968. DNA restriction enzyme from E. coli. Nature 217: 1110–1114 [DOI] [PubMed] [Google Scholar]
- Micali C. O., Smith M. L., 2006. A nonself recognition gene complex in Neurospora crassa. Genetics 173: 1991–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgroom M. G., Cortesi P., 1999. Analysis of population structure of the chestnut blight fungus based on vegetative incompatibility genotypes. Proc. Natl. Acad. Sci. USA 96: 10518–10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgroom M. G., Cortesi P., 2004. Biological control of chestnut blight with hypovirulence: a critical analysis. Annu. Rev. Phytopathol. 42: 311–338 [DOI] [PubMed] [Google Scholar]
- Nasrallah J. B., 2005. Recognition and rejection of self in plant self-incompatibility: comparisons to animal histocompatibility. Trends Immunol. 26: 412–418 [DOI] [PubMed] [Google Scholar]
- Nuss D. L., 1992. Biological control of chestnut blight: an example of virus-mediated attenuation of fungal pathogenesis. Microbiol. Rev. 56: 561–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss D. L., 2005. Hypovirulence: mycoviruses at the fungal-plant interface. Nat. Rev. Microbiol. 3: 632–642 [DOI] [PubMed] [Google Scholar]
- Nydam M. L., De Tomaso A. W., 2011. Creation and maintenance of variation in allorecognition loci: molecular analysis in various model systems. Front. Immunol. 2: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K., van Diepeningen A. D., Varga J., Hoekstra R. F., Dyer P. S., et al. , 2007. Sexual and vegetative compatibility genes in the aspergilla. Stud. Mycol. 59: 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontecorvo G., 1956. The parasexual cycle in fungi. Annu. Rev. Microbiol. 10: 393–400 [DOI] [PubMed] [Google Scholar]
- Powell W. A., 1995. Vegetative incompatibility and mycelial death of Cryphonectria parasitica detected with a pH indicator. Mycologia 87: 738–741 [Google Scholar]
- Rayner A. D. M., 1996. Interconnectedness and individualism in fungal mycelia, pp. 193–232 in A Century of Mycology, edited by Sutton B. C. University Cambridge Press, Cambridge, UK [Google Scholar]
- Robin C., Anziani C., Cortesi P., 2000. Relationship between biological control, incidence of hypovirulence, and diversity of vegetative incompatibility types of Cryphonectria parasitica in France. Phytopathology 90: 730–737 [DOI] [PubMed] [Google Scholar]
- Robin C., Capdeville X., Martin M., Traver C., Colinas C., 2009. Cryphonectria parasitica vegetative compatibility type analysis of populations in south-western France and northern Spain. Plant Pathol. 58: 527–535 [Google Scholar]
- Rosengarten R. D., Nicotra M. L., 2011. Model systems of invertebrate allorecognition. Curr. Biol. 21: R82–R92 [DOI] [PubMed] [Google Scholar]
- Saupe S. J., 2000. Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol. Mol. Biol. Rev. 64: 489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saupe S., Descampus C., Turcq B., Begueret J., 1994. Inactivation of the Podospora anserina vegetative incompatibility locus het-c, whose product resembles a glycolipid transferase protein, drastically impairs ascospore production. Proc. Natl. Acad. Sci. USA 91: 5927–5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. L., Lafontaine D., 2013. The fungal sense of nonself, pp. 9–21 in Neurospora: Genomics and Molecular Biology, edited by Kasbeker D., McClusky K. Horizon Scientific Press, Norfolk, UK [Google Scholar]
- Smith M. L., Micali O. C., Hubbard S. P., Mir-Rashed N., Jacobson D. J., et al. , 2000. Vegetative incompatibility in the het-6 region of Neurospora crassa is mediated by two linked genes. Genetics 155: 1095–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. L., Gibbs C. C., Milgroom M. G., 2006. Heterokaryon incompatibility function of barrage-associated vegetative incompatibility genes (vic) in Cryphonectria parasitica. Mycologia 98: 43–50 [DOI] [PubMed] [Google Scholar]
- Spiering M. J., Faulkner J. R., Zhang D.-X., Machado C., Grossman R. B., et al. , 2008. Role of the LolP cytochrome P450 monooxygenase in loline alkaloid biosynthesis. Fungal Genet. Biol. 45: 1307–1314 [DOI] [PubMed] [Google Scholar]
- Staben C., Yanofsky C., 1990. Neurospora crassa a mating-type region. Proc. Natl. Acad. Sci. USA 87: 4917–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Alfen N. K., Jaynes R. A., Anagnostakis S. L., Day P. R., 1975. Chestnut blight: biological control by transmissible hypovirulence in Endothia parasitica. Science 189: 890–891 [DOI] [PubMed] [Google Scholar]
- Van der Nest M. A., Olson A., Lind M., Velez H., Dalman K., et al. , 2014. Distribution and evolution of het gene homologs in the basidiomycota. Fungal Genet. Biol. 54: 45–57 [DOI] [PubMed] [Google Scholar]
- Van Diepeningen A. D., Pal K., van der Lee T. A. J., Hoekstra R. F., Debets A. J. M., 2008. The het-c heterokaryon incompatibility gene in Aspergillus niger. Mycol. Res. 113: 222–229 [DOI] [PubMed] [Google Scholar]
- Zhang D.-X., Lu H.-L., Liao X., St. Leger R. J., Nuss D. L., 2013. Simple and efficient recycling of fungal selectable marker genes with the Cre-loxP recombination system via anastomosis. Fungal Genet. Biol. 61: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]