

Abstract
“Fetal programming” is a term used to describe how early-life experience influences fetal development and later disease risk. In humans, prenatal stress-induced fetal programming is associated with increased risk of preterm birth, and a heightened risk of metabolic and neurological diseases later in life. A critical determinant of this is the regulation of fetal exposure to glucocorticoids by the placenta. Glucocorticoids are the mediators through which maternal stress influences fetal development. Excessive fetal glucocorticoid exposure during pregnancy results in low birth weight and abnormalities in a number of tissues. The amount of fetal exposure to maternal glucocorticoids depends on the expression of HSD11B2, an enzyme predominantly produced by the syncytiotrophoblast in the placenta. This protects the fetus by converting active glucocorticoids into inactive forms. In this review we examine recent findings regarding placental HSD11B2 that suggest that its epigenetic regulation may mechanistically link maternal stress and long-term health consequences in affected offspring.
Keywords: 11β-hydroxysteroid dehydrogenase type-2, epigenetic, fetal outcome, placenta, prenatal stress
Introduction
“Fetal programming” is a term used to describe how the early-life experience influences fetal development and later disease risk.1 The glucocorticoid hypothesis describes how increased fetal exposure to maternal glucocorticoids, such as cortisol, as a result of prenatal stress during pregnancy is associated with a number of poor birth outcomes and increased risk for later-in-life neurological and cardiometabolic syndromes in exposed offspring.1,2 Recent clinical and epidemiological studies have shown that maternal prenatal stress is associated with increased risk for preterm birth, reduced birth weight,3 smaller head circumference4 and later-in-life cardiometabolic5 and neuropsychiatric disorders,6 such as obesity,5,7 type 1 diabetes,8 schizophrenia,6 autism, ADHD,9 and impaired cognitive and language development.10
As early as the 11–12th week of pregnancy, profound changes in the activity of the maternal hypothalamic-pituitary axis (HPA) occur that lead to increased production of maternal cortisol. The levels of cortisol continue to rise during pregnancy to reach almost three times those of non-pregnant women by the third trimester.11-13 The HPA axis is regulated by the release of corticotrophin-releasing hormone (CRH) from the hypothalamus, which stimulates the release of adrenocorticotrophic hormone (ACTH) from the anterior pituitary gland, which in turn stimulates the production of cortisol from the adrenal glands. Normally, cortisol regulates its own secretion through a negative feedback loop that inhibits the production of CRH and ACTH.14 However, during pregnancy, the placenta begins to release CRH into the maternal bloodstream and levels reach 1000–10 000 times that of a non-pregnant woman. This further increases the production of cortisol by the adrenals.15,16 Although cortisol usually negatively regulates the secretion of hypothalamic CRH, a rise in maternal cortisol actively stimulates the release of placental CRH, which further raises the levels of maternal cortisol progressively during pregnancy.11-13 This increase in maternal glucocorticoid levels has been proposed to be required for normal fetal organogenesis17 and, in keeping with this suggestion, antenatal synthetic glucocorticoid administration to preterm infants has been shown to significantly decrease mortality and morbidity in infants born before 34 wk of gestation by accelerating lung maturation.18-20 However, excessive fetal exposure to maternal glucocorticoids has been proposed to mediate the impact of prenatal stress on birth outcomes, resulting in long-term health consequences in affected infants.21 Furthermore, elevated maternal cortisol levels during pregnancy is associated with a larger amygdala and more emotional disorders in girls aged 7, directly linking maternal cortisol to long-term programming effects.22 However, no study to date has examined if maternal stress is directly associated with elevated fetal cortisol levels during pregnancy or at term.
Normally, the fetus is protected from the high levels of maternal cortisol by molecular mechanisms in the placenta that convert active cortisol to its inactive metabolite cortisone. Consequently, fetal cortisol levels have been shown to be ten to 13 times lower than those found in maternal circulation.23,24 The primary barrier between maternal and fetal circulation is provided by two epithelial layers that cover the chorionic villi of the human placenta.25 The first of these epithelial layers is the syncytiotrophoblast, a large multi-nucleated terminally-differentiated syncytium (many nuclei in the same cytoplasmic mass), that is formed by differentiation and fusion of cells derived from the second epithelial layer, the underlying cytotrophoblast.26 Within these trophoblast layers, an enzyme called 11β-hydroxysteroid dehydrogenase type-2 (HSD11B2) is highly expressed. HSD11B2 catalyzes the conversion of active cortisol into inactive cortisone, thus protecting the fetus from excessive cortisol exposure (Fig. 1).27-29
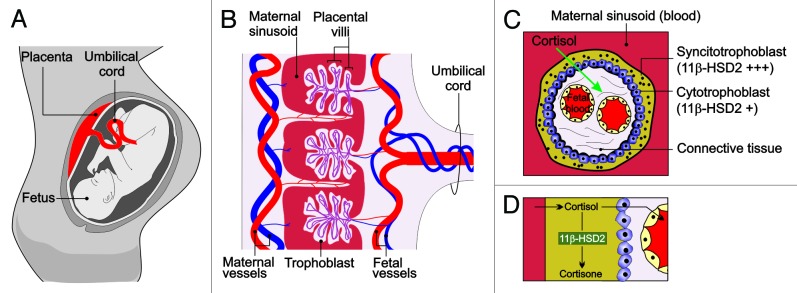
Figure 1. The placental HSD11B2 shield. (A) During pregnancy, the placenta acts as a critical regulator that limits fetal exposure to maternal glucorticoids. (B) Cross sectional image of the placenta. Blood travels from the fetus through the umbilical arteries (blue). Blood is carried to the chorionic villi, where exchange of nutrients and waste products with the mother occurs. The chorionic villi are made up of two epithelial layers, the syncytiotrophoblast and the cytotrophoblast. (C) Longitudinal section of the chorionic villi. HSD211B2 is expressed strongly in the syncytiotrophoblast and to a lesser extent in the cytotrophoblast, where it converts cortisol into inactive cortisone, thereby protecting the fetus from excessive cortisol exposure.
In this review we describe the impact of prenatal stress during pregnancy on placental HSD11B2 expression and its relationship to fetal health outcomes. We examine the available evidence that suggests that the programming effects of prenatal stress may be mediated through the regulation of placental HSD11B2 expression. We summarize recent data indicating that epigenetic regulation of placental HSD11B2 expression and/or function may be a mechanistic link between maternal stress and the resultant long-term health consequences for affected offspring. Finally, we offer a clinical perspective, highlighting the difficulties and promises of targeting placental HSD11B2 epigenetic change.
Effect of Prenatal Stress on HSD11B2 Expression
Increasing preclinical and clinical evidence suggest that maternal stress during pregnancy can lead to alterations in placental HSD11B2 expression. For example, direct cortisol infusion into pregnant mice between embryonic day (E)12 and E15 resulted in the upregulation of HSD11B2 mRNA and protein levels in the placenta at E14.5. However, by E17, placental HSD11B2 mRNA was significantly reduced, indicating an adaptive mechanism whereby acute cortisol exposure initially upregulates the placental barrier to protect the fetus, but chronic cortisol exposure reduces its expression, resulting in overexposure of the fetus to maternal glucocorticoids.30 In agreement with these findings, it has also been shown that pregnant rats subjected to acute restraint stress for one hour on gestational day 20, show increased placental HSD11B2 expression.31 In contrast, chronic restraint stress in pregnant rat dams from gestational day 14 to 20 caused a reduction of HSD11B2 protein and mRNA levels in the placenta.31,32 This was accompanied by reduced fetal birth weight, reduced circulating ACTH levels and lower adrenal weight.32 Similarly, pregnant rat dams that were food restricted from gestational day 10 to 20, which causes an increase in maternal corticosterone levels, showed reduced placental HSD11B2 mRNA expression and lower fetal birth weight.33 Furthermore, these offspring displayed reduced circulating ACTH, adrenal atrophy and decreased mineralocorticoid and glucocorticoid mRNA expression in the fetal hippocampus, indicating alterations in the fetal HPA axis.34 Similar findings have been reported in human pregnancy. Amniocentesis in pregnant women, which is associated with increased anxiety levels and is a form of acute stress in humans, is associated with increased HSD11B2 activity in the placenta.35 Prenatal anxiety and depression in humans, as determined by the Spielberger State Trait Anxiety Test and The Edinburgh postnatal depression test, is a form of chronic prenatal stress in humans. Consistent with preclinical data, chronic prenatal anxiety and depression were found to be associated with reduced activity and expression of placental HSD11B2.36
The precise mechanism(s) by which prenatal stress reduces placental expression of HSD11B2 is unclear, but it has been shown that increased circulating glucocorticoid concentrations, as a result of stress, stimulate the sympathoadrenal system, causing elevated adrenaline and noradrenaline release from the adrenal medulla.37 These stress-induced catecholamines have shown to decrease HSD11B2 mRNA in the BeWo human choriocarcinoma cell line and in primary trophoblastic cells.38 Furthermore, women experiencing high levels of stress during pregnancy also show increased circulating levels of pro-inflammatory cytokines.39 Pro-inflammatory cytokines TNF-α and IL-1β have been shown to decrease HSD11B2 mRNA levels and activity in vitro.40-42 These studies indicate that there are multiple mechanisms through which prenatal stress may affect placental HSD11B2 expression and/or activity. Collectively, these data highlight an important point: acute stress during pregnancy may not be harmful to the developing fetus due to the ability to upregulate the expression of HSD11B2; however, chronic stress can result in a significant decrease in placental HSD11B2 expression, causing the fetus to be exposed to excessive levels of maternal cortisol. Although the precise mechanisms mediating these changes in placental HSD11B2 expression remain to be fully elucidated, it has recently emerged that epigenetic regulation may play a crucial role.
Epigenetic Regulation of HSD11B2 Expression
It is now largely accepted that epigenetics can be modified by environmental exposures, particularly during development, when the epigenome undergoes profound changes.43 Recently, it was suggested that the link between prenatal stress and the reduction in placental HSD11B2 expression may involve hypermethylation of the HSD11B2 promoter, leading to a corresponding reduction in HSD11B2 expression.
The human HSD11B2 gene contains four CpG islands, two located in the promoter and exon 1 and two in exon 5 and the downstream region.44,45 Hypermethylation of normally unmethylated CpG islands is indicative of transcriptional repression of the relevant gene.46 Interestingly, hypermethylation of HSD11B2 CpG islands correlates with lower levels of HSD11B2 expression.45 Low methylation levels of HSD11B2 were observed in the placenta (were HSD11B2 expression is high), whereas in skeletal muscle (low HSD11B2 expression) the HSD11B2 promoter was hypermethylated.45 Furthermore, it has been shown that the activity of the HSD11B2 promoter is directly regulated by methylation.45 These data show that exposures that change HSD11B2 methylation status in the placenta can result in transcriptional repression of the gene, causing the fetus to be exposed to higher levels of cortisol that may adversely affect fetal development. In agreement with this idea, Marsit and colleagues determined the methylation status of HSD11B2 in 186 placentae and examined any correlation with fetal birth and neurodevelopmental milestones. The methylation status of 4 CpG islands in placental HSD11B2 was examined, showing a statistically significant, albeit moderate, negative correlation with placental HSD11B2 mRNA expression, indicating that changes in the methylation status of placental HSD11B2 can regulate its expression.44 Intriguingly, a negative correlation was found between infant birth weight and ponderal index (ratio of weight for length) and HSD11B2 methylation, demonstrating that smaller and leaner/thinner infants showed increased levels of HSD11B2 methylation.44 Furthermore, when the extent of HSD11B2 methylation was examined in intrauterine growth restricted (IUGR) infants compared with non-IUGR infants, IUGR infants showed a significantly greater extent of HSD11B2 methylation than non-IUGR infants.44 They also examined infant neurobehavioral outcomes and found that the extent of HSD11B2 methylation was greatest in infants with reduced quality of movement scores as assessed using the NICU Network Neurobehavioral Scales (NNNS) examination.44 This study is important as it is the first one to link growth, epigenetic alterations of placental genes, and early-life neurobehavioral outcomes in humans. Clearly, longer follow-up of infants is required to determine if methylation status of placental HSD11B2 can help identify those children who may develop neurobehavioral or learning difficulties later in life.44 It is possible that the epigenetic status of placental HSD11B2 could also be a useful predictor of other adverse outcomes.
New data using a rat model of prenatal anxiety has now demonstrated that prenatal maternal anxiety caused a reduction of HSD11B2 placental expression and, importantly, that this was associated with increased CpG methylation of placental HSD11B2.47 This study also attempted to assess whether methylation at certain CpG sites within placental HSD11B2 positively predicted the methylation status of HSD11B2 in the fetal cortex and hippocampus. This raises the intriguing possibility of using the epigenetic status of placenta to predict corresponding changes in the fetal brain.47 Prenatal maternal anxiety was reported to result in a small but statistically significant increase in HSD11B2 methylation at CpG sites 4, 7 and 8, and reduced expression of placental HSD11B2. When methylation at these same sites was examined in the fetal hypothalamus, significant changes in methylation were observed, but these were discordant and in the opposite direction as observed in the placenta, apparently causing no effect on HSD11B2 mRNA levels.47 However, it should be noted that significant differences in methylation at other CpG sites in the placenta were also observed that were not found in the fetal brain, suggesting that there may not be a complete direct, inverse relationship between both tissues. Furthermore, no differences in DNA methylation were observed in the fetal cortex of stressed animals compared with controls,47 suggesting that regional differences in the effects of prenatal maternal anxiety on the methylation status of HSD11B2 in the fetal brain may exist. This raises the intriguing possibility that the epigenetic status of HSD11B2 in the placenta could predict changes in the fetal brain. However, it is clear that much work remains to be done to investigate causality, and that the effects of prenatal maternal anxiety on the entire epigenome, rather than individual genes, need to be examined. Such studies are vital, given the recent demonstration that global epigenomic reconfiguration is observed during human brain development, which suggests a key role of DNA methylation in brain development and function.46
Recently, the impact of maternal prenatal anxiety on the placental epigenetic status of HSD11B2 has been reported.48 Conradt and colleagues used a structured chart review to collect information about whether a mother reported depression and/or anxiety during pregnancy. Of the 482 participants recruited for the study, 13.7% reported depression or anxiety during pregnancy, which is consistent with the estimated prevalence rates.49 In agreement with findings from animal models of prenatal anxiety,47 maternal anxiety during pregnancy resulted in a statistically significant increase in methylation of CpG site 4 of the HSD11B2 promoter.48 Furthermore, infants of mothers who were anxious during pregnancy were more hypotonic, and placentae from these pregnancies had greater methylation of CpG site 4 in HSD11B2.48 While correlations suggest that the epigenetic status of placental HSD11B2 may help identify infants at risk for poor neurodevelopmental outcomes, a key challenge that remains is to prove cause and effect. Specifically, whether the increase in HSD11B2 methylation in the placenta following prenatal anxiety in fact leads to the behavioral changes observed in affected infants needs to be further examined. One possible approach could be to examine fetal neurobehavioral outcomes following prenatal anxiety in mice that express a drug-inducible, conditional placental-specific knockdown of the methyltransferase DNMT3a, whose expression has been shown to be increased in the placenta following prenatal anxiety;47 or, more specifically, the generation of mice carrying methylation resistant CpG sites in HSD11B2.
A Clinical Perspective: Extrinsic Factors That Can Modify Placental HSD11B2 Expression
If epigenetic downregulation of placental HSD11B2 expression is the mechanistic link between the experience of maternal stress and the resultant long-term health consequences for affected offspring, then strategies that increase placental HSD11B2 expression have the potential of improving fetal outcome. Here we highlight two ways in which extrinsic factors could modify the expression of placental HSD11B2: (1) the use of pharmacological epigenetic modifiers and; (2) dietary considerations that may affect placental HSD11B2.
Effects of Pharmacological Epigenetic Modifiers on HSD11B2 Expression
A defining feature of epigenetic modifications is that they are reversible. Thus, drugs that interfere with the enzymes that produce these changes may help maintain or restore normal gene expression levels. One class of drugs that alter DNA methylation is that of DNMT inhibitors. Many types of DNMT inhibitors have been produced and some of these have been used clinically for the treatment of leukemias, myelodysplastic syndromes and hemglobinopathies.50 These compounds exert their effects by covalently trapping methyltransferases, thereby preventing full methylation of a DNA strand.51,52 A second class of molecules is that of HDAC inhibitors. These compounds inhibit the activity of HDACs, thus preventing histone deacetylation of lysine residues on histone tails and, as a consequence, enhancing gene transcription. HDAC inhibitors have also been used in epigenetic therapy and two of these compounds have recently gained FDA approval as anti-cancer therapies.53
DNMT inhibitors 5-aza-2′-deoxycytidine (5-AZA) and procainamide have been shown to demethylate the HSD11B2 promoter, enhancing both HSD11B2 expression and activity in the SW620 human colon carcinoma cell line, MCF-7 breast adenocarcinoma cell line, and JEG-3 placental carcinoma cell line.45 Furthermore, when given orally to rats for seven days, DNMT inhibitors were shown to reduce HSD11B2 methylation and to increase HSD11B2 expression,45 which highlights the potential impact of such classes of epigenetic pharmacological modifiers on HSD11B2 expression. Similar studies have also examined the effects of HDAC inhibition on HSD11B2 expression and found that the HDAC inhibitor trichostatin A had no effect by itself on these cell lines, but increased HSD211B2 expression in SW620 and MCF-7 cells when added only 48 h after 5-AZA treatment, indicating that DNA methylation is the dominant form of epigenetic modification on this gene.45
These classes of pharmacological inhibitors have been proposed to be potentially useful in reversing placental epigenetic change. However, a major challenge with these approaches will be to specifically target the placenta without affecting the developing fetus. This is particularly pertinent as recent data shows that a global reconfiguration of the fetal neural epigenome occurs during development,46 suggesting that drugs that interfere with these processes may have profound consequences for normal fetal development. Furthermore, these drugs have significant detrimental effects on both the mother and fetus. For example, intraperitoneal (ip) injection of pregnant mice with 5-AZA on gestational day 10 has been shown to reduce birth weight in affected offspring, an effect that persists in males at 5 months of age.54 This reduced weight is also associated with reduced IGF-1 levels in the serum of these offspring.54 Furthermore, males and females who were exposed to 5-AZA in utero display a 70% reduction and a 30% reduction in mating capacity and fertility, respectively, as measured by the presence of a vaginal plug and viable pregnancy.54 In a recent study by Ding and colleagues, Kunming mice were subjected to i.p. injection of 5-AZA or saline on gestational days 1, 2 and 4, followed by embryo implantation on day 4. Mice that received 5-AZA showed a significant reduction in embryo implantation, as compared with the saline group. This was associated with a cell specific reduction in mRNA levels and an increase in the methylation levels of many genes involved in endometrial change, as well as a decrease in stromal cell proliferation and differentiation.55 Furthermore, intravenous infusion of 5-AZA in patients with metastatic lung carcinomas induced hematopoiesis toxicity,56 thus limiting their use in pregnant women. Given these significant problems and challenges associated with their use, the administration of pharmacological epigenetic modifiers to modify the placental epigenome is not feasible at the present time.
Effects of Dietary Composition on Placental HSD11B2 Expression
The influence of dietary composition as a modifier of placental epigenetic machinery is becoming increasingly apparent.57,58 Mice fed a high fat diet showed reduced placental expression of a number of methyltransferases involved in epigenetic modification.57,58 This is a very important finding when one considers that, in western societies, 50% of all pregnant women are either overweight (BMI of 25–29.9 kg/m2) or obese (BMI of ≥30 kg/m2),59 have a dietary caloric excess predominantly derived from fat,60 or show a higher rate of adverse pregnancy outcomes.61,62
DNA methylation45 is dependent on the availability of methyl group donors and cofactors provided by the methionine and folate metabolisms, as well as dietary-derived vitamin B6 and B12, suggesting that dietary modifications can affect the fetal and placental epigenome.63 This is highlighted by a 12-wk dose-response choline feeding study conducted in third-trimester pregnant women that investigated the effect of a maternal choline intake of 930 mg or 480 mg per day. Higher maternal choline intake significantly increased the average methylation levels in the glucocorticoid receptor gene promoter in placenta and fetal leukocytes, with infants showing lower levels of circulating cortisol in the higher choline group.64 Although limited data exist regarding the impact of dietary composition on the epigenetic modification of placental HSD11B2, Takaya et al. examined the methylation of individual CpG dinucleotides in the promoter region of hepatic HSD11B2 in offspring of pregnant rats fed a magnesium-deficient diet from 2 wk prior to mating and throughout the gestational period.65 The average methylation of the HSD11B2 promoter in the offspring of dams fed a magnesium deficient diet was significantly higher than that from controls.65 However, it is important to note that contrary to what was expected given previous findings in the placenta,44 HSD11B2 methylation was positively correlated with HSD11B2 mRNA expression.65 However, responsiveness to environmental exposures in utero cannot be generalized across genes and tissues,66 suggesting that epigenetic modification of the HSD11B2 promoter in different tissues may result in different effects on gene expression. Together, these studies show that maternal diet can exert gene-specific epigenetic changes that can effect placental mRNA expression. The effect of diet on placental epigenetic regulation of HSD11B2 is a key question for future research. However, a number of studies have examined the effect of both dietary intake and composition on placental HSD11B2.
A recent study investigating the impact of maternal caloric restriction on placental HSD11B2 expression reported that when dietary intake was reduced by 50% in Sprague Dawley rats from gestational day 10 to 20, these rats showed higher maternal corticosterone levels than controls and a reduction in HSD11B2 expression in the labyrinth zone of the placenta.33,67 Furthermore, specific reduction in protein content of the maternal diet during early and mid, but not late, gestation in Wistar rats caused a reduction of placental HSD11B2 expression.68 These findings have been supported by some,69-71 though not all,72 studies and collectively suggest that adequate protein intake is required to maintain optimal expression of placental HSD11B2. However, an excess of calories, predominantly derived from high fat and high sugar foods largely define Western dietary habits.60 Therefore, an area that demands further investigation is the effect of such high calories, high fat/sugar diets on placental HSD11B2 expression. A recent study by Sferruzzi-Perri et al. reported altered placental phenotypes in mice fed a high fat, high sugar diet that affected placental nutrient handling, which has implications for fetal development and pregnancy outcomes.73 In addition, the high fat, high sugar diet influenced fetal growth trajectory (initial growth restriction followed by catch-up growth), which may have implications for later-life outcomes. However, one potential caveat of this well-designed study (acknowledged by its authors) was the difference in protein content between the two diets (the standard diet consisted of 26% protein compared with the high fat, high sugar diet, which consisted of 17% protein).73 It has previously been shown that even mild protein restriction can contribute to changes in placental expression of HSD211B2 and development of HPA dysfunction and hypertension.60 Therefore, the data from Sferruzzi-Perri and colleagues need to be interpreted within the context of unmatched maternal protein intakes. However, given that mice and rats fed a high fat diet during pregnancy display alterations in the placental transcriptome and have dysregulated placental epigenetic machinery,57,58 a much greater understanding of the effects of diet on placental HSD11B2 is required. When we analyzed the supplementary data available in the study by Mao et al. looking at the placental transcriptome at day 12.5 of gestation in mice fed a high fat diet, robust changes were found in the placental transcriptome however HSD11B2 was not in their list of differentially expressed genes.74 This does not mean that this gene does not change in response to a high fat diet, it only means that changes in HSD11B2 were not detected at the particular time at which they did their microarray profiling. In addition, transient changes with long-lasting effects cannot be ruled out, especially given the data from Sferruzzi-Perri and colleagues.73
Future research should focus on addressing the effects of such diets, as well as on understanding the correct balance of nutrients that may mitigate the effects of increased maternal stress. For instance, could optimal protein levels throughout pregnancy protect against the placental morphological and phenotypical changes associated with a high fat, high sugar diet? In addition, we know that a reduction in total maternal calories downregulates HSD11B2 in the labyrinth zone and preliminary evidence suggests that a high fat, high sugar diet influences labyrinth zone development; therefore, devising maternal dietary interventions that may influence both placental development and ensure optimal fetal outcomes may be an exciting area of research. Similarly, could maternal dietary manipulation represent a mechanism that could protect or counteract the effect of maternal stress (environmental or emotional) on HSD11B2 expression and fetal outcomes? These questions warrant further investigation.
Whether nutritional interventions can specifically prevent or reverse prenatal stress-induced epigenetic change in placental HSD11B2 remains to be elucidated, but it represents an exciting and, more importantly, feasible approach to potentially reverse or prevent the programming effect of prenatal stress on the placental and fetal epigenome.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors acknowledge grant support in the form of a Research Frontiers Program grant (Grant#: 10/RFP/NES2786) (G.O.’K.) and a Research Centres grant (Grant#: INFANT-12/RC/2272) (L.K.) from Science Foundation Ireland.
Glossary
Abbreviations:
- ACTH
adrenocorticotrophic hormone
- 5-AZA
5-aza-2′-deoxycytidine
- CRH
Corticotrophin-releasing hormone
- DMNT
DNA methyltransferase
- E
embryonic day
- HAT
histone acetyltransferase
- HDAC
histone deacetylases
- HSD11B2
11β-hydroxysteroid dehydrogenase type-2
- HPA
hypothalamic-pituitary axis
- IUGR
intrauterine growth restricted
- NNNS examination
NICU Network Neurobehavioral Scales examination
References
- 1.Entringer S, Buss C, Wadhwa PD. Prenatal stress, telomere biology, and fetal programming of health and disease risk. Sci Signal. 2012;5:pt12. doi: 10.1126/scisignal.2003580. [DOI] [PubMed] [Google Scholar]
- 2.Reynolds RM. Glucocorticoid excess and the developmental origins of disease: two decades of testing the hypothesis--2012 Curt Richter Award Winner. Psychoneuroendocrinology. 2013;38:1–11. doi: 10.1016/j.psyneuen.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Hobel CJ, Goldstein A, Barrett ES. Psychosocial stress and pregnancy outcome. Clin Obstet Gynecol. 2008;51:333–48. doi: 10.1097/GRF.0b013e31816f2709. [DOI] [PubMed] [Google Scholar]
- 4.Dancause KN, Laplante DP, Oremus C, Fraser S, Brunet A, King S. Disaster-related prenatal maternal stress influences birth outcomes: project Ice Storm. Early Hum Dev. 2011;87:813–20. doi: 10.1016/j.earlhumdev.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Dancause KN, Veru F, Andersen RE, Laplante DP, King S. Prenatal stress due to a natural disaster predicts insulin secretion in adolescence. Early Hum Dev. 2013;89:773–6. doi: 10.1016/j.earlhumdev.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khashan AS, Abel KM, McNamee R, Pedersen MG, Webb RT, Baker PN, Kenny LC, Mortensen PB. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch Gen Psychiatry. 2008;65:146–52. doi: 10.1001/archgenpsychiatry.2007.20. [DOI] [PubMed] [Google Scholar]
- 7.Dancause KN, Vilar M, Chan C, DeHuff C, Wilson M, Soloway LE, Tarivonda L, Regenvanu R, Kaneko A, Garruto RM, et al. Patterns of childhood and adolescent overweight and obesity during health transition in Vanuatu. Public Health Nutr. 2012;15:158–66. doi: 10.1017/S1368980011001662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hägglöf B, Blom L, Dahlquist G, Lönnberg G, Sahlin B. The Swedish childhood diabetes study: indications of severe psychological stress as a risk factor for type 1 (insulin-dependent) diabetes mellitus in childhood. Diabetologia. 1991;34:579–83. doi: 10.1007/BF00400277. [DOI] [PubMed] [Google Scholar]
- 9.Ronald A, Happé F, Dworzynski K, Bolton P, Plomin R. Exploring the relation between prenatal and neonatal complications and later autistic-like features in a representative community sample of twins. Child Dev. 2010;81:166–82. doi: 10.1111/j.1467-8624.2009.01387.x. [DOI] [PubMed] [Google Scholar]
- 10.King S, Laplante DP. The effects of prenatal maternal stress on children’s cognitive development: Project Ice Storm. Stress. 2005;8:35–45. doi: 10.1080/10253890500108391. [DOI] [PubMed] [Google Scholar]
- 11.D’Anna-Hernandez KL, Ross RG, Natvig CL, Laudenslager ML. Hair cortisol levels as a retrospective marker of hypothalamic-pituitary axis activity throughout pregnancy: comparison to salivary cortisol. Physiol Behav. 2011;104:348–53. doi: 10.1016/j.physbeh.2011.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demey-Ponsart E, Foidart JM, Sulon J, Sodoyez JC. Serum CBG, free and total cortisol and circadian patterns of adrenal function in normal pregnancy. J Steroid Biochem. 1982;16:165–9. doi: 10.1016/0022-4731(82)90163-7. [DOI] [PubMed] [Google Scholar]
- 13.Jung C, Ho JT, Torpy DJ, Rogers A, Doogue M, Lewis JG, Czajko RJ, Inder WJ. A longitudinal study of plasma and urinary cortisol in pregnancy and postpartum. J Clin Endocrinol Metab. 2011;96:1533–40. doi: 10.1210/jc.2010-2395. [DOI] [PubMed] [Google Scholar]
- 14.Myers B, McKlveen JM, Herman JP. Neural Regulation of the Stress Response: The Many Faces of Feedback. Cell Mol Neurobiol. 2012 doi: 10.1007/s10571-012-9801-y. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duthie L, Reynolds RM. Changes in the maternal hypothalamic-pituitary-adrenal axis in pregnancy and postpartum: influences on maternal and fetal outcomes. Neuroendocrinology. 2013;98:106–15. doi: 10.1159/000354702. [DOI] [PubMed] [Google Scholar]
- 16.Petraglia F, Sawchenko PE, Rivier J, Vale W. Evidence for local stimulation of ACTH secretion by corticotropin-releasing factor in human placenta. Nature. 1987;328:717–9. doi: 10.1038/328717a0. [DOI] [PubMed] [Google Scholar]
- 17.Smith ID, Shearman RP. Fetal plasma steroids in relation to parturition. I. The effect of gestational age upon umbilical plasma corticosteroid levels following vaginal delivery. J Obstet Gynaecol Br Commonw. 1974;81:11–5. doi: 10.1111/j.1471-0528.1974.tb00357.x. [DOI] [PubMed] [Google Scholar]
- 18.Block MF, Kling OR, Crosby WM. Antenatal glucocorticoid therapy for the prevention of respiratory distress syndrome in the premature infant. Obstet Gynecol. 1977;50:186–90. [PubMed] [Google Scholar]
- 19.Doyle LW, Kitchen WH, Ford GW, Rickards AL, Kelly EA. Antenatal steroid therapy and 5-year outcome of extremely low birth weight infants. Obstet Gynecol. 1989;73:743–6. [PubMed] [Google Scholar]
- 20.Eriksson L, Haglund B, Ewald U, Odlind V, Kieler H. Health consequences of prophylactic exposure to antenatal corticosteroids among children born late preterm or term. Acta Obstet Gynecol Scand. 2012;91:1415–21. doi: 10.1111/aogs.12014. [DOI] [PubMed] [Google Scholar]
- 21.Barbazanges A, Piazza PV, Le Moal M, Maccari S. Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J Neurosci. 1996;16:3943–9. doi: 10.1523/JNEUROSCI.16-12-03943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buss C, Davis EP, Shahbaba B, Pruessner JC, Head K, Sandman CA. Maternal cortisol over the course of pregnancy and subsequent child amygdala and hippocampus volumes and affective problems. Proc Natl Acad Sci U S A. 2012;109:E1312–9. doi: 10.1073/pnas.1201295109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. Dysfunction of placental glucocorticoid barrier: link between fetal environment and adult hypertension? Lancet. 1993;341:355–7. doi: 10.1016/0140-6736(93)90148-A. [DOI] [PubMed] [Google Scholar]
- 24.Gitau R, Cameron A, Fisk NM, Glover V. Fetal exposure to maternal cortisol. Lancet. 1998;352:707–8. doi: 10.1016/S0140-6736(05)60824-0. [DOI] [PubMed] [Google Scholar]
- 25.Huppertz B. The anatomy of the normal placenta. J Clin Pathol. 2008;61:1296–302. doi: 10.1136/jcp.2008.055277. [DOI] [PubMed] [Google Scholar]
- 26.Huppertz B, Borges M. Placenta trophoblast fusion. Methods Mol Biol. 2008;475:135–47. doi: 10.1007/978-1-59745-250-2_8. [DOI] [PubMed] [Google Scholar]
- 27.Brown RW, Chapman KE, Kotelevtsev Y, Yau JL, Lindsay RS, Brett L, Leckie C, Murad P, Lyons V, Mullins JJ, et al. Cloning and production of antisera to human placental 11 beta-hydroxysteroid dehydrogenase type 2. Biochem J. 1996;313:1007–17. doi: 10.1042/bj3131007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krozowski Z, MaGuire JA, Stein-Oakley AN, Dowling J, Smith RE, Andrews RK. Immunohistochemical localization of the 11 beta-hydroxysteroid dehydrogenase type II enzyme in human kidney and placenta. J Clin Endocrinol Metab. 1995;80:2203–9. doi: 10.1210/jcem.80.7.7608280. [DOI] [PubMed] [Google Scholar]
- 29.Sun K, Yang K, Challis JR. Differential expression of 11 beta-hydroxysteroid dehydrogenase types 1 and 2 in human placenta and fetal membranes. J Clin Endocrinol Metab. 1997;82:300–5. doi: 10.1210/jcem.82.1.3681. [DOI] [PubMed] [Google Scholar]
- 30.Cuffe JS, O’Sullivan L, Simmons DG, Anderson ST, Moritz KM. Maternal corticosterone exposure in the mouse has sex-specific effects on placental growth and mRNA expression. Endocrinology. 2012;153:5500–11. doi: 10.1210/en.2012-1479. [DOI] [PubMed] [Google Scholar]
- 31.Welberg LA, Thrivikraman KV, Plotsky PM. Chronic maternal stress inhibits the capacity to up-regulate placental 11beta-hydroxysteroid dehydrogenase type 2 activity. J Endocrinol. 2005;186:R7–12. doi: 10.1677/joe.1.06374. [DOI] [PubMed] [Google Scholar]
- 32.Mairesse J, Lesage J, Breton C, Bréant B, Hahn T, Darnaudéry M, Dickson SL, Seckl J, Blondeau B, Vieau D, et al. Maternal stress alters endocrine function of the feto-placental unit in rats. Am J Physiol Endocrinol Metab. 2007;292:E1526–33. doi: 10.1152/ajpendo.00574.2006. [DOI] [PubMed] [Google Scholar]
- 33.Belkacemi L, Desai M, Beall MH, Liu Q, Lin JT, Nelson DM, Ross MG. Early compensatory adaptations in maternal undernourished pregnancies in rats: role of the aquaporins. J Matern Fetal Neonatal Med. 2011;24:752–9. doi: 10.3109/14767058.2010.521870. [DOI] [PubMed] [Google Scholar]
- 34.Lesage J, Blondeau B, Grino M, Bréant B, Dupouy JP. Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo-pituitary adrenal axis in the newborn rat. Endocrinology. 2001;142:1692–702. doi: 10.1210/endo.142.5.8139. [DOI] [PubMed] [Google Scholar]
- 35.Ghaemmaghami P, Dainese SM, La Marca R, Zimmermann R, Ehlert U. The association between the acute psychobiological stress response in second trimester pregnant women, amniotic fluid glucocorticoids, and neonatal birth outcome. Dev Psychobiol. 2013 doi: 10.1002/dev.21142. Forthcoming. [DOI] [PubMed] [Google Scholar]
- 36.O’Donnell KJ, Bugge Jensen A, Freeman L, Khalife N, O’Connor TG, Glover V. Maternal prenatal anxiety and downregulation of placental 11β-HSD2. Psychoneuroendocrinology. 2012;37:818–26. doi: 10.1016/j.psyneuen.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 37.Kvetnanský R, Pacák K, Fukuhara K, Viskupic E, Hiremagalur B, Nankova B, Goldstein DS, Sabban EL, Kopin IJ. Sympathoadrenal system in stress. Interaction with the hypothalamic-pituitary-adrenocortical system. Ann N Y Acad Sci. 1995;771:131–58. doi: 10.1111/j.1749-6632.1995.tb44676.x. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar S, Tsai SW, Nguyen TT, Plevyak M, Padbury JF, Rubin LP. Inhibition of placental 11beta-hydroxysteroid dehydrogenase type 2 by catecholamines via alpha-adrenergic signaling. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1966–74. doi: 10.1152/ajpregu.2001.281.6.R1966. [DOI] [PubMed] [Google Scholar]
- 39.Coussons-Read ME, Okun ML, Schmitt MP, Giese S. Prenatal stress alters cytokine levels in a manner that may endanger human pregnancy. Psychosom Med. 2005;67:625–31. doi: 10.1097/01.psy.0000170331.74960.ad. [DOI] [PubMed] [Google Scholar]
- 40.Chisaka H, Johnstone JF, Premyslova M, Manduch Z, Challis JR. Effect of pro-inflammatory cytokines on expression and activity of 11beta-hydroxysteroid dehydrogenase type 2 in cultured human term placental trophoblast and human choriocarcinoma JEG-3 cells. J Soc Gynecol Investig. 2005;12:303–9. doi: 10.1016/j.jsgi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Kossintseva I, Wong S, Johnstone E, Guilbert L, Olson DM, Mitchell BF. Proinflammatory cytokines inhibit human placental 11beta-hydroxysteroid dehydrogenase type 2 activity through Ca2+ and cAMP pathways. Am J Physiol Endocrinol Metab. 2006;290:E282–8. doi: 10.1152/ajpendo.00328.2005. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki S, Tsubochi H, Ishibashi H, Matsuda Y, Suzuki T, Krozowski ZS, Sasano H, Kondo T. Inflammatory mediators down-regulate 11beta-hydroxysteroid dehydrogenase type 2 in a human lung epithelial cell line BEAS-2B and the rat lung. Tohoku J Exp Med. 2005;207:293–301. doi: 10.1620/tjem.207.293. [DOI] [PubMed] [Google Scholar]
- 43.Reamon-Buettner SM, Mutschler V, Borlak J. The next innovation cycle in toxicogenomics: environmental epigenetics. Mutat Res. 2008;659:158–65. doi: 10.1016/j.mrrev.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Marsit CJ, Maccani MA, Padbury JF, Lester BM. Placental 11-beta hydroxysteroid dehydrogenase methylation is associated with newborn growth and a measure of neurobehavioral outcome. PLoS One. 2012;7:e33794. doi: 10.1371/journal.pone.0033794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epigenetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest. 2004;114:1146–57. doi: 10.1172/JCI21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jensen Peña C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One. 2012;7:e39791. doi: 10.1371/journal.pone.0039791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conradt E, Lester BM, Appleton AA, Armstrong DA, Marsit CJ. The roles of DNA methylation of NR3C1 and 11β-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics. 2013;8:1321–9. doi: 10.4161/epi.26634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nylen KJ, Williamson JA, O’Hara MW, Watson D, Engeldinger J. Validity of somatic symptoms as indicators of depression in pregnancy. Arch Womens Ment Health. 2013;16:203–10. doi: 10.1007/s00737-013-0334-2. [DOI] [PubMed] [Google Scholar]
- 50.Goffin J, Eisenhauer E. DNA methyltransferase inhibitors-state of the art. Ann Oncol. 2002;13:1699–716. doi: 10.1093/annonc/mdf314. [DOI] [PubMed] [Google Scholar]
- 51.Santini V, Gozzini A, Scappini B, Grossi A, Rossi Ferrini P. Searching for the magic bullet against cancer: the butyrate saga. Leuk Lymphoma. 2001;42:275–89. doi: 10.3109/10428190109064584. [DOI] [PubMed] [Google Scholar]
- 52.Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann Intern Med. 2001;134:573–86. doi: 10.7326/0003-4819-134-7-200104030-00011. [DOI] [PubMed] [Google Scholar]
- 53.Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117–36. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cisneros FJ, Wilson R, Travlos G, Anderson LM, Branch S. Susceptibility to postnatal growth retardation induced by 5-AZA-2′-deoxycytidine in utero: gender specificity and correlation with reduced insulin-like growth factor 1. Life Sci. 2003;72:2887–94. doi: 10.1016/S0024-3205(03)00229-7. [DOI] [PubMed] [Google Scholar]
- 55.Ding YB, Long CL, Liu XQ, Chen XM, Guo LR, Xia YY, He JL, Wang YX. 5-aza-2′-deoxycytidine leads to reduced embryo implantation and reduced expression of DNA methyltransferases and essential endometrial genes. PLoS One. 2012;7:e45364. doi: 10.1371/journal.pone.0045364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J. Pilot phase I-II study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs. 1997;8:358–68. doi: 10.1097/00001813-199704000-00008. [DOI] [PubMed] [Google Scholar]
- 57.Gabory A, Ferry L, Fajardy I, Jouneau L, Gothié JD, Vigé A, Fleur C, Mayeur S, Gallou-Kabani C, Gross MS, et al. Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PLoS One. 2012;7:e47986. doi: 10.1371/journal.pone.0047986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gallou-Kabani C, Gabory A, Tost J, Karimi M, Mayeur S, Lesage J, Boudadi E, Gross MS, Taurelle J, Vigé A, et al. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS One. 2010;5:e14398. doi: 10.1371/journal.pone.0014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999-2004. JAMA. 2006;295:1549–55. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 60.Allegri C, Turconi G, Cena H. Dietary attitudes and diseases of comfort. Eat Weight Disord. 2011;16:e226–35. doi: 10.1007/BF03327465. [DOI] [PubMed] [Google Scholar]
- 61.Smith GC, Shah I, Pell JP, Crossley JA, Dobbie R. Maternal obesity in early pregnancy and risk of spontaneous and elective preterm deliveries: a retrospective cohort study. Am J Public Health. 2007;97:157–62. doi: 10.2105/AJPH.2005.074294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Triunfo S, Lanzone A. Impact of overweight and obesity on obstetric outcomes. J Endocrinol Invest. 2014 doi: 10.1007/s40618-014-0058-9. Forthcoming. [DOI] [PubMed] [Google Scholar]
- 63.West AA, Caudill MA. Applied Choline-Omics: Lessons from Human Metabolic Studies for the Integration of Genomics Research into Nutrition Practice. J Acad Nutr Diet 2014 [DOI] [PubMed] [Google Scholar]
- 64.Jiang X, Yan J, West AA, Perry CA, Malysheva OV, Devapatla S, Pressman E, Vermeylen F, Caudill MA. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012;26:3563–74. doi: 10.1096/fj.12-207894. [DOI] [PubMed] [Google Scholar]
- 65.Takaya J, Iharada A, Okihana H, Kaneko K. Magnesium deficiency in pregnant rats alters methylation of specific cytosines in the hepatic hydroxysteroid dehydrogenase-2 promoter of the offspring. Epigenetics. 2011;6:573–8. doi: 10.4161/epi.6.5.15220. [DOI] [PubMed] [Google Scholar]
- 66.Loke YJ, Galati JC, Morley R, Joo EJ, Novakovic B, Li X, Weinrich B, Carson N, Ollikainen M, Ng HK, et al. Association of maternal and nutrient supply line factors with DNA methylation at the imprinted IGF2/H19 locus in multiple tissues of newborn twins. Epigenetics. 2013;8:1069–79. doi: 10.4161/epi.25908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Belkacemi L, Jelks A, Chen CH, Ross MG, Desai M. Altered placental development in undernourished rats: role of maternal glucocorticoids. Reprod Biol Endocrinol. 2011;9:105. doi: 10.1186/1477-7827-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bertram C, Trowern AR, Copin N, Jackson AA, Whorwood CB. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11beta-hydroxysteroid dehydrogenase: potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology. 2001;142:2841–53. doi: 10.1210/endo.142.7.8238. [DOI] [PubMed] [Google Scholar]
- 69.Langley-Evans SC, Gardner DS, Jackson AA. Maternal protein restriction influences the programming of the rat hypothalamic-pituitary-adrenal axis. J Nutr. 1996;126:1578–85. doi: 10.1093/jn/126.6.1578. [DOI] [PubMed] [Google Scholar]
- 70.Whorwood CB, Donovan SJ, Wood PJ, Phillips DI. Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J Clin Endocrinol Metab. 2001;86:2296–308. doi: 10.1210/jcem.86.5.7503. [DOI] [PubMed] [Google Scholar]
- 71.Whorwood CB, Firth KM, Budge H, Symonds ME. Maternal undernutrition during early to midgestation programs tissue-specific alterations in the expression of the glucocorticoid receptor, 11beta-hydroxysteroid dehydrogenase isoforms, and type 1 angiotensin ii receptor in neonatal sheep. Endocrinology. 2001;142:2854–64. doi: 10.1210/endo.142.7.8264. [DOI] [PubMed] [Google Scholar]
- 72.Garbrecht MR, Lamb FS. Placental HSD2 Expression and Activity Is Unaffected by Maternal Protein Consumption or Gender in C57BL/6 Mice. ISRN Endocrinol. 2013;2013:867938. doi: 10.1155/2013/867938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sferruzzi-Perri AN, Vaughan OR, Haro M, Cooper WN, Musial B, Charalambous M, Pestana D, Ayyar S, Ferguson-Smith AC, Burton GJ, et al. An obesogenic diet during mouse pregnancy modifies maternal nutrient partitioning and the fetal growth trajectory. FASEB J. 2013;27:3928–37. doi: 10.1096/fj.13-234823. [DOI] [PubMed] [Google Scholar]
- 74.Mao J, Zhang X, Sieli PT, Falduto MT, Torres KE, Rosenfeld CS. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc Natl Acad Sci U S A. 2010;107:5557–62. doi: 10.1073/pnas.1000440107. [DOI] [PMC free article] [PubMed] [Google Scholar]