

Abstract
In mammalian genomes, the methylation of cytosine residues within CpG dinucleotides is crucial to normal development and cell differentiation. However, methylation of cytosines in the contexts of CpA, CpT, and CpC (non-CpG methylation) has been reported for decades, yet remains poorly understood. In recent years, whole genome bisulphite sequencing (WGBS) has confirmed significant levels of non-CpG methylation in specific tissues and cell types. Non-CpG methylation has several properties that distinguish it from CpG methylation. Here we review the literature describing non-CpG methylation in mammalian cells, describe the important characteristics that distinguish it from CpG methylation, and discuss its functional importance.
Keywords: DNA methylation, non-CpG methylation, non-CG methylation, epigenetics
Introduction
DNA methylation involves the addition of a methyl group to the 5th carbon atom of cytosine to create 5-methylcytosine. In most mammalian cells, the majority of 5-methylcytosine is found immediately preceding (5′) guanine residues and is referred to as CpG methylation. The mammalian genome is interspersed with regions of high CpG density, known as CpG islands (CGIs), which overlap the promoter regions of ~70% of human genes.1
DNA methylation has also been described in non-CpG contexts, such as CpA, CpT, and CpC, which will be collectively referred to as non-CpG methylation throughout this review. First described in the plant genome,2 non-CpG methylation has since been found independent to, or coexisting with, CpG methylation in various contexts within mammalian genomes. Non-CpG methylation occurs within various cell types and at specific stages during cell development; however, the functional significance of this in the mammalian genome is poorly understood.
In this review, we will describe evidence for the existence of non-CpG methylation, specifically in mammalian genomes, and discuss the differences between CpG and non-CpG methylation, possible mechanisms of its establishment and maintenance, and the potential functions of this DNA modification.
The function of CpG Methylation in Mammalian Cells
The function of CpG methylation is context-dependent3; however, it is most clearly understood as a mechanism of controlling gene expression. Changes in CpG methylation, histone modifications and nucleosome positioning can alter promoter DNA accessibility and regulate the recruitment of DNA binding proteins, such as transcription factors. CpG methylation can also repress transcription by directly blocking the binding of transcription activating proteins.4,5 Although CpG methylation at promoters is associated with transcriptional silencing, high levels of methylation within the gene body are associated with highly expressed genes.6 The enrichment of CpG methylation within gene bodies, particularly at exonic regions, may contribute to exonic recognition and splicing by promoting the pausing of the RNA polymerase II complex at exons and increasing the probability of co-transcriptional splicing.7,8 Other functions of CpG methylation include the regulation of interactions between enhancers and promoters through the regulation of specific DNA-protein interactions. For example, nucleosome occlusion and hypermethylation of the distal enhancers of the NANOG/OCT4 and glucocorticoid receptor promoters prevents them from activating these genes.9,10 The expression of the imprinted IGF2 gene is regulated by methylation at sites within the IGF2-H19 imprinted locus, which prevents binding of the CTCF insulator and allows interaction of the IGF2 gene promoter with its enhancer.11 CpG methylation is also important for genomic imprinting in other regions of the genome, as well as the transcriptional silencing of transposable elements, the control of tissue-specific gene expression patterns,3 and X-chromosome inactivation.12
Evidence for non-CpG Methylation in Mammalian Cells
Though reported in the literature for many years,13 it was not until the development of WGBS that accurate base pair resolution maps were generated and the extent of non-CpG methylation in mammalian cells could be fully appreciated. Non-CpG methylation has now been described in stem cells and appears to be enriched in pluripotent cell types compared with most differentiated cell types. Studies comparing human embryonic stem cells (hESCs) and human fibroblast cell lines have revealed that non-CpG methylation, primarily at CpA sites, accounts for around 25% of all methylated cytosines in hESCs but is virtually absent in fibroblast cell lines.6,14,15 The relative abundance of non-CpG methylation in stem cells compared with differentiated cells was confirmed in a subsequent study comparing 76 genome-scale methylation maps across 32 distinct pluripotent cell lines and 10 differentiated cell types.16 However, recent reports show that non-CpG methylation is not restricted to pluripotent cells. For instance, non-CpG methylation is particularly abundant in mouse and human brain tissue.17,18 Analysis of human adult brain tissue (specifically the dorsolateral prefrontal cortex of healthy donors) showed that 666/2466 non-CpG dinucleotides tested across the genome were methylated across all 24 individuals tested.18
An interesting characteristic of non-CpG methylation is that it can occur in a variety of sequence contexts, but is most frequently found at CpA dinucleotides. In the human embryonic cell line HUES48, mean global levels of CpG, CpA, CpT, and CpC methylation were 67.85%, 6.68%, 1.48%, and 0.63% of all CpG, CpA, CpT, and CpC dinucleotides tested respectively.16 This trend in the frequency of methylation at each dinucleotide (CpG > CpA > CpT > CpC) has been described in all WGBS studies to date.6,14,17,19 Surrounding sequence context and cell type may also be important in determining non-CpG methylation. For example, in human and mouse brain, CpApC sites were most frequently methylated17,18 while in hESCs it was CpApG sites.6,16,18 In mouse germinal vesicle oocytes (GVOs), 65.5% of all methylcytosines occur in a non-CpG context, with TAmCA(G/C)C (where m indicates methylcytosine) representing more than 50% of all non-CpG methylation.19
Methylation of CpG sites is usually symmetrical, meaning that a single CpG:GpC site is methylated at cytosine residues on both DNA strands. Due to sequence asymmetry, this is not the case with non-CpG methylation; for example, CpA dinucleotides are paired with complimentary TpG dinucleotides on the opposite strand (CpA:TpG). Consequently, non-CpG methylation is often only present on one DNA strand at any given site. WGBS from mouse GVOs has shown that 98% of methylated CpG sites were symmetrically methylated, whereas only 11% of methylated CpApG sites were methylated on both strands.19
Non-CpG methylation has also been found in various pathological contexts. Four independent studies analyzing breast cancer, prostate cancer, and brain lymphoma have reported methylation of non-CpG sites (mostly CpNpG sites, where N indicates A, C, T, or G) in the CDKN2A promoter.20-23 Non-CpG methylation has been reported at other important cancer-related genes, such as GSTP124 and TP5325 in endometrial and lung cancer specimens, respectively. Whether non-CpG methylation is a contributing factor or a consequence of these diseases is currently unclear.
Measurement of Non-CpG Methylation
Non-CpG methylation can be detected by either bisulphite sequencing or methylation sensitive restriction endonuclease (MSRE)-based methods. Bisulphite DNA conversion exploits the different sensitivities of cytosine and 5-methylcytosine to undergo deamination by bisulphite under acidic conditions.26 This results in the conversion of cytosine to uracil, whereas 5-methylcytosine remains non-reactive. This conversion creates non-complimentary strands that must be amplified by PCR using separate pairs of primers (Fig. 1A). Alternatively, hairpin-bisulphite PCR can identify sites of 5-methylcytosine on complementary strands of restriction enzyme-fragmented DNA by linking them with a hairpin oligonucleotide prior to bisulphite conversion.27 This method has successfully been used to investigate the symmetry of CpG and non-CpG methylation in mESCs.28 Detection of non-CpG methylation following PCR of bisulphite converted DNA and sequencing is possible, but requires careful design of primer sequences. When designing PCR primers for bisulphite DNA it is usually assumed that cytosines within non-CpG dinucleotides are converted to uracil; however, this creates a bias for amplification of DNA that is unmethylated at non-CpG sites (Fig. 1B). Also, non-CpG methylation confined to one DNA strand will not be detected if primers are specific for the unmethylated strand (Fig. 1C). Also, nested PCR amplification may cause an underestimation of non-CpG methylation29 and must be avoided. A final consideration is that bisulphite sequencing provides base pair resolution data of DNA methylation; however, incomplete bisulphite conversion is a possible source of false positive methylation. The efficiency of bisulphite conversion can be calculated after spiking samples with unmethylated lambda phage DNA.19 In addition to this control, consistent enrichment of non-CpG methylation at specific sites between technical replicates, allele-specific methylation and inducible methylation suggests the presence of genuine non-CpG methylation.
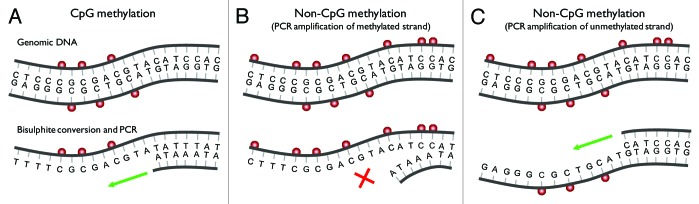
Figure 1. Bisulphite PCR can cause underestimation of the levels of non-CpG methylation. (A) A short region of genomic DNA with symmetrical CpG methylation (red spheres). Bisulphite converts cytosine to uracil (recognized as thymine during PCR), whereas 5-methylcytosine remains non-reactive. The sequence is then amplified (green arrow) using primers specific to the converted sequence. Primers are usually designed assuming that all cytosine in a non-CpG dinucleotide context are converted to uracil (and hence unmethylated). (B) The same region of DNA with additional methylation at non-CpG sites. Methylation at non-CpG dinucleotides within the binding site of the primer will result in protection from bisulphite conversion and loss of primer complementarity. Under these circumstances all molecules containing non-CpG methylation fail to amplify (red cross) and are not detected. (C) Non-CpG methylation, which is inherently asymmetrical, will remain undetected following PCR amplification (green arrow) using primers specific to the unmethylated strand.
Methylcytosine capture assays utilizing proteins such as MBD2 and MeCP2, or the use of antibodies specific for 5-methylcytidine, provide a means of enriching methylated DNA for analysis. Although currently there is a lack of information regarding the affinity of MBD2 and MeCP2 for non-CpG methylation, a previous study has shown that non-CpG methylation can be identified using antibodies specific for 5-methylcytidine.30 While the majority of methylation detected using these affinity-based methods is likely to be within a CpG context, these methods may also detect non-CpG methylation.
MSRE-based methods utilize enzymes that cannot digest DNA when specific cytosine residues within their recognition sequence are methylated. For example, MspI is inhibited by methylation of the first cytosine in its recognition sequence (mCCGG). The combination of MSRE digestion of DNA with quantitative PCR using primers that flank the restriction site allows quantification of methylation by comparing the PCR signals obtained before and after digestion.31 This relatively inexpensive method only measures methylation at the recognition sites of the restriction enzyme used. The luminometric-based assay (LUMA), which also uses MSREs, was recently adapted to detect non-CpG methylation in human and rodent tissues.29 This technique simultaneously assesses methylation at particular restriction enzyme sites across the genome; however, it is unable to identify specific positions in the genome where methylation is located.
Establishment and Maintenance of Non-CpG Methylation
In mammalian cells DNA methylation is catalyzed by a family of DNA methyltransferases (DNMTs) known as DNMT1, DNMT3A, and DNMT3B.32 DNMT1 acts as the ‘maintenance’ methyltransferase, so-called due to its preference for hemimethylated DNA, which is generated following DNA replication. This provides a mechanism by which CpG methylation is somatically heritable following cell division (Fig. 2A and B). DNMT3A and DNMT3B, often referred to as ‘de novo’ methyltransferases, are responsible for establishing patterns of DNA methylation in the early embryo.33 DNMT3L, which lacks methyltransferase activity, is essential for the establishment of genomic imprints in oocytes and for the silencing of retrotransposons within repeat sequences in male germ cells.32 In vitro substrate specificity of DNMT3A using oligonucleotides suggest this enzyme shows the substrate preference CpG > CpA > CpT,34,35 which matches the relative abundance of methylation at these sites in mammalian cells. The presence of non-CpG methylation is tightly linked to the expression of DNMT3A, DNMT3B, and DNMT3L, and it has been proposed that non-CpG methylation is a result of sustained expression of these proteins, particularly in non-dividing cells such as neurons and oocytes.17,18
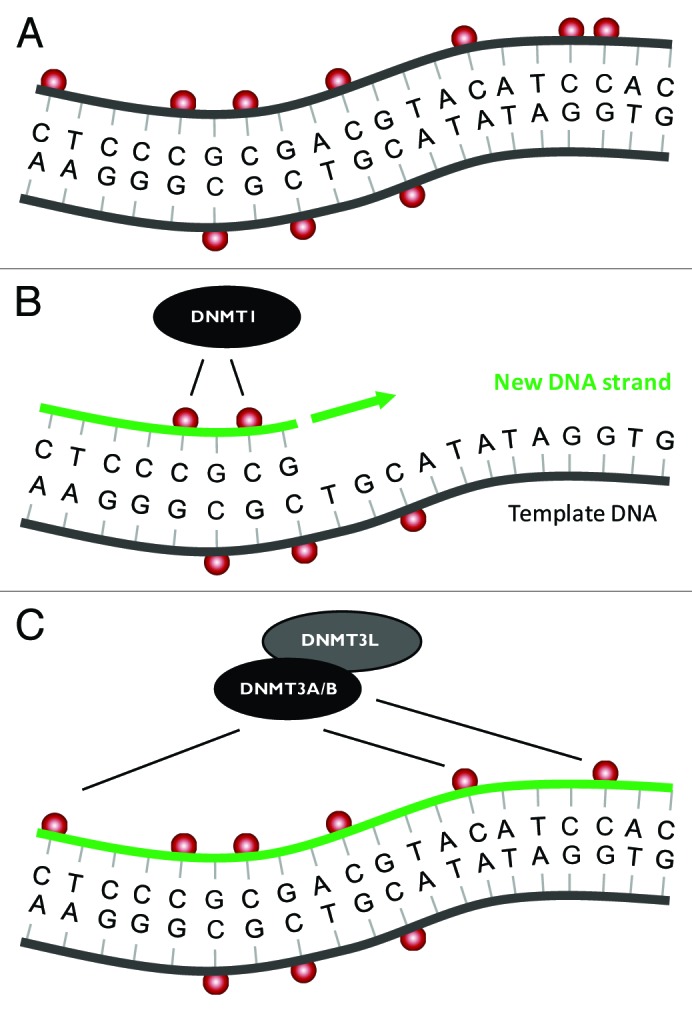
Figure 2. Maintenance of CpG and non-CpG methylation. (A) A short region of genomic DNA containing several sites of CpG methylation and non-CpG methylation (red spheres). (B) During DNA replication the ‘maintenance methyltransferase’ DNMT1 reinstates CpG methylation on the newly synthesized DNA strand (green) guided by CpG methylation on the template DNA strand. (C) Following DNA replication the ‘de novo methyltransferases’ DNMT3A and DNMT3B, in complex with DNMT3L, re-establishes non-CpG methylation.
In vivo, studies using Dnmt1 knockout mouse ESCs have shown non-CpG methylation patterns can be maintained independently of Dnmt1.35 Loss of Dnmt3l in mice reduced CpA methylation in prospermatogonia from 15.2% to 2.8% of total methylation levels.36 Barres et al. found that DNMT3B depletion caused reduced non-CpG hypermethylation at the PGC-1α promoter in human primary muscle cells.30 Mouse GVOs lacking Dnmt3a or Dnmt3l show global reductions in both CpG and non-CpG methylation.19 Finally, loss of either Dnmt3l, Dnmt3a, Dnmt3b, or both Dnmt3a and Dnmt3b in ESCs revealed that non-CpG methylation was mediated by Dnmt3a and Dnmt3b, depends on the presence of Dnmt3L and is strongly correlated with the methylation of flanking CpG positions.28 Collectively, these studies show that non-CpG methylation is attributable to the de novo activity of the DNMT3A/B-DNMT3L complex. This, coupled with the fact that non-CpG methylation is asymmetrical, indicates that there is unlikely to be a mechanism of maintenance methylation at non-CpG sites. Therefore, in contrast to CpG methylation, it is likely that non-CpG methylation would need to be re-established de novo after each cell division in order to be maintained (Fig. 2C). Consistent with this hypothesis, non-CpG methylation accumulates in non-dividing male mouse germ cells but is rapidly lost following the resumption of cell division.36
The Function of Non-CpG Methylation
Reports describing the effects of non-CpG methylation on gene expression provide the most compelling evidence that it has a functional role in mammalian cells. Methylation of CpmCpNpGpG sites within the promoter of the B29 gene has been shown to repress promoter activity in human B cells by directly blocking the binding of the early B cell factor (EBF) transcription factor.37 Non-CpG methylation of Sp transcription factor binding sites within the human SYT11 promoter region reduced the binding of Sp proteins and associated transcriptional activity.38 Other genes shown to be regulated by non-CpG methylation include PGC-1α and PDK4,30,39 which are involved in mitochondrial function and fuel utilization. A genome-scale promoter methylation analysis identified differential non-CpG methylation within the PGC-1α promoter in skeletal muscle from type II diabetes mellitus (T2DM) patients.30 This methylation negatively correlated with PGC-1α expression and was acutely inducible by exposure to systemic factors associated with insulin resistance such as tumor necrosis factor-α (TNF-α) or the free fatty acids palmitate and oleate.30 The direct effect of non-CpG methylation on promoter activity was confirmed using promoter reporter constructs containing a single methylated CpC site located -112 bp from the PDK4 TSS. Methylation of this site alone resulted in ~20% reduced promoter activity.39 In the skeletal muscle of obese individuals, non-CpG methylation levels within the PGC-1α gene promoter positively correlated with markers of obesity including body mass index, C-reactive protein and leptin hormone levels, whereas non-CpG methylation at the PDK4 promoter negatively correlated with these parameters. Furthermore, non-CpG methylation in the PGC-1α and PDK4 promoters was restored to non-obese levels following gastric bypass surgery and weight loss.39 Also, acute bouts of exercise cause transient reductions in the levels of non-CpG methylation at the PGC-1α promoter, concomitant with increased PGC-1α expression.40 Interestingly, a study of mitochondrial DNA methylation has also reported that ~50% of methylcytosine within the D-loop region, which contributes to the regulation of mitochondrial DNA replication and transcription, was within non-CpG dinucleotides.41 Collectively, these studies provide evidence that non-CpG methylation at gene promoters is associated with reduced gene expression. As discussed above, several lines of evidence implicate non-CpG methylation in the regulation of mitochondrial function. Finally, the observation that non-CpG methylation can be regulated by TNF-α, free fatty acids or exercise suggest it may represent a mechanism of acutely regulating gene expression in response to environmental changes.
Because non-CpG methylation is relatively frequent in pluripotent stem cells, several studies have investigated how levels change when cells are reprogrammed to produce induced pluripotent stem cells (iPSCs).15,16 These studies demonstrated that non-CpG methylation reappears upon reprogramming. The role of non-CpG methylation in pluripotency has important implications for regenerative medicine using stem cells. However, it is still unclear whether non-CpG methylation is a cause or a consequence of the pluripotent state. One study suggests non-CpG methylation patterns are independent from pluripotency-associated gene expression since depletion of the de novo methyltransferase DNMT3A in the hESC line HUES48 failed to cause significant changes to the expression of key pluripotency genes such as OCT4, NANOG, and SOX2.16 However, in this study CpA methylation levels were reduced by only 28% following DNMT3A depletion, and it was not determined whether DNMT3B functionally compensated for DNMT3A loss. Although studies have shown that cell reprogramming is associated with DNA methylation changes that reflect ESCs,42 Lister and colleagues have identified 22 separate megabase-scale differentially methylated regions (DMRs) that were hypomethylated in iPSCs compared with ESCs.15 These findings suggest there are hotspots of incomplete reprogramming of non-CpG methylation in iPSCs, which may have important ramifications for the validity of using these cells in clinical applications, such as regenerative medicine.
Non-CpG methylation may also play an important role in imprinting. Several studies have revealed that in mouse GVOs, non-CpG methylation is prevalent at maternally methylated imprint control regions.43,44 In mouse adult brain tissue, parent-of-origin-dependent non-CpG methylation was associated with transcriptionally repressed alleles at a further eight imprinted loci.17
There has also been a report implicating non-CpG methylation in regulating interchromosomal interactions between an enhancer element and odorant receptor (OR) genes within olfactory sensory neurons.45 The expression of a single OR gene within each olfactory sensory neuron is an essential feature in the organization and function of the olfactory system.45 Using chromosome conformation capture (3C) techniques, Lomvardas et al. identified a trans-acting enhancer region, known as the H element, on human chromosome 14 that was involved in the activation of only one OR allele in each neuron. In OR expressing cells CpA methylation was detected on one allele of the H enhancer whereas this methylation was absent in OR negative cells. The authors of this study speculated that non-CpG methylation may render one of the enhancer alleles non-functional, which would ensure the necessary expression of only one OR gene in each olfactory sensory neuron.45
Conclusions
There is now strong evidence for the existence of non-CpG methylation in specific tissues and cell types in mammals. Though the functional relevance of non-CpG methylation at most sites remains unknown, it appears to regulate the expression of at least some genes when present at promoters. However, the most intriguing characteristics of non-CpG methylation are potentially those that distinguish it from CpG methylation. Non-CpG methylation is prevalent only in specific tissues and cell types, or at particular regions of the genome. It is particularly prevalent in infrequently dividing cells such as neurons or skeletal muscle, or in cells with a long developmental hiatus such as gametes. Non-CpG methylation is inherently asymmetrical, which precludes it from the maintenance mechanisms that preserve CpG methylation through cell division. Consequently, non-CpG methylation must be established de novo after each cell division. This feature of non-CpG methylation most likely explains its accumulation in non-dividing cells and scarcity in highly proliferative cells. Acute regulation of non-CpG methylation by acute bouts of exercise or specific growth factors and lipids suggests it may be a mechanism of regulating DNA function in response to environmental factors. Finally, the prevalence of non-CpG methylation in stem cells, as well as its reinstatement upon reprogramming suggests it could be important for pluripotency, but it is currently unclear whether it is a cause or consequence of the pluripotent state.
The features of non-CpG methylation, as outlined above, have made this DNA modification difficult to study functionally. Previous advances in our understanding of the function of CpG methylation were made by generating model systems lacking functional DNMT enzymes.46,47 However, the same enzymes are required for both CpG and non-CpG methylation, therefore, it is impossible to specifically target non-CpG methylation without also significantly altering CpG methylation. In addition, CpG and non-CpG methylation are often coincident and it is difficult to extrapolate the biological effects of one vs. the other. However, the growing number of reports describing non-CpG methylation in various contexts highlights the need to uncover its functional role in mammalian cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Neil Youngson, Mathew Sloane, and Peter Zarzour for helpful discussions and careful reading of the manuscript. V.P. is supported by an Australian Postgraduate Award. R.L.W. is supported by funding from the Cancer Council NSW (http://www.cancercouncil.com.au) and Cancer Australia (http://canceraustralia.gov.au). L.B.H. is supported by a Cancer Institute New South Wales Career Development Fellowship (Grant number: 09CDF226; http://www.cancerinstitute.org.au).
Glossary
Abbreviations:
- B29
Immunoglobin β-Chain
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- CGI’s
CpG islands
- CTCF
CCCTC-binding factor
- DNMT
DNA methyltransferase
- EBF
early B-cell factor
- ESCs
embryonic stem cells
- GSTP1
glutathione S-transferase pi 1
- GVOs
germinal vesicle oocytes
- H19
imprinted maternally expressed transcript
- hESCs
human embryonic stem cells
- IGF2
insulin-like growth factor 2
- iPSCs
induced pluripotent stem cells
- LUMA
luminometric-based assay
- MSRE
methylation sensitive restriction endonuclease
- PDK4
pyruvate dehydrogenase kinase, isozyme 4
- OR
odourant receptor
- PGC-1α
peroxisome proliferator-activated receptor gamma co-activator-1 α
- SOX2
sex determining region Y-box 2
- SYT11
synaptotagmin XI
- TNF-α
tumor necrosis factor-α
- TP53
tumor protein p53
- T2DM
type 2 diabetes mellitus
- WGBS
whole genome bisulphite sequencing
References
- 1.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–7. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, Jacobsen SE. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science. 2001;292:2077–80. doi: 10.1126/science.1059745. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 4.Marx A, Kahan T, Simon I. Integrative analysis of methylome and transcriptome reveals the importance of unmethylated CpGs in non-CpG island gene activation. Biomed Res Int. 2013;2013:785731. doi: 10.1155/2013/785731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, Kerr AR, Deaton A, Andrews R, James KD, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–6. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–92. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–9. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.You JS, Kelly TK, De Carvalho DD, Taberlay PC, Liang G, Jones PA. OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc Natl Acad Sci U S A. 2011;108:14497–502. doi: 10.1073/pnas.1111309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiench M, John S, Baek S, Johnson TA, Sung MH, Escobar T, Simmons CA, Pearce KH, Biddie SC, Sabo PJ, et al. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30:3028–39. doi: 10.1038/emboj.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–5. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 12.Riggs AD. X chromosome inactivation, differentiation, and DNA methylation revisited, with a tribute to Susumu Ohno. Cytogenet Genome Res. 2002;99:17–24. doi: 10.1159/000071569. [DOI] [PubMed] [Google Scholar]
- 13.Woodcock DM, Crowther PJ, Diver WP. The majority of methylated deoxycytidines in human DNA are not in the CpG dinucleotide. Biochem Biophys Res Commun. 1987;145:888–94. doi: 10.1016/0006-291X(87)91048-5. [DOI] [PubMed] [Google Scholar]
- 14.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, Ren B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–31. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23:555–67. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirane K, Toh H, Kobayashi H, Miura F, Chiba H, Ito T, Kono T, Sasaki H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 2013;9:e1003439. doi: 10.1371/journal.pgen.1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodcock DM, Linsenmeyer ME, Doherty JP, Warren WD. DNA methylation in the promoter region of the p16 (CDKN2/MTS-1/INK4A) gene in human breast tumours. Br J Cancer. 1999;79:251–6. doi: 10.1038/sj.bjc.6690041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen NH, Roos G, Emdin SO, Landberg G. Methylation of the p16(Ink4a) tumor suppressor gene 5′-CpG island in breast cancer. Cancer Lett. 2001;163:59–69. doi: 10.1016/S0304-3835(00)00674-1. [DOI] [PubMed] [Google Scholar]
- 22.Kinoshita H, Shi Y, Sandefur C, Meisner LF, Chang C, Choon A, Reznikoff CR, Bova GS, Friedl A, Jarrard DF. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res. 2000;60:3623–30. [PubMed] [Google Scholar]
- 23.Zhang SJ, Endo S, Ichikawa T, Washiyama K, Kumanishi T. Frequent deletion and 5′ CpG island methylation of the p16 gene in primary malignant lymphoma of the brain. Cancer Res. 1998;58:1231–7. [PubMed] [Google Scholar]
- 24.Chan QK, Khoo US, Chan KY, Ngan HY, Li SS, Chiu PM, Man LS, Ip PP, Xue WC, Cheung AN. Promoter methylation and differential expression of pi-class glutathione S-transferase in endometrial carcinoma. J Mol Diagn. 2005;7:8–16. doi: 10.1016/S1525-1578(10)60003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kouidou S, Agidou T, Kyrkou A, Andreou A, Katopodi T, Georgiou E, Krikelis D, Dimitriadou A, Spanos P, Tsilikas C, et al. Non-CpG cytosine methylation of p53 exon 5 in non-small cell lung carcinoma. Lung Cancer. 2005;50:299–307. doi: 10.1016/j.lungcan.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1:2353–64. doi: 10.1038/nprot.2006.324. [DOI] [PubMed] [Google Scholar]
- 27.Laird CD, Pleasant ND, Clark AD, Sneeden JL, Hassan KM, Manley NC, Vary JC, Jr., Morgan T, Hansen RS, Stöger R. Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc Natl Acad Sci U S A. 2004;101:204–9. doi: 10.1073/pnas.2536758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A, Jenuwein T, Xu G, Leonhardt H, Wolf V, et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012;8:e1002750. doi: 10.1371/journal.pgen.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan J, Zierath JR, Barrès R. Evidence for non-CpG methylation in mammals. Exp Cell Res. 2011;317:2555–61. doi: 10.1016/j.yexcr.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Barrès R, Osler ME, Yan J, Rune A, Fritz T, Caidahl K, Krook A, Zierath JR. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009;10:189–98. doi: 10.1016/j.cmet.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Nygren AO, Ameziane N, Duarte HM, Vijzelaar RN, Waisfisz Q, Hess CJ, Schouten JP, Errami A. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005;33:e128. doi: 10.1093/nar/gni127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12:206–22. doi: 10.1002/cbic.201000195. [DOI] [PubMed] [Google Scholar]
- 33.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. doi: 10.1126/science.1111098. [DOI] [PubMed] [Google Scholar]
- 34.Gowher H, Jeltsch A. Enzymatic properties of recombinant dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-CpA sites (vol 309, pg 1201, 2001) J Mol Biol. 2001;310:951. doi: 10.1006/jmbi.2001.4837. [DOI] [PubMed] [Google Scholar]
- 35.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–42. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2013;41:738–45. doi: 10.1093/nar/gks1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malone CS, Miner MD, Doerr JR, Jackson JP, Jacobsen SE, Wall R, Teitell M. CmC(A/T)GG DNA methylation in mature B cell lymphoma gene silencing. Proc Natl Acad Sci U S A. 2001;98:10404–9. doi: 10.1073/pnas.181206898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue S, Oishi M. Effects of methylation of non-CpG sequence in the promoter region on the expression of human synaptotagmin XI (syt11) Gene. 2005;348:123–34. doi: 10.1016/j.gene.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 39.Barres R, Kirchner H, Rasmussen M, Yan J, Kantor FR, Krook A, Näslund E, Zierath JR. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013;3:1020–7. doi: 10.1016/j.celrep.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 40.Barrès R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, Caidahl K, Krook A, O’Gorman DJ, Zierath JR. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–11. doi: 10.1016/j.cmet.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Bellizzi D, D’Aquila P, Scafone T, Giordano M, Riso V, Riccio A, Passarino G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013;20:537–47. doi: 10.1093/dnares/dst029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 43.Tomizawa S, Kobayashi H, Watanabe T, Andrews S, Hata K, Kelsey G, Sasaki H. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development. 2011;138:811–20. doi: 10.1242/dev.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R. Interchromosomal interactions and olfactory receptor choice. Cell. 2006;126:403–13. doi: 10.1016/j.cell.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 46.Takebayashi S, Tamura T, Matsuoka C, Okano M. Major and essential role for the DNA methylation mark in mouse embryogenesis and stable association of DNMT1 with newly replicated regions. Mol Cell Biol. 2007;27:8243–58. doi: 10.1128/MCB.00899-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–3. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]