

Abstract
Background
Most asthma begins in the first years of life. This early onset cannot be merely attributed to genetic factors, because the prevalence of asthma is increasing. Epidemiological studies have indicated roles for prenatal and early childhood exposures, including exposure to diesel exhaust. However, little is known about the mechanisms. This is largely due to paucity of animal models.
Objective
We aimed to develop a mouse model of asthma susceptibility through prenatal exposure to diesel exhaust.
Methods
Pregnant C57BL/6 female mice were given repeated intranasal applications of diesel exhaust particles (DEP) or phosphate-buffered saline (PBS). Offspring underwent suboptimal immunization and challenge with ovalbumin (OVA) or received PBS. Pups were examined for features of asthma; lung and liver tissues were analyzed for transcription of DEP-regulated genes.
Results
Offspring of mice exposed to DEP were hypersensitive to OVA, indicated by airway inflammation and hyperresponsiveness, increased serum levels of OVA-specific IgE, and increased levels of pulmonary and systemic T-helper (Th)2 and Th17 cytokines. These cytokines were primarily produced by natural killer (NK) cells. Antibody-mediated depletion of NK cells prevented airway inflammation. Asthma susceptibility was associated with increased transcription of genes known to be specifically regulated by the aryl hydrocarbon receptor (AhR) and oxidative stress. Features of asthma were either marginal or absent in OVA-treated pups of PBS-exposed mice.
Conclusion
We created a mouse model that linked maternal exposure to DEP with asthma susceptibility in offspring. Development of asthma was dependent on NK cells and associated with increased transcription from AhR- and oxidative stress-regulated genes.
Keywords: prenatal exposure, diesel exhaust particles, asthma, mouse model, natural killer cells, aryl hydrocarbon receptor, interleukin 5, interleukin 13, interleukin 17
Introduction
Over the last several decades, the prevalence of asthma has continuously increased. In the last 10 y, the prevalence of asthma in the United States has increased from 7.3% (20.3 million persons in 2001) to 8.4% (25.7 million persons in 2010)1. This recent rise in prevalence implicates industrialization and urbanization-generated environmental exposures in disease pathogenesis. Prenatal and early childhood exposures are likely to have the highest impact as they occur in periods of intense developmental programming and thereby have the potential to induce long-term memory in cells, systems and organs. The role of early programming is underscored by the fact that asthma symptoms, in most cases, start in the first years of childhood. The argument for prenatal/maternal influences is provided by the observation on strong association of childhood asthma with maternal asthma2–4. Among children less than 5 y old, the risk of asthma is more than 3-fold greater for those with mothers with asthma than fathers with asthma.2 One of the plausible explanations is that offspring predisposition to asthma is shaped prenatally, by an altered intrauterine environment. This alteration of the intrauterine environment is imposed by maternal disease and/or disease-triggering maternal exposures. In support of the latter hypothesis, there are many epidemiological studies showing an association between various in utero exposures and asthma susceptibility5–12. Among prenatal insults with linkage to asthma, solid and consistent epidemiological evidence has been provided for exposure to traffic-related pollution, including diesel exhaust5–8. Mothers who lived near highways during pregnancy are more likely to have children with asthma5. Prenatal exposure to polycyclic aromatic hydrocarbons (PAH), which are diesel exhaust-derived toxins, is associated with increased risk of allergic sensitization and early childhood wheeze6,8.
Although epidemiological data supports the hypothesis on prenatal origins of asthma, the mechanistic understanding is still very poor. There are many obstacles to conducting mechanistic studies during pregnancy and infancy. Intentional exposures of pregnant women are unethical. Studies of infants have been limited by scant size of biological samples and ethical concerns. Last but not least, only few animal models are available. In regard to prenatal diesel exhaust exposures, the positive link to asthma has been established by three earlier models13–15. We have created a complementary mouse model and provided new mechanistic insights.
Methods
Mouse model
Time-mated C57BL/6 female mice were anaesthetized with isoflurane and given intranasal applications of diesel exhaust particles (DEP, 50 μg, National Institute of Standards and Technology, Gaithersburg, MD; SRM 2975)15–18 in 50 μl PBS (15 mice) or 50 μl of PBS alone (9 mice) on gestation days (GD) 3, 6, 9, 12, 15, and 18. The volume was delivered through 2 sequential injections of 25 μl, 15 min apart, each into a different nostril.
On postnatal day (PND) 5, pups of 10 DEP-exposed mice and 5 PBS-exposed mice (5–8 pups from each mother) were given intraperitoneal injections of 50 μl of the immunizing mixture, which contained OVA (5 μg) and Imject Alum (0.5 mg aluminum hydroxide and 0.5 mg magnesium hydroxide; Thermo Scientific, Rockford, IL) in PBS. Pups from another 5 DEP-exposed mice and 4 PBS-exposed mice were given injections of 50 μl PBS. On PNDs 20, 21, and 22, OVA-immunized offspring of three DEP-exposed mice were given injections of either the anti-NK1.1 antibody or mouse IgG2a isotype (control) (NK cell depletion experiment; see detailed method in the Online Repository). On PNDs 23, 24, and 25, all pups underwent pulmonary challenges. OVA-immunized pups were given intranasal applications of 50 μg of OVA in 15 μl of PBS and PBS-injected pups were given intranasal applications of 15 μl of PBS without OVA, all under isoflurane anesthesia. On PND 27 (22 days after immunization and 2 days after final pulmonary challenge), blood samples were collected (by tail nick) and serum was isolated. FlexiVent studies were conducted on PND 28 (3 days after final pulmonary challenge). A separate set of mice was used to obtain bronchoalveolar lavage fluid (BALF) and lung and liver tissues19,20. For each measured parameter, offspring of 3–7 mice per group were analyzed. All experiments were approved by the institutional animal care and use committee at National Jewish Health. Other methods can be found in the Online Repository.
Results
A mouse model of asthma susceptibility through prenatal exposure to DEP
To develop our mouse model of prenatally-induced asthma susceptibility, we selected the most valuable elements from existing models (protocols by Fedulov et al.13, Auten et al.14, and Reiprich et al.15; see Table EII in the Online Repository). Similar to Auten et al.14, we used C57BL/6 mice. This approach allows an immediate use of genetically-modified mice that are typically on the C57BL/6 background. We also incorporated the principle of repeated maternal exposure, similar to Auten et al.14 and Reiprich et al.15, because human exposure to diesel exhaust is chronic. For maternal challenges, we used DEP, because their pro-asthma activity was proven by all three prenatal exposure protocols and by other studies in adult mice and humans13–15, 21–31. The inflammatory activity of DEP is attributed to components such as PAH, quinones, sulfuric acid, and metal oxides32.
In our model, 50 μg of DEP was repeatedly applied to pregnant C57BL/6 mice via the intranasal route (Fig 1). Control mice received PBS. The DEP dose of 50 μg is commonly used in mouse models, including those by Auten at al. and Fedulov et al 13, 14, and 16. We considered using exposure to titanium dioxide or carbon black particles because they were used in some studies as “control” particles. 13, 33 We chose not to because these particles have specific biologic effects including the effects on fetal development13, 34, and 35. From the model of Fedulov et al., we took the idea of suboptimal immunization of offspring13. The rationale was that standard immunization protocols produce vigorous inflammation, so effects of maternal DEP exposure may become masked. The selected protocol of suboptimal sensitization included a single injection of a low dose of OVA during the neonatal period, when antigen exposure elicits a weak immune response or even immune tolerance36, 37. Intranasal challenge with OVA or PBS was done on PND 23, 24 and 25. Mice were analyzed 72h after the final challenge.
Figure 1. Experimental protocol.
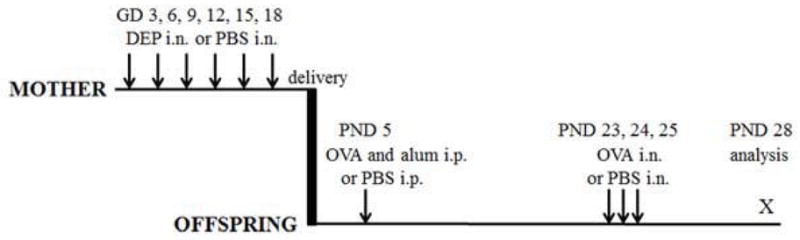
Timed pregnant C57BL/6 mice were given intranasal applications (IN) of either DEP or PBS on indicated gestation days (GD). Offspring were given intraperitoneal injections (IP) of PBS or a mixture of OVA and alum in PBS and then intranasal applications of OVA or PBS on indicated postnatal days (PND). These mice were then analyzed 3 days after the final IN application.
Prenatal exposure to DEP reduces body weight
Exposures had no effect on litter size (5–8 pups from mothers in each group). At the age of 4 weeks, mice exposed prenatally to DEP and then challenged with PBS after birth (DEP-PBS mice) or OVA after birth (DEP-OVA mice) had significantly reduced body weight (12.49±0.34 and 12.27±0.29 g, respectively) compared to mice exposed to PBS prenatally and after birth (PBS-PBS mice) (13.74±0.37 g; P<.01 for both comparisons), but not to mice exposed to PBS prenatally and OVA after birth (PBS-OVA mice) (12.82±0.46 g; P>.05 for both comparisons). There was no significant difference in the body weight between DEP-OVA and DEP-PBS mice, and, between PBS-OVA and PBS-PBS mice. Thus, postnatal exposure to OVA did not have significant effect on the body weight.
In utero exposure to DEP facilitates induction of airway inflammation
DEP-OVA pups developed peribronchial inflammatory infiltrates (Fig 2, A and B). BALF from these mice had increased numbers of cells, including eosinophils, neutrophils, and lymphocytes, compared to BALF from pups of other groups (Fig 2, C–G). Airway inflammation was absent in PBS-PBS, PBS-OVA, and DEP-PBS mice.
Figure 2. Airway inflammation and resistance.

Experiments included PBS-PBS (prenatal and postnatal PBS), PBS-OVA (prenatal PBS, postnatal OVA), DEP-PBS (prenatal DEP, postnatal PBS), DEP-OVA (prenatal DEP, postnatal OVA) mice. A, B, peribronchial inflammation (histologic analysis), mean values (± standard error of the mean - SEM) from 3–6 mice/group; C–G, mean BAL cell counts (± SEM) from 10–21 mice/group; *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; H, total lung resistance, mean values (± SEM) from 9–18 mice/group; ####, DEP-OVA vs. PBS-PBS; */****, DEP-OVA vs. PBS-OVA; ^/^^^^, DEP-OVA vs. DEP-PBS; $, PBS-OVA vs. PBS-PBS.
Prenatal exposure to DEP facilitates development of airway hyperreactivity
Airway response to methacholine is generally weak in pups/young mice, compared with adult mice. Further, baseline airway resistance in pups is high. The baseline airway resistance values for PBS-PBS, PBS-OVA, DEP-PBS, and DEP-OVA mice were high and comparable (2.22±0.22, 2.15±0.14, 2.11±0.17, and 2.02±0.15 cm H2O.s/ml, respectively; P>.05 for all comparisons). Nonetheless, DEP-OVA mice showed augmented airway resistance following exposure to methacholine (Fig 2, H). In addition, a less-pronounced but significant increase in airway resistance was observed in PBS-OVA mice. Airway hyperresponsiveness can occur in the absence of inflammation38. This hyperresponsiveness may be due to activation of airway cells such as mast cells. PBS-OVA mice had some OVA-specific IgE (discussed below; Fig 4), which might activate resident mast cells upon OVA challenge.
Figure 4. OVA-specific IgE in serum.
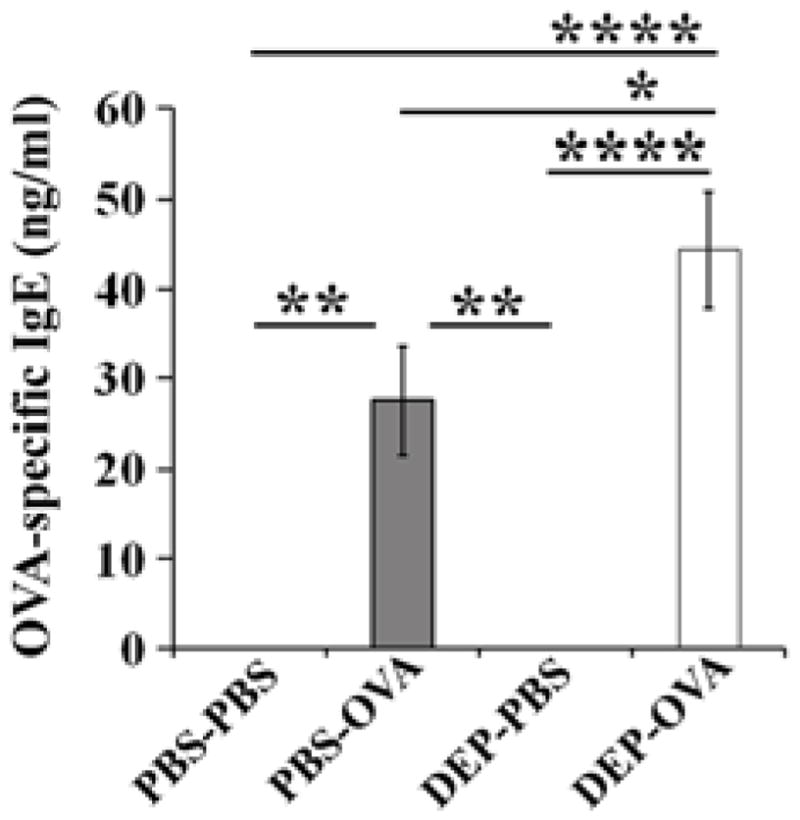
Sera from PBS-PBS, PBS-OVA, DEP-PBS, and DEP-OVA mice were analyzed for concentration of OVA-specific IgE; mean values (± SEM) shown from 20–27 mice/group
Prenatal exposure to DEP primes pulmonary T-helper (Th)2 and Th17-type responses
Lung tissues from DEP-OVA mice had higher levels of mRNAs encoding the Th2 cytokines IL4, IL5, and IL13; the Th17 cytokine IL17; and the pro-inflammatory cytokines IL6 and TNFα, compared with PBS-PBS and PBS-OVA mice (Fig 3, A and Fig E1, A–G in the Online Repository). Levels of Il4, Il5, Il13, Il17, and Tnfα transcripts corresponded with levels of proteins in BALF (Fig 3, B). DEP-OVA mice had reduced expression of the Th1 cytokine IFNγ at the protein level (Fig 3, B). PBS-OVA mice did not have any significant increases in cytokine production, compared with PBS-PBS mice.
Figure 3. Expression of cytokines in lung.

PBS-PBS, PBS-OVA, and DEP-OVA mice were analyzed for relative expression of cytokine transcripts in the lung tissue (A; mean values (± SEM) of 6–13 mice/group) and absolute level of cytokine proteins in BALF (B; mean values (± SEM) of 4–6 mice/group). Levels of each cytokine transcript were normalized to 18S rRNA and expressed as fold change in expression relative to the PBS-PBS group.
Prenatal exposure to DEP increases production of allergen-specific IgE
OVA-specific IgE was detected in serum of all mice immunized with OVA, regardless of prenatal exposure (Fig 4). Thus, our OVA exposure protocol is sufficient to induce OVA-specific IgE, but insufficient to produce pulmonary inflammation in the absence of the prenatal stimulus. Sera from DEP-OVA mice had significantly higher concentrations of the OVA-specific IgE than sera from all other groups of mice.
In utero exposure to DEP promotes production of cytokines by NK cells
The production of IL5, IL13 and IL17 was higher in splenocytes from DEP-OVA mice compared to mice from other studied groups (Fig 5, A–C). In DEP-OVA mice, the majority (>60%) of cytokine-positive cells were NK cells (Fig 5, D–F). In these pups, CD4+ T cells accounted for only 0.9%–1.5% of cytokine-positive cells (Fig 6, D–F). The percentages of NK cells expressing IL5, IL13 and IL17 were highest in DEP-OVA mice and equal to 11%, 16% and 9%, respectively (Fig 6, G–I). DEP-OVA mice had also highest percentage of NK cells expressing the activation marker CD69 (27%; Fig E2 in the Online Repository). Frequencies of NK cells in spleens of mice from studied treatment groups were similar (PBS-PBS mice, 2.65%±0.33%; PBS-OVA mice, 2.55%±0.42%; DEP-OVA mice, 2.49%±0.39%).
Figure 5. Cytokine expression in splenocyte populations.

OVA-stimulated and immunostained splenocytes (CD3, CD4, NK1.1, NKp46 and a cytokine) from PBS-PBS, PBS-OVA, and DEP-OVA mice were analyzed by flow cytometry. CD4 T cells, NK and NKT cells were defined as CD3+CD4+NK1.1−NKp46−, CD3−NK1.1+NKp46+ and CD3+NK1.1+ cells, respectively. A–C, Cytokine+ splenocytes expressed as a percentage of all splenocytes. D–F, Percentages of cytokine+ cells attributable to individual cell populations. G–I, Cytokine+ NK cells expressed as a percentage of all NK cells; mean values (± SEM) shown from 6 mice/group
Figure 6. Effect of NK cell depletion.

Pregnant mice were exposed to DEP and offspring were immunized and challenged with OVA (see Fig 1). Anti-NK1.1 or isotype control IgG were injected 72 hrs, 48 hrs, and 24 hrs before the first OVA challenge. A–B, Peribronchial inflammation (histologic analysis); C–G, BAL cell numbers; mean values (± SEM) shown from 8 mice/group
Depletion of NK cells prevents development of airway inflammation
To study the role of NK cells in our model, OVA-immunized offspring of DEP-exposed mice were given injections of the NK cell-depleting antibody anti-NK1.139–41 (or the isotype control IgG) 72 hrs, 48 hrs, or 24 hrs before the first intranasal OVA challenge. The anti-NK1.1 antibody efficiently depleted NK cells. In spleens of mice injected with the anti-NK1.1 antibody, only 0.11%±0.03% of cells were NK cells (NKp46+CD3−), whereas in spleens of mice injected with the isotype control IgG, 2.53%±0.34% of cells were NKp46+CD3− (P<.01). NK cell depletion significantly reduced airway inflammation (Fig 6, A–G).
Prenatal exposure to DEP affects expression of AhR signature transcripts
From toxicological standpoint, PAH are important components of DEP42, 43. PAH cross the placenta44. Prenatal exposure to PAH increases the risk of allergic sensitization and wheezing in children 6–8. PAH activate the transcriptional factor AhR45. Activated AhR regulates several components of immune response, including the production of IL1746–48, which we found to be increased in DEP-OVA mice. We therefore investigated AhR-dependent transcription in mice exposed to DEP in utero. We measured levels of AhR signature transcripts, encoding: the AhR repressor (AhRR); cytochrome P450, family 1, subfamily A, polypeptide 1 (Cyp1a1); and cytochrome P450, family 1, subfamily B, polypeptide 1 (Cyp1b1)45, 49. We analyzed liver in addition to the lung because liver is an important target for AhR ligands50. Levels of all 3 transcripts were increased in lungs of 4-week old DEP-PBS and DEP-OVA pups (Fig 7A and Fig E3, A–C in the Online Repository). These pups also demonstrated augmented levels of AhRR and Cyp1b1 in livers (Fig 7, B and Fig E3, D and F in the Online Repository). Interestingly, PBS-OVA pups had increased levels of Cyp1a1 mRNA in lung, compared with PBS-PBS mice (Fig 7, A and Fig E3, B in the Online Repository). This may be due to activation of the Cyp1a1 promoter by non-AhR transcription factors in response to OVA exposure. In summary, prenatal exposure to DEP was associated with increased transcription from AhR-regulated genes in the lung and the liver. Interestingly, this result was obtained using 4 week old offspring, thus roughly 4 weeks after final maternal exposure to DEP.
Figure 7. Expression of AhR signature transcripts.

Lungs (A) and livers (B) from PBS-PBS, PBS-OVA, DEP-PBS, and DEP-OVA mice were analyzed for relative levels of AhRR, Cyp1a1, and Cyp1b1 mRNA as in Fig 3; mean values (± SEM) shown from 6–13 mice/group
Prenatal exposure to DEP results in upregulation of oxidative stress signature transcripts
PAH are metabolized via AhR-inducible CYP1A1 and CYP1B1, which leads to generation of reactive oxygen species (ROS)51. ROS are also produced upon exposure to DEP-derived quinones or allergens 52. We measured levels of the ROS-sensitive transcripts heme oxygenase 1 (Hmox1) mRNA and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) mRNA52. We observed moderate upregulation of Hmox1 mRNA in lungs and livers of DEP-OVA pups, and, to a lesser extent, in lungs but not livers of PBS-OVA pups, compared with PBS-PBS mice (Fig 8 and Fig E4 in the Online Repository).
Figure 8. Levels of oxidative stress-regulated transcripts.
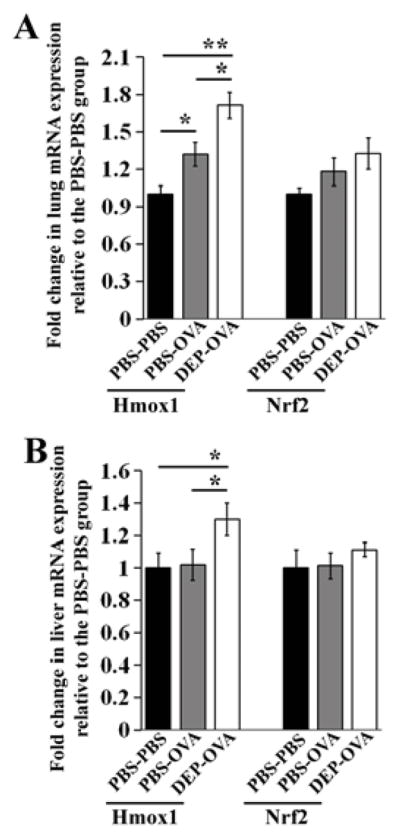
Lungs (A) and livers (B) from PBS-PBS, PBS-OVA, and DEP-OVA mice were analyzed for relative levels of Hmox1 and Nrf2 mRNAs (as in Fig 3); mean values shown from 6–13 mice/group
Discussion
We developed a new mouse model of maternal transmission of asthma susceptibility. In this model, suboptimal OVA immunization and challenge of pups born to DEP-exposed mice resulted in airway hyperresponsiveness and eosinophilic inflammation, with increased expression of Th2- and Th17-type and inflammatory cytokines. Features of asthma were either marginal or absent in OVA-treated pups of vehicle-exposed mice. Prenatal exposure to DEP therefore lowers the threshold for induction of asthma in response to allergens. The Th2- and Th17-type cytokines were mainly produced by NK cells; depletion of these cells blocked development of airway eosinophilic inflammation. Prenatal exposure to DEP upregulated expression of genes known to be controlled by AhR and oxidative stress; the upregulation persisted 1 month after birth, even though mice were no longer exposed to DEP.
We chose to expose prenatal mice to DEP because the link between DEP and asthma is well established5–8, 21–31, 53–58. Epidemiological studies associated exposure to diesel exhaust with development of new asthma and exacerbation of pre-existing asthma5–8, 53–58. The causality was determined in controlled exposure studies in humans and adult mice21–31. DEP exposure increased production of IL17, Th2-type cytokines, and specific IgEs; increased eosinophilic inflammation in the upper and lower respiratory tract; and increased vascular permeability and airway resistance21–31, 59. Although there is some understanding of how postnatal exposure to diesel exhaust triggers asthma, the mechanisms underlying asthma induction following in utero exposure have not been characterized. Through development of this mouse model, we hope to provide a tool for mechanistic studies.
Our model incorporated the most valuable elements of earlier models. Table EII in the Online Repository contains experimental details of these models. The field was pioneered by Fedulov et al.13. In their model, pregnant BALB/c mice were given a single intranasal dose of DEP on GD 14. Pups were immunized through a single intraperitoneal injection of low-dose OVA in alum during the neonatal period and challenged with aerosolized OVA when they were 2 weeks old. Asthma developed only in DEP-exposed pups. We followed this immunization strategy. The model by Auten et al. used C57BL/6 mice and combined repeated prenatal exposure to diesel exhaust (inhalation) or DEP (intranasal application) with chronic postnatal ozone exposure14. Prenatal exposures increased ozone-induced airway hyperreactivity.
A model created by Reiprich et al. used BALB/c mice and incorporated repeated prenatal co-exposure to DEP and lipopolysaccharide (LPS), postnatal exposure to LPS, and standard (optimal) postnatal immunization and challenge with OVA15. Mice were intraperitoneally immunized with an optimal dose of OVA in alum at 6 and 8 weeks of age, and then given intranasal challenge with OVA 14–16 days and 21–23 days after the initial immunization. This OVA treatment induced asthma in control pups (offspring of mice given injections of vehicle). The DEP inhibited LPS-mediated protection against asthma. As discussed earlier, we incorporated the repeated DEP exposure principle from protocols by Auten at al. and Reiprich et al., and, as described by Auten at al., utilized C57BL/6 mice.
The link between prenatal diesel exhaust exposure and asthma was examined in 2 additional studies that reached different conclusions60, 61. In the model of Sharkhuu et al., pregnant mice repeatedly inhaled diesel exhaust60. Their pups were immunized (PND 42 and 43) and challenged (PND 54, 55 and 56) with intranasal OVA. The authors reported no asthma-promoting effects of prenatal exposure to diesel exhaust. In the model of Corson et al., pregnant mice repeatedly inhaled diesel exhaust and received intranasal applications of Aspergillus fumigatus extract 61. Their pups then received several doses of Aspergillus extract, beginning at 9–10 weeks of age. In this model, the combined prenatal exposure to diesel exhaust and Aspergillus extract protected against airway eosinophilia. The discrepancies in outcomes between positive (Auten et al., Fedulov et al., and Reiprich et al.) and negative (Sharkhuu et al. and Corson et al.) studies could result from differences in methods used for prenatal exposure (instillation of the DEP suspension vs. inhalation of diesel exhaust) and postnatal exposure (allergen vs. ozone; differences in dose, route, timing, and type of postnatal allergen).
Pathogenesis of asthma involves Th2- and Th17-type cytokines. In our model, these cytokines were produced mainly by NK cells. This is in contrast to conventional mouse models of asthma, in which CD4+ T cells are primary source of Th2- and Th17-type cytokines. NK cells are believed to secrete IFNγ, TNFα, granzymes, and perforins62, to kill cells infected with viruses and tumor cells62. Less is understood about IL17+/Th2-type cytokine+ NK cells, although studies have reported that these cells emerge during development of specific disorders63–68. Th2-type cytokine+ NK cells have been detected in patients with atopic asthma, atopic dermatitis, parasitic infections, and autoimmune diseases63–66. IL17+ NK cells have been observed in mouse models of toxoplasmosis and hepatic ischemia/reperfusion injury67, 68, but there are no reports of their association with asthma or allergy.
Our data strongly indicated that NK cells were responsible for development of airway inflammation in our model. Antibody-mediated depletion of these cells before allergen challenge prevented airway inflammation. Antibody-based strategies for depletion of NK cells were previously tested in conventional models of asthma. Using these approaches, four research groups showed that NK cells positively contribute to the initiation allergic inflammation, 69–72 whereas one group reported that NK cells suppress the resolution of pulmonary inflammation.73, 74 The studies might have reached different conclusions because different experimental conditions or different phases of allergic response are associated with activation of distinct NK cell subsets. Alternatively, NK cells dynamically change phenotype during allergic response. A third possibility involves deletion of asthma-relevant non-NK cell subsets by injected antibodies.
Two antibodies are commonly used to deplete NK cells in vivo: anti-NK1.1 and anti-asialo-GM1 39–41, 73. We chose anti-NK1.1 because it is more specific for NK cells. One limitation of this approach is that anti-NK1.1 also depletes NK T cells. However, the role of NK T cells in airway inflammation in our model is probably not significant; in DEP-OVA mice, these cells accounted for only 8%–12% of cytokine-producing cells.
The AhR may provide a molecular link between maternal exposure to diesel exhaust and asthma in offspring. This receptor is activated by PAH—major components of DEP that pass through the placenta44. PAH-bound AhR activates transcription of many genes, including those that regulate the immune response45–48. Many studies have shown that AhR induces transcription of the Il17 gene in T cells and type-3 innate lymphoid cells46–48. Accordingly, DEP were shown to stimulate IL17 production by splenocytes75. The AhR is expressed in NK cells,76, 77 and it is likely that AhR regulates Il17 also in NK cells.
The effect of the AhR on Th2-type cytokines is less studied. AhR deficiency impairs production of IL5 and IL13 by splenocytes and mast cells, respectively78, 79. Conversely, the AhR ligand 6-formylindolo[3,2-b]carbazole increases production of IL13 by mast cells80. DEP-derived PAH induce IL13 production by peripheral blood mononuclear cells81. AhR-deficient CD4+ T cells increase secretion of IL4 upon stimulation through CD3 and CD2882. In contrast, other studies have shown that PAH synergize with concanavalin A and antigen to stimulate IL4 production by T cells and basophils, respectively83, 84.
The AhR also regulates the synthesis of inflammatory mediators including IL6 and TNFα85, 86. These cytokines are also induced by ROS; upregulation of transcripts that characteristic of the oxidative stress response (Hmox1 mRNA), was observed in our DEP-OVA mice. Therefore, inflammatory responses in these mice might be the result of the combined effect of AhR and ROS signaling.
The AhR- and ROS-associated transcriptional response in DEP-OVA mice was systemic, because it was observed in two different organs i.e. lung and liver. Additional evidence for the systemic effects of DEP included the lower body weight of DEP-PBS and DEP-OVA mice, compared with PBS-PBS mice. One likely scenario is that in fetuses exposed to DEP, activation of AhR and ROS pathways led to systemic priming of pro-inflammatory responses and systemic NK cell activation that ultimately facilitated postnatal induction of asthma upon lung-targeted allergen challenge.
There is evidence that AhR mediates allergic inflammation. AhR expression is increased in peripheral blood mononuclear cells from patients with allergic asthma87. The putative AhR ligand, 4-nonylphenol, enhances allergic inflammation in the lung88. The inflammation is reduced in mice carrying the AhRd allele, which is defective in ligand binding. Mice that express a constitutively active form of AhR in keratinocytes spontaneously develop skin lesions that resemble atopic dermatitis89. Repeated intranasal administration of PAH aggravates allergic rhinitis in the guinea pig model90. PAH also increase production of allergen-specific IgE91, 92. We recognize that certain AhR ligands may be anti-inflammatory. Immunotoxic 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) promotes development of T-regulatory cells, and thereby suppresses allergic sensitization93, 94. It is now believed that TCDD-induced toxicity results from inappropriate activation of AhR, leading to deregulated physiologic functions95.
In summary, we have developed a model of asthma susceptibility through prenatal exposure of mice to DEP. We provided mechanistic insight into this process, delineating the role of NK cells in asthma development, along with activation of AhR- and oxidative stress-regulated genes.
Supplementary Material
Key Messages.
Mice exposed in utero to diesel particulate matter have increased susceptibility to asthma.
Asthma in these mice is mediated by NK cells and is associated with enhanced transcription of genes known to be specifically regulated by AhR and oxidative stress.
Acknowledgments
Funding: The work was supported by the Denver Children’s Environmental Health Center Faculty Development Investigator Award - a part of NIEHS PO1 ES-018181/EPA GAD 834515010 (to M.M.G.), the NIH/NCATS Colorado CTSI KL2 TR000156 Award (to M.M.G.) and the Sheldon C. Siegel - Asthma and Allergy Foundation of America Investigator Grant Award (to M.M.G.).
Abbreviations
- AhR
aryl hydrocarbon receptor
- AhRR
aryl hydrocarbon receptor repressor
- BAL
bronchoalveolar lavage
- BALF
bronchoalveolar lavage fluid
- Cyp1a1
cytochrome P450, family 1, subfamily A, polypeptide 1
- Cyp1b1
cytochrome P450, family 1, subfamily B, polypeptide 1
- DEP
diesel exhaust particles
- GD
gestation day
- Hmox1
heme oxygenase 1
- IL
interleukin
- IN
intranasal
- IP
intraperitoneal
- LPS
lipopolysaccharide
- NK cells
natural killer cells
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- OVA
ovalbumin
- PAH
polycyclic aromatic hydrocarbons
- PBS
phosphate-buffered saline
- PND
postnatal day
- Th
T helper
- ROS
reactive oxygen species
- TNF
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moorman JE, Akinbami LJ, Bailey CM, et al. National Surveillance of Asthma: United States, 2001–2010. [Accessed June 23, 2013];National Center for Health Statistics. Vital Health Stat. 2012 3(35) Available at: http://www.cdc.gov/nchs/data/series/sr_03/sr03_035.pdf. [PubMed] [Google Scholar]
- 2.Litonjua AA, Carey VJ, Burge HA, Weiss ST, Gold DR. Parental history and the risk for childhood asthma. Does mother confer more risk than father? Am J Respir Crit Care Med. 1998;158:176–81. doi: 10.1164/ajrccm.158.1.9710014. [DOI] [PubMed] [Google Scholar]
- 3.Sears MR, Holdaway MD, Flannery EM, Herbison GP, Silva PA. Parental and neonatal risk factors for atopy, airway hyper-responsiveness, and asthma. Arch Dis Child. 1996;75:392–8. doi: 10.1136/adc.75.5.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz RG, Kemeny DM, Price JF. Higher risk of infantile atopic dermatitis from maternal atopy than from paternal atopy. Clin Exp Allergy. 1992;22:762–6. doi: 10.1111/j.1365-2222.1992.tb02816.x. [DOI] [PubMed] [Google Scholar]
- 5.Patel MM, Quinn JW, Jung KH, Hoepner L, Diaz D, Perzanowski M, et al. Traffic density and stationary sources of air pollution associated with wheeze, asthma, and immunoglobulin E from birth to age 5 years among New York City children. Environ Res. 2011;111:1222–9. doi: 10.1016/j.envres.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perzanowski MS, Chew GL, Divjan A, Jung KH, Ridder R, Tang D, et al. Early-life cockroach allergen and polycyclic aromatic hydrocarbon exposures predict cockroach sensitization among inner-city children. J Allergy Clin Immunol. 2013;131:886–93. doi: 10.1016/j.jaci.2012.12.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosa MJ, Jung KH, Perzanowski MS, Kelvin EA, Darling KW, Camann DE, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, environmental tobacco smoke and asthma. Respir Med. 2011;105:869–76. doi: 10.1016/j.rmed.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jedrychowski WA, Perera FP, Maugeri U, Mrozek-Budzyn D, Mroz E, Klimaszewska-Rembiasz M, et al. Intrauterine exposure to polycyclic aromatic hydrocarbons, fine particulate matter and early wheeze. Prospective birth cohort study in 4-year olds. Pediatr Allergy Immunol. 2010;21:e723–32. doi: 10.1111/j.1399-3038.2010.01034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke H, Leonardi-Bee J, Hashim A, Pine-Abata H, Chen Y, Cook DG, et al. Prenatal and passive smoke exposure and incidence of asthma and wheeze: systematic review and meta-analysis. Pediatrics. 2012;129:735–44. doi: 10.1542/peds.2011-2196. [DOI] [PubMed] [Google Scholar]
- 10.Spanier AJ, Kahn RS, Kunselman AR, Hornung R, Xu Y, Calafat AM, et al. Prenatal exposure to bisphenol A and child wheeze from birth to 3 years of age. Environ Health Perspect. 2012;120:916–20. doi: 10.1289/ehp.1104175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ege MJ, Bieli C, Frei R, van Strien RT, Riedler J, Ublagger E, et al. Parsifal Study team. Prenatal farm exposure is related to the expression of receptors of the innate immunity and to atopic sensitization in school-age children. J Allergy Clin Immunol. 2006;117:817–23. doi: 10.1016/j.jaci.2005.12.1307. [DOI] [PubMed] [Google Scholar]
- 12.Douwes J, Cheng S, Travier N, Cohet C, Niesink A, McKenzie J, et al. Farm exposure in utero may protect against asthma, hay fever and eczema. Eur Respir J. 2008;32:603–11. doi: 10.1183/09031936.00033707. [DOI] [PubMed] [Google Scholar]
- 13.Fedulov AV, Leme A, Yang Z, Dahl M, Lim R, Mariani TJ, et al. Pulmonary exposure to particles during pregnancy causes increased neonatal asthma susceptibility. Am J Respir Cell Mol Biol. 2008;38:57–67. doi: 10.1165/rcmb.2007-0124OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Auten RL, Gilmour MI, Krantz QT, Potts EN, Mason SN, Foster WM. Maternal diesel inhalation increases airway hyperreactivity in ozone-exposed offspring. Am J Respir Cell Mol Biol. 2012;46:454–60. doi: 10.1165/rcmb.2011-0256OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Reiprich M, Rudzok S, Schütze N, Simon JC, Lehmann I, Trump S, et al. Inhibition of endotoxin-induced perinatal asthma protection by pollutants in an experimental mouse model. Allergy. 2013;68:481–9. doi: 10.1111/all.12121. [DOI] [PubMed] [Google Scholar]
- 16.Kim J, Natarajan S, Vaickus LJ, Bouchard JC, Beal D, Cruikshank WW, et al. Diesel exhaust particulates exacerbate asthma-like inflammation by increasing CXC chemokines. Am J Pathol. 2011;179:2730–9. doi: 10.1016/j.ajpath.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arimoto T, Kadiiska MB, Sato K, Corbett J, Mason RP. Synergistic production of lung free radicals by diesel exhaust particles and endotoxin. Am J Respir Crit Care Med. 2005;171:379–87. doi: 10.1164/rccm.200402-248OC. [DOI] [PubMed] [Google Scholar]
- 18.Mundandhara SD, Becker S, Madden MC. Effects of diesel exhaust particles on human alveolar macrophage ability to secrete inflammatory mediators in response to lipopolysaccharide. Toxicol In Vitro. 2006;20:614–24. doi: 10.1016/j.tiv.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 19.Gorska MM, Liang Q, Stafford SJ, Goplen N, Dharajiya N, Guo L, et al. MK2 controls the level of negative feedback in the NF-kappaB pathway and is essential for vascular permeability and airway inflammation. J Exp Med. 2007;204:1637–52. doi: 10.1084/jem.20062621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorska MM, Goplen N, Liang Q, Alam R. Uncoordinated 119 preferentially induces Th2 differentiation and promotes the development of asthma. J Immunol. 2010;184:4488–96. doi: 10.4049/jimmunol.0903115. [DOI] [PubMed] [Google Scholar]
- 21.Muranaka M, Suzuki S, Koizumi K, Takafuji S, Miyamoto T, Ikemori R, et al. Adjuvant activity of diesel-exhaust particulates for the production of IgE antibody in mice. J Allergy Clin Immunol. 1986;77:616–23. doi: 10.1016/0091-6749(86)90355-6. [DOI] [PubMed] [Google Scholar]
- 22.Fujimaki H, Nohara O, Ichinose T, Watanabe N, Saito S. IL-4 production in mediastinal lymph node cells in mice intratracheally instilled with diesel exhaust particulates and antigen. Toxicology. 1994;92:261–8. doi: 10.1016/0300-483x(94)90182-1. [DOI] [PubMed] [Google Scholar]
- 23.Fujimaki H, Saneyoshi K, Nohara O, Shiraishi F, Imai T. Intranasal instillation of diesel exhaust particulates and antigen in mice modulated cytokine productions in cervical lymph node cells. Int Arch Allergy Immunol. 1995;108:268–73. doi: 10.1159/000237163. [DOI] [PubMed] [Google Scholar]
- 24.Miyabara Y, Takano H, Ichinose T, Lim HB, Sagai M. Diesel exhaust enhances allergic airway inflammation and hyperresponsiveness in mice. Am J Respir Crit Care Med. 1998;157:1138–44. doi: 10.1164/ajrccm.157.4.9708066. [DOI] [PubMed] [Google Scholar]
- 25.Nel AE, Diaz-Sanchez D, Ng D, Hiura T, Saxon A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol. 1998;102:539–54. doi: 10.1016/s0091-6749(98)70269-6. [DOI] [PubMed] [Google Scholar]
- 26.Salvi S, Blomberg A, Rudell B, Kelly F, Sandström T, Holgate ST, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159:702–9. doi: 10.1164/ajrccm.159.3.9709083. [DOI] [PubMed] [Google Scholar]
- 27.Diaz-Sanchez D, Jyrala M, Ng D, Nel A, Saxon A. In vivo nasal challenge with diesel exhaust particles enhances expression of the CC chemokines rantes, MIP-1alpha, and MCP-3 in humans. Clin Immunol. 2000;97:140–5. doi: 10.1006/clim.2000.4921. [DOI] [PubMed] [Google Scholar]
- 28.Nightingale JA, Maggs R, Cullinan P, Donnelly LE, Rogers DF, Kinnersley R, et al. Airway inflammation after controlled exposure to diesel exhaust particulates. Am J Respir Crit Care Med. 2000;162:161–6. doi: 10.1164/ajrccm.162.1.9908092. [DOI] [PubMed] [Google Scholar]
- 29.Nordenhäll C, Pourazar J, Ledin MC, Levin JO, Sandström T, Adelroth E. Diesel exhaust enhances airway responsiveness in asthmatic subjects. Eur Respir J. 2001;17:909–15. doi: 10.1183/09031936.01.17509090. [DOI] [PubMed] [Google Scholar]
- 30.Li N, Harkema JR, Lewandowski RP, Wang M, Bramble LA, Gookin GR, et al. Ambient ultrafine particles provide a strong adjuvant effect in the secondary immune response: implication for traffic-related asthma flares. Am J Physiol Lung Cell Mol Physiol. 2010;299:L374–83. doi: 10.1152/ajplung.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behndig AF, Mudway IS, Brown JL, Stenfors N, Helleday R, Duggan ST, et al. Airway antioxidant and inflammatory responses to diesel exhaust exposure in healthy humans. Eur Respir J. 2006;27:359–65. doi: 10.1183/09031936.06.00136904. [DOI] [PubMed] [Google Scholar]
- 32.Ristovski ZD, Miljevic B, Surawski NC, Morawska L, Fong KM, Goh F, et al. Respiratory health effects of diesel particulate matter. Respirology. 2012;17:201–12. doi: 10.1111/j.1440-1843.2011.02109.x. [DOI] [PubMed] [Google Scholar]
- 33.Caraballo JC, Borcherding J, Thorne PS, Comellas AP. Protein kinase C-ζ mediates lung injury induced by diesel exhaust particles. Am J Respir Cell Mol Biol. 2013;48:306–13. doi: 10.1165/rcmb.2012-0056OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang YJ, Jeung YS, Seo MH, Yoon JY, Kim DY, Park JW, et al. Asian dust and titanium dioxide particles-induced inflammation and oxidative DNA damage in C57BL/6 mice. Inhal Toxicol. 2010;22:1127–33. doi: 10.3109/08958378.2010.528805. [DOI] [PubMed] [Google Scholar]
- 35.Nilsen A, Hagemann R, Eide I. The adjuvant activity of diesel exhaust particles and carbon black on systemic IgE production to ovalbumin in mice after intranasal instillation. Toxicology. 1997;124:225–32. doi: 10.1016/s0300-483x(97)00150-9. [DOI] [PubMed] [Google Scholar]
- 36.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature. 1953;172:603–6. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 37.Dixon FJ, Mauer PH. Immunologic unresponsiveness induced by protein antigens. J Exp Med. 1955;101:245–57. doi: 10.1084/jem.101.3.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lommatzsch M. Airway hyperresponsiveness: new insights into the pathogenesis. Semin Respir Crit Care Med. 2012;33:579–87. doi: 10.1055/s-0032-1325617. [DOI] [PubMed] [Google Scholar]
- 39.Sun JC, Beilke JN, Bezman NA, Lanier LL. Homeostatic proliferation generates long-lived natural killer cells that respond against viral infection. J Exp Med. 2011;208:357–68. doi: 10.1084/jem.20100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geurtsvan Kessel CH, Bergen IM, Muskens F, Boon L, Hoogsteden HC, Osterhaus AD, Rimmelzwaan GF, Lambrecht BN. Both conventional and interferon killer dendritic cells have antigen-presenting capacity during influenza virus infection. PLoS One. 2009;4(9):e7187. doi: 10.1371/journal.pone.0007187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joncker NT, Shifrin N, Delebecque F, Raulet DH. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med. 2010;207:2065–72. doi: 10.1084/jem.20100570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ris C. U.S. EPA health assessment for diesel engine exhaust: a review. Inhal Toxicol. 2007;19 (Suppl 1):229–39. doi: 10.1080/08958370701497960. [DOI] [PubMed] [Google Scholar]
- 43.Diaz-Sanchez D. The role of diesel exhaust particles and their associated polyaromatic hydrocarbons in the induction of allergic airway disease. Allergy. 1997;52:52–6. doi: 10.1111/j.1398-9995.1997.tb04871.x. discussion 57–8. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava VK, Chauhan SS, Srivastava PK, Kumar V, Misra UK. Fetal translocation and metabolism of PAH obtained from coal fly ash given intratracheally to pregnant rats. J Toxicol Environ Health. 1986;18:459–69. doi: 10.1080/15287398609530885. [DOI] [PubMed] [Google Scholar]
- 45.Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology. 2009;127:299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 47.Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–9. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hahn ME, Allan LL, Sherr DH. Regulation of constitutive and inducible AHR signaling: complex interactions involving the AHR repressor. Biochem Pharmacol. 2009;77:485–97. doi: 10.1016/j.bcp.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A. 1996;93:6731–6. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xue W, Warshawsky D. Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review. Toxicol Appl Pharmacol. 2005;206:73–93. doi: 10.1016/j.taap.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 52.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–81. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 53.Ryan PH, LeMasters G, Biagini J, Bernstein D, Grinshpun SA, Shukla R, et al. Is it traffic type, volume, or distance? Wheezing in infants living near truck and bus traffic. J Allergy Clin Immunol. 2005;116:279–84. doi: 10.1016/j.jaci.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 54.McConnell R, Berhane K, Yao L, Jerrett M, Lurmann F, Gilliland F, et al. Traffic, susceptibility, and childhood asthma. Environ Health Perspect. 2006;114:766–72. doi: 10.1289/ehp.8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med. 2007;357:2348–58. doi: 10.1056/NEJMoa071535. [DOI] [PubMed] [Google Scholar]
- 56.Jacquemin B, Sunyer J, Forsberg B, Aguilera I, Bouso L, Briggs D, et al. Association between modelled traffic-related air pollution and asthma score in the ECRHS. Eur Respir J. 2009;34:834–42. doi: 10.1183/09031936.00138208. [DOI] [PubMed] [Google Scholar]
- 57.Gent JF, Koutrakis P, Belanger K, Triche E, Holford TR, Bracken MB, et al. Symptoms and medication use in children with asthma and traffic-related sources of fine particle pollution. Environ Health Perspect. 2009;117:1168–74. doi: 10.1289/ehp.0800335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McConnell R, Islam T, Shankardass K, Jerrett M, Lurmann F, Gilliland F, et al. Childhood incident asthma and traffic-related air pollution at home and school. Environ Health Perspect. 2010;118:1021–6. doi: 10.1289/ehp.0901232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diaz-Sanchez D, Tsien A, Fleming J, Saxon A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J Immunol. 1997;158:2406–13. [PubMed] [Google Scholar]
- 60.Sharkhuu T, Doerfler DL, Krantz QT, Luebke RW, Linak WP, Gilmour MI. Effects of prenatal diesel exhaust inhalation on pulmonary inflammation and development of specific immune responses. Toxicol Lett. 2010;196:12–20. doi: 10.1016/j.toxlet.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 61.Corson L, Zhu H, Quan C, Grunig G, Ballaney M, Jin X, et al. Prenatal allergen and diesel exhaust exposure and their effects on allergy in adult offspring mice. Allergy Asthma Clin Immunol. 2010;6:7. doi: 10.1186/1710-1492-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 63.Wei H, Zhang J, Xiao W, Feng J, Sun R, Tian Z. Involvement of human natural killer cells in asthma pathogenesis: natural killer 2 cells in type 2 cytokine predominance. J Allergy Clin Immunol. 2005;115:841–7. doi: 10.1016/j.jaci.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 64.Aktas E, Akdis M, Bilgic S, Disch R, Falk CS, Blaser K, et al. Different natural killer (NK) receptor expression and immunoglobulin E (IgE) regulation by NK1 and NK2 cells. Clin Exp Immunol. 2005;140:301–9. doi: 10.1111/j.1365-2249.2005.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horikawa M, Hasegawa M, Komura K, Hayakawa I, Yanaba K, Matsushita T, et al. Abnormal natural killer cell function in systemic sclerosis: altered cytokine production and defective killing activity. J Invest Dermatol. 2005;125:731–7. doi: 10.1111/j.0022-202X.2005.23767.x. [DOI] [PubMed] [Google Scholar]
- 66.Babu S, Blauvelt CP, Nutman TB. Filarial parasites induce NK cell activation, type 1 and type 2 cytokine secretion, and subsequent apoptotic cell death. J Immunol. 2007;179:2445–56. doi: 10.4049/jimmunol.179.4.2445. [DOI] [PubMed] [Google Scholar]
- 67.Passos ST, Silver JS, O’Hara AC, Sehy D, Stumhofer JS, Hunter CA. IL-6 promotes NK cell production of IL-17 during toxoplasmosis. J Immunol. 2010;184:1776–83. doi: 10.4049/jimmunol.0901843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng M, Li G, Qian X, Fan Y, Huang X, Zhang F, et al. IL-17A-producing NK cells were implicated in liver injury induced by ischemia and reperfusion. Int Immunopharmacol. 2012;13:135–40. doi: 10.1016/j.intimp.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 69.Walker C, Checkel J, Cammisuli S, Leibson PJ, Gleich GJ. IL-5 production by NK cells contributes to eosinophil infiltration in a mouse model of allergic inflammation. J Immunol. 1998;161:1962–9. [PubMed] [Google Scholar]
- 70.Korsgren M, Persson CG, Sundler F, Bjerke T, Hansson T, Chambers BJ, et al. Natural killer cells determine development of allergen-induced eosinophilic airway inflammation in mice. J Exp Med. 1999;189:553–62. doi: 10.1084/jem.189.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ple C, Barrier M, Amniai L, Marquillies P, Bertout J, Tsicopoulos A, et al. Natural killer cells accumulate in lung-draining lymph nodes and regulate airway eosinophilia in a murine model of asthma. Scand J Immunol. 2010;72:118–27. doi: 10.1111/j.1365-3083.2010.02419.x. [DOI] [PubMed] [Google Scholar]
- 72.Wang W, Hansbro PM, Foster PS, Yang M. An alternate STAT6-independent pathway promotes eosinophil influx into blood during allergic airway inflammation. PLoS One. 2011;6:e17766. doi: 10.1371/journal.pone.0017766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haworth O, Cernadas M, Levy BD. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J Immunol. 2011;186:6129–35. doi: 10.4049/jimmunol.1004007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5:174ra26. doi: 10.1126/scitranslmed.3004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakamura R, Inoue K, Fujitani Y, Kiyono M, Hirano S, Takano H. Effects of nanoparticle-rich diesel exhaust particles on IL-17 production in vitro. J Immunotoxicol. 2012;9:72–6. doi: 10.3109/1547691X.2011.629638. [DOI] [PubMed] [Google Scholar]
- 76.Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, et al. Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity. 2010;32:803–14. doi: 10.1016/j.immuni.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–5. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- 78.Rodríguez-Sosa M, Elizondo G, López-Durán RM, Rivera I, Gonzalez FJ, Vega L. Overproduction of IFN-gamma and IL-12 in AhR-null mice. FEBS Lett. 2005;579:6403–10. doi: 10.1016/j.febslet.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 79.Zhou Y, Tung HY, Tsai YM, Hsu SC, Chang HW, Kawasaki H, et al. Aryl hydrocarbon receptor controls murine mast cell homeostasis. Blood. 2013;121:3195–204. doi: 10.1182/blood-2012-08-453597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sibilano R, Frossi B, Calvaruso M, Danelli L, Betto E, Dall’Agnese A, et al. The aryl hydrocarbon receptor modulates acute and late mast cell responses. J Immunol. 2012;189:120–7. doi: 10.4049/jimmunol.1200009. [DOI] [PubMed] [Google Scholar]
- 81.Chang Y, Sénéchal S, de Nadai P, Chenivesse C, Gilet J, Vorng H, et al. Diesel exhaust exposure favors TH2 cell recruitment in nonatopic subjects by differentially regulating chemokine production. J Allergy Clin Immunol. 2006;118:354–60. doi: 10.1016/j.jaci.2006.04.050. [DOI] [PubMed] [Google Scholar]
- 82.Negishi T, Kato Y, Ooneda O, Mimura J, Takada T, Mochizuki H, et al. Effects of aryl hydrocarbon receptor signaling on the modulation of TH1/TH2 balance. J Immunol. 2005;175:7348–56. doi: 10.4049/jimmunol.175.11.7348. [DOI] [PubMed] [Google Scholar]
- 83.Kepley CL, Lauer FT, Oliver JM, Burchiel SW. Environmental polycyclic aromatic hydrocarbons, benzo(a) pyrene (BaP) and BaP-quinones, enhance IgE-mediatedhistamine release and IL-4 production in human basophils. Clin Immunol. 2003;107:10–9. doi: 10.1016/s1521-6616(03)00004-4. [DOI] [PubMed] [Google Scholar]
- 84.Bömmel H, Li-Weber M, Serfling E, Duschl A. The environmental pollutant pyrene induces the production of IL-4. J Allergy Clin Immunol. 2000;105:796–802. doi: 10.1067/mai.2000.105124. [DOI] [PubMed] [Google Scholar]
- 85.Veldhoen M, Duarte JH. The aryl hydrocarbon receptor: fine-tuning the immune-response. Curr Opin Immunol. 2010;22:747–52. doi: 10.1016/j.coi.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 86.Chen PH, Chang H, Chang JT, Lin P. Aryl hydrocarbon receptor in association with RelA modulates IL-6 expression in non-smoking lung cancer. Oncogene. 2012;31:2555–65. doi: 10.1038/onc.2011.438. [DOI] [PubMed] [Google Scholar]
- 87.Zhu J, Cao Y, Li K, Wang Z, Zuo P, Xiong W, et al. Increased expression of aryl hydrocarbon receptor and interleukin 22 in patients with allergic asthma. Asian Pac J Allergy Immunol. 2011;29:266–72. [PubMed] [Google Scholar]
- 88.Suen JL, Hsu SH, Hung CH, Chao YS, Lee CL, Lin CY, et al. A common environmental pollutant, 4-nonylphenol, promotes allergic lung inflammation in a murine model of asthma. Allergy. 2013;68:780–7. doi: 10.1111/all.12156. [DOI] [PubMed] [Google Scholar]
- 89.Tauchi M, Hida A, Negishi T, Katsuoka F, Noda S, Mimura J, et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol Cell Biol. 2005;25:9360–8. doi: 10.1128/MCB.25.21.9360-9368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mizutani N, Nabe T, Ohtani Y, Han HY, Fujii M, Yoshino S, et al. Polycyclic aromatic hydrocarbons aggravate antigen-induced nasal blockage in experimental allergic rhinitis. J Pharmacol Sci. 2007;105:291–7. doi: 10.1254/jphs.fp0071067. [DOI] [PubMed] [Google Scholar]
- 91.Kanoh T, Suzuki T, Ishimori M, Ikeda S, Ohasawa M, Ohkuni H, et al. Adjuvant activities of pyrene, anthracene, fluoranthene and benzo(a)pyrene in production of anti-IgE antibody to Japanese cedar pollen allergen in mice. J Clin Lab Immunol. 1996;48:133–47. [PubMed] [Google Scholar]
- 92.Suzuki T, Kanoh T, Kanbayashi M, Todome Y, Ohkuni H. The adjuvant activity of pyrene in diesel exhaust on IgE antibody production in mice. Arerugi. 1993;42:963–8. [PubMed] [Google Scholar]
- 93.Luebke RW, Copeland CB, Daniels M, Lambert AL, Gilmour MI. Suppression of allergic immune responses to house dust mite (HDM) in rats exposed to 2,3,7,8-TCDD. Toxicol Sci. 2001;62:71–9. doi: 10.1093/toxsci/62.1.71. [DOI] [PubMed] [Google Scholar]
- 94.Schulz VJ, Smit JJ, Willemsen KJ, Fiechter D, Hassing I, Bleumink R, et al. Activation of the aryl hydrocarbon receptor suppresses sensitization in a mouse peanut allergy model. Toxicol Sci. 2011;123:491–500. doi: 10.1093/toxsci/kfr175. [DOI] [PubMed] [Google Scholar]
- 95.Bock KW, Köhle C. Ah receptor: dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem Pharmacol. 2006;72:393–404. doi: 10.1016/j.bcp.2006.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.