

Abstract
Glioblastoma (GBM) is a very aggressive and lethal brain tumor with poor prognosis. Despite new treatment strategies, patients’ median survival is still less than 1 year in most cases. Few studies have focused exclusively on this disease in children and most of our understanding of the disease process and its clinical outcome has come from studies on malignant gliomas in childhood, combining children with the diagnosis of GBM with other pediatric patients harboring high grade malignant tumors other than GBM. In this study we investigated, using array-CGH platforms, children (median age of 9 years) affected by GBM (WHO-grade IV). We identified recurrent Copy Number Alterations demonstrating that different chromosome regions are involved, in various combinations. These observations suggest a condition of strong genomic instability. Since cancer is an acquired disease and inherited factors play a significant role, we compared for the first time the constitutional Copy Number Variations with the Copy Number Alterations found in tumor biopsy. We speculate that genes included in the recurrent 9p21.3 and 16p13.3 deletions and 1q32.1-q44 duplication play a crucial role for tumorigenesis and/or progression. In particular we suggest that the A2BP1 gene (16p13.3) is one possible culprit of the disease. Given the rarity of the disease, the poor quality and quantity of bioptic material and the scarcity of data in the literature, our findings may better elucidate the genomic background of these tumors. The recognition of candidate genes underlying this disease could then improve treatment strategies for this devastating tumor.
Keywords: Pediatric glioblastoma multiforme, central nervous tumor, copy number alterations (CNA), copy number variations (CNVs), array-CGH, minimum common regions, deletion, duplication, amplification, tumorigenesis
Introduction
Glioblastoma multiforme (GBM; WHO-grade IV), the most frequent primary malignant brain tumor in adults, accounts for approximately 7-9% of all central nervous (CNS) tumors in childhood [1,2]. It is highly invasive, poorly responsive to conventional treatments [3] and median survival for children via conventional-dose chemotherapy and radiotherapy is currently around 11-24 months. At 5 years, the overall survival rate is 5-20% [4].
In recent years, cytogenetic and molecular investigations have dramatically improved our understanding of the biology of malignant gliomas, identifying relevant molecular features. Pediatric primary GBM (pGBM), indeed, differs from its adult counterpart both in mean cumulative survival and genetic profiling [5-10].
Adult and pGBMs have distinct molecular pathways of tumorigenesis [8]. Primary adult forms present amplification of EGRF [10] and inactivation of PTEN genes [11] in about 35-50% of cases, while secondary adult GBMs, evolving from low-grade lesions, often have mutations of TP53 and, infrequently, amplification of EGFR or alteration of PTEN genes [12].
pGBM often exhibits TP53 mutations and only rarely shows EGFR amplification/overexpression [13,14] or PTEN mutations [15], suggesting that most pGBMs may be more similar to adult secondary glioblastomas than to primary ones. The frequency of mutations in TP53 is less than 40%, significantly less than those of secondary GBM of young adults that is around 65% [16].
Moreover, in 20 pGBMs with mutational inactivation of the p53 tumor suppressor gene, loss of p16 protein expression and overexpression of the EGFR protein has been described [17]. At variance with GBM adult form, no IDH1 and IDH2 mutations were found [18].
Array comparative genomic hybridization (aCGH) is a technique enabling high-resolution, genome-wide screening of genomic copy number variations (CNVs). aCGH is considered an essential and routine clinical diagnostic tool in patients with global developmental delay, intellectual disability, autism, multiple congenital anomalies and dysmorphism [19]. Moreover, several CNVs have already been associated with both complex and common disorders, including cancer. Unlike with inherited DNA variations, cancer is usually characterized by somatic copy number alterations (CNA) allowing identification of losses and/or gains crucial to the tumorigenesis process. This approach has also been applied in a wide variety of human brain tumors [20,21].
Recent studies have shown significant differences in CNA between childhood and adult GBMs. Qu et al. demonstrated that in adult GBMs the most common events are duplications/amplifications, while in pGBMs heterozygous deletions are more frequent. They demonstrated the presence of two common regions of loss of heterozygosity (LOH) in 9p24.3-9p13.1 and 17p13.3 both in pediatric and adult GBMs [22]. Moreover, in pGBMs, recurrent duplications of 1q, 3q, 2q and 17q as well as losses of chromosomal regions in 6q, 8q, 13q, and 17p have been described [23]. Paugh et al. also suggested that pediatric and adult GMBs were clearly distinguished by frequent gain of chromosome 1q and lower frequency of chromosome 7 gain and 10q loss [24].
Based on these considerations, we used aCGH platforms to query 9 pGBMs, identifying recurrent CNA and establishing their minimum common regions of duplication and deletion. In 4 cases we compared, for the first time, the tumor biopsy with blood samples of the same patient. We uncovered numerous target regions of interest and hypothesized the possible role of some genes within them, whose function seems to be crucial for tumorigenesis and/or progression.
Materials and methods
Patients
The children included in this study, approved by the institutional Ethical Committee, are affected by GBM (WHO-grade IV). Informed consent was obtained from the parents or legal guardians in all cases. We considered 9 patients, 5 females and 4 males, with a median age of 9 years (range, 1-15 years). Table 1 illustrates clinical characteristics. All received a first surgery consisting of macroscopically complete excision of tumor in two cases (P1, P7), partial excision in 6 (P2-P6, P8) and a tumor biopsy only in one children (P9). All patients underwent radiotherapy, with dose and treatment techniques being selected according to current front-line therapeutic studies of the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) and literature data [25-27].
Table 1.
Clinical characteristics of pediatric Glioblastoma Multiforme (pGBM)
| ID | Gender | Age at diagnosis (years) | Surgery | First-line Treatment | Response | FU (months) | Status |
|---|---|---|---|---|---|---|---|
| P1 | F | 1 | GTR | HDCT-RT (60 Gy) | CR | 14 | DOD |
| P2 | M | 9 | PTR | Vinorelbine+RT (60 Gy) | PR | 12 | DOD |
| P3 | M | 4 | PTR | HDCT-RT (60 Gy) | PR | 3 | DOD |
| P4 | F | 8 | PTR | Vinorelbine+RT (45 Gy) | PR | 19 | DOD |
| P5 | M | 15 | PTR | Vinorelbine+RT (60 Gy) | PR | 8 | DOD |
| P6 | F | 7 | PTR | TMZ+RT (60 Gy) | PR | 10 | DOD |
| P7 | F | 10 | GTR | TMZ+RT (60 Gy) | CR | 14 | AWD |
| P8 | F | 9 | PTR | TMZ+RT (60 Gy) | PR | 10 | DOD |
| P9 | M | 13 | By | TMZ+RT (54 Gy) | PR | 12 | DOD |
GTR: gross total removal; PTR: partial total removal; By: biopsy; HDCT: high-dose chemotherapy; RT: radiotherapy; TMZ: temozolomide; CR: complete response; PR: partial response; FU: follow-up; AWD: alive with disease; DOD: dead of disease.
Seven children were treated with fractionated-stereotactic radiotherapy at a dose of 60 Gy (P1-P3, P5-P8), one child (P4) affected by a spinal tumor with 45 Gy on the tumor site, and P9, with brainstem GBM, received 54 Gy. Two patients (P1, P3) received high dose chemotherapy with autologous stem rescue, four patients (P6-P9) adjuvant temozolomide during and after radiotherapy and three patients (P2, P4, P5) adjuvant vinorelbine concomitant to and after radiotherapy. Median follow-up of the series was 12 months (range 3 to 19 months). At the end of the study, 8 patients (P1-P6, P8, P9) had died and one (P7) was alive with severe progressive disease.
Response criteria. Extent of disease was assessed by contrast-enhanced cranial MRI scan at the time of study entry and then after every two courses. In accordance with RECIST criteria, the following radiological categories were used for evaluation of response: (a) complete response (CR) was defined as the disappearance of all known disease for at least 4 weeks; (b) partial response (PR) as at least 30% reduction in the longest diameter of measurable lesions for at least 4 weeks [28].
Histology
Surgical samples were routinely fixed in neutral buffered formol for 24 hours and embedded in paraffin for histopathological evaluation. Successively, one 5 μm thick histological section obtained from each paraffin block with hematoxylin and eosin was stained. Histological diagnosis was carried out based on 2007 WHO classification criteria [29].
Microscopic examination showed dense cellularity and composed glial cells with atypical features and brisk mytotic activity. Prominent microvascular proliferation and necrosis were present in each case. Morphological features were consistent with the diagnosis of glioblastoma.
Culture
Surgical fragments of brain tumors were collected under sterile conditions, the tissue disrupted mechanically with sterile blades and the fragments transferred into a 15-ml tube (SARSTEDT S.r.l., Italy) containing 5 ml of Dulbecco’s Modified Eagle Medium, incubated at 37°C for 5 minutes, centrifuged for 5 minutes at 1200 rpm and the pellet resuspended in DMEM/F-12 medium supplemented with 10% fetal bovine serum (Euroclone, Italy) and 1% penicillin-streptomycin antibiotics (Euroclone, Italy). Cell suspension was aliquoted in 60 mm Petri dishes and maintained at 37°C in a humidified atmosphere containing 5% CO2. After washing twice with phosphate buffered saline 1X (Euroclone, Italy), medium was replaced every 2-3 days during the 15 days of culture.
Array-CGH
We analyzed 9 pGBM specimens (P1-P9), 4 peripheral blood samples (B3, B7-B9) and one primary culture (C8) of one pGBM. We also carried out aCGH analysis of the parents of P7 and P9. Genomic tumor DNAs were extracted from GBM using QIAamp Mini Kit (QIAGEN®, Hilden, Germany) according to manufacturers’ instructions and quantified by NanoDROP 2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). aCGH was performed using the Agilent Human Genome CGH Microarray Kit 180K (Agilent Technologies, Santa Clara, California, USA). This platform is an oligonucleotide-based microarray with a resolution of about 40 kb.
Labeling and hybridization were performed following the protocols provided by Agilent. 500 ng of purified DNA of the patient and of a control of the same sex (Coriell) were double digested with RsaI and AluI enzymes (Promega) for 2 h at 37°C, obtaining products between 200 bp and 500 bp in length. Each digested sample was labeled for 2 h, minimizing light exposure, using the Agilent Genomic DNA Labeling Kit, using Cy5-dUTP for the patient DNA and Cy3-dUTP for the reference DNA. Labeled products were column purified (Amicon Ultra, Millipore) and prepared combining test and control sample according to the Agilent protocol. After probe denaturation and pre-annealing with 50 μg of Human Cot-1 DNA (Invitrogen), hybridization was performed at 65°C for 24 h in a rotating oven at 20 rpm. Images of the arrays were acquired with the Agilent C Scanner (Agilent Technologies, Santa Clara, CA, USA).
Each hybridization produced a pair of 16-bit images, which were processed using the Agilent Feature Extraction 10.5 software. Row data were analyzed using two different computational approaches: (i) the Genomic Workbench Standard Edition 5.0 software by the ADM-2 algorithm (breakpoint positions were reported according to Hg19, build 37) and (ii) a home-made pipeline based on Shifting Level Model (SLM) and FastCall algoritms [30]. This method evaluates the probability classification of each segmented region into five biologically motivated statuses (double deletion, deletion, neutral, duplication or amplification), thus permitting us to discriminate double-copy from single-copy deletions and single-copy from multiple-copy duplications. In order to take into account sample heterogeneity, for each experiment, we set the cellularity parameter c equal to 0.7, assuming 70% tumor purity.
Validation of CNVs and CNA by qPCR
Determination of CNVs and CNA by qPCR was performed using the Roche LightCycler® 480 Detection System with DNA-binding dye SYBR Green I (Roche) according to the manufacturers’ instructions. The primers were designed using Primer 3 software (http://biotools.umassmed.edu/bioapps/primer3_www.cgi).
Statistical analysis
Statistical significance of events of deletion and duplication in pGBMs was evaluated using the t-test [31].
Results
Array-CGH
Table 2 shows the results of the aCGH analysis. We also summarized the cumulative chromosomal losses and gains (CNA) of pGBMs (a) and the constitutional CNVs present in blood samples of 4 pGBMs (B3, B7-B9) (c). In two of them the parental origin of constitutional CNVs was also established (B7, B9) (c). In the absence of control DNA from the same individual allowing us to discriminate whether the observed CNA are tumor-derived or constitutional CNVs, we did not consider those CNA designated as benign in the Database of Genomic Variants (DGV: http://projects.tcag.ca/variation/).
Table 2.
Rearrangements identified by aCGH 180K
| Tumors | No CNA | CNA | |
|
| |||
|---|---|---|---|
| a) | P1 | 8 | 6q21 (del), 6q21.1 (del), 9p24.2-9p24.1 (del), 9p21.3-9p21.2 (h-del), 9p21.2-9p13.2 (del), 10q23.3 (del), 15q26.3 (del), 17p13.3 (del) |
| P2 | 27 | 1p36.12 (dup), 1p34.3 (dup), 1p31.1 (dup), 1p12 (dup), 1q21.1-1q4.1 (del), 3p14.3 (del), 4p (del-m), 4q (del), 5p (del), 5q (del), 6q12-qter (del), 9p21.3-p13.2 (del), 10p (del-m), 10q11.22 (del), 10q23.1-qter (del), 13q12.11-q31.3 (del), 13q31.3 (dup), 13q31.3-qter (del), 15q24.2 (del), 16p (del), 16q (del), 17p13.1-p12.1 (del), 17q12.1-17q12 (del), 18p (del), 18q (del), 19p13.3-19p13.2 (del), 21q (del) | |
| P3 | 4 | 1q (dup), 4q35.2 (del), 7q31.33 (dup), 16p13.3 (del) | |
| P4 | 8 | 5p14.3-pter (del-m), 6q25.1 (del), 13q (del-m), 15q13.3 (amp), 18p (del-m), 18q (del-m), Xp (del-m), Xq (del-m) | |
| P5 | 23 | 1p (del-m), 1q (dup), 3p (dup-m), 3q (dup-m), 4q11.1 (amp), 4q13.1 (del), 6p (dup-m), 6q13-q14.1 (del-m), 6q14.1-qter (dup), 7q31.1-q32.2 (del-m), 8p (dup-m), 8q (dup-m), 10p (dup-m), 10q (dup-m), 14q (del-m), 15q (del-m), 17p13.2 (del), 18p (dup-m), 18q (dup-m), 21q (dup-m), Xp (dup-m), Xq (dup-m) | |
| P6 | 25 | 1p (dup), 1q (dup), 2p21 (del), 2p12 (dup), 2p11.2 (amp), 2q22.1 (del), 2q24.2 (dup), 2q34 (dup-m), 2q34 (del), 3p21.2 (dup), 3q13.31 (dup), 3q13.31 (del-m), 7p (dup), 7q11.2-q22.2 (dup), 7q22.2-qter (amp), 9p (dup), 9q (dup), 11p11.12-pter (dup), 11q (dup), 12q24.12 (dup), 19q13.12 (amp), 20p (dup), 20q (dup), Xp21.2-cent (dup), Xq (dup) | |
| P7 | 25 | 1p36.22 (dup), 1q23.2-q23.3 (dup), 1q32.1 (amp), 2p25.3-p24.3 (dup), 2q22.1(del), 6p25.2-q26 (del-m), 7p22.2-q36.2 (dup-m), 8p23.2-q24.23 (del-m), 9p24.2-q34.2 (del-m), 9p21.3 (h-del), 10p15.2-q26.2 (del-m), 11p15.4-q24.3 (del-m), 12p11.22-p11.21 (amp), 13q (del-m), 14q (dup-m), 15q (del-m), 17p13.3 (dup), 17q11.2 (dup), 17q21.33 (dup), 17q24.3 (dup), 17q25.3 (dup), 19p13.2 (del), 19p13.2 (dup-m), 21q (del-m), 22q13.2-q13.33 (del-m) | |
| P8 | 65 | 1p (del-m), 1q25.21-qter (dup-m), 2pter-p23.2 (dup-m), 2p16.3-p12.1 (del-m), 2q21.2-q23.2 (del-m), 2q31.2-q32.3 (dup-m), 2q32.3-q34 (del-m), 2q36.3-q37.2 (dup-m), 3q13.11-q13.31 (del-m), 4p (del-m), 4q13.3-qter (del-m), 5q (del), 7pter-p21.3 (del-m), 7p11.2 (amp), 7q11.21-q21.3 (del-m), 7q22.2-qter (dup-m), 8p23.2-p21.3 (del-m), 8p21.3-p11.22 (dup-m), 8q11.22 (del-m), 8q11.23-q12 (dup-m), 8q12.1-q13.1 (del-m), 8q13.3-q21.2 (del-m), 8q21.2 (del), 8q21.3-q23.1 (del-m), 8q23.2 (dup-m), 8q24.13-qter (del-m), 9p23-p22.2 (dup-m), 9p22.21 (del-m), 9p21.3-p23.31 (h-del), 9p21.21-p21.12 (del-m), 9q21.11-q21.13 (dup-m), 9q21.12-q21.33 (del-m), 10p (del-m), 10q11.22-q21.31 (del-m), 10q22.22-q22.21 (dup-m), 10q22.21-qter (del-m), 12p (del-m), 12q12-q13.12 (del), 12q13.13-q14.31 (dup-m), 12q14.31-q21.1 (h-del), 12q21.12 (h-del), 12q21.31-qter (del), 13q12.11-q32.3 (h-del+del/-m), 14q (del), 15q (del), 16pter-p13.12 (del), 16p13.1-p11.2 (dup-m), 16p12.1-p12.11 (del-m), 17pter-p13.1 (del-m), 17q12-qter (del-m), 18q21.2 (dup-m), 18q21.32-q22.3 (del-m), 19p13.3 (dup-m), 19p13.3-p13.11 (del-m), 19q (del-m), 20p (del-m), 20q (del-m), 21q21.1-qter (del-m), 22q (del-m), Xpter-p11.3 (dup-m), Xp11.3-p11.23 (del-m), Xq11-q21.1 (del-m), Xq21.1-q21.33 (dup-m), Xq21.33-q22.1 (del-m), Xq22.1-q22.3 (dup-m), Xq22.3-q25 (del-m), Xq26.2-qter (dup-m) | |
| P9 | 18 | 1p33-p21.1 (del-m), 1q21.1-q44 (dup), 2p25.1-p21 (amp), 2p16.3-p16.1 (dup), 4q26-q35.2 (del-m), 9p23 (del-m), 10q21.2-q26.3 (del-m), 11q12.1 (dup), 14q11.2-q13.1 (del-m), 14q21.2-q21.3 (dup), 14q22.1 (del-m), 14q22.3-q24.2 (del-m), 14q24.3-q31.3 (dup-m), 14q31.3-q32.32 (del-m), 17p13.3-p11.2 (del-m), 17p11.2 (dup), 18q11.1-q23 (del-m), Yp11.32-q12 (del) | |
|
| |||
| Colture | No CNA | CNA | |
|
| |||
| b) | C8 | 65 | 1p (del-m), 1q25.21-qter (dup-m), 2pter-p23.2 (dup-m), 2p16.3-p12.1 (del-m), 2q21.2-q23.2 (del-m), 2q31.2-q32.3 (dup-m), 2q32.3-q34 (del-m), 2q36.3-q37.2 (dup-m), 3q13.11-q13.31 (del-m), 4p (del-m), 4q13.3-qter (del-m), 5q (del), 7pter-p21.3 (del-m), 7p11.2 (amp), 7q11.21-q21.3 (del-m), 7q22.2-qter (dup-m), 8p23.2-p21.3 (del-m), 8p21.3-p11.22 (dup-m), 8q11.22 (del-m), 8q11.23-q12 (dup-m), 8q12.1-q13.1 (del-m), 8q13.3-q21.2 (del-m), 8q21.2 (del), 8q21.3-q23.1 (del-m), 8q23.2 (dup-m), 8q24.13-qter (del-m), 9p23-p22.2 (dup-m), 9p22.21 (del-m), 9p21.3-p23.31 (h-del), 9p21.21-p21.12 (del-m), 9q21.11-q21.13 (dup-m), 9 q21.12-q21.33 (del-m), 10p (del-m), 10q11.22-q21.31 (del-m), 10q22.22-q22.21 (dup-m), 10q22.21-qter (del-m), 12p (del-m), 12q12-q13.12 (del), 12q13.13-q14.31 (dup-m), 12q14.31-q21.1 (h-del), 12q21.12 (h-del), 12q21.31-qter (del), 13q12.11-q32.3 (h-del+del/-m), 14q (del), 15q (del), 16pter-p13.12 (del), 16p13.1-p11.2 (dup-m), 16p12.1-p12.11 (del-m), 17pter-p13.1 (del-m), 17q12-qter (del-m), 18q21.2 (dup-m), 18q21.32-q22.3 (del-m), 19p13.3 (dup-m), 19p13.3-p13.11 (del-m), 19q (del-m), 20p (del-m), 20q (del-m), 21q21.1-qter (del-m), 22q (del-m), Xpter-p11.3 (dup-m), Xp11.3-p11.23 (del-m), Xq11-q21.1 (del-m), Xq21.1-q21.33 (dup-m), Xq21.33-q22.1 (del-m), Xq22.1-q22.3 (dup-m), Xq22.3-q25 (del-m), Xq26.2-qter (dup-m) |
|
| |||
| Blood | No CNVs | CNVs | |
|
| |||
| c) | B3 | 3 | 4q35.2 (del), 7q31.33 (dup), 16p13.3 (del) |
| B7 | 3 | 15q23 (del) (paternal), 19p13.2 (del) (maternal), 21q22.11-q22.12 (dup) (maternal) | |
| B8 | 1 | 8q21.2 (del) | |
| B9 | 2 | 11q12.1 (dup) (maternal), Yp11.32-q12 (dup-m) (de novo) | |
Rearrangements identified by aCGH using the Agilent Human Genome CGH Microarray Kit 180K. a: CNA in 9 pGBMs; b: CNA in primary colture of one pGBM; c: CNVs in four blood samples and their parental origin. P: tumor tissue; B: peripheral blood; C: primary tumor culture. “dup”: duplication (3 doses); “amp”: amplification (>4 doses); “del”: heterozygous deletion; “h-del”: homozygous deletion; -m: mosaicism.
CNA were detected in all tumors (100%) and all chromosomes were involved at least once. The range of CNA was from 4 to 65 (mean 22.55) with a single patient showing 65 CNA both in tumor (P8) (a) and in primary culture (C8) (b).
The two different computational approaches, Genomic Workbench Standard Edition 5.0 software and Shifting Level Model (SLM) and FastCall algorithms, showed overlapping results as summarized in Figure 1.
Figure 1.
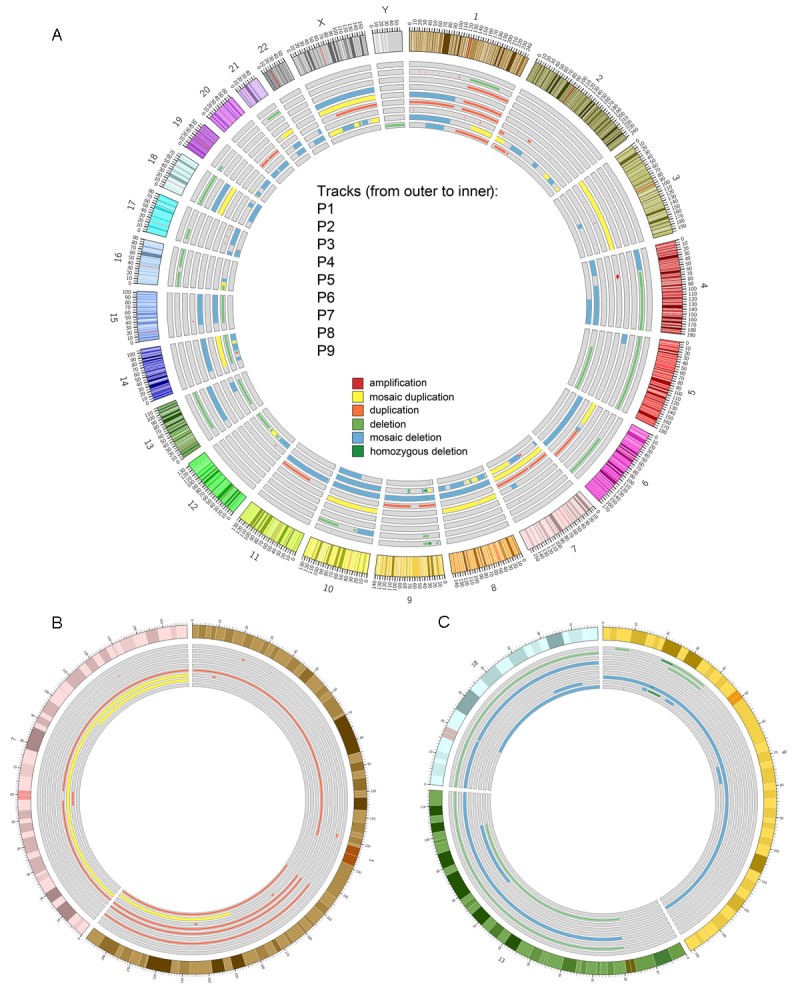
Circos plots of data obtained from the analysis of copy number alterations in pediatric glioblastoma by means of Genomic Workbench Standard Edition 5.0. Events present in the Database of Genomic Variants (DGV) were filtered out and are not reported. Tracks (from outer to inner) refer to samples P1, P2, P3, P4, P5, P6, P7, P8 and P9 while the outer ring represent chromosomes with cytogenetic bands. A: Amplifications, mosaic duplication, duplications, deletions, mosaic deletions and homozygous deletion are reported as tiles (dark red, yellow, light red, light green, light blue and dark green respectively). Mosaicisms (plotted as thicker tiles) may overlap non-mosaic events. B: Peculiar gain events, mosaic duplications (yellow) and duplications (light red) observed in chromosomes 1 and 7 for the aforementioned samples. C: Peculiar loss events (deletions, mosaic deletions and homozygous deletions in light green, light blue and dark green respectively) observed in chromosomes 9, 13 and 18.
The breakpoints of each CNA and CNVs are available upon request. The differences detected for the benign DGV not considered with the first computational approach are reported in Table 3.
Table 3.
Benign DGV
| Chr | Start | End | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| chr1 | 72768884 | 72795450 | -2 | 2 | 0 | 0 | -1 | -2 | 2 | 0 | -1 | NEGR1 |
| chr1 | 104115084 | 104211026 | 0 | 0 | 0 | -2 | -1 | 1 | 0 | 1 | -1 | AMY2B, AMY2A, AMY1A |
| chr1 | 149041962 | 149378236 | 0 | -1 | 2 | 0 | 1 | 1 | 0 | 0 | 1 | NBPF16 |
| chr1 | 248727958 | 248785532 | 0 | 0 | -2 | 0 | 1 | 1 | 0 | 1 | 1 | OR2T34, OR2T10 |
| chr1 | 248808422 | 249212638 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | OR2T27, OR14I1, SH3BP5L, ZNF672, ZNF692, PGBD2 |
| chr2 | 89163891 | 89312560 | 1 | 0 | 1 | 0 | 0 | 1 | 2 | 0 | 2 | RPIA |
| chr4 | 69414770 | 69462408 | 0 | -1 | 2 | 0 | -2 | 1 | 0 | 0 | -2 | UGT2B17 |
| chr8 | 39249381 | 39374759 | 0 | -1 | 0 | -2 | -1 | -2 | 2 | 0 | 0 | ADAM32 |
| chr11 | 55377939 | 55450758 | 2 | 2 | -2 | 0 | 0 | 1 | 0 | 2 | -1 | OR4P4, OR4S2, OR4C6 |
| chr12 | 9637352 | 9672628 | -2 | 0 | 2 | 0 | 0 | 1 | 0 | -2 | -1 | PZP |
| chr14 | 106334936 | 106370856 | 2 | 0 | 2 | 0 | 0 | 0 | 2 | 2 | 2 | TMEM121 |
| chr15 | 20886611 | 21964061 | 0 | -2 | 0 | 0 | 0 | 1 | -1 | -2 | -2 | NBEAP1, POTEB, NF1P2, CT60 |
| chr16 | 32573837 | 33651735 | 0 | -1 | 1 | 0 | 0 | -1 | -1 | 0 | -1 | TP53TG3, TP53TG3B |
Benign DGV: the first column indicates the chromosomes containing recurrent benign DGV, the second and third columns the proximal and distal breakpoints of rearrangements, the fourth to twelfth columns indicate patients P1-P9 with the respective calls: -2 (homozygous deletion); -1 (heterozygous deletion); 1 (duplication); 2 (amplification). The thirteenth column shows the genes included in the benign DGV.
CNA in loss
The most common deleted regions included: chromosome 4q13.1 (P2, P5), 6q14.1 (P2, P5, P7), 9p (P1, P2, P7-P9), 10q23.1-q26.3 (P2, P7-P9), 13q12.1-q34 (P2, P4, P7, P8), 15q (P5, P7, P8), 17p13.2 (P5, P8, P9), 18p11.32-q11.1 (P2, P4) and 18q21.31-q22.3 (P2, P4, P8, P9) (Table 2).
In particular, P4 and P7 exhibited chromosome 13 monosomy, P2 complex 13q rearrangements (deletion 13q12.11-q31.3/duplication 13q31.3/deletion 13q31.3-qter) and P8 a complex deleted region in homozygosity and heterozygosity (13q12.11-q32.3). Cases P5, P7, P8 showed chromosome 15 monosomy, P5 and P7 with mosaic status.
CNA in gain
The most common gain regions involved chromosome 1q32.1-qter (P3, P5-P9) and 7q31.32 (P3, P6-P8). In particular, patients P5, P6 and P9 had duplication of entire chromosome 1 long arm, while P7 had 1q23.2-q23.3 duplication and 1q32.1 amplification. Moreover, 1q25.21-qter mosaic duplication was observed in P8 (Table 2).
For all imbalances, gains and losses, the minimal overlapping region is shown in Figure 1A. At the bottom we indicate the most frequent gains (Figure 1B interesting 1 and 7 chromosomes) and losses (Figure 1C referred to 9, 13 and 18 chromosomes).
Real-time quantitative PCR validation
All CNVs and CNA were validated by Real Time-PCR (data available on request).
Discussion
In the present study we have investigated 9 pGBMs using aCGH and comparing constitutional CNVs and tumor CNA in 4 of them. Given the rarity of the disease, the poor quality and quantity of the bioptic material and lack of literature data regarding this comparison (CNVs and tumor CNA), our findings should better elucidate the genomic background of these tumors.
We identified in our patients a variable number of CNA from a minimum of 4 to a maximum of 65 per case (Table 2), involving different chromosome regions, in various combinations, suggesting genomic instability. The role of common CNVs, as those listed in the DGV (Table 3), has also been evaluated. Constitutional CNVs, in fact, may either represent benign polymorphic variants or be promoted to cancer. On the strength of these considerations, we decided to evaluate the benign DGVs recurring in at least 5 cases, in order to try to identify candidate genes that may be involved or in the genetic background for tumor progression or which could act together with a driver gene of cancer genesis. For example, the DGV on chromosome 16, identified both as deletion (P2, P6, P7, P9) and a duplication (P3), contains targets of p53, the TP53TG3 and TP53TG3B genes.
Despite the fact that cancer is an acquired disease caused by various factors, there is clear evidence that inherited factors play a significant role. Some of them represent loss-of-function mutations in tumor suppressor genes, resulting in a high, relative cancer risk among carriers. However, not only acquired but also inherited CNA may play a role in tumor predisposition and possibly progression. For example, a CNV at chromosome 1q21.1, which included the neuroblastoma breakpoint family gene NBPF23, was found to be associated with the disease [32].
In our P3 case, the recurrent 16p13.3 deletion including A2BP1 gene was found in both tissues (blood and tumor). A2BP1 is expressed exclusively in differentiated neurons and recently Hu J et al., using GBM as a model system, identified A2BP1 deleted in 10% of GBM cases [33]. The role of this imbalance is known to be linked to intellectual disability, but our remark suggests that a congenital CNV could actually be a driver locus promoting cancer. In fact the patient with 16p deletion (B3, P3) was the only one who died 3 months after diagnosis.
In the two cases where we also analyzed the parents of our patients (P7, P9), all constitutional CNVs were inherited from one parent except for a de novo Y chromosome rearrangement.
In 5 patients we identified rearrangements on 9p, variable in size: one region of homozygous deletion at 9p21.3 (P1, P7, P8) and another one with heterozygous 9p21.2-p21.1 (P1, P2, P7, P8) deletion.
Either homozygous or heterozygous 9p21.3 deletion, including both or one of the two genes CDKN2A and CDKN2B, had been reported in a wide variety of tumors [34] including adult GBM [35]. These two genes act as a negative controller of cell cycle progression [36]. Heterozygous deletion at 9p21.2-p21.1 includes 12 genes: TUSC1, C9orf82, LRRC19, TEK, C9orf11, LINC00032, MOB3B, IFNK, C9orf72, LINGO2, MIR876, and MIR873. We have modest information about the cellular function of these genes. TUSC1 is interesting because it was found to be underexpressed in a study on lung cancer and it has been designated as a hypothetical oncosuppressor gene [37].
We also identified recurrent large deletions of chromosomes: 13q, 18p, 18q and 15q. In regard to the 13q deletion, all of them included the RB1 gene (13q14.2), also implicated in pGBM genesis [38]. Four patients (P2, P7-P9) had LOH of 10q where the PTEN gene was also included. This gene has been identified as a tumor suppressor, mutated in a large number of cancers at high frequency [39,40]. Interestingly, its deletion/mutation has also been described in astrocytic tumors [9,41].
Three patients (P5, P8, P9) had heterozygous deletion at 17p13.2 in which 3 genes are present: ZZEF1, CYB5D2 and ANKFY1 which have heretofore never been described in association with particular tumors. No rearrangements were found involving TP53 locus.
The most frequent CNA in gain was 1q duplication (6 patients out of 9), 5 with a minimal overlapping region interesting 1q31.3-q44. This imbalance has been reported in several types of tumors [42,43]. Gain of 1q is associated with poor prognoses in various pediatric tumors as well as anaplastic astrocytomas and glioblastoma [44], ependymoma [45] and medulloblastoma [46]. In our cases P3, P5, P6, P8 and P9 the proximal breakpoint at 1q31.3 maps a region of segmental duplications, possibly making it prone to genomic rearrangements [47]. In P7 an amplification within the 1q32.1 region was observed. The PLEKHA6 gene encoding for a protein involved in intracellular signaling that can act as one of cytoskeleton components was included in the amplification.
We had four patients (P3, P6-P8) with duplication of chromosome 7 with a minimal overlapping region at 7q31.32 containing 13 candidate oncogenes: GRM8, KIAA1549, ZC3HAV1L, UBN2, LUC7L2, KLRG2, HIPK2, PARP12, JHDM1D, RAB19, MKRN1, ADCK2, BRAF. BRAF plays a role in regulating the MAP kinase/ERKs signaling pathway. Acquired alterations in the BRAF gene have been associated with various cancers, including pilocytic astrocytoma (WHO-grade I) [48,49].
Conclusions and future perspectives
The past decade of research has been invaluable in increasing our understanding of GBM. However, several outstanding questions remain. First of all, tumor heterogeneity is still poorly characterized both in adult and pediatric GBMs. The cases reported up to now are sporadic at least as regards the published literature. This situation is compatible with a de novo mutation/variation--either germline or postzygotic. In the latter case, we may assume both that the mutation occurs early in embryo and that it is limited to brain cells although the occurrence of the tumor in childhood may suggest some DNA variation present in all body cells. Moreover, some genes located in recurrent benign DGV could act as genetic background which promotes cancer. Few whole-genome studies profiling germline CNVs have been conducted in cohorts of cancer-predisposing patients negative for mutations in the major genes related to their specific cancer [50]. It is plausible that a germline variant associated with tumor development is an event increasing the probability of tumor occurrence and decreasing the time necessary for cancer manifestation. For example, a 9p21.3 germline microdeletion was recently reported in two different types of tumors, colorectal [51] and breast cancer [52]. This finding indeed supports a pathogenic role of 9p21.3 germline deletion in cancer predisposition.
All our patients had a highly overlapping clinical history, a histological picture demonstrating a severe grade IV tumor since the onset of symptoms, and a poor prognosis with an average survival of 8 months (from 3 to 19). Thus, the condition seems to be genetically homogeneous, suggesting mutation(s) in a specific driver gene acting by a dominant model, not excluding the occurrence of a second hit for initiating the tumorigenesis process.
Our study was aimed to highlight any possible recurrent genomic imbalance associated with the origin and/or the progression of the tumor. We did not find any obvious CNA common to all patients although losses are more common than gains. Although six cases (P1, P2, P4, P7-P9) presented more CNA in loss than in gain, and 3 cases (P3, P5, P6) vice versa, the t-test showed no statistically significant difference between the events of deletion and duplication.
Literature data describe copy number imbalances both in adult and pediatric GBMs and more deletions than duplications have been detected in pediatric cases than adult GBMs. Perhaps the most intriguing finding is the 1q32.1-q44 duplication detected in 6 of our patients. Of them, P3 showed only this imbalance in the tumor biopsy, a constitutional CNV (16p13.3 deletion) and died 3 months after diagnosis.
In summary, our data further define the CNA present in pGBM, showing that no specific imbalances are involved, apart from the 9p21.3 and 16p13.3 deletions and 1q32.1-q44 duplication. The focal events we detected in form of amplification/deletion at the moment suggest that the A2BP1 gene is one possible culprit of the disease. Our data also could be used to compare the constitutional CNVs and CNA acquired, because at the moment there are no works in the literature that have developed our own experimental approach. Possibly only Next Generation Sequencing will be able to solve the problem.
Acknowledgements
This work was supported by grants from AMICODIVALERIO ASSOCIAZIONE ONLUS and ASSOCIAZIONE NOI PER VOI PER IL MEYER O.N.L.U.S.
Disclosure of conflict of interest
None.
References
- 1.Arora RS, Alston RD, Eden TO, Estlin EJ, Moran A, Birch JM. Age-incidence patterns of primary CNS tumors in children, adolescents, and adults in England. Neuro Oncol. 2009;11:403–413. doi: 10.1215/15228517-2008-097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suri V, Das P, Pathak P, Jain A, Sharma MC, Borkar SA, Suri A, Gupta D, Sarkar C. Pediatric glioblastomas: a histopathological and molecular genetic study. Neuro Oncol. 2009;11:274–280. doi: 10.1215/15228517-2008-092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman GK, Spiller SE, Harrison DK, Fiveash JB, Reddy AT. Treatment of children with glioblastoma with conformal radiation, temozolomide, and bevacizumab as adjuncts to surgical resection. J Pediatr Hematol Oncol. 2013;35:e123–6. doi: 10.1097/MPH.0b013e318282cd7f. [DOI] [PubMed] [Google Scholar]
- 4.Nomura Y, Yasumoto S, Yanai F, Akiyoshi H, Inoue T, Nibu K, Tsugu H, Fukushima T, Hirose S. Survival and late effects on development of patients with infantile brain tumor. Pediatr Int. 2009;51:337–344. doi: 10.1111/j.1442-200X.2008.02760.x. [DOI] [PubMed] [Google Scholar]
- 5.Pollack IF, Finkelstein SD, Woods J, Burnham J, Holmes EJ, Hamilton RL, Yates AJ, Boyett JM, Finlay JL, Sposto R; Children’s Cancer Group. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346:420–427. doi: 10.1056/NEJMoa012224. [DOI] [PubMed] [Google Scholar]
- 6.Pollack IF, Boyett JM, Yates AJ, Burger PC, Gilles FH, Davis RL, Finlay JL Children’s Cancer Group. The influence of central review on outcome associations in childhood malignant gliomas: results from the CCG-945 experience. Neuro Oncol. 2003;5:197–207. doi: 10.1215/S1152-8517-03-00009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganigi PM, Santosh V, Anandh B, Chandramouli BA, Sastry Kolluri VR. Expression of p53, EGFR, pRb and bcl-2 proteins in pediatric glioblastoma multiforme: a study of 54 patients. Pediatr Neurosurg. 2005;41:292–299. doi: 10.1159/000088731. [DOI] [PubMed] [Google Scholar]
- 8.Pollack IF, Hamilton RL, James CD, Finkelstein SD, Burnham J, Yates AJ, Holmes EJ, Zhou T, Finlay JL Children’s Oncology Group. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children’s Cancer Group 945 cohort. J Neurosurg. 2006;105:418–424. doi: 10.3171/ped.2006.105.5.418. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura M, Shimada K, Ishida E, Higuchi T, Nakase H, Sakaki T, Konishi N. Molecular pathogenesis of pediatric astrocytic tumors. Neuro Oncol. 2007;9:113–123. doi: 10.1215/15228517-2006-036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lütolf UM, Kleihues P. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 11.Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von Deimling A. PTEN mutations in gliomas and glioneuronal tumors. Oncogene. 1998;16:2259–2264. doi: 10.1038/sj.onc.1201756. [DOI] [PubMed] [Google Scholar]
- 12.Reifenberger G, Collins VP. Pathology and molecular genetics of astrocytic gliomas. J Mol Med. 2004;82:656–670. doi: 10.1007/s00109-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 13.Di Sapio A, Morra I, Pradotto L, Guido M, Schiffer D, Mauro A. Molecular genetic changes in a series of neuroepithelial tumors of childhood. J Neurooncol. 2002;59:117–122. doi: 10.1023/a:1019697117253. [DOI] [PubMed] [Google Scholar]
- 14.Bredel M, Pollack IF, Hamilton RL, James CD. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res. 1999;5:1786–1792. [PubMed] [Google Scholar]
- 15.Kraus JA, Felsberg J, Tonn JC, Reifenberger G, Pietsch T. Molecular genetic analysis of the TP53, PTEN, CDKN2A, EGFR, CDK4 and MDM2 tumour-associated genes in supratentorial primitive neuroectodermal tumours and glioblastomas of childhood. Neuropathol Appl Neurobiol. 2002;28:325–333. doi: 10.1046/j.1365-2990.2002.00413.x. [DOI] [PubMed] [Google Scholar]
- 16.Rasheed BK, McLendon RE, Herndon JE, Friedman HS, Friedman AH, Bigner DD, Bigner SH. Alterations of the TP53 gene in human gliomas. Cancer Res. 1994;54:1324–1330. [PubMed] [Google Scholar]
- 17.Sure U, Rüedi D, Tachibana O, Yonekawa Y, Ohgaki H, Kleihues P, Hegi ME. Determination of p53 mutations, EGFR overexpression, and loss of p16 expression in pediatric glioblastomas. Neuropathol Exp Neurol. 1997;56:782–789. [PubMed] [Google Scholar]
- 18.Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, LaFramboise WA, Burger PC, Brat DJ, Rosenblum MK, Holmes EJ, Zhou T, Jakacki RI Children’s Oncology Group. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011;27:87–94. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaaf CP, Wiszniewska J, Beaudet AL. Copy number and SNP arrays in clinical diagnostics. Annu Rev Genomics Hum Genet. 2011;12:25–51. doi: 10.1146/annurev-genom-092010-110715. [DOI] [PubMed] [Google Scholar]
- 20.Cowell JK, Matsui S, Wang YD, LaDuca J, Conroy J, McQuaid D, Nowak NJ. Application of bacterial artificial chromosome array-based comparative genomic hybridization and spectral karyotyping to the analysis of glioblastoma multiforme. Cancer Genet Cytogenet. 2004;151:36–51. doi: 10.1016/j.cancergencyto.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Hui AB, Takano H, Lo KW, Kuo WL, Lam CN, Tong CY, Chang Q, Gray JW, Ng HK. Identification of a novel homozygous deletion region at 6q23.1 in medulloblastomas using high-resolution array comparative genomic hybridization analysis. Clin Cancer Res. 2005;11:4707–4716. doi: 10.1158/1078-0432.CCR-05-0128. [DOI] [PubMed] [Google Scholar]
- 22.Qu HQ, Jacob K, Fatet S, Ge B, Barnett D, Delattre O, Faury D, Montpetit A, Solomon L, Hauser P, Garami M, Bognar L, Hansely Z, Mio R, Farmer JP, Albrecht S, Polychronakos C, Hawkins C, Jabado N. Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol. 2010;12:153–163. doi: 10.1093/neuonc/nop001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rickert CH, Sträter R, Kaatsch P, Wassmann H, Jürgens H, Dockhorn-Dworniczak B, Paulus W. Pediatric high-grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol. 2001;158:1525–1532. doi: 10.1016/S0002-9440(10)64103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG, Baker SJ. Integrated Molecular Genetic Profiling of Pediatric High-Grade Gliomas Reveals Key Differences With the Adult Disease. J. Clin. Oncol. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Massimino M, Gandola L, Luksch R, Spreafico F, Riva D, Solero C, Giangaspero F, Locatelli F, Podda M, Bozzi F, Pignoli E, Collini P, Cefalo G, Zecca M, Casanova M, Ferrari A, Terenziani M, Meazza C, Polastri D, Scaramuzza D, Ravagnani F, Fossati-Bellani F. Sequential chemotherapy, high-dose thiotepa, circulating progenitor cell rescue, and radiotherapy for childhood highgrade glioma. Neuro Oncol. 2005;7:41–48. doi: 10.1215/S1152851704000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 27.Biassoni V, Casanova M, Spreafico F, Gandola L, Massimino M. A case of relapsing glioblastoma multiforme responding to vinorelbine. J Neurooncol. 2006;80:195–201. doi: 10.1007/s11060-006-9176-3. [DOI] [PubMed] [Google Scholar]
- 28.Galanis E, Buckner JC, Maurer MJ, Sykora R, Castillo R, Ballman KV, Erickson BJ. Validation of neuroradiologic response assessment in gliomas: measurement by RECIST, two-dimensional, computer-assisted tumor area, and computer-assisted tumor volume methods. Neuro Oncol. 2006;8:156–165. doi: 10.1215/15228517-2005-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magi A, Benelli M, Marseglia G, Nannetti G, Scordo MR, Torricelli F. A shifting level model algorithm that identifies aberrations in array-CGH data. Biostatistics. 2010;11:265–280. doi: 10.1093/biostatistics/kxp051. [DOI] [PubMed] [Google Scholar]
- 31.Box JF. Guinness, Gosset, Fisher, and Small Samples. Statist Sci. 1987;2:1–104. [Google Scholar]
- 32.Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mossé YP, Wood A, Lynch JE, Pecor K, Diamond M, Winter C, Wang K, Kim C, Geiger EA, McGrady PW, Blakemore AI, London WB, Shaikh TH, Bradfield J, Grant SF, Li H, Devoto M, Rappaport ER, Hakonarson H, Maris JM. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459:987–991. doi: 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu J, Ho AL, Yuan L, Hu B, Hua S, Hwang SS, Zhang J, Hu T, Zheng H, Gan B, Wu G, Wang YA, Chin L, DePinho RA. From the Cover: Neutralization of terminal differentiation in gliomagenesis. Proc Natl Acad Sci U S A. 2013;110:14520–7. doi: 10.1073/pnas.1308610110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novara F, Arcaini L, Merli M, Passamonti F, Zibellini S, Rizzi S, Rattotti S, Rumi E, Pascutto C, Vetro A, Astori C, Boveri E, Lucioni M, Paulli M, Zuffardi O, Lazzarino M. High-resolution genomewide array comparative genomic hybridization in splenic marginal zone B-cell lymphoma. Hum Pathol. 2009;40:1628–1637. doi: 10.1016/j.humpath.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 35.Riemenschneider MJ, Jeuken JW, Wesseling P, Reifenberger G. Molecular diagnostics of gliomas: state of the art. Acta Neuropathol. 2010;120:567–584. doi: 10.1007/s00401-010-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soto JL, Cabrera CM, Serrano S, López-Nevot MA. Mutation analysis of genes that control the G1/S cell cycle in melanoma: TP53, CDKN1A, CDKN2A, and CDKN2B. BMC Cancer. 2005;5:36. doi: 10.1186/1471-2407-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shan Z, Shakoori A, Bodaghi S, Goldsmith P, Jin J, Wiest JS. TUSC1, a Putative Tumor Suppressor Gene, Reduces Tumor Cell Growth In Vitro and Tumor Growth In Vivo. PLoS One. 2013;8:e66114. doi: 10.1371/journal.pone.0066114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agamanolis DP, Malone JM. Chromosomal abnormalities in 47 pediatric brain tumors. Cancer Genet Cytogenet. 1995;81:125–134. doi: 10.1016/0165-4608(94)00123-s. [DOI] [PubMed] [Google Scholar]
- 39.Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, Ittmann M. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Nat Acad Sci U S A. 2001;98:11563–11568. doi: 10.1073/pnas.201167798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nature Genet. 2002;32:355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 41.Ichimura K, Schmidt EE, Miyakawa A, Goike HM, Collins VP. Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Genes Chromosomes Cancer. 1998;22:9–15. doi: 10.1002/(sici)1098-2264(199805)22:1<9::aid-gcc2>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 42.Balcárková J, Urbánková H, Scudla V, Holzerová M, Bacovský J, Indrák K, Jarosová M. Gain of chromosome arm 1q in patients in relapse and progression of multiple myeloma. Cancer Genet Cytogenet. 2009;192:68–72. doi: 10.1016/j.cancergencyto.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 43.Szponar A, Zubakov D, Pawlak J, Jauch A, Kovacs G. Three genetic developmental stages of papillary renal cell tumors: duplication of chromosome 1q marks fatal progression. Int J Cancer. 2009;124:2071–2076. doi: 10.1002/ijc.24180. [DOI] [PubMed] [Google Scholar]
- 44.Faria C, Miguéns J, Antunes JL, Salgado D, Nunes S, Barroso C, Martins Mdo C, Nunes VM, Roque L. Pediatric brain tumors: genetics and clinical outcome. J Neurosurg Pediatr. 2010;5:263–70. doi: 10.3171/2009.10.PEDS09240. [DOI] [PubMed] [Google Scholar]
- 45.Hirose Y, Aldape K, Bollen A, James CD, Brat D, Lamborn K, Berger M, Feuerstein BG. Chromosomal abnormalities subdivide ependymal tumors into clinically relevant groups. Am J Pathol. 2001;158:1137–1143. doi: 10.1016/S0002-9440(10)64061-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lo KC, Ma C, Bundy BN, Pomeroy SL, Eberhart CG, Cowell JK. Gain of 1q is a potential univariate negative prognostic marker for survival in medulloblastoma. Clin Cancer Res. 2007;13:7022–7028. doi: 10.1158/1078-0432.CCR-07-1420. [DOI] [PubMed] [Google Scholar]
- 47.Sawyer JR, Tricot G, Lukacs JL, Binz RL, Tian E, Barlogie B, Shaughnessy J Jr. Genomic Instability in Multiple Myeloma: Evidence for Jumping Segmental Duplications of Chromosome Arm 1q. Genes Chromosomes Cancer. 2005;42:95–106. doi: 10.1002/gcc.20109. [DOI] [PubMed] [Google Scholar]
- 48.Jones DT, Kocialkowski S, Liu L, Pearson DM, Bäcklund LM, Ichimura K, Collins VP. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones DW, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549: BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–2123. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krepischi AC, Pearson PL, Rosenberg C. Germline copy number variations and cancer predisposition. Future Oncol. 2012;8:441–450. doi: 10.2217/fon.12.34. [DOI] [PubMed] [Google Scholar]
- 51.Venkatachalam R, Verwiel ET, Kamping EJ, Hoenselaar E, Görgens H, Schackert HK, van Krieken JH, Ligtenberg MJ, Hoogerbrugge N, van Kessel AG, Kuiper RP. Identification of candidate predisposing copy number variants in familial and early-onset colorectal cancer patients. Int J Cancer. 2011;129:1635–1642. doi: 10.1002/ijc.25821. [DOI] [PubMed] [Google Scholar]
- 52.Krepischi AC, Achatz MI, Santos EM, Costa SS, Lisboa BC, Brentani H, Santos TM, Gonçalves A, Nóbrega AF, Pearson PL, Vianna-Morgante AM, Carraro DM, Brentani RR, Rosenberg C. Germline DNA copy number variation in familial and early-onset breast cancer. Breast Cancer Res. 2012;14:R24. doi: 10.1186/bcr3109. [DOI] [PMC free article] [PubMed] [Google Scholar]