

Significance
The characterization of rare monogenic human disorders can and has yielded unique biological insights. Our phenotypic description and functional characterization of human loss-of-function mutations in PCYT1A is both clinically important for patients and their families afflicted with this rare but serious metabolic disease and biologically helpful in advancing understanding of the physiological consequences of impaired PCYT1A activity in humans.
Abstract
Phosphatidylcholine (PC) is the major glycerophospholipid in eukaryotic cells and is an essential component in all cellular membranes. The biochemistry of de novo PC synthesis by the Kennedy pathway is well established, but less is known about the physiological functions of PC. We identified two unrelated patients with defects in the Kennedy pathway due to biallellic loss-of-function mutations in phosphate cytidylyltransferase 1 alpha (PCYT1A), the rate-limiting enzyme in this pathway. The mutations lead to a marked reduction in PCYT1A expression and PC synthesis. The phenotypic consequences include some features, such as severe fatty liver and low HDL cholesterol levels, that are predicted by the results of previously reported liver-specific deletion of murine Pcyt1a. Both patients also had lipodystrophy, severe insulin resistance, and diabetes, providing evidence for an additional and essential role for PCYT1A-generated PC in the normal function of white adipose tissue and insulin action.
All living cells are surrounded by a lipid membrane. Eukaryotic cells also contain several internal membrane-bound organelles, which enable them to compartmentalize related biological functions and thereby to enhance the efficiency of these processes. Phospholipids are the predominant component of these membranes. Their hydrophilic head groups interact with the cytosol, whereas their hydrophobic side chains are either buried within the hydrophobic interior of a typical membrane bilayer or interact with the hydrophobic neutral lipid core of lipoproteins and lipid droplets (LDs). Phospholipids are generally defined by their organic head group with phosphatidylcholine (PC) constituting over 50% of all membrane phospholipids. PC was first isolated in the 19th century and the major enzymatic pathway involved in its synthesis was revealed by Kennedy and Weiss (1) in the 1950s. Cells synthesize PC in three consecutive steps (Fig. 1A): choline kinase phosphorylates choline before choline phosphate cytidylyltransferase 1 α (encoded by the PCYT1A gene) generates the high-energy donor CDP-choline in the rate-limiting step of the pathway. In the last step, DAG:CDP-choline cholinephosphotransferase (CPT) uses CDP-choline and diacylglycerol (DAG) to form PC (2, 3).
Fig. 1.

Cosegregation of biallelic PCYT1A mutations with fatty liver, low HDL cholesterol levels, lipodystrophy, insulin-resistant diabetes, and short stature. (A) Schematic illustration of the Kennedy PC synthesis pathway. CK, choline kinase; CPT, CDP-choline:1,2-diacylglycerol cholinephosphotransferase; PCYT1A, choline-phosphate cytidylyltransferase A, CTP:phosphocholine-cytidylyltransferase. (B) Family pedigrees of both probands demonstrating that only compound heterozygous carriers of PCYT1A mutations manifest fatty liver (red), low HDL cholesterol (blue), lipodystrophy (yellow), and insulin resistance/type 2 diabetes (T2DM) (green). PCYT1A mutation status, height (Ht.), and body mass index (BMI) are indicated below each individual’s symbol. ND, not determined; WT, wild type. (C) The location of PCYT1A mutations E280del, V142M, and 333fs in relation to known functional domains of PCYT1A. Domain M, membrane binding domain; domain P, phosphorylated region. (D) Conservation around the V142(red*) and E280(red*) mutation sites. Sequence alignment of representative metazoan sequences in the region surrounding the mutated residues. Hydrophobic (blue) and polar (green) residues interacting with V142 are highlighted. Only residues different from the human sequence are shown. Sequence IDs: human (Homo sapiens) P49585, zebrafish (Danio rerio) F1QEN6, sea squirt (Ciona intestinalis) XP_002130773.1, sea urchin (Strongylocentrotus purpuratus) H3I3V9, water flee (Daphnia pulex) E9G1P5, Drosophila (D. melanogaster) Q9W0D9, Caenorhabditis (C. elegans) P49583, Trichoplax (T. adherens) B3RI62. (E and F) Structure of the catalytic domain of PCYT1A highlighting the role of V142M in the core packing. The two chains in the dimer are shown in yellow and gray; the residues and the secondary structure units are highlighted in color in the yellow monomer A: loop L3 with V142, red; α-helix, green; and the interacting β-sheet, blue. The residues packing with V142 are shown in ball-and-stick and space-filling representations, the dimer stabilizing R140 is shown in ball-and-stick colored according to the atom type. E is a global view, and F is a zoomed-in view of the catalytic core.
Membrane phospholipids are a defining feature of advanced life-forms so it is perhaps not surprising that the pathways involved in their synthesis are ancient, and mutations affecting them are rarely tolerated in evolution. Here, we describe the identification and characterization of pathogenic human loss-of-function mutations affecting the eponymous Kennedy pathway.
Case Histories
We studied two female patients with similar metabolic syndromes characterized by childhood presentation with severe nonalcoholic fatty liver disease, lipodystrophy, dyslipidemia in which the dominant feature was very low HDL cholesterol levels, insulin-resistant diabetes, and short stature (Table 1) (4, 5).
Table 1.
Metabolic characteristics, adipose tissue distribution, and liver steatosis in patients with mutations in PCYT1A
| Proband 1 | Proband 2 | ||
| PCYT1A mutations | |||
| p.Glu280del p.Val142Met | p.Glu280del. p.333fs | Reference ranges | |
| Age, y | 18 | 10.9 | |
| Sex | Female | Female | |
| BMI, kg/m2 | 17.9 | 17.9 | 18–25 |
| BMI Z score | −1.48 | 0.18 | |
| Glucose, mg/dL | 126 | 109 | 80–101 |
| Insulin, pM | 114 | 451 | 13.9–62.5 |
| Triglyceride, mg/dL | 301 | 416 | 35–135 |
| Cholesterol, mg/dL | 85 | 255 | 160–240 |
| HDL cholesterol, mg/dL | 14 | 33 | 50–80 |
| NEFA, µmol/L | 101 | ND | 100–600 |
| Leptin, µg/L | 1.7 | 0.95 | * |
| Adiponectin, mg/L | 0.2 | ND | 3.9–12.9 |
| Total fat, % | 5.0 | 21.2 | 27.2–33.3 (4) |
| Upper limb fat, % | 4.3 | 23.8 | 26.5–33.9 (4) |
| Trunk fat, % | 5.2 | 24.7 | 25.7–32.3 (4) |
| Lower limb fat, % | 4.4 | 12.6 | 30.1–36.1 (4) |
| Total lean mass, % | 91.5 | 76.4 | 60.5–70.7 (4) |
| Fatty liver, % | 76.7 | Yes† | <8 |
| DNL, % | 6.7 | ND | 2.1 ± 1.3‡ |
Body composition was assessed using dual energy X-ray absorptiometry. Fatty liver was assessed by magnetic resonance spectroscopy in proband 1 and ultrasound and biopsy in proband 2. BMI, body mass index; DNL, hepatic de novo lipogenesis; ND, not determined.
Leptin BMI-adjusted reference ranges for women: BMI, <25 kg/m2 → 2.4–24.4 μg/L.
Grade 3/3 steatosis, 3/4 fibrosis, 2/2 ballooning injury on biopsy using NASH Clinical Research Network scoring system (5).
Reference presented as mean ± SD from eight healthy controls.
Proband 1 (P1) was the only affected child of unrelated unaffected parents and had four unaffected sisters (Fig. 1B). She initially presented, aged 18 y, with type 2 diabetes. Her body mass index (BMI) was 17.9 kg/m2, and her total fat mass (6.5%) was very low in keeping with lipodystrophy (Fig. S1A). However, in contrast to most patients with congenital lipodystrophy, her height was significantly less than predicted (height SD score, −2.8), and her lean mass was not increased. At presentation, she was extremely insulin resistant (requiring very high-dose insulin therapy to control her diabetes) hypertriglyceridemic, and had severe hepatic steatosis (Table 1). Her HDL cholesterol level was particularly low and remained low even after normalization of her triglyceride levels. We used stable isotopes to measure her rate of hepatic de novo lipogenesis and found this to be significantly elevated (Table 1).
Proband 2 (P2) was also the only affected member of a smaller family with one unaffected brother (Fig. 1B). She presented at age 3 mo with pneumonia and was noted to have abnormal liver enzyme levels. A liver biopsy revealed hepatic steatosis. Her height was below the third centile for age throughout childhood, and near-adult height was 148 cm (at age 16.8 y, bone age, 15.5 y, suggesting growth 99.6% complete). At age 9 y, progressive limb fat loss developed. Subsequent evaluation at the National Institutes of Health revealed lipodystrophy (Fig. S1B), hypertriglyceridemia, low HDL cholesterol, severe insulin resistance, impaired glucose tolerance, and nonalcoholic steatohepatitis (Table 1).
Results
Genetic and Bioinformatic Analysis.
Neither proband was found to have mutations in genes previously linked to fatty liver disease (PNPLA3) or lipodystrophy (LMNA, PPARG, BSCL2, AGPAT2, AKT2, CIDEC, PLIN1, and CAV1). The fact that both P1 and P2 were born to unrelated parents and were the sole affected family member, lead us to hypothesize the presence of either biallelic or de novo mutations. To test this, we sequenced the exome of the probands and their parents and analyzed the genomic data for de novo variants, as well as for compound heterozygous mutations in the probands. Resulting variant lists were filtered to exclude variants unlikely to alter protein function or those common in control populations. None of the genes with de novo mutations in P1 appeared likely to account for the observed phenotype (Table S1) and no rare de novo variants remained in P2 after filtering for common variation. There were nine genes containing rare “functional” (as defined in SI Methods) compound heterozygous mutations; however, only one of these, PCYT1A, was common to both patients (Table S2). P1 was compound heterozygous for [c.838_840delCTC] p.E280del, inherited from her father, and [c.424G > A] p.V142M, inherited from her mother. P2 shared the p.E280del mutation, also inherited from her father, and inherited a second heterozygous mutation ([c.996delC] p.S333Lfs*164) from her mother (Fig. 1B). All three mutations were absent from 1,092 individuals from the 1000 Genomes Project Consortium (6), 6,250 individuals from the National Heart, Lung, and Blood Institute (NHLBI) Grand Opportunity (GO) Exome Sequencing Project, and 4,190 internal exome and genome controls, suggesting that these mutations arose recently. Although the two families have no known shared ancestry, genotyping of the Illumina HumanOmniExpress BeadChip array confirmed that each inherited the p.E280del from a common ancestral haplotype >713 kb long, arising 42–51 generations (∼1,260–1,530 y) ago, estimated from the local recombination rate.
The mutation found in both kindreds, referred to herein as E280del, was a trinucleotide deletion, predicted to result in the in-frame omission of a single glutamine (E) residue (Fig. 1C). E280 lies within the amphipathic membrane binding helical region of the protein and is conserved in all available deuterostomian sequences (Fig. 1D). None of the sequences contains an insertion or deletion over the whole sequence of the amphipathic region, suggesting that the exact sequence length is functionally important. The conservation surrounding E280 is much higher than that within the overall helical region. P1 was also heterozygous for p.V142M. V142 is located within a loop (L3, Fig. 1E) between an α-helix (green in Fig. 1E) and a β-strand (blue in Fig. 1E) of the catalytic domain of PCYT1A (7). Its backbone is involved in dimerization of the catalytic domain through a hydrogen bond with the side chain of R140, whereas its side chain lies within the highly conserved hydrophobic core of the catalytic domain. Here, it forms hydrophobic interactions with L108, V110, and V136, stabilizing the enzyme’s core (Fig. 1F). All of these residues are conserved in metazoa (Fig. 1D). Mutation of V142 to methionine is expected to destabilize the entire structural domain. The second mutation found in P2 was a single-nucleotide deletion (c.996delC) resulting in a frameshift (herein referred to as 333fs) leading to a considerably elongated C terminus missing several phosphorylation sites (Fig. 1C). The precise function of this highly phosphorylated C terminus remains unclear.
Functional Studies.
To determine the effect of these mutations on protein expression, we immunoblotted lysates from EBV-transformed lymphocytes (EBVLs) from each proband with a PCYT1A antibody that targets an N-terminal epitope. In both cases, PCYT1A protein was barely detectable (Fig. 2A). These observations were confirmed by immunoblotting of lysates from primary fibroblasts (Fig. 2B). PCYT1A protein expression was also reduced in EBVL lysates from the heterozygous parents of P2 (Fig. 2A). PCYT1A mRNA expression was similar to control levels in cells derived from the patients (Fig. S2 A and B). Although reduced expression of the V142M and 333fs mutants is consistent with the expected destabilizing effects of these mutations, we were surprised by the near absence of the E280del mutant. When expressed in heterologous COS7 cells, expression of myc-tagged E280del was similar to wild-type protein levels, whereas expression of the other mutants was more significantly reduced (Fig. S2C) (6). [35S]Methionine/cysteine labeling confirmed that the E280del mutant was expressed in skin fibroblasts but then more rapidly degraded than wild-type protein (Fig. 2C).
Fig. 2.

Functional consequences of the mutations on PCYT1A expression in primary cells. (A and B) Lysates were prepared from EBVLs (A) or skin fibroblasts (B) from each proband (P1 and P2) and three healthy controls (C1–3). EBVL lysates from the unaffected parents (R1–R2) of P2 are also included in A. These were immunoblotted for PCYT1A and β-Actin (loading control) expression. (C) Primary skin fibroblasts from proband 2 were l-methionine/l-cysteine starved for 30 min and pulse labeled with [35S]methionine/cysteine for 4 h before they were chased in fibroblast culturing medium for 0–4 h. PCYT1A was then immunoprecipitated from cell lysates before being subjected to SDS/PAGE and autoradiography. (D) mCherry-PCYT1A constructs [wild type (WT) and E280del] were expressed in Drosophila S2 cells in the absence (−) or presence (+) of oleic acid (OA)-supplemented culture medium. Lipid droplets (LDs) were stained with Bodipy. (Scale bars: 5 µm.) (E) Normalized fluorescence intensity/recovery of mCherry-PCYT1A constructs [wild type (WT), black symbols, and E280del, blue symbols]. Signal of the bleached lipid droplet (LD) was normalized to that of an unbleached LD in the same cell. Values are means ± SD of three experiments. Apparent off-rates (koff) were calculated after curve fitting of the data with exponential functions (red trace). See Movies S1 and S2 for representative images of these data. (F) Helical projections of the membrane binding amphipathic helical domain of wild-type (WT) and E280del PCYT1A (amino acids 241–294). Amino acid one-letter code sizes are proportional to amino acid volumes. Cationic amino acids are shown in blue, anionic amino acids in red, and hydrophobic amino acids in yellow. The putative hydrophobic faces of the amphipathic helices are indicated by yellow dotted lines. N and C termini of the helices are indicated with red letters. E280, the amino acid deleted by the E280del mutation, is shown by a black arrow.
Because deletion of E280 results in the loss of a negative charge within an amphipathic helix, which is involved in the association of PCYT1A with phospholipid membranes and with the phospholipid monolayer surrounding LDs deficient in PC (8), we next examined the impact of E280del in the latter experimental paradigm. When human E280del mutant PCYT1A was expressed in Drosophila S2 cells, it localized to LDs in contrast to a previously reported artificial W397E mutant in this domain (8) (Fig. 2D). However, fluorescence recovery after photobleaching analyses showed that LD localized E280del mutant rapidly equilibrates with a cytosolic pool compared with stably bound wild-type PCYT1A, implying a faster “off” rate (Fig. 2E and Movies S1 and S2). These altered protein–membrane interactions presumably reflect the predicted change in direction and absolute value of the hydrophobic moment of the PCYT1A helix (Fig. 2F) and may contribute to the significant reduction in expression of this mutant in vivo, as was previously reported for artificial mutants with impaired membrane binding affinity studied in vitro (9).
Mammals generally express an α (PCYT1A) and β (PCYT1B) isoform of this enzyme (2). These are expressed at similar levels in the brain, but in most other tissues PCYT1A is expressed at ∼10-fold higher levels, likely explaining why Pcyt1a-null mice die in utero (10). To assess the impact of the PCYT1A mutations on PC synthesis, we used [3H]choline to measure Kennedy pathway PC synthesis in EBVLs and fibroblasts from both patients. PC synthesis was very significantly reduced, although not undetectable, in both cell types from both patients (Fig. 3 A and B), resulting in a reduction in the levels of PC relative to those of phosphatidylethanolamine (PE) (Fig. 3C). These data highlight the importance of the PCYT1A isoform in these cell types and suggest that neither PCYT1B nor phosphatidylethanolamine N-methyltransferase (PEMT), which can catalyze PC synthesis via an alternative pathway, is able to compensate fully for the loss of PCYT1A. PCYT1B and PEMT mRNA expression were similar in patient and control cells (Fig. S2 A and B).
Fig. 3.
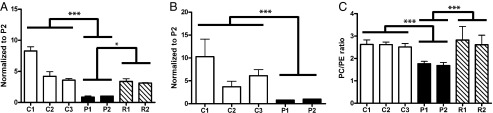
Metabolic consequences of reduced Pcyt1a expression. (A and B) Phosphatidylcholine (PC) synthesis via the Kennedy pathway was determined in EBVLs (A) and in primary skin fibroblasts (B) from both patients (P1–2) and, in the case of EBVLs, from the unaffected parents (R1–2) of P2. These data were compared with measurements in cell lines from three healthy controls (C1–3). Data represent mean ± SE of three separate experiments and are normalized to P2 for consistency. **P < 0.01; ***P < 0.001, P1–2 versus C1–3. (C) PC and phosphatidylethanolamine (PE) levels were assessed in lipid extracts from EBVLs (grown in lipid-depleted serum containing medium for 48 h) using TLC. The data were then quantified using Image laboratory 4.1 software (Bio-Rad). Data represent mean ± SE of three experiments.
Lipodystrophy was very prominent in both affected patients, highlighting for the first time (to our knowledge) the importance of PCYT1A in adipose tissue. To better understand the role of PCYT1A in adipogenesis, we next evaluated the impact of PCYT1A knockdown during the course of 3T3L1 adipocyte differentiation. PCYT1A protein expression does not change significantly during 3T3L1 differentiation (Fig. 4A). PCYT1A siRNA led to a >90% reduction in PCYT1A expression (Fig. 4B), reduced expression of proteins typically expressed in differentiated adipocytes (perilipin1 and aP2), and substantially impaired neutral lipid accumulation (Fig. 4 B and C).
Fig. 4.

Pcyt1a knockdown in cultured adipocytes. (A) 3T3L1 preadipocytes were grown to a postconfluent state (2 d) before being induced to differentiate into mature adipocytes using standard protocols. Cell lysates were harvested at the indicated time points during adipocyte differentiation and immunoblotted with PCYT1A, perilipin1, or calnexin antibodies. (B) Scrambled or Pcyt1a siRNA were transfected during cell differentiation. The substantial reduction in PCYT1A expression in cells transfected with Pcyt1a siRNA (KD, knockdown) is demonstrated in this representative immunoblot. Perilipin1 and aP2 expression was also significantly reduced in the Pcyt1a knockdown cells. (C) At day 10, cells were fixed and stained with oil red O (E) to assess neutral lipid accumulation. The image is representative of n = 4 repeat experiments in triplicate.
Discussion
PC is the most abundant phospholipid in mammalian cells, and perturbation of its synthesis has dramatic effects on membrane and cellular function (11). In addition to the Kennedy pathway, PC can also be synthesized by a second pathway, the PEMT pathway. PEMT catalyses three repeated methylation reactions converting PE to PC. Under normal circumstances, the PEMT pathway is primarily active in the liver, where the demand for PC is particularly high due to the production and secretion of very-low–density lipoproteins (VLDLs) and PC secretion in bile, in addition to the normal cellular requirement for the synthesis of membranes. It contributes ∼30% of PC produced in the liver, when choline supply is adequate to maintain PC synthesis through the CDP-choline pathway. However, when choline is limiting in the diet, the PEMT pathway is critical for maintaining the supply of PC in the liver. The fact that Pcyt1a-null mice die in utero attests to the primary importance of the Kennedy pathway in most cell types (12) and has also necessitated the need to generate tissue specific knockouts in an attempt to gain more insight into the physiological roles of this enzyme in postnatal life. Our observations, which stem from the identification and characterization of a rare Mendelian disorder, provide a unique insight into the physiological consequences of a defect in PC synthesis in humans and draw attention to the essential role of PCYT1A-mediated PC biosynthesis in the liver and adipose tissue.
Links between reduced PC levels and hepatic steatosis were first recognized in rats and dogs in the 1930s (13, 14) before being widely confirmed in mice and then in humans in the 1990s (15). Li et al. (16) later suggested that a reduction in the PC/PE ratio affects hepatocyte membrane integrity and plays a role in the progression of steatosis to steatohepatitis in humans. Mechanistic insight was recently provided by Walker et al. (17) in a series of experiments suggesting that PC levels act as a rheostat for the activation of SREBP1c, a transcription factor known to induce expression of all of the key enzymes involved in fatty acid synthesis. Knockdown of PCYT1A expression in Caenorhabditis elegans and in human hepatocytes was shown to reduce PC levels, activate SREBP1c, and increase de novo lipogenesis and LD/triacylglycerol accumulation (17). These findings are consistent with the presence of steatosis in liver-specific Pcyt1a knockout mice (18) and in our patients. Lipodystrophy is a well-established cause of nonalcoholic fatty liver disease as ectopic lipid deposition results from the absence of the normal site of triglyceride storage. It is therefore very likely to be a significant contributor to the liver phenotype in our patients. Nevertheless, the extreme prematurity of steatosis in P2 and the extreme severity of steatosis (76%) in P1 is greater than that normally seen in lipodystrophy and is consistent with additional direct involvement of altered hepatic PC metabolism. In addition to its roles in bilayer membranes, PC is also a major component of the phospholipid monolayer surrounding LDs and lipoproteins, which might explain the fact that lipoprotein export was reduced in liver specific Pcyt1a-null mice (12). However, how and why this potential “lack” of surface PC does not then seem to impair the development of intrahepatic steatosis (LDs) remains unclear. The probands’ relatively modest hypertriglyceridemia would also be consistent with reduced lipoprotein particle synthesis, although this will require identification and characterization of additional affected patients.
Mammalian adipocytes characteristically form a huge unilocular LD in each cell surrounded by a phospholipid monolayer coated with perilipin1 and other coat proteins involved in the formation and turnover of the LD. We have recently reported the fact that loss-of-function mutations in two of these LD coat proteins are associated with smaller LDs and lipodystrophy (19, 20). We have now also shown that siRNA-mediated PCYT1A knockdown impairs adipogenesis in cultured adipocytes. Although we have yet to formally examine PCYT1A expression and activity in vivo in our patients, these data, together with the predicted and observed impact of the PCYT1A mutations on PCYT1A expression in other primary cell types, lead us to infer that this is a likely explanation for the observed lipodystrophy. Whether this change in neutral lipid accumulation is a consequence of changes in the phospholipid monolayer surrounding the LD or a more general result of changes in phospholipid levels or both remains unclear at this stage. Severe lipodystrophy is consistently associated with insulin resistance in humans and so is certain to be a significant factor in the pathogenesis of insulin resistance in both probands. However, it is also possible that changes in the membrane composition of cells in other key insulin target tissues such as skeletal muscle and the liver, could compound this effect.
During the course of our study, two independent manuscripts described the identification of biallellic PCYT1A mutations in eight unrelated probands with severe short stature due to spondylometaphyseal dysplasia and visual impairment due to cone-rod retinal dystrophy (21, 22). All affected individuals presented with severe skeletal manifestations early in childhood, whereas the retinal phenotype was considerably more variable in onset. Most of their patients were not carefully assessed for the presence of lipodystrophy and/or metabolic disease, but in a few cases liver ultrasounds did not reveal significant hepatic steatosis (22). Neither report formally characterized the biochemical impact of the mutations but many of the mutations, which spanned all of the known functional domains of PCYT1A, besides the extreme N-terminal nuclear localization domain, were strongly predicted to be damaging. Given these compelling genetic findings, P1 in our study was recalled for formal retinal assessment and a skeletal survey. Her retinal assessment was normal and the skeletal survey did not reveal any features of spondylometaphyseal dysplasia. P2’s vision is also normal and her height SD score of −2.6 is considerably greater than even the mildest case (height SD score, less than −5) reported by Hoover-Fong et al. (21).
Why the phenotype we report should be so different to that described in these genetic studies remains unclear. Phenotypic heterogeneity is widely acknowledged in human monogenic disease including other lipodystrophies such as that associated with LMNA mutations, where mutations in a single gene have been linked to 12 apparently different diseases (23). Nevertheless, this sort of phenotypic diversity is more typically explained by mutations affecting particular protein domains. In the case of PCYT1A, the mutations described to date do not appear to be confined to specific protein domains. Possibilities that will require consideration in future studies include (i) the presence of additional genetic modifiers, (ii) dietary/environmental modifiers, and/or (iii) differences in the degree to which the biochemical activity of PCYT1A is impaired by the mutations in vivo. Mutations in the INSR gene provide a germane precedent for the latter possibility. In this case, biallellic mutations resulting in near total loss of insulin receptor activity result in a characteristic phenotype known as Donohue syndrome, milder loss-of-function mutations are associated with a distinct phenotype known as Rabson–Mendenhall syndrome, and heterozygous mutations cause an even milder syndrome without any dysmorphic features known as type A insulin resistance (24). Future studies aimed at characterizing the different mutations as well as the identification of further affected patients ought to provide answers to these intriguing questions.
In summary, our data clearly demonstrate that the biallellic mutations identified in each of two patients with a similar metabolic phenotype result in near-total lack of PCYT1A expression and significantly reduce PC synthesis via the Kennedy pathway. Yet compelling genetic data suggest that biallellic PCYT1A mutations predicted to perturb PCYT1A, although this is still to be directly demonstrated, cause an apparently distinct phenotype epitomized by skeletal and retinal disease, raising fascinating questions about the apparent human phenotypic diversity associated with mutations affecting such a fundamental biochemical pathway.
Methods
Genetic Analysis.
The study was conducted in accordance with the Declaration of Helsinki and approved by the relevant research ethics committees. Each participant, or a parent in the case of minors, provided written informed consent; minors provided oral consent. The study was approved by the UK National Research Ethics Committee and the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Genomic DNA was extracted from peripheral blood leukocytes. Sample preparation was performed as described previously (25), and the captured samples were eluted, amplified, and sequenced on the Illumina Hi-Seq platform (Illumina) as 75-bp paired-end reads. The full methods used for whole exome sequencing and mutation analysis, confirmatory Sanger sequencing, and haplotype analysis are described in SI Methods.
Phenotyping Studies.
Biochemical analyses were performed on blood samples collected after a standardized overnight fast. Insulin, leptin, and adiponectin were measured using customized autoDELFIA immunoassays as previously described (26). Body composition, liver fat, and hepatic de novo lipogenesis were also measured as described previously (26).
Functional Analysis of the PCYT1A Mutations.
Details regarding the methods used to predict and then to determine the functional consequences of the mutations in cells from the probands are described in SI Methods.
Supplementary Material
Acknowledgments
We are very grateful to the patients who consented to and facilitated detailed studies of themselves, as well as to Dr. Ben Challis, Julie Harris, and the staff of the Wellcome Trust Clinical Research Facility in Cambridge for assistance with metabolic studies of P1. We thank Prof. Paul Lehner, Prof. David Ron, Dr. Heather Harding, Prof. Sadaf Farooqi, and Dr. Eleanor Wheeler for technical advice. We are grateful for access to exome sequence data from the CoLaus cohort, which was sequenced as part of a partnership between the Wellcome Trust Sanger Institute, the CoLaus principal investigators, and the Quantitative Sciences Department of GlaxoSmithKline. We thank the NHLBI GO Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women's Health Initiative Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). This study makes use of data generated by the UK10K Consortium, derived from samples from TwinsUK and Avon Longitudinal Study of Parents and Children. A full list of the investigators who contributed to the generation of the data is available from www.UK10K.org. We also thank Emma Gray, David Jones (Sample Management), Danielle Walker (Sequencing Pipeline), Carol Scott and Jillian Durham (Variation Informatics), and the staff of the Wellcome Trust Sanger Institute. Clinical studies of P2 were supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases. R.K.S., S.O., I.B., and D.B.S. were supported by the Wellcome Trust (Grants WT098498, WT095515, WT098051, and WT091551, respectively), the Medical Research Council Metabolic Diseases Unit, and the United Kingdom National Institute for Health Research Cambridge Biomedical Research Centre. S.O. was supported by the European Union/European Federation of Pharmaceutical Industries and Associations Innovative Medicines Initiative Joint Undertaking (European Medical Information Framework Grant 115372). N.K. and T.C.W. were supported by National Institute of General Medical Sciences Grant R01 GM09719. Funding for UK10K was provided by the Wellcome Trust under Award WT091310.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1408523111/-/DCSupplemental.
References
- 1.Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem. 1956;222(1):193–214. [PubMed] [Google Scholar]
- 2.Gibellini F, Smith TK. The Kennedy pathway—de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life. 2010;62(6):414–428. doi: 10.1002/iub.337. [DOI] [PubMed] [Google Scholar]
- 3.Vance DE. Boehringer Mannheim Award Lecture. Phosphatidylcholine metabolism: Masochistic enzymology, metabolic regulation, and lipoprotein assembly. Biochem Cell Biol. 1990;68(10):1151–1165. doi: 10.1139/o90-172. [DOI] [PubMed] [Google Scholar]
- 4.Mazess RB, Barden HS, Bisek JP, Hanson J. Dual-energy x-ray absorptiometry for total-body and regional bone-mineral and soft-tissue composition. Am J Clin Nutr. 1990;51(6):1106–1112. doi: 10.1093/ajcn/51.6.1106. [DOI] [PubMed] [Google Scholar]
- 5.Kleiner DE, Brunt EM. Nonalcoholic fatty liver disease: Pathologic patterns and biopsy evaluation in clinical research. Semin Liver Dis. 2012;32(1):3–13. doi: 10.1055/s-0032-1306421. [DOI] [PubMed] [Google Scholar]
- 6.Abecasis GR, et al. 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J, Johnson J, Ding Z, Paetzel M, Cornell RB. Crystal structure of a mammalian CTP: Phosphocholine cytidylyltransferase catalytic domain reveals novel active site residues within a highly conserved nucleotidyltransferase fold. J Biol Chem. 2009;284(48):33535–33548. doi: 10.1074/jbc.M109.053363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krahmer N, et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011;14(4):504–515. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craig L, Johnson JE, Cornell RB. Identification of the membrane-binding domain of rat liver CTP:phosphocholine cytidylyltransferase using chymotrypsin proteolysis. J Biol Chem. 1994;269(5):3311–3317. [PubMed] [Google Scholar]
- 10.Wang L, Magdaleno S, Tabas I, Jackowski S. Early embryonic lethality in mice with targeted deletion of the CTP:phosphocholine cytidylyltransferase alpha gene (Pcyt1a) Mol Cell Biol. 2005;25(8):3357–3363. doi: 10.1128/MCB.25.8.3357-3363.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fagone P, Jackowski S. Phosphatidylcholine and the CDP-choline cycle. Biochim Biophys Acta. 2013;1831(3):523–532. doi: 10.1016/j.bbalip.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs RL, Devlin C, Tabas I, Vance DE. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J Biol Chem. 2004;279(45):47402–47410. doi: 10.1074/jbc.M404027200. [DOI] [PubMed] [Google Scholar]
- 13.Best CH, Hershey JM, Huntsman ME. The effect of lecithine on fat deposition in the liver of the normal rat. J Physiol. 1932;75(1):56–66. doi: 10.1113/jphysiol.1932.sp002875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Best CH, Ferguson GC, Hershey JM. Choline and liver fat in diabetic dogs. J Physiol. 1933;79(1):94–102. doi: 10.1113/jphysiol.1933.sp003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeisel SH. A brief history of choline. Ann Nutr Metab. 2012;61(3):254–258. doi: 10.1159/000343120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, et al. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006;3(5):321–331. doi: 10.1016/j.cmet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Walker AK, et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell. 2011;147(4):840–852. doi: 10.1016/j.cell.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vance DE, Vance JE. Physiological consequences of disruption of mammalian phospholipid biosynthetic genes. J Lipid Res. 2009;50(Suppl):S132–S137. doi: 10.1194/jlr.R800048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubio-Cabezas O, et al. LD Screening Consortium Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1(5):280–287. doi: 10.1002/emmm.200900037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gandotra S, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364(8):740–748. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoover-Fong J, et al. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94(1):105–112. doi: 10.1016/j.ajhg.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto GL, et al. Mutations in PCYT1A cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94(1):113–119. doi: 10.1016/j.ajhg.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119(7):1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev. 2011;32(4):498–514. doi: 10.1210/er.2010-0020. [DOI] [PubMed] [Google Scholar]
- 25.Lindhurst MJ, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44(8):928–933. doi: 10.1038/ng.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semple RK, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119(2):315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.