


Abstract
Robustness and completion of DNA replication rely on redundant DNA replication origins. Reduced efficiency of origin licensing is proposed to contribute to chromosome instability in CDK-deregulated cell cycles, a frequent alteration in oncogenesis. However, the mechanism by which this instability occurs is largely unknown. Current models suggest that limited origin numbers would reduce fork density favouring chromosome rearrangements, but experimental support in CDK-deregulated cells is lacking. We have investigated the pattern of origin firing efficiency in budding yeast cells lacking the CDK regulators Cdh1 and Sic1. We show that each regulator is required for efficient origin activity, and that both cooperate non-redundantly. Notably, origins are differentially sensitive to CDK deregulation. Origin sensitivity is independent on normal origin efficiency, firing timing or chromosomal location. Interestingly, at a chromosome arm, there is a shortage of origin firing involving active and dormant origins, and the extent of shortage correlates with the severity of CDK deregulation and chromosome instability. We therefore propose that CDK deregulation in G1 phase compromises origin redundancy by decreasing the number of active and dormant origins, leading to origin shortage and increased chromosome instability.
INTRODUCTION
DNA replication and chromosome segregation occur once per cell cycle and are mutually exclusive in eukaryotic cell cycles to prevent genome variations in progeny. Maintaining intact genomes also requires complete DNA duplication, contributed by redundancy amongst the many origins of DNA replication on each chromosome which vary in their efficiency and timing of firing during S phase (1–3). Origins are activated in two steps (for review see (4)). Firstly, in G1 phase during the origin-licensing step, the Orc1-6, Cdc6 and Cdt1 proteins are recruited to origins and load a double hexameric ring of the Mcm2-7 proteins around dsDNA, to form pre-replicative complexes (pre-RC). The Mcm2-7 proteins form the catalytic core of the essential DNA helicase at replication forks, but are inactive when loaded at origins during G1 phase. Secondly, in S phase the GINS (Go, Ichi, Ni, San; Sld5, Psf1, Psf2, Psf3) complex and the Cdc45 protein are recruited to Mcm2-7 to form the active CMG (Cdc45-Mcm2-7-GINS) helicase (5,6) allowing further recruitment of polymerases and other factors. This leads to the formation of a replication bubble with two polar replisomes that progress in opposite directions, until they either reach telomeres at chromosome ends or else meet a converging fork to terminate replication. Some of the licensed origins remain dormant and are either passively replicated by progressing forks from neighbouring origins, or else provide a backup in case of inactivity of flanking origins or failure or stalling of neighbouring forks (7,8). This flexible pattern of redundant origin usage favours replication completion by the end of S phase.
During the initiation of DNA replication, cyclin-dependent kinase (CDK)/cyclin complexes regulate both steps of origin activation. For instance in the budding yeast Saccharomyces cerevisiae (S. cerevisiae), CDK activity inhibits origin licensing which is thus restricted to G1 phase when CDKs are low. Subsequently, CDK activation is necessary for origin firing during S phase. This double control allows time to license sufficient origins in G1 phase while avoiding origin re-licensing and re-replication (9–11). The low CDK activity in G1 phase is achieved by many levels of control. In addition to repressed cyclin expression, B-type cyclins are ubiquitylated and degraded by the Anaphase Promoting Complex/Cyclosome (APC/C)-Cdh1 E3 ligase (12–16), and any remaining CDK/cyclin complexes are bound by CDK inhibitors (CKIs), such as the CIP/KIP families that include in mammals p21Cip1/Waf1 (17,18), and p27Kip1 (19). The budding yeast Sic1 protein performs a similar role as a CKI (20,21). Proliferative stimuli through G1-CDKs commit cells to terminate the licensing period and enter the cell cycle by APC/C-Cdh1 inhibition and CKI degradation (for review see (9,22)). Hence, the activation of a normal set of replication origins relies on the scheduled regulation of CDK both in G1 phase and also during the transition to S phase.
Deregulation of CDK is frequent in cancer cells (23) and leads to unscheduled CDK activity even in G1 phase. This can drive cells into a premature G1 exit and an aberrant replication leading to replicative stress and DNA damage, and the activation of DNA damage repair (DDR) pathways as barriers to genome instability (24–26). Continued aberrancies in repeated cell cycles would impose a selective pressure in favour of DDR defective clones that are unable to deal properly with damage on DNA, leading to genome instability (27,28).
Although it seems clear that CDK deregulation leads to DNA replication abnormalities that drive the acquisition of genome instability, the molecular nature of such abnormalities and how can they mechanistically cause genome instability is largely unknown. Diminished origin activity is likely to be important, since mutation of budding yeast pre-RC components, or reduced origin density, cause chromosomal instability (29–31). Similarly, decreased levels of Mcm2-7 components in metazoans promote minimal licensing and limit dormant origin numbers, increase DNA damage, impair chromosome stability especially in conditions of replicative stress and increase the occurrence of cancer in mammals (32,33). Moreover, in budding yeast and mammalian cells, CDK deregulation in G1 phase reduces origin activity and firing efficiencies, correlating with increased chromosomal rearrangements (34–38). Insertion of multiple origins reduces the local rates of gross chromosomal rearrangements (GCR) in S. cerevisiae cells that overexpress the G1-cyclin Cln2, presumably by increasing the efficiency of origin firing, and fork density (35). However, important questions remain unanswered regarding the links between CDK deregulation, origin licensing and genome instability. Firstly, the licensing and firing activity of few individual origins have been studied, and only in yeast (34), so that the general applicability of the model remains unclear. Secondly, it remains unaddressed if reduced origin activity spontaneously concentrates at unstable chromosome regions in CDK-deregulated cells, and if the extent of origin inefficiency reflects instability. Thirdly, reduction of origin firing efficiency is not uniform amongst the studied origins, posing the question of which origins are most sensitive to CDK deregulation (34). Indeed, not all chromosome regions are equally prone to genome instability, and oncogenic cell cycles show increased instability at fragile sites in the form of loss of heterozigosity (24–26). Moreover, common fragile sites display origin paucity in correlation with increased fragility (39,40). Finally, origin-licensing inefficiency may not be limited to active origins, and the behaviour of dormant origins upon CDK deregulation remains to be explored.
Here, we model CDK deregulation in G1 phase and analyse origin activity in budding yeast cells. Whereas previous studies focused on cells lacking the CDK inhibitor Sic1, we have also analysed cells lacking the APC/C activator Cdh1. We report the firing efficiency of a variety of origins upon CDK deregulation to address if particular subtypes are more sensitive than others to CDK deregulation. We also study if reduced origin usage colocalises with increased GCRs at a chromosome region. We conclude that the CDK regulators Cdh1 and Sic1 cooperate in promoting origin redundancy to prevent a regional shortage of active origins and maintain chromosome stability.
MATERIALS AND METHODS
Yeast strains and growth conditions
The yeast strains in this study are based on W303-1a or S288C backgrounds (Supplementary Table S1), and derive from the parental YAC36 and RDKY3615, respectively. Cells were routinely grown at 25ºC in YP media supplemented with 40 mg/L adenine and 2% glucose (YPAD). GAL1,10p-SIC1 cells were always grown in 1% raffinose, 0.3% galactose YP media supplemented with 40 mg/L adenine (YPARG), and before analysis were transferred to YPAD for 4 h to repress Sic1 expression (the minimal time required by an asynchronous cell culture to stabilise the cell cycle). Cell cycle arrests were performed in nocodazole (15 μg/mL for 210 min) or alpha-factor (40 ng/mL for 3 h in bar1Δ strains). Samples for flow cytometry were fixed and processed as described previously (41), analysed using a FACSCalibur flow cytometer (BD Biosciencies), and plotted using the Cell Quest software (BD Biosciencies). The GAL1,10p-SIC1 strain was constructed by replacing 19 bp immediately before the SIC1 ATG with a TRP1-GAL1,10p cassette, so the only SIC1 copy in the cell is under the control of the GAL1,10 promoter. HphNT was inserted in the parental strains at coordinates 71495–71533 of chromosome VI (according to the Saccharomyces Genome Database, SGD, http://www.yeastgenome.org) to construct YAC177 and YAC188; the remaining strains derive from these two and are isogenic for this insertion except YAC571. To construct the SIC1p-SIC1 strains, the SIC1 gene together with both flanking intergenic regions (SGD, coordinates 286446–287933, chromosome XII) was amplified by PCR, cloned into the pGEM-T Easy system I vector (Promega) and fully sequenced. The SacII-SalI fragment containing SIC1p-SIC1 was subcloned into pRS305 (42) to generate the plasmid pAC738 that linearised with NarI was targeted to LEU2, and single integrants were selected by Southern analysis. The ars507Δ::ARS305 strains were constructed by first deleting the 234 bp of ARS507 (SGD, coordinates 59283–59516, chromosome V) with the URA3 cassette from pRS306 (42), what completely abolishes origin firing activity (data not shown), followed by a replacement of URA3 with 234 bp of ARS305 (43) (SGD, coordinates 39382–39615, chromosome III).
Two-dimensional DNA gel electrophoresis
Samples of chromosomal DNA for 2D gels were prepared as previously described (41) and from 5 to 15 μg of digested DNA was employed per gel. DNA was digested with the following enzymes to generate the indicated origin-containing fragments: MluI-ClaI (4.84 kb ARS504.2, 6.33 kb ClaI-ClaI proARS506), ClaI-EcoRI (5.49 kb ARS607, 3.85 kb ARS522-501, 5.36 kb ARS302-ARS303-ARS320, 5.93 kb ARS503-ARS504, 1.81 kb EcoRI-ClaI ARS608, 4.23 kb ClaI-ClaI ARS306), NcoI (5,12 kb ARS305, 4.67 kb ARS416-1, 2.61 kb ARS603.5) and ClaI-SacI-KpnI (2.64 kb ClaI-KpnI ARS432, 5.82 kb ClaI-KpnI ARS1014, 2.47 kb SacI-KpnI ARS507, 2.38 kb KpnI-ClaI ARS508, 2.47 kb SacI-KpnI ars507Δ::ARS305). The specific probe for each origin corresponds to the following coordinates of the indicated chromosomes (according to the SGD): ARS306 (73001–73958, chromosome III), ARS305 (39073–40557, chromosome III), ARS607 (198958–199845, chromosome VI), ARS432 (1158713–1159194, chromosome IV), ARS1014 (417974–418715, chromosome X), ARS416-1 (462772–463329, chromosome IV), ARS522-501 (548689–549857, chromosome V), ARS603.5 (118090–118962, chromosome VI), ARS302-ARS303-ARS320 (14040–14991, chromosome III), ARS608 (215755–216554, chromosome VI), ARS503-ARS504 (8919–9432, chromosome V), ARS504.2 (10744–11272, chromosome V), proARS506 (18123–18663, chromosome V), ARS507 (59965–60438, chromosome V) and ARS508 (92964–93372, chromosome V). After hybridisation, detection was performed exposing against BAS-MS imaging plates (Fujifilm), and read with a Personal Molecular Imager FX (Bio-Rad). Quantitative determination of origin efficiency was determined as previously described (44,45), by measuring the signal of the whole bubble-arc and the Y-arc for each blot by free-hand contouring of specific signals above the background, employing the Quantity One software (Bio-Rad). Origin efficiency is the frequency of active initiation (bubble arc/(bubble arc + Y arc)). We discarded from the measurement the region of large Ys where passive Ys comigrate with active Ys resulting from bubble resolution, so is a region of mixed active and passive signals. The distinct quality of blots may influence the accuracy of quantification, but our results were largely reproducible in independent repeats and in distinct backgrounds.
Western blot analysis
Yeast proteins were extracted by the TCA method (46) from 2.5×108 cells. Sic1 was detected with the rabbit polyclonal anti-Sic1 FL-284 antibody (Santa Cruz Biotechnology Inc.), and Pgk1 with the 22C5D8 mouse monoclonal antibody (Invitrogen). Secondary antibodies employed were the HRP donkey anti-rabbit, and sheep anti-mouse (GE Healthcare), respectively.
GCR assay
The estimation of GCR rates was performed by fluctuation analysis as described (35,47). At least five independent colonies were inoculated from YPAD (control and Δcdh1 cells) or YPARG (GAL1,10p-SIC1) plates into liquid YPAD or YPARG, as indicated in each experiment, at a density of 2.5×105 cells/mL and grown at 25ºC until stationary phase (usually 32 to 40 h). Cells were collected, counted in a Neubauer counting chamber (Brand), and 5×108 cells were spread per 150 mm Petri dish of synthetic minimal FOA-Can media (1.1 g/L of 5-FOA, and 60 mg/L of sulphate salt L-canavanine) supplemented with glucose except for GAL1,10p-SIC1 strains where raffinose-galactose was employed. The number of colonies was counted and the median employed to estimate the mutation (GCR) rates, as described previously (35,48).
Pulse field gel electrophoresis
Cells were collected by centrifugation in log phase and prepared employing the CHEF Yeast Genomic DNA Plug Kit (Bio-Rad), following manufacture's instructions. Plugs were loaded into a 1% Megabase (Bio-Rad) agarose gel in 0.5x TBE and run at 14ºC in a Gene Navigator System (GE Healthcare Life Sciences) at 180 V during 24 h with pulses of 90, 105 and 120 s for 9:36, 6 and 8:20 h, respectively. DNA was transferred to Hybond-XL membranes (GE Healthcare Life Sciences) and Southern analysis, exposure, and image processing was performed as for 2D gels.
Fork direction analysis
DNA samples were prepared as for 2D gels and digested with the following enzymes to generate the fragments: NheI-NheI (5.01 kb, chr. III, coordinates 68091–73104 ‘upstream ARS306’), BamHI-HindIII (3.63 kb, chr. VI, coordinates 195438–199068 ‘upstream ARS607’) and HindIII-HindIII (3.80 kb, chr. IV, coordinates 1161197–1165004 ‘downstream ARS432’). Electrophoresis, DNA transfer, hybridisation and signal detection were identical to 2D gels except for the in-gel digestion between both dimensions that was performed as indicated (49) using PstI (chr. 3, 71542), XbaI (chr. 6, 198128) and KpnI (chr. 4, 1164170), respectively. The specific probes correspond to the following coordinates and chromosomes (SGD): upstream ARS306 (68661–69685, chr. III), upstream ARS607 (196158–196887, chr. VI) and downstream ARS432 (1161848–1162567, chr. IV). Signal quantification was performed as described (57) employing the same software as for 2D gels.
RESULTS
Cdh1 is important for efficient initiation of DNA replication
Inefficient origin activity has been suggested to contribute to genome instability in cells lacking Cdh1 (50–52), and Cdh1−/− mouse embryonic fibroblasts (MEFs) show less efficient DNA replication and reduced levels of Mcm4 and Mcm5 at chromatin, as well as increased chromosome aberrations (38). However, no direct analysis of origin activity has been reported previously for cells lacking Cdh1, and plasmid loss in Δcdh1 yeast cells is not reverted with extra origins (50) as is often the case in mutants with defective origin activation.
Therefore, we made a direct comparison of origin firing and genome instability in yeast cells lacking either Cdh1 or Sic1. We deleted the CDH1 gene in budding yeast, and Δcdh1 cells displayed the known cell cycle distribution and synthetic interaction with cells lacking Sic1 (Supplementary Figure S1). Given the reported elevated GCR rates of Δsic1 cells (34,53), we instead employed cells conditionally expressing Sic1 under the control of the GAL1,10 promoter to minimise the undesired selection of genomically modified clones. Repression of the promoter mimics the deletion of SIC1 in cell viability (20,34), although with milder phenotypes (Supplementary Figure S1). GAL1,10p repression largely removes Sic1 (Supplementary Figure S2), but not completely (residual amounts are detectable in Western blots after very sensitive detection (data not shown) consistent with its milder phenotypes) so hereinafter we refer to GAL1,10p-SIC1 repression as Sic1-depletion or sic1 cells. Accordingly, in contrast to the lethality of Δcdh1 Δsic1 cells (15,16), sic1 Δcdh1 cells were viable although very sick (Supplementary Figure S1). However, the Sic1-depletion was sufficient to render Δ47cdc6 Δcdh1 sic1 cells inviable (54,55) (data not shown).
We measured the firing efficiency (frequency of firing in the population of cells) of individual origins in their natural loci by 2D ‘neutral–neutral’ agarose gel electrophoresis of replication intermediates (56) in isogenic control, Δcdh1, sic1 and double mutant sic1 Δcdh1 cells (Figure 1). The relative intensities of the external active bubble-arc and the internal passive fork-arc determine the firing efficiency of the origin (Figure 1A). Control and Δcdh1 cells were collected after asynchronous growth to log phase in YPAD, and compared with asynchronous sic1 cells after repressing Sic1 expression for 4 h in YPAD (the minimum time cells require to display and maintain the cell cycle distribution of cells lacking Sic1; Supplementary Figure S1). We firstly analysed three well-known early efficient origins, ARS306, ARS305 and ARS607, and confirmed in control cells the reported high efficiency (45,57,58) (Figure 1B, upper panels). Sic1-depleted cells showed differential loss of firing efficiency amongst origins regarding control cells, with ARS306 losing around more than 15% of efficiency while ARS305 was not noticeably affected (Figure 1B, second row). This non-uniform origin sensitivity was previously described for Δsic1 cells for ARS305 and ARS306, although with higher efficiency losses in the complete absence of Sic1 (34), consistent with the stronger severity of Δsic1 cells, and was reproduced by ourselves (Supplementary Figure S3). To generalise this observation we extended this analysis to two more origins, ARS432 and ARS1014, randomly chosen amongst the earliest in their chromosomes and not studied previously by 2D gels (as compiled in the DNA Replication Origin Database, OriDB, (59)). Similar to ARS305, ARS1014 also displayed normal efficiency in sic1 cells, whereas ARS432 lost around 30% of efficiency (Figure 1B). These results were confirmed in independent repeats (Supplementary Figure S4A) and also in other strain backgrounds (Supplementary Figure S4B and see below).
Figure 1.
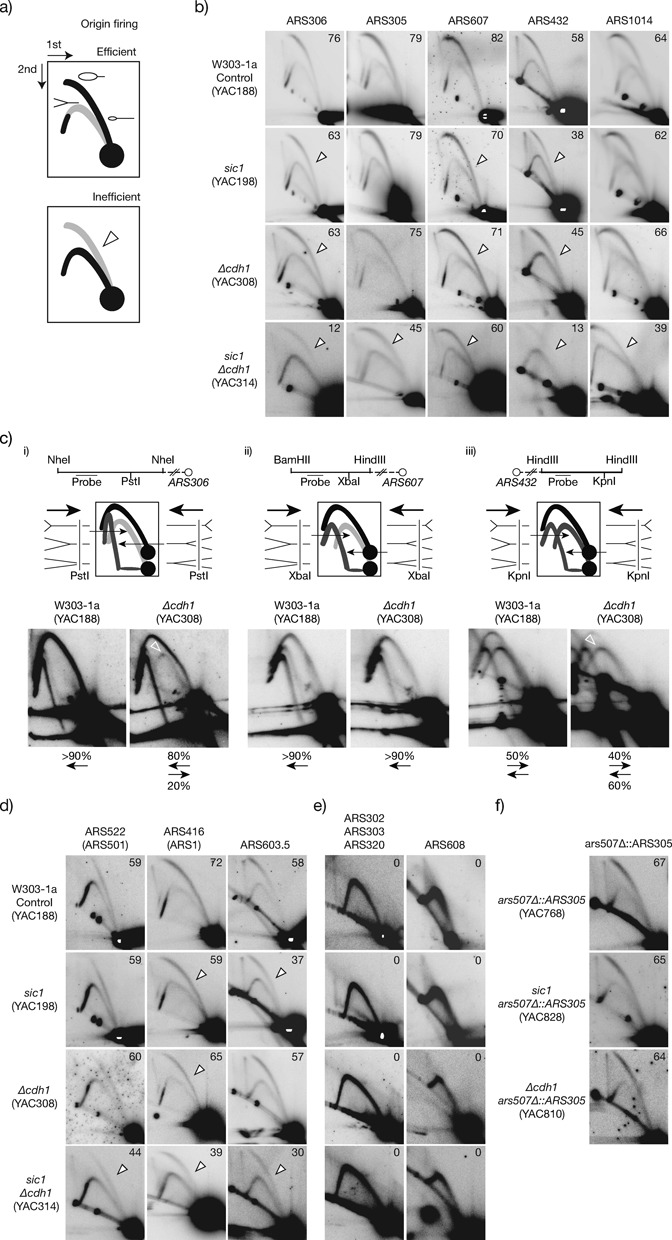
Differential origin firing sensitivity in cells lacking the CDK regulators Sic1 and Cdh1. (a) Scheme of efficient and inefficient origin firing observed by 2D gels. The external arc is formed by fragments containing a bubble and reveals origin firing activity, and is termed active- or bubble-arc. The internal arc is formed by forks (Ys) that passively replicate the fragment and indicates passive replication, so is referred as passive-, or Y-arc. The relative intensity of both arcs (open arrowhead) denote origin efficiency. (b) 2D blots of early firing origins in control, sic1, Δcdh1 and double mutant sic1 Δcdh1 cells. Origin efficiencies are shown in the right-upper corners. (c) Analysis of fork direction at origin-adjacent regions upstream to ARS306 (i) and ARS607 (ii), and downstream to ARS432 (iii). Upper panels: maps of the analysed restriction fragments containing the enzymes employed before the 1st dimension (above) and for in-gel digestion (below the chromosome lane), the region recognised by the hybridisation probe and the relative position of ARSs. Middle panels: diagrams of rightward- and leftward-moving forks, the expected products after the in-gel digestion, and the observed migration patterns. Lower panels: hybridisation blots of control and Δcdh1 cells, including the percentage of signal of forks moving in each direction (when detected). (d) Origin firing activity of mid-S phase, and (e) dormant origins. (f) Efficiency of ARS305 when replacing ARS507 at chromosome V. Open arrowheads show loss of efficiency relative to control cells. See Material and Methods for details of chromosome coordinates, restriction enzymes and probes employed for each origin. Diverse spots appear below the internal arc by unspecific probe hybridisation, or are remaining signals from preceding hybridisations.
Importantly, Δcdh1 cells also showed loss of firing efficiency of ARS306, ARS607 and ARS432 compared to control cells, whereas ARS305 and ARS1014 were unaffected (Figure 1B, third row). Origin inefficiencies in Δcdh1 cells were more modest than in sic1 cells. To confirm further the observed inefficiencies in origin activity in Δcdh1 cells, we analysed the direction of replication fork movement at origin-adjacent regions by using a modification of the 2D gel electrophoresis that includes an in-gel digestion of replication intermediates between both dimensions (49). This approach differentiates the amount of forks progressing in both directions (departing from or arriving to the origin) therefore reflecting origin activity. Hence, reductions in origin activity can be evaluated by the increase of forks moving toward the origin (60). We analysed the direction of fork movement at fragments immediately upstream ARS306 or ARS607, and downstream of ARS432 in wt and Δcdh1 cells, and the results are shown in Figure 1(C). In control cells only forks departing from the origin are detected close to ARS306 and ARS607, as previously reported and consistent with their elevated firing efficiency (49,58). ARS432 has more moderate origin activity, and the amount of replication forks adjacent to this origin was similar in both directions in control cells. In contrast, in Δcdh1 cells an arc of forks moving toward ARS306 was now visible (Figure 1C(i), right panel) consistent with the reduced ARS306 activity seen in our 2D gels. Similarly at the site adjacent to ARS432, we observed a decreased intensity of the arc of forks departing from the origin (Figure 1C(iii), right panel). Consistent with our earlier finding that ARS607 is less affected than the above two origins in Δcdh1 cells (Figure 1B), we did not observe variations in fork direction between wt and Δcdh1 cells for this origin (Figure 1C(ii)). Hence, the direction of fork progression at regions adjacent to origins in Δcdh1 cells is altered only when the origin efficiency is altered compared to control cells, and accordingly no variation is observed at a region adjacent to ARS305 (Supplementary Figure S5). This is consistent with previous reports of trinucleotide repeat instability in association with decreased origin usage and changes in fork direction (61).
Altogether these results indicate that differential sensitivity of origins is a common response of cells lacking CDK regulators. We conclude that cells require Cdh1 to promote efficient firing of a subset of origins during the initiation of DNA replication.
The differential origin sensitivity to CDK deregulation is independent of normal origin efficiency, firing timing and chromosomal location
Origins differ in their time of firing during S phase, ranging from very early to mid or late, and in their efficiency from very efficient to dormant (45,57,58,62). In control cells, the five early origins analysed in this study range in efficiency (Figure 1B; 20–40% higher in ARS305, ARS306 and ARS607 regarding ARS432 and ARS1014) and firing timing (Supplementary Table S2), with some being sensitive to CDK deregulation and others resistant (Figure 1B). We also analysed a variety of other origins with varying efficiency that fire in mid-S or late-S phase, or that are dormant (Supplementary Table S2). ARS1 (ARS416), ARS501 (ARS522) and ARS603.5 fire with diverse efficiencies in mid-S phase in control cells. As for the early origins studied above, they showed differential sensitivity in Δcdh1 and sic1 cells, with ARS522 being resistant to CDK deregulation, and ARS603.5 and ARS416 being sensitive (Figure 1D). Similarly, sensitivity in mid-S origins is highly variable from 14 to 35% in sic1 cells (ARS607 and ARS603.5), and from 10 to 21% (ARS416 and ARS432) in Δcdh1 cells (Figure 1D). The tested dormant origins remained inactive in both mutants (Figure 1E). Despite similar origin-sensitivity patterns in both mutants, origin efficiency was reduced to a greater degree in sic1 compared to Δcdh1 cells. In addition, ARS603.5 was sensitive to CDK deregulation in sic1 cells but normal in Δcdh1 cells. Overall, these data indicate that Cdh1 contributes less than Sic1 to the regulation of origin activity.
Chromosomal location can affect origin efficiency and also the timing of firing (reviewed in (63)). Two consecutive origins separated about 35 kb, ARS305 and ARS306, behave differently (resistant and sensitive, respectively) in Δcdh1 or sic1 cells, despite normally having similar efficiency and timing (Supplementary Table S2). However, their chromosomal position and particular cis-acting elements are unique, so we investigated whether sensitivity was influenced by these particular origin characteristics. The known ARS305 cis elements are contained in a sequence of 234 bp (43,64). We tested whether moving this region to a sensitive locus would change the resistance of the origin to CDK deregulation, by making an exact replacement of the 234 bp ARS507 origin sequence on chromosome V (Supplementary Table S2 and Figure 2 show is sensitive to CDK deregulation) with ARS305 (see Material and Methods). In this way we found that ars507Δ::ARS305 maintains similar efficiency as the endogenous ARS305 on chromosome III (Figure 1F; compare with the ARS305 panel in B) and notably, efficiency of ars507Δ::ARS305 was not altered in Δcdh1 or sic1 cells. These data indicate that ARS305 resistance to CDK deregulation is independent of its location, but rather is inherent to its cis-acting elements present in the 234 bp region. From these results overall we conclude that origin sensitivity to CDK deregulation is independent of normal origin efficiency, firing timing, or chromosomal location, suggesting that is influenced by the specific origin configuration of cis elements.
Figure 2.
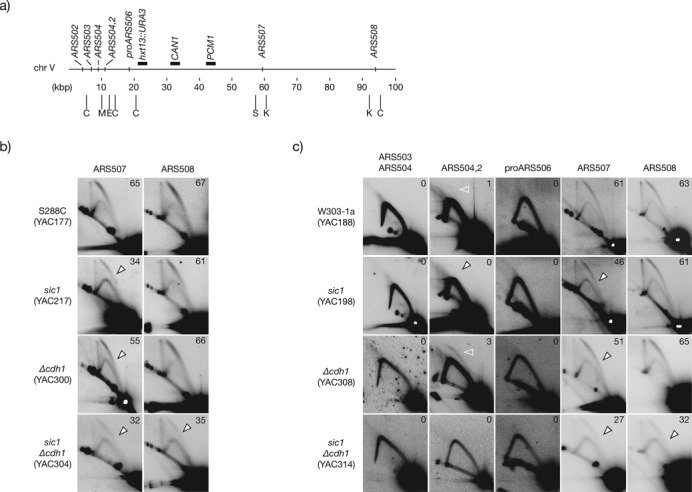
Overall origin firing efficiency at the left arm of chromosome V in CDK-deregulated cells. (a) Map of the first 100 kbp of chromosome V including the relative position of all confirmed and likely origins (according to the OriDB and the SGD). Horizontal black bars above the chromosome denote the relative position of markers for the GCR assay (URA3 and CAN1), and the first essential gene from the left telomere (PCM1) relevant for the GCR assay. Below the chromosome, vertical bars indicate the position of the restriction enzymes employed to digest DNA for 2D gel analysis: C, ClaI; M, MluI; E, EcoRI; S, SacI; K, KpnI. (b) Firing efficiency analysis of ARS507 and ARS508 in S288C control, sic1, Δcdh1 and sic1 Δcdh1, double mutant cells. (c) Firing efficiency of the indicated active, dormant and likely origins at the left arm of chromosome V in W303–1a control and CDK-deregulated cells. Open arrowheads indicate loss of efficiency relative to control cells. Open white arrowheads mark the bubble arc at ARS504.2. See Material and Methods for details of chromosome coordinates and probes employed for each origin.
Origin firing shortage and GCR rates coincide at the left arm of chromosome V in Δcdh1 and sic1 cells
In human cells, fragile site instability is associated with a paucity of initiation events (39). Previous work with budding yeast showed that elevated Cln2, lack of Sic1, or lack of Cdh1 increased the rate of GCRs at the left arm of chromosome V (34,35,47,50,53). We observed similar GCR rates, indicating once again that Cdh1 is important but less critical than Sic1 to prevent chromosomal rearrangements (Table 1). Moreover, Sic1-depleted cells displayed reduced GCR rates than Δsic1 cells (12 and 30 times the control, respectively). We reasoned that if reduced origin activity in CDK-deregulated cells favours chromosome rearrangements, then both should coincide spatially. Hence, we analysed the pattern of origin usage on the first 100 kb of the left arm of chromosome V (Figure 2A). The efficiency of these origins has not been previously analysed by 2D gels (see OriDB), but two early and active origins were identified by genome-wide studies in this chromosome arm, ARS507 and ARS508 (62,65). We firstly analysed their efficiency in the same cells employed to estimate the rate of GCRs. In control cells, both origins were active and similarly highly efficient, however, while ARS508 remained unaffected in Δcdh1 and sic1 cells, ARS507 had reduced efficiency (Figure 2B). Notably, ARS507 was less affected in Δcdh1 cells, correlating with the observed GCR rates. We extended the analysis to W303–1a cells with similar results (Figure 2C), confirming that differential origin tolerance to CDK deregulation is origin specific, independently on the strain background.
Table 1. The lack of Sic1 and Cdh1 increase GCR rate.
| Relevant genotype (Strain) | Cells analysed | GCR rate | (*) | (**) |
|---|---|---|---|---|
| Control (YAC177) | 8.21 × 1010 | 2.70 × 10−9 | (1.0) | |
| Control (YAC177) | 4.16 × 1010 | 5.84 × 10−9 | (1.0) | |
| sic1 (YAC217) | 5.03 × 109 | 3.44 × 10−8 | (12.7) | |
| GAL1-SIC1 (YAC217) | 1.28 × 1010 | 1.27 × 10−8 | (2.2) | |
| Δsic1 (YAC571) | 2.35 × 109 | 8.31 × 10−8 | (30.8) | |
| Δcdh1 (YAC300) | 1.28 × 1010 | 5.64 × 10−9 | (2.1) |
The rates shown are the probability of mutation per cell per division.
*Fold increase versus control cells in glucose media.
**Fold increase versus control cells in raffgal media.
All strains are derivative of RDKY3615 (47).
See Supplementary Table S1 for detailed genotypes.
The remaining origins in the arm are located near the telomere (Figure 2A). Telomere-adjacent regions in budding yeast chromosomes generally have origins that fire late with low efficiency, or that are dormant origins that likely function as a backup if replication of adjacent origins is delayed (7). We did not detect origin activity by 2D gels for the telomere proximal origins ARS503, ARS504 and proARS506 in any of the strains, consistent with them being dormant (Figure 2C; lack of specific probes prevented analysis of ARS502). In contrast, we detected a weak origin firing at ARS504.2 in control cells indicating that it is an active origin, but the activity was lost in sic1 cells, showing that ARS504.2 is sensitive to CDK deregulation in these cells (Figure 2C). Significantly, firing at ARS504.2 actually increased in Δcdh1 cells (Figure 2C). This is probably due to the milder CDK deregulation reducing efficiency of ARS507 but not blocking licensing of ARS504.2, which thus is able to fire in at least some cells due to delayed arrival of forks from internal origins. Altogether, these results show qualitative and quantitative differences in the pattern of origin usage that would be predicted to affect the stability of this chromosome arm. The pattern of origin usage in the chromosome arm correlates well with the rate of GCR in the region, with the greatest GCR defect in sic1 cells that have the lowest origin firing in the chromosome arm, and a much milder effect in Δcdh1 cells that retain more origin activity overall. We conclude that shortage of active origins coincides spatially with increased chromosome rearrangements in CDK-deregulated cells.
Cdh1 and Sic1 cooperate in promoting efficient origin firing at all origins
As CDK overexpression or premature CDK activation in G1 phase blocks origin licensing and the initiation of DNA replication (66,67), the sensitivity of only a subset of origins in cells lacking Sic1 or Cdh1 might reflect the fact that cells only experience moderate CDK upregulation. Worsening CDK deregulation should thus reduce origin firing efficiency further, and accordingly increase chromosomal instability. In contrast to Δsic1 Δcdh1 cells that are lethal (15,16), we analysed double mutant Sic1-depleted Δcdh1 cells that are alive although very sick due to strong CDK deregulation, with a greatly decreased length of G1 (54) (Supplementary Figure S1A). When assessing the pattern of origin usage on the left arm of chromosome V we observed that ARS508 was less efficient than in control or single mutant cells, ARS507 inefficiency increased compared to the single mutants and, similar to sic1 cells, there was no detectable firing of the telomeric proximal origins (Figure 2C). This meant that the density of origin firing in the left arm of chromosome V was severely reduced. The efficiency of all other origins examined, including those resistant to CDK deregulation in single mutants, was now reduced (Figure 1B, D and E, lower panels), a result reproduced also in the S288C strain background (Figure 2B and Supplementary Figure S4B). However, differential origin inefficiency was maintained, supporting the conclusion that origins are differentially tolerant to inhibitory CDK. The decreased firing density at the left arm of chromosome V should increase the rate of GCRs accordingly, but the compromised viability of the double mutants (Supplementary Figure S1) precluded an accurate estimation of GCR rates (data not shown). Instead, we measured the incidence of chromosome breakage by pulse field gel electrophoresis (PFGE) and Southern analysis to detect chromosome fragments (Figure 3A). We collected control, single mutant and double mutant cells grown to log phase in glucose-based media to repress SIC1 in GAL1,10p-SIC1 cells, and observed that single mutants showed increased breakage at chromosomes III and V relative to control cells, consistent with their higher instability. Moreover, breakage was further increased in the double mutant (Figure 3B and C). We conclude that the level of CDK deregulation in G1 phase directly influences the extent of reduced origin efficiency and chromosomal instability. Moreover, Cdh1 and Sic1 cooperate non-redundantly for efficient firing of origins and the prevention of chromosome breakage.
Figure 3.
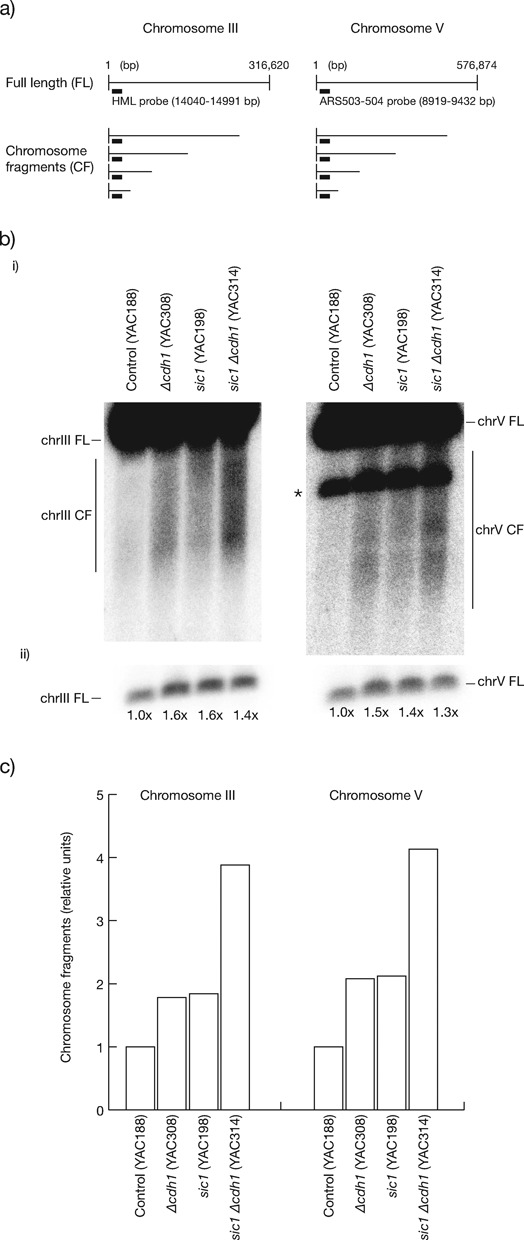
Breakage at chromosomes III and V increase in Δcdh1 sic1 cells over single mutants. (a) Maps of chromosomes III (left panel) and V (right panel) indicating the expected chromosome fragments recognised by the showed probes. (b, panel (i)) PFGE and Southern analysis of chromosomes III and V, including the band corresponding to the full-length (FL) chromosomes, and chromosome fragments (CF), as developed by the specific probes indicated in (a). *, indicates in the PFGE of chromosome V the residual signal of FL chromosome III not fully removed after stripping of the previous hybridisation with the HML probe. (Panel (ii)) Short exposures of the FL bands of blots in (i) showed as loading controls, and quantification of signals relative to control cells. (c) Graph representing the quantification of chromosome fragments of PFGEs in (b) (quantified as described in Material and Methods), in units relative to control cells.
Origin inefficiency and gross chromosomal instability in Δcdh1 cells is suppressed by increased SIC1 gene dosage
Defects in origin activity and increased GCR rates in Δcdh1 cells are likely to result from elevated levels of mitotic cyclins and M-Cdk1 activity (15,16), but the accumulation of the many other APC/C-Cdh1 substrates raised the question of the relative contribution of CDK deregulation to those defects. To determine if origin inefficiency in Δcdh1 cells is dependent on increased B-cyclin associated CDK activity in G1 phase, we duplicated the SIC1 dosage under the control of its promoter (SIC1p-SIC1). The duplicated SIC1 expression will preserve in every cell cycle the normal regulation of the Sic1 protein, which accumulates in G1 phase and is degraded outside of G1 phase, thus allowing the analysis of GCR rates. In control cells, the SIC1 duplication does not significantly modify the GCR rate (Table 2), cell viability, nor the cell cycle profile that display a normal length of G1 phase (Figure 4A and B). In addition, the SIC1p-SIC1 integration suppressed the GCRs and other defects associated with the depletion of Sic1 (Figure 4; Table 2). Significantly, Δcdh1 SIC1p-SIC1 cells also reverted the rate of GCRs (Table 2), cell viability, length of G1 phase and origin firing efficiency (Figure 4), consistent with previous findings (54,68). These results strongly suggest that increased Cdk1/Clb activity causes most of the origin firing inefficiency and GCR rates in Δcdh1 cells. We cannot, however, exclude that increased Sic1 levels are compensating the stability of certain Cdh1 substrate(s) indirectly through CDK regulation.
Table 2. The GCR rate of Δcdh1 cells is suppressed by one extra copy of SIC1.
| Relevant genotype (Strain) | Cells analysed | GCR rate | (*) |
|---|---|---|---|
| SIC1p-SIC1 (YAC800) | 9.25 × 1010 | 2.19 × 10−9 | (0.8) |
| sic1 SIC1p-SIC1 (YAC801) | 9.95 × 1010 | 5.16 × 10−9 | (1.9) |
| Δcdh1 SIC1p-SlC1 (YAC803) | 1.07 × 1011 | 2.26 × 10−9 | (0.8) |
Figure 4.
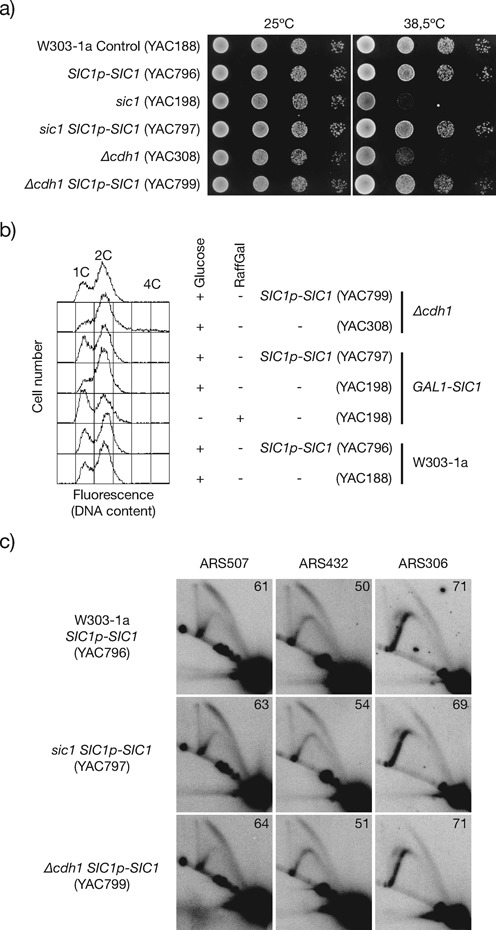
Duplicating the gene dosage of SIC1 restores origin firing efficiency in Δcdh1 cells. (a) Dilution spotting of W303–1a control, sic1 and Δcdh1 cells carrying a SIC1 allele under the control of its own promoter (SIC1p-SIC1), at 25ºC and 38.5ºC, growing in glucose-based rich media plates. (b) FACS analysis of asynchronous cells at 25ºC in glucose or raffinose-galactose (RaffGal). (c) 2D analysis of origin firing efficiency at ARS507, ARS432 and ARS306.
DISCUSSION
Our approach of studying in parallel the firing efficiency of a variety of origins in budding yeast cells lacking the CDK regulators Cdh1 and Sic1 allowed us to analyse the patterns of origin usage in CDK-deregulated cells and, at the same time, to directly address the relationships between the overall origin firing and the rates of chromosomal rearrangements in a chromosome arm.
We show that Cdh1 is required for optimal origin firing efficiency during initiation of DNA replication in budding yeast. The APC/C-Cdh1-dependent degradation of licensing-inhibitory factors including cyclin A, cyclin B and geminin in metazoans and M-cyclins in yeasts (9,10,69,70) supports the relevance of Cdh1 for origin licensing and efficient origin use in S phase. Previous studies suggested that loss of Cdh1 compromises licensing leading to genome instability (38,50,52,54,71,72), but no direct analysis of origin activity had been reported in cells lacking Cdh1. Here, we show that origin firing efficiency is reduced in Δcdh1 cells and, importantly, that the degree of origin sensitivity to the lack of Cdh1 is not uniform amongst origins. Despite the many APC/C-Cdh1 substrates, we show that duplicating the SIC1 dosage in Δcdh1 cells suppresses firing inefficiency and GCR rates, strongly supporting the idea that Cdk1/Clb inactivation is the major Cdh1 target for chromosome rearrangements and origin activity in budding yeast. This is consistent with CDK inactivation being the only essential function of APC/C for origin resetting (68). Cdh1 can also be relevant in metazoans for normal origin activity given the conserved APC/C and Cdh1 roles in targeting the origin-licensing inhibitors cyclin A and B and geminin. However, we imagine that this scenario will not be as simple in metazoans where Cdh1 also targets the licensing proteins Cdc6, Orc1 and Cdt1 in G1 phase for proteolysis (73–75).
Importantly, differential origin sensitivity is not a particularity of the lack of Cdh1. Previous studies showed that Δsic1 cells display differential origin sensitivity (34), and we show that the pattern of origin sensitivity is mostly the same in cells lacking either Cdh1 or Sic1, although Sic1 contributes more than Cdh1 for efficient origin activity in budding yeast. Origin usage is a reflect of the extent of CDK deregulation. All origins are sensitive to more severe CDK deregulation in the double mutant sic1 Δcdh1, consistent with previous studies of strong CDK deregulation (76), although differential sensitivities are maintained amongst origins. Hence, in budding yeast the CDK regulators in G1 phase, Cdh1 and Sic1, cooperate to activate exceeding origin numbers for efficient origin activation. We presume that the differential tolerance of origins to CDK deregulation adds more redundancy and robustness to replication initiation.
Differential origin responses have been previously reported upon mutation of CDK-inhibitory controls in pre-RC components, resulting in re-replication (77–79), but the patterns of origin sensitivity are different to those observed in our study. We see that sensitivity is independent on either the timing of origin firing, the origin efficiency, or the chromosomal location. We hypothesise that origin sensitivity to CDK deregulation is instead dependent on unique origin characteristics as the cis-acting origin elements that, apart from the ARS consensus site, are highly variable amongst origins and modulate origin activities (80,81). Hence, particular configurations of cis elements may result in differential recruitment abilities amongst origins for licensing and/or firing factors in budding yeast, as observed in fission yeast origins (82), influencing the origin tolerance to CDK deregulations.
Origin redundancy is likely to be a determinant factor for the stability of a chromosome region in CDK-deregulated cells. For chromosome maintenance, normal cells tolerate the loss of a few active origins, but not the additional loss of flanking efficient origins and particularly the loss of dormant origins at chromosome ends (29,60). In CDK-deregulated cells, the coincidence of sensitive origins at a chromosome region can lead to reduce firing to levels compromising chromosome stability. We found, at the left arm of chromosome V, that origin firing shortage increases with the severity of CDK deregulation, in correlation with higher rates of GCRs. Furthermore, the extent of efficiency loss, the number of origins losing activity and the activity of a dormant origin reflect the instability of the chromosome.
Fragile site expression is linked to origin paucity in human cells (39) and reduced Mcm2-7 subunits in primary MEFs decrease the licensing of dormant origins, leading to genome instability and increased cancer incidence (33). Presumably, in CDK-deregulated cells origin firing shortage can compromise the timing and the completion of DNA replication regionally within S phase, agreeing with previous suggestions (24,34–36,83). In mammalian cells, the activity of individual origins upon CDK deregulation remains largely unaddressed. Our preliminary data indicates that origin efficiency is altered in p27Kip1-/- MEFs (J. Sequeira-Mendes, C. Ávila-Zarza, M. Gómez and A. Calzada, unpublished results), but it remains to be established whether this reflects regional changes in origin density or in the timing of firing. The lengthening of S phase in CDK-deregulated cells might also reflect an altered DNA replication dynamics, consistent with increased arrested forks in human cell lines overexpressing cyclin E (84), and with slower replication fork progression and reduced frequency of termination events in Cdh1-depleted cells (85). Addressing the dynamics of DNA replication along the genome in time and space in CDK-deregulated oncogenic cells, and whether it contributes to chromosome instability by untimely termination, are interesting open questions that will require further analysis in the future.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including reference [86].
Acknowledgments
We are thankful to A. Bueno, J. Pérez-Martín, J.A. Tercero and F. Prado for helpful discussions, advice, and encouraging support, and K. Labib and J. P-M for critically reading the manuscript. We thank S. Tanaka, K. Myung and R. Kolodner for strains and protocols for the GCR assay. We also thank A.B., K.L. and D. Koshland for plasmids, and yeast strains. P.A-D received a predoctoral fellowship (University of Salamanca), F.D. is supported by a predoctoral FPI fellowship (Spanish Ministry of Economy and Competitiveness) and F.G. receives a FCT doctorate fellowship for research abroad (Portuguese Ministry of Education and Science).
FUNDING
Institute of Health Carlos III, Fondo de Investigaciones Sanitarias, and Fondo Europeo de Desarrollo Regional [PI061409 to A.C.]; Spanish Ministry of Economy and Competitiveness [BFU2010-18225 to A.C., BFU2010-18992 to M.G.]. Funding for open access charge: Spanish Ministry of Economy and Competitiveness.
Conflict of interest. None declared.
REFERENCES
- 1.Hyrien O., Marheineke K., Goldar A. Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. Bioessays. 2003;25:116–125. doi: 10.1002/bies.10208. [DOI] [PubMed] [Google Scholar]
- 2.Schwob E. Flexibility and governance in eukaryotic DNA replication. Curr. Opin. Microbiol. 2004;7:680–690. doi: 10.1016/j.mib.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Rhind N. DNA replication timing: random thoughts about origin firing. Nat. Cell Biol. 2006;8:1313–1316. doi: 10.1038/ncb1206-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Remus D., Diffley J. Eukaryotic DNA replication control: lock and load, then fire. Curr. Opin. Cell Biol. 2009;21:771–777. doi: 10.1016/j.ceb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Gambus A., Jones R.C., Sanchez-Diaz A., Kanemaki M., van Deursen F., Edmondson R.D., Labib K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006;8:358–366. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- 6.Ilves I., Petojevic T., Pesavento J.J., Botchan M.R. Activation of the MCM2–7 helicase by association with Cdc45 and GINS proteins. Mol. Cell. 2010;37:247–258. doi: 10.1016/j.molcel.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vujcic M., Miller C.A., Kowalski D. Activation of silent replication origins at autonomously replicating sequence elements near the HML locus in budding yeast. Mol. Cell Biol. 1999;19:6098–6109. doi: 10.1128/mcb.19.9.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santocanale C., Sharma K., Diffley J.F. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999;13:2360–2364. doi: 10.1101/gad.13.18.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diffley J.F.X. Regulation of early events in chromosome replication. Curr. Biol. 2004;14:R778–R786. doi: 10.1016/j.cub.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 10.Blow J.J., Dutta A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 2005;6:476–486. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diffley J.F.X. The many faces of redundancy in DNA replication control. Cold Spring Harb. Symp. Quant. Biol. 2011;75:135–142. doi: 10.1101/sqb.2010.75.062. [DOI] [PubMed] [Google Scholar]
- 12.Sudakin V., Ganoth D., Dahan A., Heller H., Hershko J., Luca F.C., Ruderman J.V., Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell. 1995;6:185–197. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King R.W., Peters J.M., Tugendreich S., Rolfe M., Hieter P., Kirschner M.W. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- 14.Irniger S., Piatti S., Michaelis C., Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–278. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- 15.Schwab M., Lutum A.S., Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- 16.Visintin R., Prinz S., Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- 17.Harper J.W., Adami G.R., Wei N., Keyomarsi K., Elledge S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 18.el-Deiry W.S., Tokino T., Velculescu V.E., Levy D.B., Parsons R., Trent J.M., Lin D., Mercer W.E., Kinzler K.W., Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 19.Polyak K., Kato J.Y., Solomon M.J., Sherr C.J., Massague J., Roberts J.M., Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 20.Nugroho T.T., Mendenhall M.D. An inhibitor of yeast cyclin-dependent protein kinase plays an important role in ensuring the genomic integrity of daughter cells. Mol. Cell Biol. 1994;14:3320–3328. doi: 10.1128/mcb.14.5.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwob E., Böhm T., Mendenhall M.D., Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 22.Reed S.I. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat. Rev. Mol. Cell Biol. 2003;4:855–864. doi: 10.1038/nrm1246. [DOI] [PubMed] [Google Scholar]
- 23.Sherr C.J. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- 24.Bartkova J., Hořejší Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C., et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 25.Gorgoulis V.G., Vassiliou L.-V.F., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R.A., Kastrinakis N.G., Levy B., et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 26.Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre’ M., Nuciforo P.G., Bensimon A., et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 27.D’adda Di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer. 2008;8:512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 28.Halazonetis T.D., Gorgoulis V.G., Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 29.Dershowitz A., Newlon C.S. The effect on chromosome stability of deleting replication origins. Mol. Cell Biol. 1993;13:391–398. doi: 10.1128/mcb.13.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruschi C.V., McMillan J.N., Coglievina M., Esposito M.S. The genomic instability of yeast cdc6–1/cdc6–1 mutants involves chromosome structure and recombination. Mol. Gen. Genet. 1995;249:8–18. doi: 10.1007/BF00290230. [DOI] [PubMed] [Google Scholar]
- 31.Stirling P.C., Bloom M.S., Solanki-Patil T., Smith S., Sipahimalani P., Li Z., Kofoed M., Ben-Aroya S., Myung K., Hieter P. The complete spectrum of yeast chromosome instability genesidentifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet. 2011;7:e1002057. doi: 10.1371/journal.pgen.1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodward A.M., Göhler T., Luciani M.G., Oehlmann M., Ge X., Gartner A., Jackson D.A., Blow J.J. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawabata T., Luebben S.W., Yamaguchi S., Ilves I., Matise I., Buske T., Botchan M.R., Shima N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell. 2011;41:543–553. doi: 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lengronne A., Schwob E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1) Mol. Cell. 2002;9:1067–1078. doi: 10.1016/s1097-2765(02)00513-0. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka S., Diffley J.F.X. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002;16:2639–2649. doi: 10.1101/gad.1011002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ekholm-Reed S., Méndez J., Tedesco D., Zetterberg A., Stillman B., Reed S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004;165:789–800. doi: 10.1083/jcb.200404092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong A., Narbonne-Reveau K., Riesgo-Escovar J., Fu H., Aladjem M.I., Lilly M.A. The cyclin-dependent kinase inhibitor Dacapo promotes replication licensing during Drosophila endocycles. EMBO J. 2007;26:2071–2082. doi: 10.1038/sj.emboj.7601648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.García-Higuera I., Manchado E., Dubus P., Cañamero M., Méndez J., Moreno S., Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 2008;10:802–811. doi: 10.1038/ncb1742. [DOI] [PubMed] [Google Scholar]
- 39.Letessier A., Millot G.A., Koundrioukoff S., Lachagès A.-M., Vogt N., Hansen R.S., Malfoy B., Brison O., Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 40.Ozeri-Galai E., Lebofsky R., Rahat A., Bester A.C., Bensimon A., Kerem B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell. 2011;43:122–131. doi: 10.1016/j.molcel.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 41.Calzada A., Hodgson B., Kanemaki M., Bueno A., Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–1919. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sikorski R.S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crampton A., Chang F., Pappas D.L., Frisch R.L., Weinreich M. An ARS element inhibits DNA replication through a SIR2-dependent mechanism. Mol. Cell. 2008;30:156–166. doi: 10.1016/j.molcel.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 44.Theis J.F., Irene C., Dershowitz A., Brost R.L., Tobin M.L., Di Sanzo F.M., Wang J.-Y., Boone C., Newlon C.S. The DNA damage response pathway contributes to the stability of chromosome III derivatives lacking efficient replicators. PLoS Genet. 2010;6:e1001227. doi: 10.1371/journal.pgen.1001227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita M., Hori Y., Shinomiya T., Obuse C., Tsurimoto T., Yoshikawa H., Shirahige K. The efficiency and timing of initiation of replication of multiple replicons of Saccharomyces cerevisiae chromosome VI. Genes Cells. 1997;2:655–665. doi: 10.1046/j.1365-2443.1997.1530351.x. [DOI] [PubMed] [Google Scholar]
- 46.Foiani M., Marini F., Gamba D., Lucchini G., Plevani P. The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol. Cell Biol. 1994;14:923–933. doi: 10.1128/mcb.14.2.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen C., Kolodner R.D. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat. Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 48.Myung K., Chen C., Kolodner R.D. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–1076. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 49.Friedman K.L., Brewer B.J. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 1995;262:613–627. doi: 10.1016/0076-6879(95)62048-6. [DOI] [PubMed] [Google Scholar]
- 50.Ross K.E., Cohen-Fix O. The role of Cdh1p in maintaining genomic stability in budding yeast. Genetics. 2003;165:489–503. doi: 10.1093/genetics/165.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lehman N.L., Verschuren E.W., Hsu J.Y., Cherry A.M., Jackson P.K. Overexpression of the anaphase promoting complex/cyclosome inhibitor Emi1 leads to tetraploidy and genomic instability of p53-deficient cells. Cell Cycle. 2006;5:1569–1573. doi: 10.4161/cc.5.14.2925. [DOI] [PubMed] [Google Scholar]
- 52.Engelbert D., Schnerch D., Baumgarten A., Wäsch R. The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene. 2008;27:907–917. doi: 10.1038/sj.onc.1210703. [DOI] [PubMed] [Google Scholar]
- 53.Enserink J.M., Hombauer H., Huang M.-E., Kolodner R.D. Cdc28/Cdk1 positively and negatively affects genome stability in S. cerevisiae. J. Cell Biol. 2009;185:423–437. doi: 10.1083/jcb.200811083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wäsch R., Cross F.R. APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature. 2002;418:556–562. doi: 10.1038/nature00856. [DOI] [PubMed] [Google Scholar]
- 55.Archambault V., Li C.X., Tackett A.J., Wasch R., Chait B.T., Rout M.P., Cross F.R. Genetic and biochemical evaluation of the importance of Cdc6 in regulating mitotic exit. Mol. Biol. Cell. 2003;14:4592–4604. doi: 10.1091/mbc.E03-06-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brewer B.J., Fangman W.L. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- 57.Friedman K.L., Brewer B.J., Fangman W.L. Replication profile of Saccharomyces cerevisiae chromosome VI. Genes Cells. 1997;2:667–678. doi: 10.1046/j.1365-2443.1997.1520350.x. [DOI] [PubMed] [Google Scholar]
- 58.Poloumienko A., Dershowitz A., De J., Newlon C.S. Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol. Biol. Cell. 2001;12:3317–3327. doi: 10.1091/mbc.12.11.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siow C.C., Nieduszynska S.R., Muller C.A., Nieduszynski C.A. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res. 2012;40:D682–D686. doi: 10.1093/nar/gkr1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dershowitz A., Snyder M., Sbia M., Skurnick J.H., Ong L.Y., Newlon C.S. Linear derivatives of Saccharomyces cerevisiae chromosome III can be maintained in the absence of autonomously replicating sequence elements. Mol. Cell Biol. 2007;27:4652–4663. doi: 10.1128/MCB.01246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerhardt J., Tomishima M.J., Zaninovic N., Colak D., Yan Z., Zhan Q., Rosenwaks Z., Jaffrey S.R., Schildkraut C.L. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell. 2014;53:19–31. doi: 10.1016/j.molcel.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raghuraman M.K., Winzeler E.A., Collingwood D., Hunt S., Wodicka L., Conway A., Lockhart D.J., Davis R.W., Brewer B.J., Fangman W.L. Replication dynamics of the yeast genome. Science. 2001;294:115–121. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- 63.Aparicio O.M. Location, location, location: it's all in the timing for replication origins. Genes Dev. 2013;27:117–128. doi: 10.1101/gad.209999.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Knott S.R.V., Peace J.M., Ostrow A.Z., Gan Y., Rex A.E., Viggiani C.J., Tavaré S., Aparicio O.M. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell. 2012;148:99–111. doi: 10.1016/j.cell.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wyrick J.J., Aparicio J.G., Chen T., Barnett J.D., Jennings E.G., Young R.A., Bell S.P., Aparicio O.M. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science. 2001;294:2357–2360. doi: 10.1126/science.1066101. [DOI] [PubMed] [Google Scholar]
- 66.Dahmann C., Diffley J.F., Nasmyth K.A. S-phase-promoting cyclin-dependent kinases prevent re-replication by inhibiting the transition of replication origins to a pre-replicative state. Curr. Biol. 1995;5:1257–1269. doi: 10.1016/s0960-9822(95)00252-1. [DOI] [PubMed] [Google Scholar]
- 67.Piatti S., Böhm T., Cocker J.H., Diffley J.F., Nasmyth K. Activation of S-phase-promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- 68.Noton E., Diffley J.F. CDK inactivation is the only essential function of the APC/C and the mitotic exit network proteins for origin resetting during mitosis. Mol. Cell. 2000;5:85–95. doi: 10.1016/s1097-2765(00)80405-0. [DOI] [PubMed] [Google Scholar]
- 69.Sivaprasad U., Machida Y.J., Dutta A. APC/C–the master controller of origin licensing. Cell Div. 2007;2:8. doi: 10.1186/1747-1028-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hook S.S., Lin J.J., Dutta A. Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 2007;19:663–671. doi: 10.1016/j.ceb.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wäsch R., Engelbert D. Anaphase-promoting complex-dependent proteolysis of cell cycle regulators and genomic instability of cancer cells. Oncogene. 2005;24:1–10. doi: 10.1038/sj.onc.1208017. [DOI] [PubMed] [Google Scholar]
- 72.Wäsch R., Robbins J.A., Cross F.R. The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene. 2010;29:1–10. doi: 10.1038/onc.2009.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Narbonne-Reveau K., Senger S., Pal M., Herr A., Richardson H., Asano M., Deak P., Lilly M. APC/CFzr/Cdh1 promotes cell cycle progression during the Drosophila endocycle. Development. 2008;135:1451–1461. doi: 10.1242/dev.016295. [DOI] [PubMed] [Google Scholar]
- 74.Petersen B.O., Wagener C., Marinoni F., Kramer E.R., Melixetian M., Lazzerini Denchi E., Gieffers C., Matteucci C., Peters J.M., Helin K. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000;14:2330–2343. doi: 10.1101/gad.832500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sugimoto N., Kitabayashi I., Osano S., Tatsumi Y., Yugawa T., Narisawa-Saito M., Matsukage A., Kiyono T., Fujita M. Identification of novel human Cdt1-binding proteins by a proteomics approach: proteolytic regulation by APC/CCdh1. Mol. Biol. Cell. 2008;19:1007–1021. doi: 10.1091/mbc.E07-09-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Detweiler C.S., Li J.J. Ectopic induction of Clb2 in early G1 phase is sufficient to block prereplicative complex formation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1998;95:2384–2389. doi: 10.1073/pnas.95.5.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nguyen V.Q., Co C., Li J.J. Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature. 2001;411:1068–1073. doi: 10.1038/35082600. [DOI] [PubMed] [Google Scholar]
- 78.Green B.M., Morreale R.J., Ozaydin B., Derisi J.L., Li J.J. Genome-wide mapping of DNA synthesis in Saccharomyces cerevisiae reveals that mechanisms preventing reinitiation of DNA replication are not redundant. Mol. Biol. Cell. 2006;17:2401–2414. doi: 10.1091/mbc.E05-11-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanny R.E., MacAlpine D.M., Blitzblau H.G., Bell S.P. Genome-wide analysis of re-replication reveals inhibitory controls that target multiple stages of replication initiation. Mol. Biol. Cell. 2006;17:2415–2423. doi: 10.1091/mbc.E05-11-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin S., Kowalski D. Functional equivalency and diversity of cis-acting elements among yeast replication origins. Mol. Cell Biol. 1997;17:5473–5484. doi: 10.1128/mcb.17.9.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rao H., Marahrens Y., Stillman B. Functional conservation of multiple elements in yeast chromosomal replicators. Mol. Cell Biol. 1994;14:7643–7651. doi: 10.1128/mcb.14.11.7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu P.-Y.J., Nurse P. Establishing the program of origin firing during S phase in fission Yeast. Cell. 2009;136:852–864. doi: 10.1016/j.cell.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Spruck C.H., Won K.A., Reed S.I. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 84.Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L., Kolettas E., Niforou K., Zoumpourlis V., et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 85.Yuan X., Srividhya J., De Luca T., Lee J.-H.E., Pomerening J.R. Uncovering the role of APC-Cdh1 in generating the dynamics of S-phase onset. Mol. Biol. Cell. 2014;25:441–456. doi: 10.1091/mbc.E13-08-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yabuki N., Terashima H., Kitada K. Mapping of early firing origins on a replication profile of budding yeast. Genes to cells: devoted to molecular & cellular mechanisms. 2002;7:781–789. doi: 10.1046/j.1365-2443.2002.00559.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.