


Abstract
Nucleosomes play important roles in a cell beyond their basal functionality in chromatin compaction. Their placement affects all steps in transcriptional regulation, from transcription factor (TF) binding to messenger ribonucleic acid (mRNA) synthesis. Careful profiling of their locations and dynamics in response to stimuli is important to further our understanding of transcriptional regulation by the state of chromatin. We measured nucleosome occupancy in human hepatic cells before and after treatment with transforming growth factor beta 1 (TGFβ1), using massively parallel sequencing. With a newly developed method, SuMMIt, for precise positioning of nucleosomes we inferred dynamics of the nucleosomal landscape. Distinct nucleosome positioning has previously been described at transcription start site and flanking TF binding sites. We found that the average pattern is present at very few sites and, in case of TF binding, the double peak surrounding the sites is just an artifact of averaging over many loci. We systematically searched for depleted nucleosomes in stimulated cells compared to unstimulated cells and identified 24 318 loci. Depending on genomic annotation, 44–78% of them were over-represented in binding motifs for TFs. Changes in binding affinity were verified for HNF4α by qPCR. Strikingly many of these loci were associated with expression changes, as measured by RNA sequencing.
INTRODUCTION
Nucleosomes form the basal units for condensation of deoxyribonucleic acid (DNA) into higher order chromatin through inter-nucleosomal interactions of histones (1). These interactions are, however, dependent on post-translational modifications (PTMs) of histones (2) and high nucleosome occupancy alone is therefore not a reliable indicator of condensed chromatin. Rather, intragenic regions are more nucleosome dense than intergenic regions (3), suggesting that nucleosomes play an important role beyond their basal functionality in chromatin compaction. In fact, the locations of nucleosomes have been suggested to follow distinct patterns around transcription start sites (TSSs) (4,5), internal exons (6) and enhancers (7,8). At these loci, PTMs of histones reflect the transcriptional (6,9–12) or regulatory status (9,13).
Not only do PTMs of histones affect gene transcription, the mere placement of nucleosomes may also play a regulatory role. For instance, the nucleosome-free region (NFR) upstream of a TSS may be crucial for recruiting the transcriptional machinery to a gene, while the first nucleosome downstream of the TSS may be involved in pre-initiation complex formation and ribonucleic acid (RNA) polymerase II (RNAPII) promoter-proximal pausing (4,14). Along the DNA, histones and other DNA-binding proteins, such as transcription factors (TFs), competitively bind to the DNA. The binding of TFs is thus affected by the placement and stability of nucleosomes along DNA and also by enzymatic activities that modify, reposition, reconfigure or even remove nucleosomes. Stably positioned nucleosomes may block the binding of some TFs (15), while other TFs may reposition nucleosomes with the involvement of other partners, such as histone modifiers or coactivators (14). Hence, nucleosome positioning and repositioning play important roles in the transcriptional regulation of a cell.
Current knowledge about the nucleosomal landscapes in humans is limited to a few cell types and their dynamics in response to intracellular events or external stimuli are even less explored (5,16). There is thus a great need to profile the nucleosomal landscapes of various cell types and to systematically analyze the repositioning and removal of nucleosomes to further our understanding of chromatin regulation of transcription.
Several methods for processing the resulting data from sequencing of nucleosomal or TF-bound DNA have been developed. Some early methods transformed the data either by extending sequence reads to the average length of sequenced DNA fragments (10,17) or through strand-directed shifts of reads to a putative nucleosome center (18,19). Calling of interaction sites was then based on peak shapes or on significance of counts or kernel density estimates when compared to the genomic distribution of transformed data. To remove unreliable predictions, one method used thresholds of the minimum number of reads from each strand (17). Strand-specific requirements were also adopted in other methods (20,21), yielding more reliable predictions. However, in the majority of suggested methods only non-overlapping predictions of nucleosomes are considered. Since nucleosome sequencing measures an average over millions of cells, such a limitation assumes that all cells have a nucleosome at the same or at non-conflicting locations. This is, however, not the case for a large fraction of nucleosomes (4), calling for careful considerations of the data and the heterogeneity of nucleosome positioning.
Here, we introduce SuMMIt, a Bayesian strategy for positioning of nucleosomes or other protein–DNA interactions that require support from both ends of sequenced fragments for accurate positioning. For precise placement of interaction sites unaffected by discrepancies in localization between cells in the same sequenced sample, we model their mid-positions as well as background noise in the data. SuMMIt also allows for investigating differences in e.g. nucleosome positioning under different conditions. Using SuMMIt, we profile the nucleosomal landscape of the human hepatocellular HepG2 genome before and after 1 h of stimulation with transforming growth factor beta 1 (TGFβ1). The TGFβ pathways have been extensively studied and several TFs have been shown to regulate the expression of specific genes in TGFβ stimulated cells (22). In addition, HepG2 is one of the cell lines studied by the ENCODE consortium (23) and several data sets of TF binding in this cell line exist. Taken together, monitoring the nucleosomal landscape in HepG2 cells in response to TGFβ1 stimuli offers the opportunity to study nucleosome dynamics in a well-characterized system. As a first approach, we impose strict criteria for the identification of depleted nucleosomes in response to TGFβ1 treatment. We further investigate their possible relation to transcription and transcriptional regulation.
MATERIALS AND METHODS
Cell culture and TGFβ1 treatment
HepG2 cells were cultured to confluence in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). Cells were serum-starved overnight (1% FBS) before stimulation with TGFβ1 to a final concentration of 2.5 ng/ml. TGFβ1-treated and control cells were harvested after 1 h of treatment.
Preparation and sequencing of mononucleosomes
HepG2 control and TGFβ1-treated cells were washed with phosphate buffered saline and resuspended in ice-cold buffer A [0.32-M sucrose, 15-mM Hepes pH 7.9, 60-mM KCl, 2-mM ethylenediaminetetraacetic acid (EDTA), 0.5-mM ethyleneglycoltetraaceticacid (EGTA), 0.5% bovine serum albumin, 0.5-mM spermidine, 0.15-mM spermine and 0.5-mM dithiothreitol (DTT)]. After a short incubation on ice, cells were homogenized with a Dounce homogenizer. The nuclear suspension was slowly layered on an equal volume of ice-cold buffer B (30% sucrose, 15-mM Hepes pH 7.9, 60-mM KCl, 2-mM EDTA, 0.5-mM EGTA, 0.5-mM spermidine, 0.15-mM spermine and 0.5-mM DTT) and centrifuged at 3000 revolutions per minute for 15 min to pellet the nuclei. Collected nuclei were washed once and resuspended in buffer N (0.34-mM sucrose, 15-mM HEPES pH 7.5, 60-mM KCl, 15-mM NaCl, 0.5-mM spermidine, 0.15-mM spermine and 0.15-mM β-mercaptoethanol). Samples were adjusted to 3-mM CaCl2 and the nuclear suspension was adjusted to 30 million nuclei per milliliter and incubated at 37°C for 5 min. Micrococcal nuclease (MNase, 300 U) was added to each aliquot and incubated for another 5 min at 37°C. Buffer S, containing 90-mM HEPES pH 7.9, 220-mM NaCl, 10-mM EDTA, 2% Triton X-100, 0.2% Na-deoxycholate, 0.2% sodium dodecyl sulphate (SDS), 0.5-mM phenylmethanesulfonylflouride (PMSF) and 2-μg/ml aprotinin, was added to stop the reaction. The solution was centrifuged and the supernatant was used to extract the DNA by phenol/chloroform/ethanol precipitation. DNA was loaded onto a 2% agarose gel and the mononucleosome size (147 bp) fragments were excised. DNA from excised gel pieces was purified with a Qiagen gel extraction kit. Libraries of nucleosomal DNA were prepared from 1-μg DNA according to the SOLiD fragment library protocol. After ligation and nick-translation, two emulsion polymerase chain reactions (PCRs) were done for each sample, one without pre-amplification of the library and one after three sounds of PCR. Sequencing was done using v3 chemistry with one slide per emulsion PCR.
RNA-seq
Total RNA was prepared using the TRIzol-chloroform method (Invitrogen) according to the manufacturer's protocol. We used a BioAnalyzer (Agilent) to quantify the RNA integrity (RIN 9.8) before library construction. The SOLiD whole transcriptome library kit (Ambion) was used to produce strand-specific libraries. This procedure includes two rounds of ribosomal RNA depletion (RiboMinus, Invitrogen) and ligation of adaptors to RNAse-III fragmented RNA before reverse transcription and library amplification (AmpliTaq, 18 cycles). Each library was split after emulsion PCR and sequenced on two slides (SOLiD v3 chemistry) with TGFβ1 and control libraries sequenced in parallel on the same instruments to avoid instrument-related biases.
Validation of RNA-seq using Taqman real-time quantitative reverse transcriptase-PCR
RNA from HepG2 cells with and without TGFβ1-stimulation was isolated as described above. The RNA was treated with DNase I (Qiagen) to degrade any genomic DNA. Reverse transcription was performed with 1.0 μg of RNA per 20-μl reaction using the iScript complementary DNA (cDNA) Synthesis Kit (Bio-Rad). Triplicate 25-μl reactions were prepared containing 1.0 μl of cDNA, TaqMan Gene Expression Master Mix (Applied Biosystems) and TaqMan Gene Expression Assay Mix (Applied Biosystems) specific for the transcript investigated according to the instructions of the manufacturer. PCR was performed on an Applied Biosystems 7000 Real-Time PCR System (Applied Biosystems) with SDS software 1.2.3, using the following conditions: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Control reactions to demonstrate the specificity of the reactions were for each gene expression assay: use of cDNA synthesized without reverse transcriptase and without template RNA, respectively, and replacing the cDNA with water altogether. All controls passed and are not included in the figure. Expression levels were determined with the comparative Ct method using GAPDH as reference, related to the RNA sequencing expression levels and presented in Figure 8C.
Figure 8.
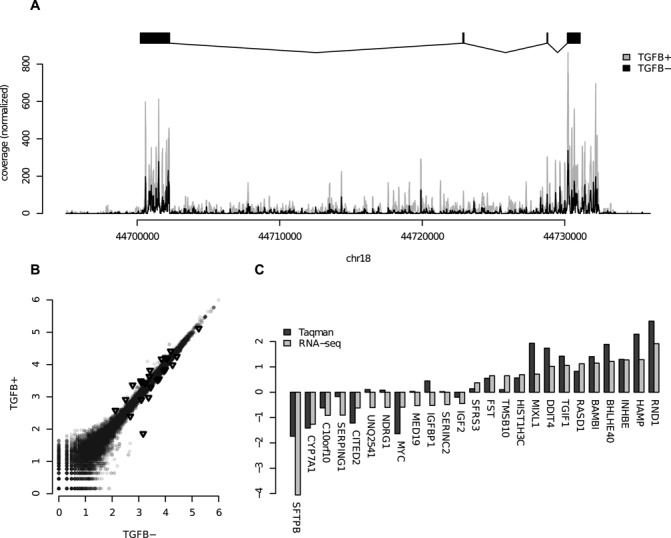
(A) Coverage plots of RNA-seq data over the SMAD7 gene. The TGFβ stimulated (TGFB+) data have been normalized to the same sequencing depth as the unstimulated (TGFB−). (B) Scatter plot of RPKM-counts over genes for TGFB+ and TGFB−. The genes selected for Taqman validation are marked with black triangles. (C) Taqman validation results for 25 selected genes. The RNA-seq is represented as log2 of sequence depth normalized fold change between TGFB+ and TGFB−, and the Taqman values are also log2 of fold change between TGFB+ and TGFB−.
HNF4α ChIP and quantiative polymerase chain reaction
ChIP on control and TGFβ1-stimulated cells was performed as described in (24) using anti HNF4α (SC-6556) antibody. qPCR was then performed in triplicates at 10 (10) candidate sites where HNF4α binding was predicted to have changed due to nucleosomal relocation. The qPCR was performed using SYBR green and fold enrichment between TGFβ stimulated and control cells was calculated with average qPCR values obtained from three (3) negative sites. Locations and primer sequences are available from Supplementary Table S4.
SuMMIT rationale
To accurately position sequenced nucleosomes, we developed a Bayesian method that requires support from both ends of sequenced DNA fragments (see Supplementary Methods for details). In summary, SuMMIt (Strand-based Mixture Modeling of protein–DNA Interactions) infers the a posteriori most probable model from the data describing nucleosome mid-positions and background noise. Information about size selection of sequenced DNA fragments is incorporated in the model, determining the expected distance from the boundaries to the mid-position of a nucleosome. The a priori expected distance between reads defining a nucleosome is 147 bp (25). Hence, in theory, start positions of sequenced reads are expected to be located 73 bp from a nucleosome mid-position, i.e. from the translational setting of a nucleosome. However, due to heterogeneity in nucleosome positioning among cells and biased cleavage of nucleosomal DNA, the positions may vary. To deal with such positional variation, SuMMIt considers a size range of sequenced fragments, 130–180 bp for nucleosomal DNA, when determining where start positions of reads supporting a nucleosome mid-position will be located (Supplementary Figure S1). The lower and upper boundaries of the size range are used to determine the size of two flanking windows. In these windows, we use the counts of reads, in favorable direction only, for training Poisson mixture models to separate true positioning data from background noise. Using these models, log-odds of nucleosome mid-position against the background are calculated for each position in the genome separately for each strand. Nucleosome mid-positions are called whenever support (positive log-odds) is given from both strands. The necessary parameters for the Poisson mixture models are learnt from the data using a Gibbs sampling approach. The fuzziness, i.e. level of concordance among cells, for each called mid-position is also determined and calculated as the positional spread (standard deviation) of surrounding reads.
Training and prediction using sequencing data
SuMMIt was trained separately on HepG2 TGFβ unstimulated and stimulated nucleosome sequencing data. Size selection of sequenced DNA fragments determined the minimum (mind) and maximum (maxd) fragment lengths to 130 and 180, respectively, which were used in the model. Gibbs sampling was run with 1000 iterations after 100 burn-in iterations. Manual inspection of the parameter values after training ensured convergence after 1000 iterations. Each predicted nucleosome mid-position with support from both sense and antisense read data was assigned the sum of log-odds values LO+ and LO−. The predicted locations were combined into regions with consecutive positive log-odds values allowing no gaps. Regions were subsequently merged with adjacent ones if the center-to-center distance was less than 65 bp.
Fuzziness of nucleosomes
The fuzziness of nucleosome positions was calculated from the average of standard deviations of sense and antisense reads falling into windows [i−(θf,1+θf,2), i−1] and [i+1, i+θr,1+θr,2] of a nucleosome mid-position i, respectively. Using these windows, no reads defining adjacent non-overlapping nucleosomes will affect the fuzziness score. Nucleosomes with a fuzziness score less than or equal to 60, between 60 and 80, and greater than 80 were considered phased, intermediate and fuzzy, respectively. These numbers were motivated by the distribution of fuzziness scores (Supplementary Figure S3).
Nucleosomal depletions
The models of nucleosome mid-positions and background noise allowed us to calculate log-odds of nucleosomal changes between samples. For each putative position in the genome, we calculated the odds of having a nucleosome mid-position in one sample and no mid-position in the other sample against all other combinations, separately for sense read data and antisense read data. Summation over strands yielded the resulting change log-odds score.
To systematically identify nucleosomes present in unstimulated cells that were depleted in TGFβ stimulated cells and to avoid any biases in sequencing depth between samples possibly not dealt with by SuMMIt modeling of data, we imposed strict filters on identified nucleosomes with non-overlapping positions between samples. Firstly, we required that the regions called in unstimulated cells did not overlap any region called in TGFβ stimulated cells with LO+ > 0 or LO− > 0 within flanking regions of 65 bp from the interior mid-positions. Secondly, we required log-odds of change above 10 to pass the filtering. Finally, we required that the flanking 65-bp regions of unstimulated mid-positions did not overlap any nucleosome interior regions.
Annotations
Gene and exon annotations were extracted from the ENSMBL database (26) (release 54, NCBI 36). Only known protein-coding genes were considered. For partitioning of loci, we applied a sequential approach similar to a previous partitioning scheme of ENCODE data (27) in which loci were associated with the following characteristics, in prioritized order:
| Exonic | Overlapping with an exon |
| Intronic proximal | Intronic and no more than 5 kb away from an exon |
| Intergenic proximal | Intergenic and no more than 5 kb away from an exon |
| Intronic distal | Intronic and more than 5 kb away from an exon |
| Intergenic distal | Intergenic and more than 5 kb away from an exon |
Partitioning of loci into these categories was done using BEDTools (28).
Motif finding
For the identification of over-represented TF motifs in sequences of loci with depleted nucleosomes, we ran the program clover (29) on 130-bp sequences centered at center positions of nucleosome interior regions in TGFβ− cells associated with depleted nucleosomes in TGFβ+ cells. This was performed separately for each annotation category (described above). Twice as many DNA sequences were randomly selected from nucleosome loci falling into the same partitioning category and with no associated depletion in TGFβ stimulated cells as background. TF motifs with raw scores above 6 and P-values less than 0.05 were considered for further analyses, as recommended by the authors of the program Clover. Position weight matrices were collected from the JASPAR database (30), clade vertebrates.
Data handling
All sequencing data were represented using a binary file format (31). The nucleosome data were represented by the position of the 5′ end of the aligned reads for nucleosome mid-position predictions by SuMMIt. Full-length alignments, 147-bp extended sense and antisense reads and combined signal, were used for plotting purposes and comparison with log-odds values. All footprints were produced using the SICTIN software suite (31).
Implementation
SuMMIt was implemented in C++ relying on functions from the GNU Scientific Library (http://www.gnu.org/software/gsl/) (32) and is platform independent. The source code has been successfully compiled and run on single Mac OSX Server (leopard) computational node with only 2 GB of RAM. The current implementation of SuMMIt traverses one chromosome at a time in large chunks, resulting in low memory requirements.
RESULTS
Data
We used massively parallel sequencing-by-ligation (SOLiD System, Applied Biosystems, Foster City, CA, USA) to sequence size-selected mono-nucleosomal MNaseI cleaved DNA fragments from HepG2 cells before and after stimulation with TGFβ1 (see the Materials and Methods section for details). In brief, 396M 50-bp reads were uniquely aligned to the reference genome in the unstimulated cells and 301M reads in the TGFβ stimulated sample. In addition, we also performed strand-specific RNA sequencing (see Materials and Methods section for details) resulting in 195.3M uniquely aligned reads for the unstimulated cells and 146.5M reads for TGFβ stimulated cells (Supplementary Table S1).
Nucleosome positioning revealed in detail
We applied SuMMIt on nucleosome data from unstimulated cells and the resulting log-odds scores provided clear indications of which positions had good support from both strands. The positioning was made at a very high resolution since only nucleosome mid-positions were called. Plotting these values gave a crisp view of the nucleosome landscape revealing details that were hidden using mere counts of read alignments. This is apparent around the TSS of gene HEY2 (Figure 1A) where log-odds values indicate several nucleosomes positioned at loci where peak shapes of read counts indicate only one nucleosome. Exploiting the 5′ ends of read alignments rather than their genome coverage proved very useful in detecting differences in positioning among cell populations in the same sample. This is clearly visible downstream of the TSS of gene HEY2 (Figure 1A), where seemingly three different proximal preferential positions of a nucleosome were detected.
Figure 1.
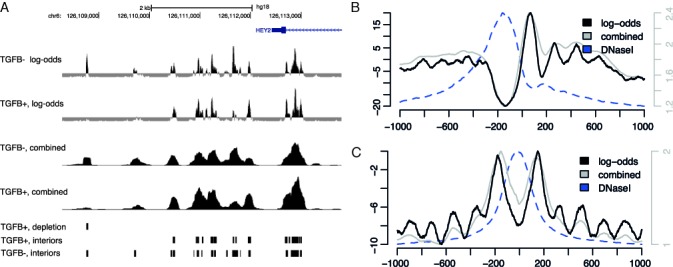
(A) Log-odds values around the TSS of gene HEY2 for nucleosome mid-positions in HepG2 unstimulated (labeled TGFB−) cells (Nucl Interior log-odds TGFβ−) and TGFβ stimulated (labeled TGFB+) cells (Nucl Interior log-odds TGFβ+). For comparison, counts of reads extended to the average fragment length of sequenced DNA, combined signal, are shown (Nucleosome TGFβ− and Nucleosome TGFβ+). Locations of inferred nucleosome interior regions in TGFβ− cells (Nucleosome interiors TGFβ−) and TGFβ+ cells (Nucleosome interiors TGFβ+) as well as locations of inferred nucleosome depletions in TGFβ+ cells (Nucleosome depletion TGFβ+) are depicted in the bottom panels. Data were uploaded as custom tracks to the UCSC Genome Browser where the graphics were produced. (B) and (C) Average signal footprints of log-odds (black lines, left vertical axes), counts of strand-directed fragment-length extended reads (gray lines, right vertical axes) and DNaseI hypersensitivity (blue dashed lines, scaled to fit) around TSSs of the top 5000 high-expressed protein-coding genes (B) and 25 651 JUND binding sites (C) .
We then created average footprints (31), centered on the TSSs of known protein coding genes with high expression (top 5000) as determined by gene RPKM counts from our RNA-seq data (Figure 1B) (see the Materials and Methods section for details). The log-odds values were compared with the combined counts of strand-directed extensions of reads to the average length of sequenced fragments (31), hereafter referred to as the combined signal. We observed the classical nucleosome pattern around the TSS, e.g. a well-positioned nucleosome downstream of the TSS, preceded by an NFR and yet another well-positioned nucleosome upstream of the TSS. The peaks are more defined using the log-odds values than the combined signal. The results of clustering the inferred nucleosome mid-positions derived from the log-odds values around these loci did, however, display a much more fragmented pattern than the suggested pattern from averaging over many loci (Figure 2). These patterns are clearly visible both from the log-odds representation and from the combined signal. Only a small minority of TSS regions agreed with the average pattern. Rather, preferential nucleosome positioning around TSSs was rarely accompanied by other well-positioned nucleosomes. Surprisingly many TSSs (∼33%) lacked flanking nucleosomes. This is in agreement with similar analysis performed on nucleosomal sequencing data from human CD4+ T-cells (31), K562 and GM12878 (16).
Figure 2.
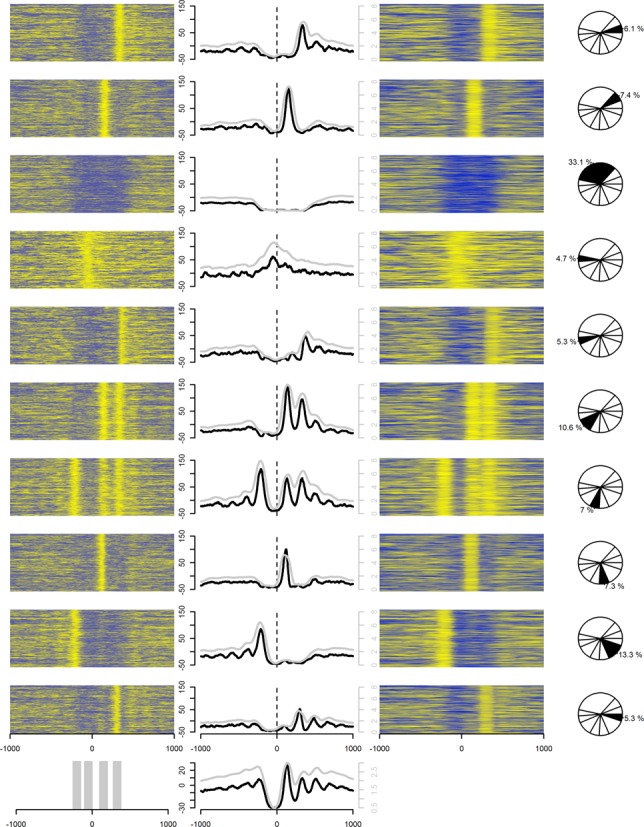
Heat maps of log-odds values (left column) and counts of fragment-length extended reads, combined signal, (right column) in 2-kb windows around TSS of highly expressed genes (top 5000). Their average values are also shown (middle column) in 10 K-means clusters (rows) inferred from discretized (0 or 1) log-odds data. The clusters were inferred using the data within the gray-marked regions in the bottom panel, leftmost figure. The middle figure in the bottom panel shows the combined average of all 10 clusters. Black lines (left vertical axes) and gray lines (right vertical axes) in the middle column depict the average of log-odds values and the combined signals, respectively, within each cluster. Yellow colors indicate high values while blue colors indicate low values.
We further examined the average log-odds values around 25 651 ENCODE JUND binding sites (23,33) (Figure 1C). Several peaks were visible around the JUND binding sites in the log-odds footprint with a sharp double peak surrounding the binding sites. This fits well with previously described patterns of nucleosomes around functional binding sites for TFs (34–36). The average peaks were, however, at log-odds levels below zero indicating positioned nucleosomes only at a subset of JUND locations. These peaks did not stand out using the combined signal alone, demonstrating that SuMMIt leads to detection of important, although weak, patterns that are largely hidden due to background enrichment. Further inspection of individual binding sites did, in fact, show that only a small minority (5.7%) of sites had flanking nucleosomes at the same distance from the central position of interaction (Figure 3) and 39.9% (10 234 of 25 651) did not have any indication of nucleosomes at all. Notably, the double peak surrounding the sites of interaction was rarely present but merely an artifact of averaging over many loci.
Figure 3.
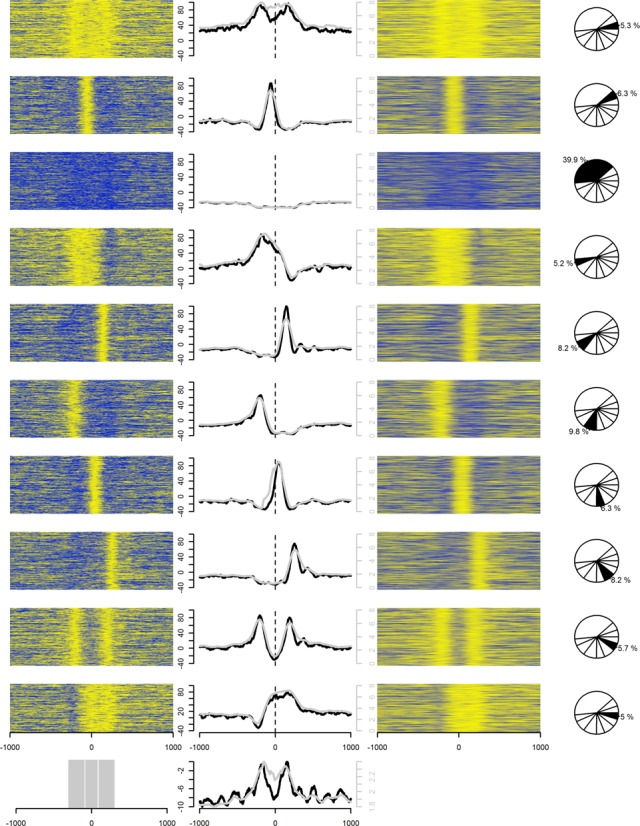
Heat maps of log-odds values (left column) and counts of fragment-length extended reads, combined signal, (right column) in 2-kb windows around 25 651 JunD binding sites. Their average values are also shown (middle column) in 10 K-means clusters (rows) inferred from discretized (0 or 1) log-odds data. The clusters were inferred using the data within the gray-marked regions in the bottom panel, leftmost figure. The middle figure in the bottom panel shows the combined average of all 10 clusters. within the gray-marked regions in the bottom panel. Black lines (left vertical axes) and gray lines (right vertical axes) in the middle column depict the average of log-odds values and the combined signals, respectively, within each cluster. Yellow colors indicate high values while blue colors indicate low values.
Due to positional heterogeneity among cells, a nucleosome is rarely indicated by only one position with positive log-odds, apparent around the TSS of HEY2 (Figure 1A). Rather, stretches of consecutively called positions of nucleosome mid-positions will be detected. We denoted such regions as nucleosome interiors. Here, we identified 5 231 084 regions of nucleosome interiors in unstimulated HepG2 cells. Since the aim of SuMMIt was to identify the genomic locations of nucleosome mid-positions, no consideration of adjacent nucleosomes is made. Interiors may thus locate closely if the data support a mixture of positions, e.g. overlapping nucleosome locations. Consequently, for subsequent analyses, we merged proximal nucleosome interiors (center distance <65 bp) to ease interpretation of results (see the Materials and Methods section for details).
This resulted in 3 335 364 merged regions of nucleosome interiors. Out of these, ∼44% represented non-conflicting adjacent nucleosome positions, i.e. with interior center positions separated by more than 130 bp. The majority (∼56%) of inferred nucleosome positions were thus not separated far enough to be considered as distinct nucleosomes, but rather as indications of discrepancies in localizations between cells in the same sample.
In accordance with our previous findings in CD4+ T-cells (6), a clear preference for exonic positioning was observed (Figure 4) when comparing the inferred nucleosomal locations to the genomic sequence coverage. We also found a preference for intragenic over intergenic positioning (51% of regions in 37% of sequence). In fact, the center-to-center distance between adjacent non-conflicting nucleosome interiors in exons, introns and intergenic regions (median values of 194 bp, 253 bp and 292 bp, respectively) indicated higher density of well-supported nucleosomes in exons than in introns and in intragenic than in intergenic regions (Supplementary Figure S2).
Figure 4.
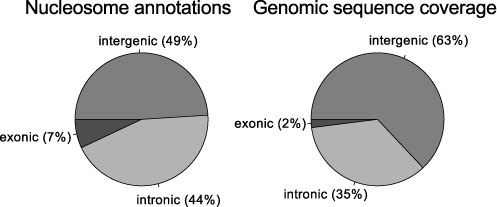
Annotations of nucleosomes in HepG2 unstimulated cells. Left pie chart shows the distribution of nucleosomes in exonic, intronic and intergenic regions. For comparison (right pie chart), the genomic sequence coverage of these regions is shown.
We next investigated the level of positional discordance by means of positional spread of nucleosome-supporting reads and calculated a fuzziness score for each inferred nucleosome (Supplementary Figure S4; see Methods for details). We separated nucleosomes into those with low, medium and high fuzziness score, yielding groups of phased, intermediate and fuzzy nucleosomes, respectively. Notably, exonic regions contained a much higher fraction of fuzzy nucleosomes than phased ones (Figure 5A). In contrast, intergenic regions contained an equal fraction of phased, intermediate and fuzzy nucleosomes. Although marginal, we found a higher fraction of fuzzy nucleosomes at exons shorter than 200 bp compared to nucleosomes at longer exons (Figure 5B). The observed discrepancy between exonic and intergenic nucleosomes may be explained by a higher guanine/cytocine (GC)-content in exons, since adenine/thymine (AT)-content has nucleosome-disfavoring properties (8,37). At boundary regions of phased nucleosomes, we observed lower AT-content in exonic regions than in intronic or intergenic regions (Supplementary Figure S5). The difference in boundary AT-content was, however, prominent between phased and fuzzy nucleosomes (Supplementary Figure S5A and C), further strengthening the importance of nucleosome-disfavoring sequences in nucleosome positioning. We also examined if the fuzziness could be explained by transcriptional activity but found no difference (P > 0.1, Mann–Whitney) in distribution of RPKM values between the low, medium and high fuzziness score classes.
Figure 5.
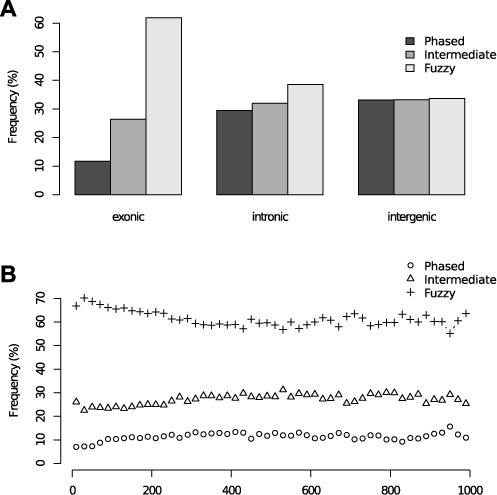
(A) Distribution of phased, intermediate and fuzzy nucleosomes in unstimulated cells in exonic, intronic and intergenic regions. (B) The fraction of phased, intermediate and fuzzy nucleosomes in exons of lengths within intervals ranging between 0 and 1000 bp. Length intervals of 20 bp in size were used.
SuMMIt performance
We compared the performance of SuMMIt to two previously published nucleosome positioning methods: PING 2.0 (38) and NORMAL (39). As a test case, we analyzed all 37.3M reads aligned to chromosome 1 in unstimulated cells. In brief, SuMMIt required less computing time and less memory compared to the other methods and resulted in more predicted nucleosome positions. The positions uniquely predicted by SuMMIt showed a more balanced support with reads from both stands as compared to the positions unique to the other methods. A detailed account of the comparisons can be found in Supplementary Figure S7 and Supplementary Tables S6 and S7.
Nucleosome dynamics in response to TGFβ1 stimuli
To investigate the regulatory role of TGFβ1 and its impact on nucleosome dynamics, we applied and trained SuMMIt on our data from TGFβ stimulated cells resulting in 4 679 107 predicted nucleosome interior regions of consecutive positive log-odds values. Merging of interior regions, as with the regions called in the unstimulated sample, resulted in 2 964 738 regions. As expected, the distribution of nucleosomes falling into exonic, intronic and intergenic regions (Supplementary Figure S6) was very similar to the distribution in unstimulated cells.
We calculated the shortest distance between the center position of each inferred nucleosome interior in unstimulated cells to those in TGFβ stimulated cells. This indicated that a large majority of nucleosomes (∼81%) had matching TGFβ stimulated nucleosomes (center-to-center distance <65 bp). A stunning 11% of nucleosomes called in the unstimulated sample were not called in TGFβ stimulated HepG2 cells. A fraction of those sites may, however, be unreliable indications of depleted nucleosomes in TGFβ stimulated cells due to e.g. uneven sequencing depths between samples.
To systematically identify nucleosomes present in unstimulated cells that were reliably depleted in TGFβ stimulated cells, we imposed strict filtering of the above identified nucleosomes with non-overlapping positions between samples (see the Materials and Methods section for details). Firstly, only non-conflicting nucleosomes were considered. Secondly, we required that the nucleosome interiors in unstimulated cells did not have any proximal strand call of TGFβ stimulated nucleosome interiors. Finally, we imposed strict cutoffs on log-odds of nucleosomal depletion in TGFβ stimulated cells. As a result, 24 318 nucleosomes were found to be depleted in TGFβ stimulated cells. One of the loci is visible 3.5 kb upstream of the TSS of gene HEY2 (Figure 1A). The average profiles of log-odds and the combined signal over all identified loci of nucleosome depletions are shown in Figure 6. The reverse analysis, aimed at identifying nucleosomes enriched in TGFβ stimulated cells but not present in the unstimulated cells, resulted in 8595 regions.
Figure 6.

Average signal footprints of log-odds values [(A) and (B)] and counts of reads extended to the average fragment length of sequenced DNA [(C) and (D)] around loci of nucleosome depletion in TGFβ stimulated cells using data from unstimulated [(A) and (C)] and TGFβ stimulated cells [(B) and (D)]. Combined signals refer to the summation of signals from both ends of sequenced fragments (Start and End for log-odds and Sense and Antisense for counts of extended reads).
Nearly 50% of depleted nucleosome loci were located within or in close proximity to genes (Figure 7A). RNA-seq of unstimulated and TGFβ stimulated HepG2 cells provided information regarding changed expression levels (log2 fold change > 1 or < −1) of genes and exons in response to TGFβ treatment. Out of all gene-associated loci, with nucleosome signal removed, nearly 20% were associated with changed gene expression (Table 1). A third of the exon-associated loci were coupled with exon expression changes suggesting either positions of importance in regulating exon usage or RNAPII-directed eviction for passage.
Figure 7.
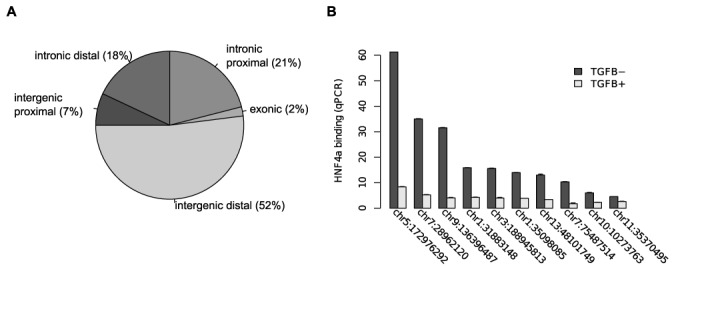
(A) Distribution of loci with nucleosome depletion in TGFβ stimulated cells according to genomic annotation. (B) qPCR validation in unstimulated (TGFB−) and TGFβ stimulated (TGFB+) cells of HNF4α binding in regions with ejected nucleosomes.
Table 1. Genomic distribution of inferred loci with nucleosomal depletion in TGFβ stimulated cells and their association with genes and expression changes.
| Exonic | Intronic proximal | Intronic distal | Intergenic proximal | Intergenic distal | |
|---|---|---|---|---|---|
| Number of loci | 454 | 5157 | 4290 | 1725 | 12692 |
| Number of associated genes | 437 | 4158 | 2235 | 1811 | |
| Number of loci associated with gene expression change | 68 | 599 | 976 | 242 | |
| Number of associated genes with expression change | 108 | 1737 | 397 | 239 | |
| Number of associated exons | 553 | 16883 | |||
| Number of loci associated with exon expression change | 84 | 1772 | |||
| Number of associated exons with expression change | 99 | 2659 |
To further explore the role of nucleosome positioning or repositioning in the regulation of TGFβ signaling, we searched the DNA sequences of loci with depleted nucleosomes for TF motifs, as defined by the JASPAR database (30). The program Clover (29) was used to search for over-represented TF motifs in those sequences with sampled nucleosome loci as background (see the Materials and Methods section for details). The same analysis was conducted in the DNA sequences of loci with gained nucleosomes. As many as 78% (depleted intergenic distal) of the loci contained an over-representation of TF motifs (Table 2 and Supplementary Tables S2 and S5). The highest proportion was found in depleted intergenic distal regions (one of more motifs found in 78% of the regions, Table 2) and the lowest in depleted intergenic proximal (14%). Apart from the intergenic proximal category (14% versus 34%), the depleted regions contained consistently higher proportion of regions with an over-representation of TFG motifs; exonic regions (44% versus 22%), intronic proximal (54%, 46%) and intronic distal (58%, 41%). Mere occurrence of a TF motif in the sequence of an identified region does not guarantee actual binding of that TF, but is still an indicator of its aggregate relevance for nucleosomal depletion.
Table 2. Genomic distribution of inferred loci with nucleosomal depletion in TGFβ stimulated cells with associated over-represented TF binding motifs in defined categories according to distance from exons and genes and their association with genes and expression changes.
| Exonic | Intronic proximal | Intronic distal | Intergenic proximal | Intergenic distal | |
|---|---|---|---|---|---|
| Number of loci | 199 | 2802 | 2469 | 245 | 9859 |
| Percentage of total loci | 43.8 | 54.3 | 57.6 | 14.2 | 77.7 |
| TFs | SPI1, ELF5, FEV, CTCF, ETS1, Hltf | Hand1::Tcfe2a, SPI1, NFE2L2, SOX10, NFATC2, FEV, Mafb, EBF1, CTCF | MZF1_1–4, PLAG1, MZF1_5–13, SPI1, RUNX1, NFATC2, EBF1, SOX10, FEV, BRCA1 | SPI1, FEV | NFATC2, EBF1, Myf, SPIB, SOX10, PPARG::RXRA, Hand1::Tcfe2a, NR2F1, Gfi, INSM1, SPI1, FEV, MZF1_1–4, Mafb, HNF4A, RREB1, En1, BRCA1 |
| Number of associated genes | 182 | 2503 | 1539 | 279 | |
| Number of loci associated with gene expression change | 45 | 607 | 1173 | 56 | |
| TFs associated with gene expression change | SPI1, Hltf, ELF5, FEV, CTCF, ETS1 | FEV, SPI1, Hand1::Tcfe2a, Mafb, EBF1, NFE2L2, SOX10, NFATC2, CTCF | MZF1_5–13, MZF1_1–4, PLAG1, EBF1, SPI1, FEV, NFATC2, RUNX1, SOX10, BRCA1 | SPI1, FEV | |
| Number of associated genes with expression change | 21 | 290 | 306 | 38 | |
| Number of associated exons | 225 | 9229 | |||
| Number of loci associated with exon expression change | 64 | 1711 | |||
| TFs associated with exon expression change | SPI1, ELF5, ETS1, CTCF, FEV, Hltf | NFE2L2, FEV, SOX10, Hand1::Tcfe2a, SPI1, Mafb, EBF1, CTCF, NFATC2 | |||
| Number of associated exons with expression change | 30 | 1498 |
Among the suggested TFs in the depleted regions we found the transcriptional activator and splicing regulator SPI1 (also known as PU.1), which is regulated by TGFβ1 (40), its related factor SPIB as well as MAFB, which may interact with SPI1 (41). In addition to SPI1, we also found motifs for other factors belonging to the TF family with ETS domains, namely ETS1 that acts as a cofactor with SMAD2/3/4 (22), key players in TGFβ signaling, FEV and ELF5. RUNX1, BRCA1 and HNF4α, all which may interact or cooperate with SMADs (22,42,43), also had over-represented binding motifs in the sequences of loci with depleted nucleosomes in TGFβ stimulated cells. Coupling these results with expression changes observed by RNA-seq after TGFβ1 treatment revealed that many displaced nucleosomes associated with TF motifs were also related to gene or exon expression changes (Table 2). A striking 61% of intronic proximal depleted nucleosomes with over-represented TF motifs were associated with exon expression changes. Surprisingly, as many as 1173 out of 2469 depleted intronic distal loci were associated with gene expression changes in 306 genes. These loci may reflect temporary ejection of nucleosomes due to RNAPII passage. We cannot, however, rule out that these loci are related to alternative TSSs not considered in our analysis.
Nucleosome depletion allows for HNF4α binding
We have previously generated ChIP-seq data for HNF4α in unstimulated HepG2-cells (44). Starting from regions where we had evidence of nucleosome depletion in unstimulated cells compared to TGFβ stimulated cells, we identified 37 candidate regions of HNF4α binding based on the HNF4α ChIP-seq signal and the presence of the canonical HNF4α binding motif. For 10 of these regions we validated the change in HNF4α occupancy using qPCR (Methods; Figure 7B). This validates not only the nucleosomal changes at these sites, but also that the rapidly evicted TF is subsequently replaced by a nucleosome.
Differentially expressed genes after TGFβ stimulation
We calculated RPKM values and fold change after 1 h of TGFβ1 treatment and defined a list of 590 up-regulated and 195 down-regulated genes (Supplementary Table S3). Fewer down-regulated genes than up-regulated are in agreement with previous data from genome-wide microarray-based studies and reflect the fact that the gene repression is a relatively late response to TGFβ1 that peaks after longer time than 1 h. The up-regulated genes include many with a neuronal function, including 31 ion channels and ion transporters, 25 olfactory receptors, 12 neurotransmitters and 17 G-protein coupled receptors. Many TFs previously known to be in the TGFβ pathway were among the most up-regulated genes with the highest fold change observed for JUND. Our total-RNA approach also allowed us to detect significant changes for genes without poly-A tails, including those for four histone proteins, and significant changes outside of exons as is shown for SMAD7 in Figure 8A. We found that in many cases a large number of members of different protein families were co-regulated. Examples are three members of the Inhibitors of differentiation family (ID1/2/3) and 21 secreted polypeptides including members of the Wnt, FGF, EGF and GDF families of growth factors previously linked to TGFβ.
We selected 25 genes with different levels of change in expression for TaqMan qPCR validations (Figure 8B). All up-regulated genes had a positive fold change also in the qPCR, but some genes with a relatively low level of down-regulation according to the RNA-seq data did not replicate (Figure 8C).
DISCUSSION
Careful assessments of the chromatin landscape of a cell and the dynamics of chromatin in response to external stimuli are essential in order to understand the involvement of chromatin in transcriptional regulation. To this end, we have developed SuMMIt, an accurate method for the precise placement of nucleosomes or other protein–DNA interactions in a genome from large-scale sequencing data. We observed superior capability of positioning in terms of resolution when compared with a simpler procedure using aggregations of strand-directed extensions of reads to expected fragment length which is commonly used when visualizing ChIP-seq data and identifying interaction sites.
Assessment of nucleosomal positioning around TSSs of highly transcribed genes in HepG2 cells showed that the commonly presented positioning pattern was merely an artifact of averaging over many loci. Surprisingly many TSSs lacked well-supported positioned nucleosomes at loci suggested from such average profiles. These results need to be considered in models of transcription and transcriptional regulation that rely on nucleosome positioning.
Investigating the nucleosomal locations showed that the majority of positions were not in agreement between cells in the same sample indicating distinct, albeit possibly phased, placements within different cell populations. Examination of nucleosome phasing among cells revealed that exonic regions contained a much higher fraction of fuzzy, i.e. non-phased, nucleosomes than phased ones. This was not the case for intronic or intergenic nucleosomes. High fuzziness was clearly related to the absence of boundary disfavoring sequence characteristics such as AT-content. The high GC-content of exons may thus explain the elevated fuzziness of contained nucleosomes.
However, exonic regions were more nucleosome dense than intronic ones. This is in agreement with our previous findings in resting CD4+ T-cells (6), further supporting our observation that exonic nucleosome positioning is maintained across cell types as well as organisms. Our finding that nucleosome occupancy in intergenic regions was even less abundant may be attributed to the presence of sequencing gaps in the reference genome or to repetitive sequences, calling for further investigations.
Treatment of HepG2 cells with TGFβ1 for 1 h did not affect the large majority of nucleosomes. Still, surprisingly many nucleosomes were depleted after TGFβ1 treatment suggesting that chromatin remodeling is an important factor in TGFβ signaling. This novel finding agrees with recent reports on the role of chromatin remodeling factors as regulators of Smad protein binding to regulatory sequences of target genes of TGFβ (45). Many inferred depletions were also associated with expression changes. Searching the sequences at depleted nucleosomes for TF motifs revealed many putative binding sites for TFs and changes in HNF4α binding over such sites were demonstrated using ChIP-qPCR. The apparent release of HNF4α binding sites after nucleosomal depletion caused by TGFβ signaling is in full agreement with recent genome-wide ChIP experiments that defined HNF4α sites as the major locations where Smad complexes associate with chromatin in HepG2 cells (46). Many of the other TFs are also relevant for TGFβ signaling and were also related to changes in gene or exon expression. A large fraction of loci were found in intergenic regions and were thus difficult to associate with possible target genes, but may indicate possible novel functions of TGFβ signaling at loci distant from RNA-coding genes. High-throughput extensions of chromatin conformation capture will be required for assessing their functional roles.
CONCLUSION
The nucleosome landscape of HepG2 cells has been characterized and was found to display the same fundamentals as in other cell types and organisms. In addition, we have found that chromatin itself is very dynamic, as observed after only 1 h of stimulus, and plays a major role in transcriptional regulation in general and in the interpretation of signaling networks such as TGFβ by the genome .
AVAILABILITY
Raw nucleosome and RNA-seq reads have been deposited in the Array Express/European Nucleotide Archive under accession numbers E-MTAB-1750 and E-MTAB-1819. The source code for SuMMIt is available from GitHub under project name ‘summit’ (https://github.com/rhentofs/summit) as is the latest version of SICTIN under project name ‘sictin’ (https://github.com/rhentofs/sictin).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [1, 2].
Acknowledgments
Sequencing was performed at the Applied Biosystems R&D laboratories in Beverly, MA, USA. The authors would also like to acknowledge Catalin Barbacioru, Heather Peckham, Kevin McKernan and Clarence Lee for their assistance in the sequencing of the samples. Additional sequencing was performed at Uppsala Genome, Science for Life Laboratory at Uppsala University, a national infrastructure supported by the Swedish Research Council (VR-RFI) and the Knut and Alice Wallenberg Foundation. Taqman gene expression assays were performed with assistance from Yoshitaka Nishikawa.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
Present addresses:
Stefan Enroth, Science for Life Laboratory, Department of Immunology, Genetics and Pathology, Rudbeck Laboratory, Uppsala University, SE-75185 Uppsala, Sweden.
Robin Andersson, The Bioinformatics Centre, University of Copenhagen, Ole Maaloes Vej 5, DK-2200 Copenhagen N, Denmark.
Ola Wallerman, Department of Medical Biochemistry and Microbiology, Uppsala University, SE-75124 Uppsala, Sweden.
Brian B. Tuch, Translational Genomics, Onyx Pharmaceuticals, 249 E. Grand Ave., South San Francisco, CA 94080, USA.
Francisco M. De La Vega, Department of Genetics, Stanford School of Medicine, Stanford, CA 94305-5120, USA.
Stefan Termén, BioOutsource Ltd, 1 Technology Terrace, Todd Campus, West of Scotland Science Park, Glasgow, G20 0XA, UK.
Jan Komorowski, Department of Cell and Molecular Biology, Science for Life Laboratory, Uppsala University, BMC, Box 596, SE-75124 Uppsala, Sweden.
FUNDING
Swedish Research Council [521-2010-3505 and 621-2011-6052]; Diabetes Foundation, Sweden; Ernfors Family Foundation; Diabetes Wellness Network, Sverige; Network of Excellence ‘ENFIN’ [LSHG-CT-2005-518254], European Union FP6 Program; Polish Ministry of Science and Higher Education [N301 239536 to J.K., in part]; eSSENCE Program [to J.K., in part].
Conflict of interest statement. None declared.
REFERENCES
- 1.Zhou J., Fan J.Y., Rangasamy D., Tremethick D.J. The nucleosome surface regulates chromatin compaction and couples it with transcriptional repression. Nat. Struct. Mol. Biol. 2007;14:1070–1076. doi: 10.1038/nsmb1323. [DOI] [PubMed] [Google Scholar]
- 2.Shogren-Knaak M., Ishii H., Sun J.M., Pazin M.J., Davie J.R., Peterson C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 3.Campos E.I., Reinberg D. Histones: annotating chromatin. Annu. Rev. Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 4.Jiang C., Pugh B.F. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schones D.E., Cui K., Cuddapah S., Roh T.Y., Barski A., Wang Z., Wei G., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersson R., Enroth S., Rada-Iglesias A., Wadelius C., Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009;19:1732–1741. doi: 10.1101/gr.092353.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He H.H., Meyer C.A., Shin H., Bailey S.T., Wei G., Wang Q., Zhang Y., Xu K., Ni M., Lupien M., et al. Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segal E., Widom J. From DNA sequence to transcriptional behaviour: a quantitative approach. Nat. Rev. Genet. 2009;10:443–456. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z., Zang C., Rosenfeld J.A., Schones D.E., Barski A., Cuddapah S., Cui K., Roh T.Y., Peng W., Zhang M.Q., et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enroth S., Bornelov S., Wadelius C., Komorowski J. Combinations of histone modifications mark exon inclusion levels. PLoS ONE. 2012;7:e29911. doi: 10.1371/journal.pone.0029911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cairns B.R. The logic of chromatin architecture and remodelling at promoters. Nature. 2009;461:193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- 15.Beato M., Eisfeld K. Transcription factor access to chromatin. Nucleic Acids Res. 1997;25:3559–3563. doi: 10.1093/nar/25.18.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kundaje A., Kyriazopoulou-Panagiotopoulou S., Libbrecht M., Smith C.L., Raha D., Winters E.E., Johnson S.M., Snyder M., Batzoglou S., Sidow A. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res. 2012;22:1735–1747. doi: 10.1101/gr.136366.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Shin H., Song J.S., Lei Y., Liu X.S. Identifying positioned nucleosomes with epigenetic marks in human from ChIP-Seq. BMC Genomics. 2008;9:537–547. doi: 10.1186/1471-2164-9-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albert I., Mavrich T.N., Tomsho L.P., Qi J., Zanton S.J., Schuster S.C., Pugh B.F. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 19.Zang C., Schones D.E., Zeng C., Cui K., Zhao K., Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jothi R., Cuddapah S., Barski A., Cui K., Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kharchenko P.V., Tolstorukov M.Y., Park P.J. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moustakas A., Heldin C.H. The regulation of TGFbeta signal transduction. Development. 2009;136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 23.Encode Project Consortium. Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Motallebipour M., Ameur A., Bysani M.S.R., Patra K., Wallerman O., Mangion J., Barker M.A., McKernan K.J., Komorowski J., Wadelius C. Differential binding and co-binding pattern of FOXA1 and FOXA3 and their relation to H3K4me3 in HepG2 cells revealed by ChIP-seq. Genome Biol. 2009;10:R129. doi: 10.1186/gb-2009-10-11-r129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richmond T.J., Davey C.A. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 26.Hubbard T.J., Aken B.L., Ayling S., Ballester B., Beal K., Bragin E., Brent S., Chen Y., Clapham P., Clarke L., et al. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blankenberg D., Taylor J., Schenck I., He J., Zhang Y., Ghent M., Veeraraghavan N., Albert I., Miller W., Makova K.D., et al. A framework for collaborative analysis of ENCODE data: making large-scale analyses biologist-friendly. Genome Res. 2007;17:960–964. doi: 10.1101/gr.5578007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frith M.C., Fu Y., Yu L., Chen J.F., Hansen U., Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004;32:1372–1381. doi: 10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Portales-Casamar E., Thongjuea S., Kwon A.T., Arenillas D., Zhao X., Valen E., Yusuf D., Lenhard B., Wasserman W.W., Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–D110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enroth S., Andersson R., Wadelius C., Komorowski J. SICTIN: rapid footprinting of massively parallel sequencing data. BioData Min. 2010;3:4. doi: 10.1186/1756-0381-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galassi M., Davies J., Theiler J., Gough B., Jungman G., Alken P., Booth M., Rossi F. GNU Scientific Library Reference Manual. 3rd edn (v1.12) UK: Network Theory Ltd; 2009. [Google Scholar]
- 33.Rosenbloom K.R., Dreszer T.R., Pheasant M., Barber G.P., Meyer L.R., Pohl A., Raney B.J., Wang T., Hinrichs A.S., Zweig A.S., et al. ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010;38:D620–D625. doi: 10.1093/nar/gkp961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee C.K., Shibata Y., Rao B., Strahl B.D., Lieb J.D. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat. Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 35.Bernstein B.E., Liu C.L., Humphrey E.L., Perlstein E.O., Schreiber S.L. Global nucleosome occupancy in yeast. Genome Biol. 2004;5:R62. doi: 10.1186/gb-2004-5-9-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J., Zhuang J.L., Iyer S., Lin X.Y., Whitfield T.W., Greven M.C., Pierce B.G., Dong X.J., Kundaje A., Cheng Y., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segal E., Widom J. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 2009;19:65–71. doi: 10.1016/j.sbi.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woo S., Zhang X., Sauteraud R., Robert F., Gottardo R. PING 2.0: an R/Bioconductor package for nucleosome positioning using next-generation sequencing data. Bioinformatics. 2013;29:2049–2050. doi: 10.1093/bioinformatics/btt348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polishko A., Ponts N., Le Roch K.G., Lonardi S. NORMAL: accurate nucleosome positioning using a modified Gaussian mixture model. Bioinformatics. 2012;28:i242–i249. doi: 10.1093/bioinformatics/bts206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinz L.X., Platzer B., Reisner P.M., Jorgl A., Taschner S., Gobel F., Strobl H. Differential involvement of PU.1 and Id2 downstream of TGF-beta1 during Langerhans-cell commitment. Blood. 2006;107:1445–1453. doi: 10.1182/blood-2005-04-1721. [DOI] [PubMed] [Google Scholar]
- 41.Bakri Y., Sarrazin S., Mayer U.P., Tillmanns S., Nerlov C., Boned A., Sieweke M.H. Balance of MafB and PU.1 specifies alternative macrophage or dendritic cell fate. Blood. 2005;105:2707–2716. doi: 10.1182/blood-2004-04-1448. [DOI] [PubMed] [Google Scholar]
- 42.Li H., Sekine M., Seng S., Avraham S., Avraham H.K. BRCA1 interacts with Smad3 and regulates Smad3-mediated TGF-beta signaling during oxidative stress responses. PLoS ONE. 2009;4:e7091. doi: 10.1371/journal.pone.0007091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y., Derynck R. Transcriptional regulation of the transforming growth factor-beta -inducible mouse germ line Ig alpha constant region gene by functional cooperation of Smad, CREB, and AML family members. J. Biol. Chem. 2000;275:16979–16985. doi: 10.1074/jbc.M001526200. [DOI] [PubMed] [Google Scholar]
- 44.Wallerman O., Motallebipour M., Enroth S., Patra K., Bysani M.S.R., Komorowski J., Wadelius C. Molecular interactions between HNF4a, FOXA2 and GABP identified at regulatory DNA elements through ChIP-sequencing. Nucleic Acids Res. 2009;37:7498–7508. doi: 10.1093/nar/gkp823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Massague J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizutani A., Koinuma D., Tsutsumi S., Kamimura N., Morikawa M., Suzuki H.I., Imamura T., Miyazono K., Aburatani H. Cell type-specific target selection by combinatorial binding of Smad2/3 proteins and hepatocyte nuclear factor 4alpha in HepG2 cells. J. Biol. Chem. 2011;286:29848–29860. doi: 10.1074/jbc.M110.217745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.