


Abstract
Cyclodipeptide synthases (CDPSs) use two aminoacyl-tRNA substrates in a sequential ping-pong mechanism to form a cyclodipeptide. The crystal structures of three CDPSs have been determined and all show a Rossmann-fold domain similar to the catalytic domain of class-I aminoacyl-tRNA synthetases (aaRSs). Structural features and mutational analyses however suggest that CDPSs and aaRSs interact differently with their tRNA substrates. We used AlbC from Streptomyces noursei that mainly produces cyclo(l-Phe-l-Leu) to investigate the interaction of a CDPS with its substrates. We demonstrate that Phe-tRNAPhe is the first substrate accommodated by AlbC. Its binding to AlbC is dependent on basic residues located in the helix α4 that form a basic patch at the surface of the protein. AlbC does not use all of the Leu-tRNALeu isoacceptors as a second substrate. We show that the G1-C72 pair of the acceptor stem is essential for the recognition of the second substrate. Substitution of D163 located in the loop α6–α7 or D205 located in the loop β6–α8 affected Leu-tRNALeu isoacceptors specificity, suggesting the involvement of these residues in the binding of the second substrate. This is the first demonstration that the two substrates of CDPSs are accommodated in different binding sites.
INTRODUCTION
Cyclodipeptide synthases (CDPSs) form a family of tRNA-dependent enzymes that catalyse the synthesis of various cyclodipeptides, which are the precursors of various secondary metabolites with important biological activities (1–3). CDPSs use two aminoacyl-tRNA (aa-tRNA) substrates in a sequential ping-pong mechanism to form the two peptide bonds of cyclodipeptides (4–7). The first catalytic steps involve the binding of the first aa-tRNA to the CDPSs and the subsequent transfer of the aminoacyl moiety onto an active-site serine residue to form an acyl-enzyme intermediate. The acyl-enzyme then reacts with the aminoacyl moiety of the second aa-tRNA substrate to form a dipeptidyl intermediate that undergoes intramolecular cyclisation, yielding the final cyclodipeptide product.
Crystal structures are available for three CDPSs (4–6). The three CDPSs share an architecture that is very similar to the catalytic domain of class-I aminoacyl-tRNA synthetases (aaRSs), especially class-Ic TyrRSs and TrpRSs. Conventional aaRSs catalyse the formation of aa-tRNAs in a two-step reaction consisting of activation of the amino acid by formation of an aminoacyl-adenylate intermediate, followed by esterification at the 3’-hydroxyl of a cognate tRNA. CDPSs would have diverged from conventional aaRSs and acquired new active site residues, converting them into cyclodipeptide-forming enzymes, for which aa-tRNAs are the substrates instead of the final products (8). This evolutionary event would have entailed significant divergence between CDPSs and class-Ic aaRSs (4–6). First, class-Ic aaRSs are homodimers that have interdigitation of the two active sites at the dimer interface whereas CDPSs are active as monomers. Second, the specific signature motifs involved in ATP binding are absent in CDPSs, consistent with the absence of ATP-dependent activation by CDPSs. Third, the tRNA-binding domain involved in interaction with the anti-codon loop of tRNA in aaRSs is absent in CDPSs. CDPSs possess instead a large patch of positively charged residues that are absent in aaRSs. Finally, unlike aaRSs, CDPSs exhibit a large tRNA substrate specificity. Although each characterized CDPS preferentially synthesizes one predominant cyclodipeptide, most of these enzymes are promiscuous and produce several cyclodipeptides. For example, AlbC from Streptomyces noursei synthesizes cyclo(l-Phe-l-Leu) (cFL) and Ndas_1148 from Nocardiopsis dassonvillei synthesizes cyclo(l-Phe-l-Tyr) (cFY), but both enzymes produce appreciable amounts of other phenylalanyl-containing cyclodipeptides (cyclo(l-Phe-l-X), in which X indicated any of the incorporated amino acids) (cFX). Rv2275 from Mycobacterium tuberculosis mostly produces cYY but also cYX cyclodipeptides, and the YvmC enzymes from Bacillus species mainly produce cLL and cLX compounds (2,9).
No crystal structure has yet been obtained for a CDPS in complex with a ligand representative of its natural substrates. Nevertheless, the structural similarity between CDPSs and aaRSs, and extensive mutagenesis analyses of CDPSs have provided insight into the interaction of these enzymes with their aa-tRNA substrates (4–6). CDPSs have a surface-accessible pocket containing the catalytic residues that superimposes well with the amino acid-binding pocket of class-Ic aaRSs. This pocket accommodates the aminoacyl moiety of one of the two aa-tRNA substrates (5). The cyclodipeptides formed by a given CDPS almost invariably have one amino acid in common. This suggests that only the aminoacyl group of the first substrate, probably the ‘common’ aminoacyl group, binds and remains in the catalytic pocket, whereas the second substrate is probably accommodated at a different site with less stringent recognition (6). The involvement of the tRNA moiety of either substrate in substrate binding and selectivity is poorly documented. Mutagenesis studies and tRNA-binding assays identified two regions of CDPSs that are likely to interact with tRNA: the positively charged patch mentioned above and a loop located between helices α6 and α7 (5,6).
Here, we studied the interaction of CDPSs with their two aa-tRNA substrates. To discriminate the interaction of either substrate with the enzyme, we worked with AlbC that uses two different substrates, Phe-tRNAPhe and Leu-tRNALeu, to catalyse cFL synthesis. We show that Phe-tRNAPhe is specifically bound in the first step of the reaction. We also show the involvement of the nucleotide sequence of the acceptor stem in the recognition of the tRNA moiety of the second substrate and identify key bases involved in substrate specificity. Finally, we explored the role of residues predicted to interact with the tRNA moiety of the substrates and demonstrated that AlbC possesses different binding sites for its two aa-tRNA substrates.
MATERIALS AND METHODS
Mutagenesis and purification of AlbC variants
Genes coding the six new single point variants of AlbC (R80A, R102A, N159A, R160A, D163A, D205A) were obtained via polymerase chain reaction (PCR) mutagenesis of the plasmid pQE60-AlbC encoding C-terminal His6-tagged AlbC (5) according to the QuikChangeTM site-directed mutagenesis method (Stratagene). Sequences were verified by DNA sequencing. Plasmids encoding the other AlbC variants used in this study (R91A, K94A, R98A, R98A-R99A) were constructed previously (5).
The C-terminal His6-tagged proteins were produced and purified as described previously (2,5). Purified proteins were quantified by UV spectrophotometry. Protein molecular weights were verified by electrospray ionisation mass spectrometry (Esquire HCT ion trap mass spectrometer (Bruker Daltonik, GmbH)).
In vitro transcription/semi-synthesis
tRNAPhe, tRNALeu isoacceptors and tRNALeuTAA mutants were obtained by in vitro transcription of double-stranded DNA templates (10). Double-stranded DNA templates were synthesized by PCR using three synthetic DNA oligonucleotides purchased from Eurogentec. For site-directed mutagenesis of the acceptor stem, the first oligonucleotide (Matrix) includes complementary sequence of the tRNA from positions 8 to 65. To avoid mis-transcription products, a second oligonucleotide (Forward) containing the T7 promoter sequence is fused to the tRNA sequence from positions 1 to 26. Finally, a third oligonucleotide (Reverse) includes the complementary sequence of the tRNA from positions 47 to 76. Sequences of the oligonucleotides used for synthesis of tRNAPhe and tRNALeu isoacceptors are given in Supplementary Table S1. Amplified double-stranded DNA templates are used for in vitro transcription with T7 RNA polymerase as previously described (11) except for A1-U72 tRNALeuTAA where guanosine monophosphate (GMP) is replaced by adenosine monophosphate (AMP). Transcripts of tRNA were separated from the reaction mixture by extraction with phenol/chloroform and precipitation with ethanol. The tRNAs were purified by size exclusion chromatography (Superdex® 75 HR 10/30, GE Healthcare). The concentrations of tRNAs were determined by absorption at 260 nm (1 OD260 unit equivalent to 40 μg/ml) and were further corrected by plateau charging (see below).
Cloning, mutagenesis and purification of Escherichia coli tRNA
The plasmid encoding tRNAPhe, pBSTNAV2/tRNAPhe, was a gift from Y. Mechulam (Ecole Polytechnique, Palaiseau, France) (12). The plasmid encoding tRNALeuCAG was constructed as follows: tRNALeuCAG was amplified from a lysate of E. coli K12 using Thermo Scientific Phusion High-Fidelity DNA polymerase according to the manufacturer's guidelines. The primers used included EcoRI and PstI restriction sites (underlined) as follows: CTTGTAACGCTGAATTCGCGAAGGTGGCGGAATTG (forward) and CGCTAAGGATCTGCAGTGGTGCGAGGGGGGGG (reverse). Amplified fragments were digested by FastDigest EcoRI and PstI (Thermo Scientific) and ligated into the digested pBSTNAV2 vector using T4 DNA ligase (Thermo Scientific) according to the manufacturer's protocol.
The two double mutants C1-G72 tRNALeuCAG and C1-G72 tRNAPhe were obtained from wild-type sequences cloned into a pBSTNAV2 vector by amplification with oligonucleotides containing the desired mutation (in bold): LeuG1C (GTAACGCTGAATTCCCGAAGGTGGCGGAATTG), LeuC72G (CTAAGGATCTGCAGTGGTCCGAGGGGGG), PheG1C (CTTGTAACGCTGAATTCCCCCGGATAGCTCAGTC), PheC72G (CGCTAAGGATCTGCAGTGGTCCCCGGACTCGGAATC). Amplified fragments were treated as described above for wild-type sequences. All constructions were verified by DNA sequencing.
Production and purification of tRNAs were performed as previously described (13). Briefly, cells overexpressing tRNAs were harvested by centrifugation and tRNAs were extracted by phenol followed by centrifugation steps and precipitation with ethanol. The pellet was suspended in 20 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 8 mM MgCl2, 0.2 M NaCl and tRNAs were purified by Q-Sepharose Fast Flow column with a linear gradient of 0.4 M to 0.7 M NaCl. The fractions containing tRNAs were precipitated by isopropanol and stored at −20°C after centrifugation and suspension in water.
Acylation of tRNAs
The concentration of aminoacylated tRNA was determined from plateau charging experiments. The assay was performed in buffer A (50 mM HEPES-KOH pH 7.5, 150 mM KCl, 15 mM MgCl2, 0.1 mM EDTA, 2 mM ATP, 10 mM ß-mercaptoethanol) with 1 μM PheRS or LeuRS and 50 μM of radiolabelled amino acids (l-[14C]Leu, 324 mCi/mmol (12.0 GBq mmol−1), or l-[14C]Phe, 487 mCi/mmol (18.0 GBq mmol−1), Perkin-Elmer). The reaction was incubated for 10 min at 30°C and aa-tRNAs were precipitated with 5% TCA and 0.5% casamino acids, filtered on Whatman GF/C filters and quantified by liquid scintillation counting. Mutated tRNAs were aminoacylated for different times; 15 min was enough to ensure complete acylation.
AlbC coupled assay
The CDPS activity of AlbC was determined in a coupled assay containing aaRSs to generate in situ the aa-tRNA substrates as described previously (7). The standard assay was performed in buffer A, with 50 μM Phe and Leu, 50 nM AlbC or AlbC variant, 1 μM PheRS and LeuRS, and aa-tRNAs at the concentrations specified in the text. The reaction was carried out at 30°C with a preincubation of 15 min with the aaRSs prior to the addition of AlbC for the synthesis of aa-tRNAs. Under these conditions, the tRNAs were completely acylated at the beginning of the AlbC reaction and remained acylated during the entire reaction (Supplementary Figure S1). The enzymatic reaction was initiated by the addition of AlbC. Aliquots were withdrawn at various times, acidified with 2% TFA to stop the reaction and mixed with known concentrations of stable isotope internal standards (13C9,15N-labelled cFF and cFL solutions), prior to cFF and cFL quantification by liquid chromatography coupled to mass spectrometry (LC-MS) as described previously (2,7).
Detection of acyl-enzyme intermediates
Purified AlbC wild-type, pSHaeC06 (positive controls), AlbC S37A (negative control) and AlbC variants were incubated with [14C]Phe-tRNAPhe or [14C]Leu-tRNALeuCAG (0.5 μM). The labelled substrates were obtained as described previously (5). The enzyme was added at a final concentration of 1 μM. After 30 s of incubation, the reaction was quenched and analysed. Radioactivity was detected on polyvinylidene difluoride (PVDF) membranes by a Beta-ImagerTM 2000 (Biospace) (5).
RESULTS
AlbC discriminates Phe-tRNAPhe and Leu-tRNALeu
Post-transcriptional modification of tRNAs is not required for AlbC activity
We previously showed that AlbC is active when it is produced in E. coli. AlbC mainly synthesizes cFL but also produces appreciable amounts of other L-Phe-containing cyclodipeptides, especially cFF (2). We also showed that AlbC is active in vitro. The purified enzyme can use aminoacylated tRNAPhe purified from E. coli as the substrates to produce cFF (2,5,7). Based on these findings, we performed all experiments described herein using the sequences of E. coli tRNAs, in particular that of the unique tRNAPhe isoacceptor. To determine the importance of post-transcriptional modifications of tRNAPhe on AlbC activity, we compared its cFF-synthesizing activity using tRNAPhe either purified from E. coli or obtained by in vitro transcription. We used a coupled PheRS-AlbC assay to generate Phe-tRNAPhe in situ and measure the time course for the synthesis of cFF at various concentrations of Phe-tRNAPhe (Supplementary Figure S2) (7). The rates of formation of cFF were deduced from individual kinetics and plotted against Phe-tRNAPhe concentrations (Figure 1A). The rate of formation of cFF was similar, regardless of the Phe-tRNAPhe used. This result shows that post-transcriptional modification of the tRNAPhe is not essential for enzymatic activity. The tRNAs used in subsequent experiments were obtained by in vitro transcription, except where otherwise stated.
Figure 1.

(A) cFF-synthesizing activity of AlbC using either tRNAPhe purified from E. coli (blue) or tRNAPhe obtained by in vitro transcription (orange). Enzymatic measurements were performed as described in ‘Materials and Methods’ with 50 nM AlbC. The points reported are the result of three independent experiments. Error bars show the uncertainty on measurement. (B) Rate of formation of cFL (left panel) or cFF (right panel) by AlbC for different tRNALeu isoacceptors. Kinetics of synthesis were determined with 0.2 μM Phe-tRNAPhe and three concentrations of Leu-tRNALeu. Isoacceptors are identified by the following colours: tRNALeuCAA (light blue), tRNALeuTAG (dark blue), tRNALeuGAG (yellow), tRNALeuTAA (orange), tRNALeuCAG* (grey), tRNALeuCAG (black). Curves are drawn for clarity and are not representative of kinetic models. (C) Covalent labelling of AlbC, S37A and pSHaeC06 (pSH) by [14C]Phe transferred from [14C]Phe-tRNAPhe or [14C]Leu transferred from [14C]Leu-tRNALeuCAG. Enzymes were incubated with labelled aa-tRNA, as described in ‘Materials and Methods’, separated on SDS-PAGE, then transferred onto a PVDF membrane that was analysed with a radioimager.
AlbC does not use all Leu-tRNALeu isoacceptors
We tested the cyclodipeptide-synthesizing activity of AlbC using both Phe-tRNAPhe and Leu-tRNALeu as substrates. While the tRNAPhe sequence is unique, E. coli possesses six different tRNALeu isoacceptors, two of which differ from each other only by one base in the variable loop (tRNALeuCAG/tRNALeuCAG*) (Supplementary Figure S3). We determined the kinetics of cFL, cFF and cLL synthesis by the coupled aaRS-AlbC assay for a fixed concentration of Phe-tRNAPhe (0.2 μM) and various concentrations of each of the six tRNALeu isoacceptors. Formation of cLL was not detected in our experimental conditions, regardless of the sequence and concentration of tRNALeu isoacceptor. The rates of formation of the cyclodipeptides cFL and cFF were deduced from individual kinetics and plotted against Leu-tRNALeu concentrations for each isoacceptor (Figure 1B). Leu-tRNALeu isoacceptors were not used with the same catalytic efficiency by AlbC. The rates of cFL formation (Figure 1B, left panel) were the highest for Leu-tRNALeuCAG or Leu-tRNALeuCAG*, two times lower for Leu-tRNALeuCAA or Leu-tRNALeuTAG, and close to zero for Leu-tRNALeuGAG or Leu-tRNALeuTAA. AlbC synthesized higher quantities of cFF than cFL for equivalent concentrations of Phe-tRNAPhe and Leu-tRNALeu (i.e. 0.2 μM), regardless of the Leu-tRNALeu isoacceptor used (Figure 1B). Thus, AlbC used more efficiently Phe-tRNAPhe than any of the Leu-tRNALeu. The efficiency of cFF synthesis was not affected by the presence of Leu-tRNALeuGAG or Leu-tRNALeuTAA indicating that these molecules do not compete with Phe-tRNAPhe for binding to AlbC (Figure 1B, right panel). In contrast, the other four Leu-tRNALeu isoacceptors inhibited cFF synthesis revealing a competition with Phe-tRNAPhe.
Phe-tRNAPhe is the first substrate accommodated by AlbC
The CDPSs AlbC, Rv2275, YvmC-Blic and Nvec-CDPS2 have a common first catalytic step. This step involves the binding of the first aa-tRNA substrate to the enzyme and the subsequent transfer of its aminoacyl moiety onto the conserved serine residue of the catalytic pocket to form an acyl-enzyme intermediate (4–7). In all four cases, the formation of the acyl-enzyme was tested using a unique aa-tRNA as a substrate since these enzymes synthesize homocyclodipeptides (cYY for Rv2275 and cLL for YvmC-Blic; cFF for AlbC and Nvec-CDPS2). We used the property of AlbC to synthesize the heterocyclodipeptide cFL to investigate substrate-binding order. AlbC was incubated with either [14C]Phe-tRNAPhe or [14C]Leu-tRNALeuCAG and the formation of an acyl-enzyme intermediate was determined by autoradiography following sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transfer onto a PVDF membrane (5). Formation of [14C]AlbC was detected with [14C]Phe-tRNAPhe but not with [14C]Leu-tRNALeuCAG (Figure 1C), indicating that Phe-tRNAPhe is the first substrate of AlbC. As a negative control, we showed that an inactive AlbC variant carrying a S37A substitution (5) was not labelled upon incubation with [14C]Phe-tRNAPhe in the same conditions. As a positive control for the formation of an acyl-enzyme with [14C]Leu-tRNALeuCAG, we used CDPS pSHaeC06 from Staphylococcus haemolyticus, which mainly synthesizes cLL (2). Finally, the LS-MS/MS coupled assay with unlabelled substrates was used to check that the AlbC and pSHaeC06 enzyme preparations used for these experiments were fully functional.
Specificity determinants in aa-tRNAs substrates of AlbC
The pair G1-C72 of the second substrate is essential for cFL synthesis
Comparison of the sequences of the six tRNALeu isoacceptors of E. coli revealed that a wobble base pair G1•U72 is present in the distal position of the acceptor arm of the two tRNALeu isoacceptors that are not used as substrates by AlbC, i.e. Leu-tRNALeuGAG and Leu-tRNALeuTAA. In contrast, the four other Leu-tRNALeu isoacceptors and Phe-tRNAPhe possess a canonical G1-C72 base pair at this position (Figures 1B and 2A). We introduced a U to C base substitution at position 72 of tRNALeuTAA to restore a G1-C72 Watson–Crick base pair in the acceptor arm of this tRNA. A second tRNALeuTAA mutant was constructed by introducing an A1-U72 base pair at the same position. tRNALeuTAA was preferred to tRNALeuGAG for this analysis since the acceptor stems tRNALeuTAA and tRNAPhe only differ by U and C at position 72 (Figure 2A). This allowed comparison of Phe-tRNAPhe with a Leu-tRNALeuTAA variant containing identical acceptor stems. The mutants were used as substrates in the coupled aaRS-AlbC assay. This assay was carried out as described above for the screening of Leu-tRNALeu isoacceptors, except that the KCl concentration was 50 mM instead of 150 mM, to maximize enzymatic activity. Leu-tRNALeuTAA and Leu-tRNALeuCAG were analysed in the same experimental conditions for comparison. The results are presented in Figure 2B. The U72C mutation in Leu-tRNALeuTAA resulted in an enzymatic activity close to that measured for the best isoacceptor, Leu-tRNALeuCAG. The G1A mutation in Leu-tRNALeuTAA did not enable the significant formation of cFL, as observed for its wild-type counterpart, Leu-tRNALeuTAA. Our results clearly show that the pair G1-C72 is a key identity determinant for the second substrate of AlbC.
Figure 2.

(A) Sequences of the acceptor arm of the natural and mutant tRNAs used in this study. (B) Rate of formation of cFL (left panel) and cFF (right panel) by AlbC using tRNALeuTAA mutants: G1-C72 tRNALeuTAA (grey) or A1-U72 tRNALeuTAA (light blue). Wild-type tRNALeuTAA (orange) was used as a control and tRNALeuCAG (black) as a reference of substrate recognized by AlbC. Kinetics of synthesis were determined as for experiments shown in Figure 1, except for KCl concentration (see results). Curves are drawn for clarity and are not representative of kinetic models.
In addition, we investigated the capacity of G1-C72 tRNALeuTAA, which has an acceptor stem identical to that of tRNAPhe, to bind AlbC as a first substrate. We did not detect any signal using [14C]Leu G1-C72 tRNALeuTAA as a substrate in the acyl-enzyme formation assay (Supplementary Figure S4). Formation of cLL was also not detected using G1-C72 tRNALeuTAA as the sole substrate. Thus, the sequence of the tRNAPhe acceptor stem is not the key identity determinant for the first substrate of the reaction.
Recognition of the sequence G1-C72 versus C1-G72 for the two substrates of AlbC
The last base pair of the acceptor stem of tRNATyr is a key element for the recognition of this tRNA by TyrRSs. This base pair is responsible for the absence of cross reactivity between archeal or eukaryotic TyrRS-tRNATyr and bacterial TyrRS-tRNATyr (recognition of C1-G72 and G1-C72, respectively) (14,15). To investigate whether AlbC can also discriminate G1-C72 from C1-G72, we constructed the C1-G72 tRNAPhe and C1-G72 tRNALeuCAG mutants (tRNALeuCAG being the best tRNALeu isoacceptor substrate). These two mutants could not be obtained by in vitro transcription. They were constructed by mutagenesis from natural tRNAs and produced by overexpression in E. coli. The cFF-synthesizing activity of AlbC was more than 10-fold lower with the C1-G72 tRNAPhe mutant than for the wild-type tRNAPhe (Figure 3A) whereas similar amounts of phenylalanyl-enzyme were formed (Figure 3B). These results show that AlbC does not discriminate G1-C72 from C1-G72 for the first substrate but does for the second one. This conclusion was also supported by comparison C1-G72 and wild-type tRNALeuCAG as the second substrate. The formation of cFL was strongly decreased by the C1-G72 mutation and inhibition of cFF was also reduced (Figure 3C). As a wild-type tRNALeuCAG reference, we used tRNALeuCAG produced by overexpression in E. coli. The rates of cFL or cFF synthesis were similar to those observed with tRNALeuCAG obtained by in vitro transcription (Figure 3C), indicating that post-transcriptional modification of the second aa-tRNA substrate is not essential for enzymatic activity.
Figure 3.

(A) cFF-synthesizing activity of AlbC using either Phe-tRNAPhe purified from E. coli (black/grey) or Phe C1-G72 tRNAPhe (orange/yellow). The points reported are the result of two independent experiments. Error bars show the uncertainty on measurement. Curves are drawn for clarity and are not representative of kinetic models. Enzymatic measurements were performed as described in ‘Materials and Methods’ with 50 nM AlbC. (B) Covalent labelling of AlbC and S37A by [14C]Phe-tRNAPhe or [14C]Phe C1-G72 tRNAPhe. Enzymes were incubated with labelled aa-tRNA, as described in ‘Materials and Methods’, separated on SDS–PAGE, then transferred onto a PVDF membrane that was analysed with a radioimager. (C) Rate of formation of cFL (left panel) and cFF (right panel) by AlbC using tRNALeuCAG obtained by in vitro transcription (black), tRNALeuCAG purified from E. coli (grey) or Leu C1-G72 tRNALeuCAG (orange). Kinetics of synthesis were determined as described in Figure 1. Curves are drawn for clarity and are not representative of kinetic models.
Regions of AlbC involved in interaction with tRNAs
Interaction of aa-tRNAs with the basic patch of AlbC
We previously substituted each of the basic residues located in helix α4 with alanine. When expressed in E. coli, most of the resulting variants produced lower amounts of cFL than the wild-type enzyme, suggesting that these basic residues interact with the tRNA moiety of the substrates (5). We selected and purified AlbC variants encompassing residues distributed across the whole basic patch, namely R80A, R91A, K94A, R98A, R102A and R98A/R99A (Figure 5). We first investigated the propensity of the variants to form the acyl-enzyme intermediate with either Phe-tRNAPhe or Leu-tRNALeuCAG. Similar to the wild-type enzyme, labelled intermediates were only detected in the presence of [14C]Phe-tRNAPhe, indicating that Phe-tRNAPhe was also the first substrate accommodated by each of the variants (Figure 4A and Supplementary Figure S5). The amount of phenylalanyl-enzyme detected was decreased for all variants. In particular, substitutions of residues R98 and R99 located in the C-terminal part of helix α4 prevented acylation. These results show that the basic patch of helix α4 of AlbC is involved in the binding of the first substrate Phe-tRNAPhe.
Figure 5.
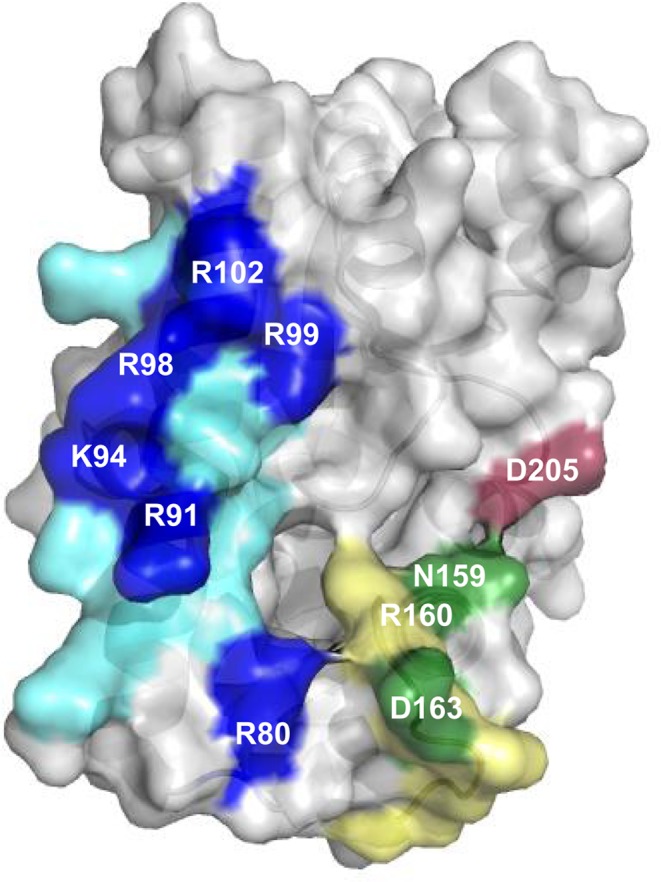
AlbC regions involved in interaction with tRNA substrates. The overall structure of AlbC (PDB, 3OQV) is shown in surface mode. The patch of basic residues located on helix α4 is coloured in blue; the residues substituted in this study are in dark blue. The loop α6–α7 is coloured in green; the residues substituted in this study are in dark green. The residue D205 belonging to the loop β6- α8 is coloured in dark red.
Figure 4.

Covalent labelling of AlbC and variants by [14C]Phe transferred from [14C]Phe-tRNA Phe. AlbC was used as positive control for the formation of phenylalanyl-enzyme and S37A was used as a negative control in this experiment (not shown). Enzymes were incubated with labelled aa-tRNA, as described in ‘Materials and Methods’, separated on SDS–PAGE, then transferred onto a PVDF membrane that was analysed with a radioimager. Detection of potential leucyl-enzyme intermediates is shown in Supplementary Figure S5. (A) Variants of the basic patch. (B) Variants of the loops α6–α7 and β6–α8. Proteins were analysed on different gels. AlbC was loaded onto each gel as a standard.
We wondered if information regarding the binding of the second substrate could be obtained from measuring the activity of these different variants. We used Phe-tRNAPhe and three different Leu-tRNALeu isoacceptors representative of the different activity patterns observed for wild-type AlbC (Leu-tRNALeuCAG, Leu-tRNALeuCAA and Leu-tRNALeuTAA, see Figure 1B). In standard assay conditions (50 nM AlbC or AlbC variant, 0.25 μM Phe-tRNAPhe and 0.22 μM Leu-tRNALeu isoacceptor), the amounts of cyclodipeptides produced by the variants were under the detection threshold (25–30 nM). We modified the experimental conditions so that enzyme/substrate ratios were set at values similar to those used in the acyl-enzyme detection assay (i.e. 1.5 μM AlbC or AlbC variant, 0.75 μM Phe-tRNAPhe and 0.66 μM Leu-tRNALeu isoacceptor). All of the variants showed lower cyclodipeptide-synthesizing activity than the wild-type enzyme; the more affected being the variant R98A/R99A (Table 1A). For each variant, the cFL- and cFF-synthesizing activities were decreased by the same factor, regardless of the tRNALeu isoacceptor used. As none of the substitutions changed the pattern of use of the second substrate, it is likely that the basic residues of the helix α4 are not involved in its binding.
Table 1. cFF and cFL synthesis activity of AlbC variants with different tRNALeu isoacceptors.
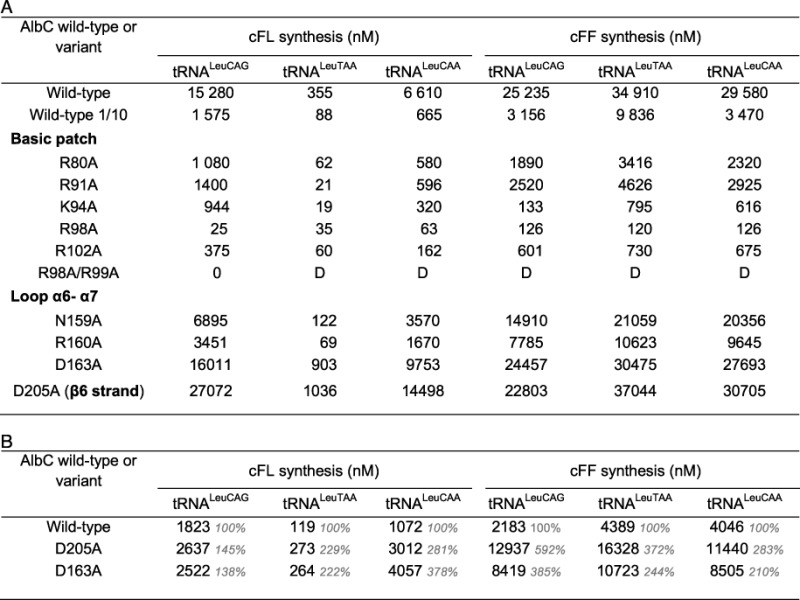 |
Activities are expressed in concentration of cyclodipeptides (nM) after 21 min of reaction; D, detectable. (A) The standard enzymatic assay was performed with 0.75 μM Phe-tRNAPhe, 0.66 μM Leu-tRNALeu isoacceptor and 1.5 μM AlbC or AlbC variant (50 mM KCl). Under these conditions, wild-type AlbC catalysed the complete incorporation of Phe into cFL and cFF. An additional assay was performed in non-limiting conditions using 150 nM AlbC (1/10). (B) The most active variants were tested with diluted concentrations of enzyme and both tRNA substrates. The enzymatic assay was performed with 0.25 μM Phe-tRNAPhe, 0.22 μM Leu-tRNALeu isoacceptor and 300 nM AlbC or AlbC variant (50 mM KCl). Residual activity (% of wild-type activity) is in grey.
Implication of other AlbC residues in tRNA interaction
Bonnefond et al. showed that deletion of the R158-V166 region that corresponds to the loop α6–α7 abolished non-acylated tRNALeu binding to CDPS YvmC (PDB, 3OQH) (6). To evaluate the role of this region of AlbC in tRNA binding, we substituted polar residues N159, R160, D163 and D205 with alanine (D205 belongs to the loop β6–α8 but is close to N159; Figure 5). Incubation of the variants with [14C]Phe-tRNAPhe indicated that substitutions N159A, R160A and D163A did not affect phenylalanyl-enzyme formation (Figure 4B). No labelled enzyme was detected with [14C]Leu-tRNALeu (Supplementary Figure S5). These results show that this region of AlbC is not involved in the binding of the first substrate. For the variant D205A, more labelled phenylalanyl-enzyme was detected (Figure 4B). This was the only variant for which a significant amount of leucyl-enzyme intermediate was also detected (Supplementary Figure S5).
We next determined the activity of these variants in the presence of the different Leu-tRNALeu isoacceptors (Table 1A). The N159A and R160A substitutions decreased cyclodipeptide production. Synthesis of cFL was more affected than that of cFF. Furthermore, the decrease in activity of the variant N159A varied according to the tRNALeu isoacceptor used. These results suggest an involvement of both residues in the binding of the second substrate. For the variants D163A and D205A, as for the wild-type enzyme, the phenylalanine present in the enzymatic assay was completely incorporated into cFF and cFL, preventing a comparison of their activities. A new measurement was performed with reduced concentrations of enzyme and substrates (i.e. 300 nM enzyme, 0.25 μM Phe-tRNAPhe and 0.22 nM Leu-tRNALeuCAG) (Table 1B). Both variants synthesized much more cFF and cFL than the wild-type enzyme. In addition, the pattern of use of the different isoacceptors tRNALeu changed. Both variants used preferentially tRNALeuCAA, followed by tRNALeuCAG, and then by tRNALeuTAA. These results show that modification of residues D163 and D205 changed tRNALeu isoacceptor specificity, indicating their involvement in the binding of the second substrate.
DISCUSSION
Here, we show that AlbC discriminates between its two substrates and possesses a different binding site for each of them.
AlbC first specifically interacts with Phe-tRNAPhe, forming a phenylalanyl-enzyme intermediate. Phenylalanine is also the amino acid preferentially incorporated into cyclodipeptides synthesized by AlbC. This confirms the previous assumption that the preferred amino acid is first bound to the CDPS and remains in the catalytic pocket as an acyl-enzyme intermediate (3,5,6). We did not detect a covalent leucyl-enzyme intermediate when leucylated G1-C72 tRNALeuTAA was used as a substrate. The acceptor arm sequence of G1-C72 tRNALeuTAA is identical to that of tRNAPhe. This indicates that the nature of the amino acid loaded onto tRNA is a key determinant of the specificity of AlbC for its first substrate.
We analysed the interaction of AlbC with Leu-tRNALeu as a second substrate by measuring its enzymatic activity with each of the six different tRNALeu isoacceptors. AlbC does not use all of the Leu-tRNALeu isoacceptors as a second substrate, implying the involvement of the tRNA moiety in the recognition of the second substrate. The two tRNAsLeu not used as substrates (tRNALeuTAA and tRNALeuTAG) are the only ones to have a U base at position 72. Mutation of this position (U72C) in tRNALeuTAA was sufficient for high AlbC activity. This mutation restores a Watson–Crick base pair G1-C72. However, despite the presence of Watson–Crick base pairing, the A1-U72 tRNALeuTAA mutant is not a substrate of AlbC, and the C1-G72 tRNALeuCAG mutant is a poor substrate of AlbC. The base pair G1-C72 of the acceptor arm is hence a key determinant for the interaction of AlbC with its second aa-tRNA substrate. The preference of AlbC for tRNALeuCAG/tRNALeuCAG* versus tRNALeuCAA/ tRNALeuTAG may also be related to particular sequences in the acceptor arm. The best Leu-tRNALeu substrates (tRNALeuCAG/tRNALeuCAG*) are the only substrates possessing the base pairs A4-U69 and A5•C68. The less efficient Leu-tRNALeu substrates (tRNALeuCAA/ tRNALeuTAG) share the sequences G4-C69 and A6-U67. Systematic mutational analysis will be required to clarify the involvement of these base pairs in the efficiency of substrate usage. The aminoacyl moiety of the second aa-tRNA also appears to be important for its interaction with the CDPS. Indeed, AlbC prefers Phe-tRNAPhe to Leu-G1-C72 tRNALeuTAA as a second substrate, despite the fact that these two substrates have the same acceptor arm sequence. In addition, more cFF was synthesized than cFL for equivalent concentrations of Phe-tRNAPhe and Leu-tRNALeu. In vivo, AlbC produces more cFL than cFF (2). Dong and collaborators determined the abundances of tRNA in E. coli at different growth rates (16). Whatever the growth rate, there is about 9-fold more tRNALeu than tRNAPhe in E. coli cells (about 7-fold more if we only consider tRNALeu isoacceptors that are efficiently used by AlbC). For a tRNALeu/tRNAPhe ratio representative of in vivo conditions, we indeed observed in vitro higher amounts of cFL than cFF.
In E. coli, all amino acids except Asn, Gln, Ile, Pro and Trp can be loaded onto tRNAs that contain a G1-C72 pair in the acceptor arm (http://gtrnadb.ucsc.edu/Esch_coli_K12/Esch_coli_K12-align.html). However, only a few of them can be incorporated into cyclodipeptides by AlbC as a second substrate, as shown by the cyclodipeptide production profile of recombinant E. coli producing AlbC (2). This clearly indicates that other determinants are important for the selection of the second substrate by AlbC. Phe, Leu, Tyr, Met and Ala amino acids are all incorporated into cyclodipeptides by AlbC. Closer examination of the sequences encoding tRNAs that are associated with these amino acids shows that these sequences all contain the G1-C72 pair as well as A76C75C74A73 in the single strand extremity. The rest of the sequence of these tRNAs, including the acceptor arm, is highly variable. Other E. coli tRNAs that possess the sequence A76C75C74A73C72-G1 can be charged with Arg, Lys or Val, but these amino acids are not incorporated by AlbC into cyclodipeptides. This definitely indicates that the selectivity of AlbC for its second substrate involves tRNA sequences but also the nature of the amino acid.
AlbC originates from S. noursei, therefore, we examined the tRNA sequences in this host. The sequences are not available for S. noursei but can be found for many other Streptomyces species. All the Streptomyces tRNA sequences contain A76C75C74A73C72-G1, suggesting that all tRNAPhe and tRNALeu are substrates for AlbC. The acceptor arm of one Streptomyces tRNALeu isoacceptor, tRNALeuTAA, is identical to that of E. coli tRNAPhe. The sequence of this tRNALeuTAA is highly conserved in Streptomyces species and this tRNA recognizes a rare codon. tRNALeuTAA has been thoroughly studied: it is not required for growth but is required for some aspects of secondary metabolism and morphological development (17). This tRNA would be particularly involved in regulatory pathways of biosynthesis of several antibiotics. Its potential relationship with AlbC remains to be determined.
In view of these results with AlbC, one may wonder if other CDPSs share the same key determinants for the first and the second aa-tRNA substrate. Like AlbC, most of the biochemically characterized CDPSs are promiscuous and synthesize several cyclodipeptides. One exception to this rule is the CDPS Amir_4627 from Actinosynnema mirum, which synthesizes exclusively cWW when this enzyme is expressed in E. coli (18). The sequence of the E. coli tRNATrp contains the combination of an A1-U72 base pair and a G73 residue, which is a unique feature of tRNATrp in E. coli. This could contribute to the selectivity of Amir_4627 for its second amino acid substrate. The tRNA sequence of the second substrate may also be critical for the determination of the activity of eukaryotic CDPSs in E. coli. The CDPS Nvec-CDPS2 from the eukaryotic organism Nematostella vectensis mainly synthesizes cWX cyclodipeptides when it is expressed in E. coli (7). The base at position 73 of the tRNATrp acceptor arm is largely responsible for the very low cross reactivity between archeal or eukaryotic and bacterial TrpRS-tRNATrp pairs: this base is generally adenine in archeal or eukaryotic tRNATrp and guanine in bacterial tRNATrp (19). Expression of this CDPS in E. coli results in the synthesis of very small amounts of cWM, cWF and cWL. A search for tRNATrp sequences in the genome available for N. vectensis (http://www.ncbi.nlm.nih.gov) revealed five different sequences possessing characteristics of eukaryotic tRNATrp. However, the acceptor arm sequences of these five candidates are more similar to E. coli tRNAPhe or tRNAMet than to E. coli tRNATrp (Supplementary Figure S6). Thus, an experimental system that uses eukaryotic tRNAs would probably give a more accurate view of N. vectensis CDPS activity. It follows that care must be taken when interpreting the results of activities of CDPSs expressed in E. coli: discrepancies between E. coli tRNA sequences and tRNA sequences in the species from which the CDPSs originates can give a misleading view of the activity of these enzymes in their natural environment.
The second important result of our work concerns the identification of two different regions of AlbC that interact with either the tRNA moiety of the first or that of the second substrate. We show that the basic patch of helix α4 is important for the binding of the first substrate. All the substitutions involving amino acids in this patch dramatically affected enzymatic activity in vitro, probably by perturbing the binding of the tRNA moiety. This indicates that all these residues, in particular R98 and R99, participate in the interaction. The basic patch does not belong to a clearly defined class of RNA-binding motif. It nevertheless shares features with some of them (20). In particular, the drastic decrease of enzymatic activity that is associated with substitutions of any residues of the motif has also been described for double-stranded RNA-binding motifs (20,21). Arginines are essential residues for RNA binding. They are involved in non-specific interactions mediated by their positive charge. However, they also participate in specific hydrogen bonding networks with RNA bases, especially guanines (20,22,23). The occurrence of arginine•guanine pairs in specific RNA–protein interactions is very high in complex structures (24). The sequences of all the acceptor stems of aa-tRNAs used as substrates by AlbC have a high GC content, which is consistent with RNA–protein interactions involving arginines. Nonetheless, many new putative CDPSs that have been identified by Basic Local Alignment Search Tool searches (3) are devoid of the basic patch that is found in AlbC and other characterized CDPSs (2,5). These putative CDPSs possess a large number of aspartic and glutamic acids, asparagine, glutamine, serine and threonine at a similar position to the basic patch of AlbC. These residues are known to interact specifically with nucleotide bases other than guanine. Thus, residues of helix α4 of these putative CDPSs may in part determine substrate specificity by interacting only with a limited number of tRNA sequences.
We found that the binding of the second substrate was affected when residues belonging to the loop α6–α7 or loop β6–α8 were substituted for alanine. In particular, binding was strongly affected in D163A (in loop α6–α7) and D205A (in loop β6–α8). In both TrpRSs and TyrRSs, the loop equivalent to this loop α6–α7 is involved in tRNA binding (25–28). Furthermore, during the binding of the second substrate, AlbC discriminates between G1-C72 and C1-G72, similar to TyrRSs and TrpRSs (14,15,19). This discrimination can be only achieved if the interaction of the enzyme with its substrate occurs from the major groove side of the acceptor stem (22,23), which is also a distinctive feature of class-Ic aaRSs (27,28), apparently shared by AlbC. This suggests that CDPSs may have retained some characteristics of class-Ic aaRSs regarding their interaction with their second substrate. In TyrRSs, a second cluster of amino acids belonging to the Rossmann-fold is involved in N1 recognition (27–30). In AlbC, this region corresponds to residues of helix α8 (P207-L212) and part of the following loop. Although AlbC has none of the consensus sequences that allow TyrRSs to specifically recognize G1-C72 or G1-C72 (31), mutational analysis of this region should be carried out to test its possible involvement in tRNA binding.
Both residues D205, which neighbours helix α8, and D163 from the loop α6–α7 appear to have a negative effect on catalytic activity, because substitution of these residues with alanine creates variants with higher catalytic activity. Such a phenomenon is not uncommon in enzymatic world (32). Both residues may be incorporated to improve enzyme specificity at the expense of efficiency, perhaps by preventing the binding of undesired tRNA sequences. Bedouelle et al. (33) identified such a residue in TyrRS from Bacillus stearothermophilus: the primary function of an acidic residue, E152, was to inhibit the use of non-cognate tRNAs by electrostatic and steric repulsions, and hence ensure enzyme specificity for tRNATyr. Another possibility is that D205 and D163 residues may reduce the affinity of cognate aa-tRNAs to AlbC so that the major fractions of Phe-tRNAPhe and Leu-tRNALeu are used for ribosomal protein synthesis.
In conclusion, we demonstrate that AlbC handles differently its two substrates. The binding of its first substrate is highly dependent on the aminoacyl moiety of the tRNA, whereas both the aminoacyl moiety and the tRNA sequence itself are essential for the specific recognition of the second substrate. This first and second substrates bind to the enzyme at different sites: binding of the first substrate involves the basic patch centred on helix α4 whereas binding of the second substrate involves the loop α6–α7. These regions modulate the specificity of AlbC though specific interactions with appropriate tRNA sequences. Noted that the two binding sites could be simultaneously present on the enzyme or the second binding-site could result from a structural reorganisation induced by the formation of the aminoacyl-enzyme intermediate. The interaction of AlbC with its second aa-tRNA substrate shares features with the interaction of class-Ic aaRSs with their aa-tRNA substrates. Complementary biophysical analyses will be required to precisely define the outlines of these two binding sites and determine how many features CDPSs have retained from their aaRS ancestors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Cédric Masson for the purification of AlbC variants, Robert Thai for helpful advice about mass spectrometry, and Bertrand Czarny for use of the Beta-imager. We thank Fabrice Beau for useful discussions about enzymatic analyses. We are grateful to Emmanuelle Schmitt and Yves Mechulam for helpful discussions about aminoacyl-tRNA synthetases.
FUNDING
Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA); French National Research Agency (ANR). The Service d’Ingénierie Moléculaire des Protéines is a member of the Laboratory of Excellence LERMIT. Funding for open access charge: [ANR 2010/BLAN 1501 01].
Conflict of interest statement. None declared.
REFERENCES
- 1.Lautru S., Gondry M., Genet R., Pernodet J.L. The albonoursin gene Cluster of S. noursei: biosynthesis of diketopiperazine metabolites independent of nonribosomal peptide synthetases. Chem. Biol. 2002;9:1355–1364. doi: 10.1016/s1074-5521(02)00285-5. [DOI] [PubMed] [Google Scholar]
- 2.Gondry M., Sauguet L., Belin P., Thai R., Amouroux R., Tellier C., Tuphile K., Jacquet M., Braud S., Courcon M., et al. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat. Chem. Biol. 2009;5:414–420. doi: 10.1038/nchembio.175. [DOI] [PubMed] [Google Scholar]
- 3.Belin P., Moutiez M., Lautru S., Seguin J., Pernodet J.L., Gondry M. The nonribosomal synthesis of diketopiperazines in tRNA-dependent cyclodipeptide synthase pathways. Nat. Prod. Rep. 2012;29:961–979. doi: 10.1039/c2np20010d. [DOI] [PubMed] [Google Scholar]
- 4.Vetting M.W., Hegde S.S., Blanchard J.S. The structure and mechanism of the Mycobacterium tuberculosis cyclodityrosine synthetase. Nat. Chem. Biol. 2010;6:797–799. doi: 10.1038/nchembio.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sauguet L., Moutiez M., Li Y., Belin P., Seguin J., Le Du M.H., Thai R., Masson C., Fonvielle M., Pernodet J.L., et al. Cyclodipeptide synthases, a family of class-I aminoacyl-tRNA synthetase-like enzymes involved in non-ribosomal peptide synthesis. Nucleic Acids Res. 2011;39:4475–4489. doi: 10.1093/nar/gkr027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnefond L., Arai T., Sakaguchi Y., Suzuki T., Ishitani R., Nureki O. Structural basis for nonribosomal peptide synthesis by an aminoacyl-tRNA synthetase paralog. Proc. Natl Acad. Sci. U.S.A. 2011;108:3912–3917. doi: 10.1073/pnas.1019480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seguin J., Moutiez M., Li Y., Belin P., Lecoq A., Fonvielle M., Charbonnier J.B., Pernodet J.L., Gondry M. Nonribosomal Peptide synthesis in animals: the cyclodipeptide synthase of Nematostella. Chem. Biol. 2011;18:1362–1368. doi: 10.1016/j.chembiol.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Aravind L., de Souza R.F., Iyer L.M. Predicted class-I aminoacyl tRNA synthetase-like proteins in non-ribosomal peptide synthesis. Biol. Direct. 2010;5:48. doi: 10.1186/1745-6150-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giessen T.W., von Tesmar A.M., Marahiel M.A. Insights into the generation of structural diversity in a tRNA-dependent pathway for highly modified bioactive cyclic dipeptides. Chem. Biol. 2013;20:828–838. doi: 10.1016/j.chembiol.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villet R., Fonvielle M., Busca P., Chemama M., Maillard A.P., Hugonnet J.E., Dubost L., Marie A., Josseaume N., Mesnage S., et al. Idiosyncratic features in tRNAs participating in bacterial cell wall synthesis. Nucleic Acids Res. 2007;35:6870–6883. doi: 10.1093/nar/gkm778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meinnel T., Mechulam Y., Fayat G. Fast purification of a functional elongator tRNAMet expressed from a synthetic gene in vivo. Nucleic Acids Res. 1988;16:8095–8096. doi: 10.1093/nar/16.16.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mechulam Y., Guillon L., Yatime L., Blanquet S., Schmitt E. Protection-based assays to measure aminoacyl-tRNA binding to translation initiation factors. Methods Enzymol. 2007;430:265–281. doi: 10.1016/S0076-6879(07)30011-6. [DOI] [PubMed] [Google Scholar]
- 14.Tsunoda M., Kusakabe Y., Tanaka N., Ohno S., Nakamura M., Senda T., Moriguchi T., Asai N., Sekine M., Yokogawa T., et al. Structural basis for recognition of cognate tRNA by tyrosyl-tRNA synthetase from three kingdoms. Nucleic Acids Res. 2007;35:4289–4300. doi: 10.1093/nar/gkm417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnefond L., Giege R., Rudinger-Thirion J. Evolution of the tRNA(Tyr)/TyrRS aminoacylation systems. Biochimie. 2005;87:873–883. doi: 10.1016/j.biochi.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Dong H., Nilsson L., Kurland C.G. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 17.Chater K.F., Chandra G. The use of the rare UUA codon to define “expression space” for genes involved in secondary metabolism, development and environmental adaptation in Streptomyces. J. Microbiol. 2008;46:1–11. doi: 10.1007/s12275-007-0233-1. [DOI] [PubMed] [Google Scholar]
- 18.Giessen T.W., von Tesmar A.M., Marahiel M.A. A tRNA-Dependent Two-Enzyme Pathway for the Generation of Singly and Doubly Methylated Ditryptophan 2,5-Diketopiperazines. Biochemistry. 2013;52:4274–4283. doi: 10.1021/bi4004827. [DOI] [PubMed] [Google Scholar]
- 19.Shen N., Guo L., Yang B., Jin Y., Ding J. Structure of human tryptophanyl-tRNA synthetase in complex with tRNATrp reveals the molecular basis of tRNA recognition and specificity. Nucleic Acids Res. 2006;34:3246–3258. doi: 10.1093/nar/gkl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burd C.G., Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 21.Masliah G., Barraud P., Allain F.H. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell. Mol. Life Sci. 2013;70:1875–1895. doi: 10.1007/s00018-012-1119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frugier M., Schimmel P. Subtle atomic group discrimination in the RNA minor groove. Proc. Natl Acad. Sci. U.S.A. 1997;94:11291–11294. doi: 10.1073/pnas.94.21.11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seeman N.C., Rosenberg J.M., Rich A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl Acad. Sci. U.S.A. 1976;73:804–808. doi: 10.1073/pnas.73.3.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lustig B., Arora S., Jernigan R.L. RNA base-amino acid interaction strengths derived from structures and sequences. Nucleic Acids Res. 1997;25:2562–2565. doi: 10.1093/nar/25.13.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia J., Xu F., Chen X., Chen L., Jin Y., Wang D.T. Two essential regions for tRNA recognition in Bacillus subtilis tryptophanyl-tRNA synthetase. Biochem. J. 2002;365:749–756. doi: 10.1042/BJ20020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia J., Chen X.L., Guo L.T., Yu Y.D., Ding J.P., Jin Y.X. Residues Lys-149 and Glu-153 switch the aminoacylation of tRNA(Trp) in Bacillus subtilis. J. Biol. Chem. 2004;279:41960–41965. doi: 10.1074/jbc.M401937200. [DOI] [PubMed] [Google Scholar]
- 27.Yaremchuk A., Kriklivyi I., Tukalo M., Cusack S. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 2002;21:3829–3840. doi: 10.1093/emboj/cdf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi T., Nureki O., Ishitani R., Yaremchuk A., Tukalo M., Cusack S., Sakamoto K., Yokoyama S. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Biol. 2003;10:425–432. doi: 10.1038/nsb934. [DOI] [PubMed] [Google Scholar]
- 29.Bedouelle H. Recognition of tRNA(Tyr) by tyrosyl-tRNA synthetase. Biochimie. 1990;72:589–598. doi: 10.1016/0300-9084(90)90122-w. [DOI] [PubMed] [Google Scholar]
- 30.Nair S., Ribas de Pouplana L., Houman F., Avruch A., Shen X., Schimmel P. Species-specific tRNA recognition in relation to tRNA synthetase contact residues. J. Mol. Biol. 1997;269:1–9. doi: 10.1006/jmbi.1997.1025. [DOI] [PubMed] [Google Scholar]
- 31.Bonnefond L., Frugier M., Giege R., Rudinger-Thirion J. Human mitochondrial TyrRS disobeys the tyrosine identity rules. RNA. 2005;11:558–562. doi: 10.1261/rna.7246805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen H.M., Tawfik D.S., Griffiths A.D. Altering the sequence specificity of HaeIII methyltransferase by directed evolution using in vitro compartmentalization. Protein Eng. Des. Sel. 2004;17:3–11. doi: 10.1093/protein/gzh001. [DOI] [PubMed] [Google Scholar]
- 33.Bedouelle H., Guez-Ivanier V., Nageotte R. Discrimination between transfer-RNAs by tyrosyl-tRNA synthetase. Biochimie. 1993;75:1099–1108. doi: 10.1016/0300-9084(93)90009-h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.