


Abstract
Myotonic dystrophy type 1 (DM1) is caused by CUG triplet expansions in the 3′ UTR of dystrophia myotonica protein kinase (DMPK) messenger ribonucleic acid (mRNA). The etiology of this multi-systemic disease involves pre-mRNA splicing defects elicited by the ability of the CUG-expanded mRNA to ‘sponge’ splicing factors of the muscleblind family. Although nuclear aggregation of CUG-containing mRNPs in distinct foci is a hallmark of DM1, the mechanisms of their homeostasis have not been completely elucidated. Here we show that a DEAD-box helicase, DDX6, interacts with CUG triplet-repeat mRNA in primary fibroblasts from DM1 patients and with CUG–RNA in vitro. DDX6 overexpression relieves DM1 mis-splicing, and causes a significant reduction in nuclear DMPK-mRNA foci. Conversely, knockdown of endogenous DDX6 leads to a significant increase in DMPK-mRNA foci count and to increased sequestration of MBNL1 in the nucleus. While the level of CUG-expanded mRNA is unaffected by increased DDX6 expression, the mRNA re-localizes to the cytoplasm and its interaction partner MBNL1 becomes dispersed and also partially re-localized to the cytoplasm. Finally, we show that DDX6 unwinds CUG-repeat duplexes in vitro in an adenosinetriphosphate-dependent manner, suggesting that DDX6 can remodel and release nuclear DMPK messenger ribonucleoprotein foci, leading to normalization of pathogenic alternative splicing events.
INTRODUCTION
Myotonic dystrophy type 1 (DM1) is a multi-systemic disease and represents the most common muscular dystrophy among adults. It affects about 1/8000 in most populations and is inherited in an autosomal dominant manner [recently reviewed in (1–3)]. It is seen both in a congenital form (cDM1) and an adult form and symptoms include muscle wasting, myotonia, cardiac conduction defects, cataracts and insulin resistance (1–3). DM1 is caused by an expansion of a tri-nucleotide CTG-repeat in the gene encoding myotonic dystrophy protein kinase (DMPK) (4). While the DMPK-messenger ribonucleic acid (mRNA) of unaffected individuals contains between 5 and 38 CUG-repeats in their 3′ UTRs, disease severity increases with the number of repeats (5); where symptoms have been reported from 50 repeats and severely affected individuals can have several thousand repeats (1–3). Studies using dmpk-knockout mice suggested that DMPK insufficiency could contribute to DM1 etiology but fail to explain all DM1 features, since these mice only developed mild myopathies and cardiac conduction defects in older animals (6–8). More recent studies have defined a ‘gain-of-function’ role for the CUG-expanded repeats by their ability to sequester splicing factors including muscleblind-like protein 1 (MBNL1), which in turn leads to mis-splicing of numerous target pre-mRNAs, allowing for expression of otherwise embryonally restricted isoforms in adults (9–16). This phenotype is augmented by a protein kinase C-dependent hyper-phosphorylation and stabilization of an MBNL1-antagonizing splicing factor CUG-BP1, which also plays important roles in the regulation of protein translation and mRNA decay (16–20). Supporting a mechanism that sequestered MBNL1 functions as the main cause of aberrant splicing in DM1, ∼80% of all CUG repeat-induced mis-splicing events can be correlated to a similar splicing pattern following ablation of MBNL1 in a mouse knockout model (10). Interestingly, the mutant DMPK allele produces mRNAs, which despite their functional 5′ cap and 3′ poly(A) tails, fail to efficiently escape the nucleus and is sequestered in messenger ribonucleoprotein (mRNP) aggregates containing MBNL-proteins (21). These mRNPs can be visualized as distinct nuclear foci by RNA-fluorescent in situ hybridization (RNA-FISH) and have been shown to rely on the expression of MBNL1 (9,21–25). Several studies have described distinct cytoplasmic foci in cells expressing CUG-expanded mRNAs although the potential function of these remains unknown (17,26). In addition, the mechanisms of nuclear CUG-foci assembly and homeostasis remain largely unknown although in vitro pull-down experiments using CUG-repeat oligonucleotides as bait, have identified several protein-interactors aside from MBNL1, including DEAD-box RNA helicases (DDX17, DDX5), hnRNP-proteins (hnRNP L, M, A2/B2) and splicing factors (27). Interestingly, a number of these factors, including hnRNP L, A2/B1, DDX5 and DDX17, have been shown to directly interact with MBNL1 in a RNA-independent manner (13). Recently, the double-stranded RNA-binding protein Staufen 1 (Stau1) was shown to interact with the 3′ UTR of the DMPK-mRNA to increase DMPK-mRNA nuclear export/translation and rescued DM1-specific mis-splicing events (28), suggesting a central role for Stau1 in diminishing DM1 pathogenesis. DEAD-box helicases, or superfamily two helicases, function in all aspects of mRNP-metabolism and govern regulated nuclear and cytoplasmic events including transcription, RNA splicing, nuclear export, translation and mRNA turnover [recently reviewed in (29)]. These proteins use ATP-hydrolysis to allow for regulated interactions with mRNA substrates and to remodel RNA-binding proteins within complex mRNPs (29). DDX6 is a predominantly cytoplasmic localized DEAD-box helicase, which is necessary for numerous steps in regulated mRNA turnover and translation (30–33). In mammalian cells, DDX6 is necessary for assembly of processing bodies (PBs), which harbor repressed mRNPs, a large number of mRNA decay factors and proteins central to the miRNA-machinery (30–35). Here we show that DDX6 is able to remodel and reduce nuclear CUG-mRNP foci and facilitate an elevated cytoplasmic abundance of the mutant DMPK-mRNA and MBNL1 protein in fibroblasts isolated from DM1 patients. We show that DDX6 associates strongly with DMPK-mRNA in a CUG-repeat-dependent manner, both in vitro and in vivo, suggesting that the modulatory effect on CUG-foci number and localization is mediated by a direct interaction and unwinding of secondary mRNA structure and/or displacement of MBNL1 from the mRNP. In line with this, we demonstrate that DDX6 is able to unwind CUG–RNA duplexes in an ATP- and DEAD-box-dependent manner. Importantly, the DDX6-mediated dissemination of nuclear CUG-foci correlates with a restoration of select DM1-specific alternative splicing events, including that of insulin receptor 2 (IR2) pre-mRNA. Taken together, our results suggest that DDX6 although previously being implicated in post-transcriptional events in the cytoplasm is able to regulate nuclear-accumulated DMPK-mRNA, remodel its structure/mRNP composition and reduce the pathogenic features of DM1.
MATERIALS AND METHODS
Plasmids
pcDNA3-FLAG-DDX6 was cloned by standard polymerase chain reaction (PCR) protocols using a HeLa cell complementary deoxyribonucleic acid (cDNA) library with forward primer encoding a FLAG-tag: AAGGATCCGCCACCATGGACTACAAGGACGACGATGACAAGATGAGCACGGCCAGAACAG-3′ and reverse primer: 5′-ATCGGCGGCCGCCTCGAGTTAAAGGTTTCTCATCTTCTAC-3′. The PCR product was digested and inserted into the BamHI and NotI restriction sites of pcDNA3 (Invitrogen). For cloning lentiviral vectors, pCCL-PGK-EGFP (36) was digested with BamHI–XhoI and used for insertion of the BamHI–XhoI fragment excised from pcDNA3-FLAG-DDX6. The DEAA mutant was generated using standard PCR site-directed mutagenesis techniques with primers: 5′-AGATGATAGTATTGGGATGAGGCAGCTAAGTGGCTGTCACA-3′ and TGTGACAGCAACTTAGCTGCCTCATCCAATACTATCCATCT-3′. To generate pcNEGFP-DDX6 (and DEAA version), an isolated BamHI–NotI pcDNA3-FLAG-DDX6/DEAA fragment was inserted into the BamHI–NotI sites of pcNEGFP-C1 (37). To generate pcDNA5-3XFLAG-DDX6 (and DEAA version) an isolated BamHI–NotI pcDNA3-FLAG-DDX6/DEAA fragment was inserted into the BamHI–NotI sites of pcDNA5-3XFLAG. To generate pGEX-4TI-DDX6 (and DEAA version) an isolated BamHI–NotI pcDNA3-FLAG-DDX6/DEAA fragment was inserted into the BamHI-NotI sites of pGEX-4TI. All constructs were verified by sequencing.
Cell culture and transfection
Human fibroblast cells; normal human dermal fibroblast (NHDF) (wild-type, WT), GM03132 (DM1), GM04602 (DM1′) (Coriell) and DM2 fibroblasts (DM2) (38), were maintained in Dulbecco's Modified Eagle Medium (DMEM), 10% fetal bovine serum (FBS), 1% penicillin/streptomycin at 37°C, 5% CO2. Human embryonic kidney Flp-In (HEK293S) cells were used to establish stable cell lines expressing either 3XFLAG-tagged DDX6 or DEAA helicase mutant according to a previously described protocol (39). Knockdown experiments were basically conducted as described earlier (37). Briefly, cells were seeded on collagen-coated coverslips in 12-well plates at an ∼60% confluency. Twenty four hours later, cells were transfected using Silent-Fect (Bio-Rad) according to the manufacturers directions at a final siRNA concentration of 20 nM and incubated for 24 h. Cells were then washed twice in phosphate buffered saline (PBS) and replenished with DMEM, 10% FBS, 1% penicillin/streptomycin and incubated for another 24 h. The second transfection was done using Lipofectamine 2000 (Life Technologies) according to manufacturers directions with siRNA at 20 nM. After another 48 h cells were washed and fixed for RNA-FISH, immunofluorescence or lysed for preparation of whole-cell lysates for western blotting. Plasmid transfection for exogenous expression of Green Fluorescent Protein-tagged (GFP-tagged) DDX6 WT or DDX6 DEAA was done using 0.1–2 μg plasmid in six-well plates using Lipofectamine LTX according to manufacturer's directions.
siRNAs used:
| Target gene | Target sequence |
|---|---|
| Human DDX6 (siRNA #1) | GAGUUACUGAUGGGAAUUU |
| Human DDX6 (siRNA #2) | CUAUUCCGAGCAACAUUGA |
| hXRN1 | AGAUGAACUUACCGUAGAA |
| hDCP2 | GGACUGGCUUUCUCGAAGA |
Antibodies and western blotting
Rabbit anti-hDCP1a, anti-hDCP1b, anti-hEDC3 and anti-GW182 antibodies were generously provided by Professor Jens Lykke-Andersen, University of California San Diego, USA. Goat anti-DDX6 (sc-51416) from Santa Cruz Biotechnology (SCBT) was used for western blotting and rabbit anti-DDX6 (#9407) from Cell Signaling Technologies was used for immunofluorescence and western blotting. Mouse anti-MBNL1 (3A4) (sc-47740) and rabbit anti-myogenin (5FD) (sc-12732) were purchased from SCBT. Mouse anti-CUG-BP1 antibody (4A3) was obtained from Millipore. Rabbit anti-Staufen antibody (ab73478), mouse anti-DDX5 monoclonal antibody 2257C3a (ab53216) and rabbit anti-PABP antibody (ab153930) were purchased from Abcam. Western was performed according to standard protocols using PVDF (Millipore) membrane and antibody dilutions at 1:1000 for anti-FLAG, anti-DDX6 and anti-CUG-BP1 and at 1:200 for anti-MBNL1.
RNA-fluorescent in situ hybridization (RNA-FISH) and immunofluorescence
For RNA-FISH experiments NHDF or DM1 cells kept in DMEM/10% FBS were seeded at ∼50% confluency in 12-well plates containing collagen-coated coverslips and incubated overnight. Cells were fixed in 4% paraformaldehyde for 15 min, washed twice in PBS and stored at 4°C in 70% EtOH until used for RNA-FISH. RNA-FISH was performed essentially as described previously (40). Briefly, cells were rehydrated in PBS for 5 min and then pre-equilibrated in 2× SSC, 50% formamide (Sigma; BioUltra >99.5%) at RT for 5–10 min. Hybridizations were performed in a humidified chamber for 3 h at 37°C using a 30-mer Cy5- or Cy3-labeled DNA oligo containing 10 CAG-repeats at 10 ng probe per hybridization containing 50% formamide (Sigma; BioUltra >99.5%), 2× SSC, 1 mg/ml bovine serum albumin (BSA) (Ultrapure Roche), 0.2 μg/ml yeast transfer RNA (tRNA), 0.2 μg/ml salmon sperm DNA. Cells were then washed twice in 2× SSC, 50% formamide for 30 min (1 ml) followed by one 5-min wash in 2× SSC (1 ml) at RT and another wash in PBS (1 ml). For mounting cells, nuclei were counterstained using PBS containing 0.5 μg/ml 4',6-diamidino-2-phenylindole (DAPI) . Cells were then washed once in PBS (1 ml) and then nuclease-free water (Ambion) prior to mounting in ProLong GOLD (Invitrogen) and left in the dark at RT overnight. When RNA-FISH was combined with immunofluorescence, cells were instead of DAPI counterstaining blocked with PBS containing, 0.1% Triton X-100 (Invitrogen), 1% BSA (Roche), 200 U/ml Ribolock (Fermentas) for 10 min at RT. Cells were then washed twice with PBS/1% BSA (1 ml) and then incubated with primary antibody in PBS/1% BSA (20–200 μl) for 1 h at RT. After three 5-min washes in PBS (1 ml) cells were incubated with Alexa488-coupled secondary antibodies (Molecular Probes) for an additional 1 h at RT. Cells were washed twice in PBS (1 ml) and mounted as described above. Unless otherwise stated all antibodies were used at a dilution of 1:500.
Quantification and statistics
RNA foci were counted and quantified using ImageJ software by applying an identical threshold of pixel intensity within frames acquired with identical exposure time and scaling or by visual inspection of foci. Visual counting was performed on unidentified samples to avoid any bias. We observed no significant difference between the two different methods of counting on identical samples (data not shown). Unless otherwise noted distribution of foci per cell are represented as boxplots after pooling of three independent experiments after two-way ANOVA analysis (P < 0.01). Number of cells counted is noted as n. Mean values (foci per cell) are given in the text along with standard error of mean (SEM), n > 3. P-values presented above boxplots are based on Student's t-test unless otherwise is noted, where *P < 0.05, **P < 0.01 and ***P < 0.001. Integrated densities of pixels (quantified in ImageJ) in indicated nuclear or cytoplasmic areas from representative cells are where relevant presented as surface plots and their distribution as boxplots.
Lentiviral vector-production and transduction
293T cells were seeded in 15 cm plates. Twentyfour hours later, the cells were transfected with 7.3 μg pRSV-Rev, 9 μg pMD.2G, 31.5 μg pMDGP-Lg/RRE and 31.5 μg of pCCL-PGK vector plasmid using the calcium phosphate co-precipitation method. The viral particles were harvested 48 h post-transfection and filtered through a 45 μm filter (Sarstedt, Nümbrecht, Germany) before ultracentrifugation through a 20% sucrose cushion at 25 000 rpm for 2 h. Virus pellets were suspended in PBS in a volume of 1/300 of the original supernatant volume, and the resulting titer was measured by HIV-1 gag p24 antigen ELISA kit (ZeptoMetrix, Buffalo, NY) according to manufactures protocol. The concentrated virus was stored at −80°C. Thawed virus (2.5 μg p24 units) was diluted growth media in the presence of 8 μg/ml polybrene to transduce a 10 cm plate of fibroblasts. Media was renewed 24 h later.
Splicing analyses—RT-PCR
Total RNA was prepared from separately non-, GFP- or FLAG-DDX6-transduced DM1–2 cells (10 cm plates) using Trizol (Invitrogen) according to manufacturer's protocol. An oligo-dT-primed cDNA library was obtained using 5 μg total RNA and SuperScript II (Invitrogen) according to manufacturers protocol. Primers used to amplify Insulin receptor 2 cDNA; IR2 F: CCAAAGACAGACTCTCAGAT, IR2 R: AACATCGCCAAGGGACCTGC and ppp2r5c cDNA; ppp2r5c F: GGGAAGAAGCATGGGTTAAA, ppp2r5c R: CTTCCAAGGCTTTCTTGGTG. 10 pmol of each primer was 32P-5′ end-labeled prior to a standard Taq-polymerase 18-cycle PCR (62°C annealing temperature) to semi-quantitatively detect splice-variant amplicons in triplicates. Reactions were ethanol precipitated, redissolved in formamide load buffer (95% formamide, 5 mM EDTA and 0.1% xylene cyanol/bromphenol blue) and run on a 6% polyacrylamide gel, which was dried exposed and quantified by phosphorimager analysis. Primers used for qRT-PCR of IR2 alternatively spliced transcripts were as follows: exon 10/11: AATGCTGCTCCTGTCCAAAG, AGAGGTTTTTCTGGGGACGA. Exon 10/12: AATGCTGCTCCTGTCCAAAG, AGATGGCCTGGGGACGA. cDNA libraries were prepared using 1 μg total RNA from non-transduced or transduced cells (iScript cDNA synthesis, Bio-Rad) and ratios between exon 11 inclusion/exclusion was quantified in triplicate.
RNA immunoprecipitation
GFP- or FLAG-DDX6-transduced DM1′ cells were used for FLAG-immunoprecipitation (IP) as described previously (37). Briefly, ∼5 × 106 cells (p10 plates) were harvested per IP 5 days post-transduction by lysis in 1 ml hypotonic lysis buffer (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 2 mM EDTA, 0.1% Triton X-100 and 1 pill Complete (Roche) protease inhibitor per 10 ml lysis buffer). Five minutes later, NaCl was added at a final concentration of 150 mM and lysates were cleared by 20 000 × g centrifugation at 4°C for 10 min. The ‘input’ was collected (10%) from the cleared lysate prior to IP. Each IP was conducted by incubating 20 μl of pre-equilibrated anti-FLAG-M2-Agarose beads (Sigma-Aldrich) with 900 μl cleared lysate at 4°C for 2 h followed by 8 × 1.5 ml washes in NET-2 buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Triton X-100). Protein and bound RNA was eluted by addition of 1 ml Trizol.
Recombinant protein expression
GST-DDX6 or GST-DDX6(DEAA) was expressed in Escherichia coli. BL21Rosetta2 grown in 2 X TY medium until an OD600 0.8 and subsequently induced with 0.1 mM IPTG overnight at 22°C. Purification was performed according to a standard protocol as previously described (41). 3XFLAG-tagged DDX6 or DDX6(DEAA) was expressed in 5 × 107 stable HEK293S cells by a 36 h induction with 500 ng/ml tetracycline. FLAG-tagged protein was immunoprecipitated according to previously described protocols (37) with the following modifications. Captured protein was washed in 400 × bead volumes of NET500 buffer [20 mM Tris-HCl, pH 7.4, 500 mM NaCl, 0.1% Triton X-100, 1 mM EDTA], prior to elution in 100 μl 200 μg/ml 3XFLAG-peptide in NET2 buffer [20 mM Tris-HCl, pH 7.4, 120 mM NaCl, 0.1% Triton X-100, 1 mM DTT] at 10°C overnight with gentle agitation.
In vitro transcription and band shift analysis
A template for T7 polymerase in vitro transcription was generated by standard PCR amplifications using primers containing T7 and SP6 polymerase tags at either end with a cDNA library from DM1 fibroblasts as template. Size-selected PCR products (220–240 nucleotides) were gel-purified and re-amplified and gel-purified using short T7- and SP6-specific primers. In vitro transcription using 32P-UTP, RNA purification and bandshift analyses were essentially performed as described elsewhere (41). Briefly, 5 fmol renatured CUG–RNA was incubated in 10 μl binding reactions containing 10 mM HEPES/KOH, pH 7.4, 100 mM KCl, 2 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, 5% glycerol, 100 ng tRNA, 40 units RiboLock (Fermentas) and either 100 ng (125 nM), 200 ng (250 nM), 800 ng (1 μM) or 1.6 μg (2 μM) GST-DDX6. The reactions were mixed and incubated for 20 min at room temperature and run on a native 6% polyacrylamide gel containing 100 mM Tris/borate, pH 8.3 and 1 mM EDTA (1 × TBE).
Helicase assay
Five picomoles 45-mer 5′-biotinylated CUG-oligo was hybridized to 1 pmol 45-mer CUG-oligo 32P-labeled by T4 polynucleotide kinase by manufacturers protocol. Hybridization was performed in 1 × anneal buffer [10 mM HEPES/KOH, pH 7.4, 1 mM EDTA, 75 mM KCl] by incubating the oligos at 80°C for 4 min and slowly cooled to RT for 15 min. The complex was subsequently captured using 60 μl Dynabeads® MyOne™ Streptavidin C1 and washed twice in 1 ml 1× unwinding buffer [25 mM HEPES/KOH, pH 7.4, 1 mM MgCl2, 100 mM KCl, 1 mM DTT] to remove any unbound radiolabeled CUG-oligo. Captured duplex was divided in to 12 × 20 μl aliquots and beads were incubated with either 5 μl of 3xFLAG-tagged DDX6 (∼50 ng) purified from stable HEK293S cells or 3XFLAG peptide for 10–60 min at 37°C in the presence or absence of 1 mM ATP as indicated. Beads were immobilized on a magnet and supernatant was collected and release of radiolabeled CUG-oligo was monitored by denaturing polyacrylamide gel electrophoresis and exposed to phosphor-Imager.
RESULTS
Cytoplasmic DM1 CUG-foci are not associated with processing bodies
Previous studies using stable cell-lines expressing reporter mRNAs, which contain various numbers of CUG-repeats in their 3′ UTRs, have revealed both nuclear and cytoplasmic CUG-foci when visualized by RNA-FISH (26,42,43). We speculated whether these relatively rare cytoplasmic foci are also present in primary fibroblasts isolated from DM1 patients. To test this, we performed RNA-FISH using either WT fibroblasts containing 12 and 13 CTG repeats in their two respective dmpk alleles or DM1 patient-derived human fibroblasts (DM1) containing 5 and 2250 CTG repeats, respectively (Figure 1 and data not shown). Indeed, in DM1 cells, we observed both distinct cytoplasmic and nuclear foci (0.63 ± 0.10 and 4.14 ± 0.22 foci per cell, respectively), while WT fibroblasts displayed no detectable foci (Figure 1A). The observed nuclear foci represent classical DM1-specific CUG-expanded mRNP aggregates, since they co-localize with MBNL1 protein when assessed by combined RNA-FISH/immunofluorescence (RNA-FISH/IF) (Figure 1B). Interestingly, the relatively rare cytoplasmic foci also co-localized with MBNL1 in all cells, suggesting that the cytoplasmic occurrence of CUG-foci may redirect MBNL1 to the cytoplasmic compartment (Figure 1B, insert labeled ‘C’). To address the generality of this observation we performed RNA-FISH/IF on another DM1 patient-derived fibroblasts (DM1′), which harbors 5 and 3000 CTG repeats in their two dmpk alleles, respectively, and obtained very similar results (Supplementary Figure S1A). We conclude that cytoplasmic CUG-foci are found in primary DM1 fibroblasts and are not limited to cell lines expressing synthetic CTG-repeats in reporter genes. Many regulated mRNAs particularly those with short half-lives accumulate in PBs known to harbor common cellular RNases and RNA-binding modulators (44). To test if the cytoplasmic CUG-expanded DMPK-mRNAs reside in PBs, we performed combined RNA-FISH/IF to co-stain for endogenous PB-factors; DEAD-box helicase DDX6 (Figure 1C), decapping factors hDCP1a, hDCP1b, EDC3 or miRNA factor GW182 (Supplementary Figure S1B). The cytoplasmic CUG-foci did not significantly overlap with PBs, as evidenced by the lack of RNA co-localization with DDX6- (Figure 1C), hDCP1a-, hDCP1b-, GW182- or EDC3-positive foci (Supplementary Figure S1B), suggesting that the distinct cytoplasmic CUG-foci do not co-localize with PBs in DM1 cells. Similar results were obtained using the other DM1 fibroblast cell line (DM1′) (data not shown). Upon longer exposures of DDX6 signal, we consistently observed cells with nuclear signal surrounding CUG-foci (Supplementary Figure S1C). We also did not observe any strong co-localization between DDX6 and MBNL1 in the cytoplasmic compartment of DM1 cells (Supplementary Figure S2). To fully rule out a role of PBs in cytoplasmic CUG-foci homeostasis, we exploited the fact that some mRNAs can be forced to accumulate in PBs by reducing the levels of the 5′-3′ mRNA decay machinery (hDCP2 or XRN1), reviewed in (31), we next manipulated the level of these proteins by RNAi. We predict that if CUG-expanded DMPK-mRNAs do not enter PBs, hDCP2 or hXRN1 knockdown should also not increase the CUG RNA-FISH signal, whereas knockdown of DDX6 would remove any detectable low frequency CUG-mRNA from PBs, since DDX6 is required for PB-integrity in mammalian cells (32,45). Knockdown of either XRN1 or hDCP2 significantly increased both the number and size of PBs as measured by hDCP1a immunofluorescence (Supplementary Figure S3A, compare two upper panels to the lower panel), without changing the cytoplasmic CUG-foci number or intensity (Supplementary Figure S3A). However, DDX6 knockdown decreased the already low abundance of cytoplasmic CUG-foci significantly, which could only very rarely be observed (Supplementary Figure S3B and quantified in C). We conclude that cytoplasmic CUG-foci are robustly detectable at steady-state in primary DM1 fibroblasts and that they are distinct from PBs. Importantly, the rare cytoplasmic CUG-foci become highly reduced upon knockdown of DDX6.
Figure 1.

Cytoplasmic CUG-expanded DMPK-mRNA foci co-localize with MBNL1 outside processing bodies. (A) RNA-FISH using CAG10 fluorescent probes showing both nuclear and cytoplasmic foci within DM1 patient-derived fibroblasts (left panel). White arrows indicate cytoplasmic foci. Right panel shows frequency of cytoplasmic foci (‘n’ indicates number of cells scored for foci; quantified using ImageJ). Scale bar = 10 μm. (B) Combined RNA-FISH/immunofluorescence (FISH/IF) analysis detecting CUG-foci (left panel) and endogenous MBNL1 (middle panel) in DM1 fibroblasts (merged in right panel). Below each panel are inserts with enlarged areas indicated as stippled squares (two nuclear; ‘N’ and one cytoplasmic ‘C’). (C) Combined RNA-FISH/immunofluorescence (FISH/IF) analysis detecting CUG-foci and endogenous DEAD-box helicase DDX6 as a processing body (PB) marker in either DM1 fibroblasts (top three panels) or wild-type fibroblasts (lower three panels). Arrows in top panels indicate cytoplasmic foci (enlarged within insert in the merged panel), which are absent in wild-type cells. Scale bar = 10 μm.
DDX6 depletion increases the number of nuclear CUG-foci in DM1 patient cells
Since DDX6 does not co-localize with cytoplasmic CUG-foci, yet lowers the frequency of cytoplasmic CUG-foci, the helicase could potentially act at an earlier step in the life of the CUG-expanded DMPK-mRNP. To address whether DDX6 may affect nuclear CUG-mRNP homeostasis, we transfected DM1 or WT fibroblasts with siRNAs targeting DDX6 and performed combined RNA-FISH/IF analyses using DDX6 antibodies (Figure 2A). Knockdown of DDX6 was efficient as judged by IF-signal (Figure 2A; upper second panel) and by western blotting (Figure 2C, upper) and highly functional as seen by a significant drop in number of PBs per cell, based on hDCP1a immunostaining (Figure 2C, lower). Interestingly, we found a highly significant (P < 0.001) increase in nuclear CUG-foci frequency in DM1 fibroblasts derived from two different patients (DM1 and DM1′) upon knockdown of DDX6 (foci elevated from 4.4 ± 0.28 and 7.9 ± 0.34 foci per cell respectively, to 7.1 ± 0.48 and 9.7 ± 0.37 per cell, respectively) (Figure 2A and quantified in Figure 2D). To rule out an siRNA ‘off-target’ effect, we repeated the experiment using an siRNA targeting a different region within the DDX6 mRNA. We observed an even more pronounced increase in the number of nuclear CUG-foci upon DDX6 knockdown (nuclear foci were elevated from 4.9 ± 0.30 to 10.6 ± 0.49 foci per nucleus (Figure 2E), likely correlating with the apparently higher efficiency of DDX6 knockdown (Supplementary Figure S4A). To test the specificity of the observed increase we tested DDX6 knockdown in fibroblasts from DM2 patients (myotonic dystrophy type 2) containing CCUG-repeats within intron 1 of the ZNF9 gene (38,46). Interestingly, we observed no significant change in CCUG-foci upon efficient knockdown of DDX6 (Supplementary Figure S4B and quantified in 4C), suggesting that DDX6 might act at a step downstream of pre-mRNA splicing. To further strengthen these observations and to test whether the phenotype could be recapitulated in DM1 muscle cells, we induced myogenesis in DM1′ fibroblasts by stable transduction with a lentiviral vector encoding the muscle-specific transcription factor MyoD as previously described (47) and monitored CUG-foci frequency with or without DDX6 knockdown (Figure 2B). When monitoring only multinuclear cells, which also express the MyoD-induced transcription factor myogenin (Supplementary Figures S5 and 6), we indeed observed a highly significant increase (P < 0.001) in the frequency of nuclear CUG-foci upon DDX6 knockdown (Figure 2B and quantified in Figure 2F). We conclude that DDX6 knockdown significantly and specifically increases the accumulation of CUG-expanded DMPK-mRNA in distinct nuclear foci in both patient-derived DM1 fibroblasts and MyoD-differentiated muscle cells.
Figure 2.

Knockdown of DDX6 leads increases CUG-foci frequency in DM1 cells. (A) Representative pictures of FISH/IF experiments with DM1 fibroblasts using either DDX6 siRNA (DDX6 siRNA; upper panels) or control (control siRNA; lower panels). The two pictures to the right are enlarged areas originating from the indicated outlined boxes. Scale bar = 10 μm. (B) FISH/IF experiments with differentiated multinuclear muscle cells using either DDX6 siRNA (DDX6 siRNA; upper panels) or control (control siRNA; lower panels). The two pictures to the right are enlarged areas originating from the indicated outlined boxes. Scale bar = 10 μm. (C) Upper panel: western blot detecting endogenous DDX6 and a control (HuR) with or without transfected control or DDX6 siRNAs. Lower panel: functional quantification of knockdown efficiency by measuring hDCP1a positive PBs after DDX6 knockdown. Significance was calculated by a two-sided Student's t-test, where ‘***’ denotes P < 0.001). (D) Boxplots showing distribution of CUG-foci frequencies counted in DM1 or DM1′ cells transfected with either control or DDX6 siRNA. The experiments were performed in triplicate and significance was determined by two-sided Student's t-test, where ‘***’ denotes P < 0.001). (E) Boxplots of nuclear foci frequency in DM1 cells using a DDX6 siRNA (DDX6#2) targeting a different region within the mRNA. (F) Boxplots of nuclear foci frequency in MyoD-differentiated DM1′ cells using a DDX6#2 siRNA targeting a different region within the mRNA.
DDX6 expression reduces nuclear CUG-foci in DM1 fibroblasts
To provide further mechanistic evidence for a possible function of DDX6 in CUG-foci homeostasis, we overexpressed DDX6 in either wildtype or DM1 fibroblasts, which normally express similar levels of DDX6 (Supplementary Figure S5A), by stably introducing either a DDX6- or GFP-expression cassette facilitated by lentiviral transduction. This allows for a homogenous and modest overexpression, when compared to transient and very inefficient transfection approaches (Supplementary Figure S5B). If DDX6 plays a role already in the nuclear compartment of DM1 fibroblasts and possibly unwinds or remodels the CUG-expanded DMPK-mRNP, we predict that raising the level of DDX6 would potentially reduce the number of CUG-foci. Indeed, when performing RNA-FISH on DDX6 transduced DM1 fibroblasts, we observed a significant (P < 0.001) reduction in nuclear foci frequency (nuclear foci dropped from 8.3 ± 0.57 in control cells to 5.2 ± 0.24 foci per nucleus in DDX6-transduced cells) (Figure 3A and quantified in Figure 3C). Interestingly, we observed an increased RNA-FISH signal in the cytoplasm of DM1 cells indicating that the partial loss of nuclear CUG-foci could be a consequence of enhanced nuclear release/nuclear export and accumulation in defined cytoplasmic foci (Figure 3A). When quantifying this, we observed a highly significant increase (P < 0.001) in the frequency of cytoplasmic CUG-foci upon DDX6 overexpression (cytoplasmic foci elevated from 2.8 ± 0.35 in GFP-transduced cells to 7.2 ± 0.89 in DDX6 transduced DM1′ cells) (Figure 3C). Notably, the frequency of cytoplasmic foci in DM1′ is reproducibly higher than that of DM1 (Figure 1), which is potentially due to the increased number repeats in the CUG-expansion of DMPK gene in the former. When carefully quantifying DDX6-transduced cells for cytoplasmic CUG-foci by integrating pixel densities using ImageJ software (exemplified in Figure 3B), we observed a significantly higher level of CUG-expanded mRNA compared to control transduced cells. Importantly, we did not observe any significant changes in the expression levels of MBNL1, CUG-BP1, Staufen or DDX5 upon exogenous expression of DDX6 (Supplementary Figure S5B) ruling out an indirect effect of DDX6 potentially regulating at least the level of these known DM1 factors. Taken together with the knockdown experiments, these results demonstrate that DDX6 either directly or indirectly regulates the ability of the CUG-expanded DMPK-mRNA, but not CCUG-expanded ZNF9 pre-mRNA in DM2 cells, to accumulate in nuclear foci, which suggests that high DDX6 levels allows an increased CUG-expanded DMPK-mRNA escape of nuclear retention in DM1 patient fibroblasts.
Figure 3.

DDX6 expression modulates nuclear and cytoplasmic CUG-foci homeostasis. (A) RNA-FISH experiment of DM1′ cells transduced with vectors expressing either FLAG-tagged DDX6 (upper panels) or GFP (lower panels). RNA-FISH was performed 5 days post-transduction and RNA signal was merged with DAPI stain in a single channel with arrows indicating cytoplasmic CUG-foci. Stippled boxes are enlarged in the right panels with arrows marking cytoplasmic foci in the top right panel. Scale bar = 10 μm. (B) Quantification of RNA-FISH signal from the right panels in (A) using identical exposure and scaling (ImageJ). (C) Boxplot showing distribution of CUG-foci within the nuclear (left panel) or cytoplasmic (right panel) compartments. ‘n’ indicates number of cells scored for cytoplasmic and nuclear foci within one representative experiment. Note that DM1′ fibroblasts display a higher average number of cytoplasmic foci than DM1 cells. Significance was determined by two-sided Student's t-test, where ‘***’ denotes P < 0.001.
DDX6 expression partially relieves insulin receptor 2 mis-splicing
Since DDX6 overexpression reduced the nuclear CUG-foci formation, we speculated that this potentially could reduce the reported mis-splicing events, which are hallmarks of DM1. To test this, we utilized RNA purified from either GFP- or DDX6-transduced cells and performed semi-quantitative hot RT-PCR and quantified the level of mis-splicing of mRNAs encoding human IR2, GNAS, PPP2R5C, SPAG9 and NFIX (10). We observed a modest but significant defect in splicing of IR2 (P < 0.01) and ppp2r5c (P < 0.05) pre-mRNA in DM1 fibroblasts compared to untreated WT fibroblasts (Figure 4A and B, compare lanes 5–8 and lanes 3–4, respectively). Most importantly, fibroblasts expressing FLAG-DDX6 allowed for a partial rescue of the IR2 (P < 0.01) and ppp2r5c mis-splicing events (P < 0.05), suggesting that the decrease in nuclear CUG-foci translates into functional relief of DM1 specific mis-splicing, whereas splicing in WT cells remained unaffected. The defective IR2 splicing phenotype in DM1 cells and the restoration to ‘near-normal’ splicing pattern by DDX6 overexpression was confirmed by qRT-PCR, by which we quantified the individual transcripts either containing or lacking alternative exon 11 (Supplementary Figures S7A and B). Deregulation of ppp2r5c alternative splicing in DM1 cells was modest but similar to previously published phenotypes (10). Alternative splicing of pre-mRNAs encoding GNAS, SPAG9 or NFIX was not significantly changed between WT and DM1 fibroblasts (unpublished observations) in contrast to previous reports using DM1 muscle biopsies (10). This was not an effect of non-quantitative PCR conditions as evidenced by the linear range of amplification observed for the IR2 (Figure 4A lanes 1–4) and by qRT-PCR (Supplementary Figure S7). This is consistent with previous reports, stating that DMPK-mRNA expression in undifferentiated fibroblasts is much lower than that of differentiated muscle cells (21), which suggests that robust mis-splicing is evident only in cells of higher CUG-mRNA levels. Although we cannot formally exclude the possibility that higher DDX6 expression may change the mRNA stability of specific IR2/pp2r5c splice-variants, our data indicates that DDX6 overexpression partially relieves DM1 specific mis-splicing of IR2 pre-mRNA in human DM1 fibroblasts.
Figure 4.
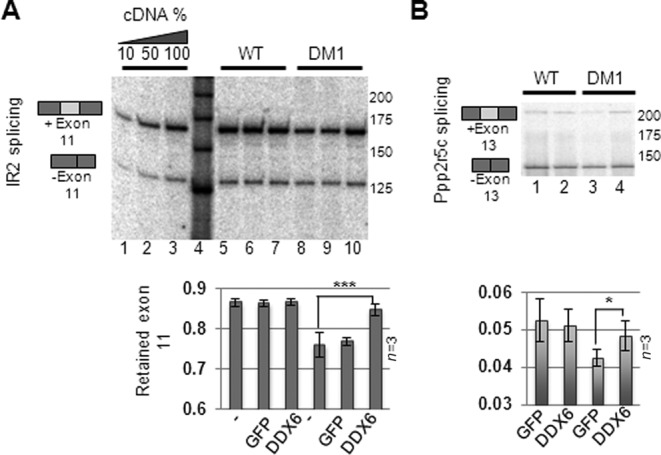
DDX6 expression partially rescues DM1-specific mis-splicing. (A) Total RNA from FLAG-DDX6- or GFP-expressing cells was isolated and subjected to semi-quantitative RT-PCR amplifying Insulin Receptor 2 (IR2) amplicons either lacking (bottom bands) or retaining exon 11 (top bands) and these were quantified from three independent experiments (quantified in lower panel). (B) Same as (A) but for ppp2r5c cDNA. Error bars indicate standard deviation from triplicate experiments. Significance was determined by two-sided Student's t-test, where ‘*’ denotes P < 0.05 and ‘***’ denotes P < 0.001.
DDX6 interacts with CUG-expanded DMPK-mRNA and binds to CUG-repeats in vitro
Given the fact that DDX6 regulates nuclear CUG-foci homeostasis and also DM1-specific alternative splicing events, one prediction is that the helicase activity of DDX6 is able to directly bind, unwind and change the protein composition of the CUG-mRNP, which might allow for nuclear release and increased export to the cytoplasm. Alternatively, DDX6 could regulate the expression of other key factors involved in DM1 pathogenesis. The finding that we do not observe a robust co-localization of endogenous DDX6 with nuclear (unless DDX6 signal is highly exposed, Figure 1C) or cytoplasmic CUG-foci, suggests that a possible interaction within the nucleus must be very transient. To test if DDX6 can be detected in an mRNP complex with CUG-expanded DMPK-mRNA, we performed FLAG-specific immunoprecipitations of lentivirally delivered and stably expressed FLAG-DDX6 in either WT or DM1 cells, followed by RT-PCR using a DMPK-specific amplicon to measure the co-precipitated DMPK-mRNA (GAPDH mRNA was used as control). DDX6 purified from WT cells specifically pulled-down WT DMPK-mRNA, whereas the expressed GFP control-protein did not (Figure 5A, compare lane 10 and 12; quantified in Figure 5B). When extracting DDX6 from DM1 cells we observed a strong and highly significant enrichment of DMPK-mRNA (>4-fold; P < 0.001) in the DDX6 pull-down (Figure 5A, compare lanes 12 and 13; quantified in Figure 5B), even though DMPK expression levels are similar to those in the WT cells (Figure 5A, compare lanes 7 and 9), which strongly indicates that the enhanced association is dependent on expanded CUG-repeats. Western blots of protein fractions demonstrate an equal expression (input) and IP-efficiency of FLAG-DDX6 in both cell lines (Figure 5C). To investigate the possibility that the intrinsic binding properties of DDX6 could be changed between WT and DM1 cells, i.e. by post-translational modification events specific to either cell type, we quantitatively assessed the association with GAPDH mRNA by qRT-PCR and found it to be associated as efficiently as WT DMPK-mRNA (Figure 5B, light gray bars). The rather efficient association of DDX6 with GAPDH mRNA in lysates from both WT and DM1 cells (Figure 5B), indicates that the helicase likely interacts with numerous mRNAs, and that the intrinsic binding activity of DDX6 remains similar in the two cell lines. To test whether DDX6 interacts directly with CUG-repeat RNAs, we performed a band shift assay using recombinant GST-DDX6 expressed in E. coli. and a radiolabeled 200-nucleotide in vitro-transcribed CUG-repeat RNA as a substrate (Figure 5D). Indeed GST-DDX6 binds the CUG-repeat RNA directly in a concentration dependent manner (apparent kD < 250 nM) (Figure 5D, left panel, lanes 1–4). To test whether the helicase activity of DDX6 is required for CUG–RNA binding we introduced a mutation in the DEAD-box motif (D249A or DEAA mutant), which reportedly affects ATP hydrolysis in the yeast DDX6 homologue (Dhh1) (48). As seen from Figure 5D, left panel, lanes 5–7, the DEAA mutant retained its RNA-binding capability and radioactive complexes formed by both WT and DEAA mutant could efficiently be competed by an excess of non-labeled CUG–RNA (Figure 5D, right panel, lanes 1–4). Interestingly, although DEAD-box helicases are thought to interact rather non-specifically with RNA via phosphate backbone interactions (49), we did observe significant specificity for CUG-repeats as compared to bacterial ribosomal RNA (Supplementary Figure S8). Taken together, these results strongly indicate that the functional impact of manipulating the DDX6 levels in DM1 fibroblasts (CUG-foci homeostasis and mis-splicing events) is mediated through a direct interaction of DDX6 with DMPK CUG-repeats.
Figure 5.

DDX6 interacts with CUG-expanded DMPK-mRNA in vivo and in vitro. (A) RNA immunoprecipitation assay followed by semi-qRT-PCR analysis of FLAG-DDX6 immunoprecipitates reveals a strong binding preference of DDX6 for CUG-expanded DMPK-mRNA. Lanes 1–4 represents cDNA dilutions showing that PCR is conducted within the linear area of amplification. Lane 5 is DNA size marker. Lanes 6–9 show products of PCR reactions performed on the input cDNA before immunoprecipitation, which both demonstrates comparable loading and that DMPK-mRNA levels are virtually unaffected by either GFP (lane 6–7) or FLAG-DDX6 (lane 8–9) expression in both WT or DM1 fibroblasts. Lanes 10–13 shows the products from immunoprecipitated DMPK-mRNA for GFP negative control (lanes 10–11) and DDX6 (lanes 12–13). (B) Experiment was performed in triplicate and significance was determined by two-sided Student's t-test, where ‘***’ denotes P < 0.001). DDX6 IP-efficiency of GAPDH mRNA was included as a control and was quantified qRT-PCR, revealing that DDX6 precipitates GAPDH mRNA to similar levels in both WT and DM1 fibroblasts. (C) Western blots showing expression profile (INPUT—lanes 1 and 2) and IP-efficiency of FLAG-tagged DDX6 (lanes 3 and 4) used in the RIP experiment, demonstrating similar expression and IP-profiles between WT and DM1 cells. (D) Left panel: Band shift analysis using recombinant GST-DDX6 and CUG-200 RNA in binding reactions with increasing amounts of either DDX6 or DDX6(DEAA) mutant. Right panel: Competition experiment using high concentration of DDX6 or DDX6(DEAA) and cold CUG–RNA at 20-fold (+) or 500-fold (++) excess compared to radiolabeled CUG–RNA.
MBNL1 relocalizes from nuclear foci upon DDX6 overexpression
Given the fact that MBNL1 levels were not significantly affected by manipulating DDX6 levels as judged from whole cell lysates subjected to western blotting (Supplementary Figure S5B), we speculated whether MBNL1 localization might be affected. To test this we first immuno-stained MBNL1 and DDX6 in DM1 cells, which had been transfected with either control- or DDX6-siRNA (Figure 6A). While cells transfected with control siRNA displayed several faint nuclear foci and only a few highly intense foci (likely corresponding to the number of CUG-mRNA foci), DDX6 knockdown significantly increased the number of highly intense foci (ImageJ-applied threshold and counting) (Figure 6B; quantified in Figure 6C). Taken together with the demonstrated increase in the frequency of nuclear CUG-mRNA foci (Figure 2), we conclude that DDX6 knockdown also augments MBNL1 sequestration in these nuclear domains. Next, we tested whether DDX6 overexpression, which increases the frequency of CUG-mRNA foci in the cytoplasmic compartment (Figure 3), also perturbs normal MBNL1 localization (Figure 6D and E. Indeed, MBNL1 staining was significantly altered to a more diffuse staining in most cells (>50%) when overexpressing DDX6 (Figure 6D; middle three panels) in DM1 cells, whereas GFP-transduced cells did not alter localization significantly (Figure 6D; upper three panels—integrated signal density in the displayed nuclei from Figure 6D are quantified in Figure 6E). Importantly, DDX6 overexpression did not lead to a non-specific re-localization of MBNL1 in WT cells (Figure 6D, lower panel), lending support to a CUG-specific phenotype. We conclude that manipulation of DDX6 levels alters not only nuclear and cytoplasmic CUG-foci but also the sub-cellular localization of MBNL1.
Figure 6.

MBNL1 re-localizes upon manipulation of DDX6 levels. (A) Co-immunofluorescence analysis using MBNL1 (left) and DDX6 (middle) antibodies reveals increased focal accumulation of MBNL1 in the nucleus upon DDX6 knockdown (compare top panels to lower panels). (B) Integrated pixel densities of the nuclei displayed in (A) (ImageJ). Insert next to Z-axis indicate quantified area. (C) Boxplot showing distribution of integrated intensities from three cells randomly chosen with efficient knockdown (low DDX6 signal). D) Co-immunofluorescence analysis using MBNL1 (left) and DDX6 (middle panel) antibodies reveals decreased focal accumulation of MBNL1 in the nucleus upon DDX6 overexpression. (E) Integrated pixel densities of the nuclei displayed in (D) (ImageJ). Insert next to Z-axis indicate quantified area.
Exogenous DDX6 co-localizes with CUG-repeat mRNA and unwinds CUG–CUG duplexes in vitro
Since the partial relief of mis-splicing observed with IR2 and ppp2r5c pre-mRNA substrates and the DDX6/CUG-repeat interaction is observed upon DDX6 overexpression, we speculated whether this forces a DDX6-CUG-repeat interaction, which can be visualized as DDX6 co-localization with CUG-expanded mRNA both in the nuclear and cytoplasmic compartment of the cell, where we observed a significant CUG-foci accumulation. To test this we performed combined RNA-FISH-immunofluorescence experiments on DDX6- or GFP-transduced WT or DM1 fibroblasts (Figure 7A). As predicted, we observed a significant proportion of DDX6 protein overlapping with nuclear CUG-foci signal suggesting co-localization (Figure 7A, left panel and merged insert). In addition, we observed numerous cytoplasmic DDX6-foci, which upon longer exposure of the CUG-specific RNA-FISH signal co-localized in a high number of cells (>50%) (Figure 6A, right panel). Next, we tested whether the helicase activity of DDX6 is necessary for the reduction in nuclear CUG-foci by expressing the previously used DEAA mutant in DM1 cells. Counting nuclear CUG-foci after RNA-FISH analysis revealed that the DEAA-mutant failed to efficiently reduce nuclear CUG-foci frequency in contrast to wild-type DDX6 upon transient transfection of GFP-DDX6 expression plasmids (Figure 7B and Supplementary Figure S9A for DDX6 levels). To decide whether DDX6 may actively participate in CUG-mRNP remodeling of sequestered CUG-foci, we devised a biotin/streptavidin-based helicase assay (Figure 7C) to monitor the activity of 3XFLAG-tagged DDX6 purified from human embryonic kidney cells. By incubating equal amounts of purified WT DDX6 or DEAA mutant with a pre-annealed, Dynabead-tethered CUG-duplex, we observed specific release of the radioactively labeled CUG-strand by WT DDX6, an activity that was much less efficient when using equal amounts of the DEAA mutant (Figure 7D, compare lanes 5–8 with lanes 9–12 and Supplementary Figure S9B and C for DDX6 levels). This was confirmed using three independent preparations of FLAG-tagged DDX6 and the results are quantified in Figure 7E. Importantly, the unwinding is dependent on the presence of biotinylated CUG-oligo (lane 2), DDX6 (lane 3) and ATP (Figure 7D). Taken together, we conclude that DDX6 can localize to focal locales of CUG-mRNPs in both the nucleus and cytoplasmic compartment of DM1 fibroblasts upon overexpression and that an intact DEAD-box motif is required for the observed CUG-foci remodeling by ATP-dependent unwinding and perhaps even displacement of MBNL1.
Figure 7.

CUG-foci release to the cytoplasm correlates with co-localization of overexpressed DDX6 and CUG-expanded DMPK-mRNA in DM1 cells. (A) Representative FISH/IF analysis of DDX6 and CUG-expanded DMPK-mRNA in transduced (FLAG-DDX6) DM1 cells with ‘normal’ exposure of the FISH signal (left panel) and ‘longer’ exposure of FISH signal (right panel). Note that nuclear FISH signal is saturated. (B) Boxplot showing quantification of nuclear foci between GFP-DDX6 WT and GFP-DDX6 DEAA expressing cells. Significance was tested using Student's t-test where ‘***’ denotes P < 0.001. (C) RNA helicase assay. A biotinylated 45-mer CUG-oligo was annealed to a radioactively labeled 45-mer CUG-oligo prior to capture on Dynabeads. Immunopurified 3XFLAG-DDX6 from HEK293S cells was incubated with the RNA-duplex and unwinding and release of the radiolabeled strand from dynabeads was monitored. (D) RNA helicase assay. Release of radiolabeled CUG-strand was monitored over time by denaturing acrylamide PAGE. Lane 1: 25% of input RNA. Lane 2: no biotinylated CUG-oligo was added to the annealing reaction prior to capture. Lane 3: no DDX6 was added to the reaction. Lane 4: no ATP was added to reaction. Lanes 5–8: timecourse experiment incubating wild type DDX6 with RNA duplex for 0 min, 10 min, 25 min and 60 min. Lanes 9–12: timecourse experiment incubating DDX6(DEAA) with RNA duplex for 0 min, 10 min, 25 min and 60 min. (E) Quantification of helicase assay using three independent preparations of DDX6 [error bars represent standard deviation (n = 3)].
DISCUSSION
Despite major advances in understanding the role of CUG-expanded DMPK-mRNA and massive mis-splicing as a consequence of MBNL1 sequestration in affected cells, DM1 pathogenesis has remained unclear for decades. We have assigned a novel function to a classical DEAD-box helicase, DDX6 with known cytoplasmic localization and well-described functions from yeast to man in regulated mRNA decay, translation and as an integral PB-factor necessary for miRNA-mediated silencing (31–34,50–52). We have shown that manipulation of DDX6 levels in primary fibroblasts and MyoD-induced muscle cells from two different DM1 patients regulates nuclear and cytoplasmic CUG-foci homeostasis, along with a partial rescue of mis-splicing events specific to DM1 upon DDX6 overexpression. In DM1 cells, CUG-expanded DMPK-mRNA remains highly stable and most is retained in CUG-foci (21), reportedly in the periphery of para-speckles (53), suggesting that the CUG-expansions, with the potential to form an extended hairpin structure, stalls the mRNA in a repressed state that may require helicase/PnPase activity for release and further processing/nuclear export. In line with this, we observed a rare but consistent immunostaining of DDX6 in the periphery of nuclear CUG-foci, suggesting that a fraction of the cellular DDX6 may be ‘moonlighting’ in the nucleus. The finding that not all DM1 cells exhibit a pronounced nuclear DDX6-staining in the periphery of the CUG-foci, indicates that an interaction between DDX6 and CUG-expanded DMPK-mRNA is likely very transient. Importantly however, co-localization was strongly augmented upon overexpression of DDX6 with a concurrent significant reduction in the mean number of nuclear CUG-foci per cell. Consistent with a nuclear role of DDX6, the Xenopus homologue (Xp54), which is a known nucleo-cytoplasmic shuttling protein that contains conserved nuclear localization and nuclear export signals, exerts important nuclear functions in early stages of oocyte development, where it is an integral part of maternally deposited mRNPs (52,54). Although Xp54 at later stages turns predominantly cytoplasmic, similar to our observations in WT and DM1 fibroblasts, increased transcription in Xenopus oocytes leads to accumulation of Xp54 at active transcription sites indicating that it is able to interact, perhaps transiently, with nascent transcripts and facilitate their downstream processing (52). Interestingly, the cytoplasmic occurrence of CUG-expanded DMPK-mRNA upon DDX6 overexpression suggests a release of nuclear CUG-foci and enhanced export to the cytoplasm. The importance of the helicase activity in this process is supported by expression of a DEAD-box mutant, which fails to decrease the frequency of nuclear CUG-foci in contrast to its WT counterpart and the finding that only an intact DEAD-box motif is able to unwind CUG-duplexes in an ATP-dependent manner in vitro. These phenotypes likely reflect direct effects since DDX6 strongly associates with CUG-expanded DMPK-mRNA in RNA-immunoprecipitations and binds CUG-repeat RNA directly and with rather high affinity in vitro. This suggests that CUG-foci homeostasis may indeed be directly linked to DDX6-dependent unwinding/remodeling of the CUG-hairpin and perhaps modulation of associated proteins like MBNL1. In line with this, we observed a significant re-distribution of MBNL1 upon manipulation of DDX6 (knockdown or overexpression). Importantly, we did not observe any significant differences in the overall expression levels of MBNL1, CUG-BP1, Staufen or DDX5 during overexpression of DDX6, arguing against an indirect effect of DDX6 on the levels of these important DM1 factors. The modulation of CUG-foci by DDX6 was not observed in DM2 cells containing thousands of similar CCUG-repeats within the intron of ZNF9 pre-mRNA, which argues for a highly DM1-specific process. Aside from the single nucleotide difference between the DM1 and DM2 repeats, one plausible explanation for this phenotypic discrepancy, could be that DDX6 acts on CUG-expanded DM1 repeats as a post-splicing event, and DDX6 will therefore not encounter DM2 repeats before they are excised from the ZNF9 pre-mRNA, which likely occurs co-transcriptionally. In addition, the CCUG-foci were previously shown not to localize to the periphery of para-speckles (53), as opposed to the CUG-foci, indicating diverse fates of these toxic RNAs. Another DEAD-box helicase, DDX5, was recently shown to interact with CUG-repeats in vitro and furthermore to be able to modulate the general RNA-binding activity of MBNL1 in vitro (27). In marked contrast to DDX6, knockdown of DDX5 decreases nuclear CUG-foci formation when using artificial CUG-expanded reporters in HeLa cells (27). In vitro binding assays furthermore suggest that DDX5 facilitates MBNL1 RNA binding through transient unwinding of C-G basepairs within a CUG-hairpin structure (27). Since DDX5 interacts with MBNL1 in an RNA independent manner (13), one possibility is that DDX5 functions as a MBNL1 co-factor and affects its binding capacity toward multiple pre-mRNA substrates aside from CUG-expanded repeats (27). Conversely, our data suggest that DDX6 may compete with or even remove MBNL1 from CUG-repeats. Taken together, these results strongly implicate DEAD-box helicases as intimate regulators of CUG-expanded foci homeostasis and in turn the mis-splicing events occurring in DM1. Staufen 1 (Stau1) was recently demonstrated to be upregulated in muscle tissue from a DM1 mouse model and DM1 patients (28), which is consistent with our observations with MyoD-induced myogenesis. Interestingly, Stau1 overexpression shares some of the phenotypes of DDX6 overexpression, including CUG-repeat binding and rescue of DM1 mis-regulated splicing (28). On the contrary, increasing Stau1 levels does not robustly recruit the protein to nuclear CUG-foci nor does it affect CUG-foci frequency and MBNL1 distribution in reporter-expressing cells (28). One possibility is that DDX6 acts upstream of Stau1 in a synergistic fashion to remodel MBNL1-containing CUG-foci, which allows for their release and subsequent enhanced nuclear export and translation facilitated by Stau1 (28).
At normal steady state levels of DDX6 in DM1 fibroblasts, we did not observe co-localization between rare cytoplasmic CUG-expanded DMPK-mRNA and any of the tested PB factors (hDCP1a, hDCP1b, hEDC3, GW182 or DDX6). Furthermore, these factors were not found to strongly accumulate in visible PBs in WT cells as observed in a number of mammalian cancer cell lines including HeLa, HEK293, U2OS, COS-7 and N1E115 (unpublished observations), suggesting that these primary fibroblasts display important differences in PB-formation and homeostasis. Since we were unable to force CUG-repeat mRNA into enlarged PBs by reducing the levels of either hDCP2 or XRN1, we conclude that the function of DDX6 in CUG-expanded DMPK-mRNA homeostasis is independent of its functions related to PBs. In conclusion, our data is consistent with a model in which DDX6 is able to remodel CUG-expanded repeats leading to their nuclear dissemination, a re-distribution of MBNL1 with a concurrent accumulation of the CUG-expanded DMPK-mRNA on the cell cytoplasm. Whether or not increased DDX6 expression leads to increased translation and expression of the DMPK kinase remains to be resolved.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are thankful to Professor Benedikt G. Schoser and the Muscle Tissue Culture Collection (MTCC) Friedrich-Baur Institute, Ludwig-Maximilians University, Munich, Germany for generously providing DM2 cells and Professor Jens-Lykke Andersen, University of California San Diego, USA for supplying antibodies for PB-factors. Inger Juncker and Anni Aggerholm, Department of Clinical Genetics, Aarhus University Hospital, are thanked for CTG repeat measurements in the WT, DM1 and DM1′ cell lines. Birgit Holm Hansen is thanked for technical assistance.
FUNDING
The Karen Elise Jensen Foundation; the Danish Council for Independent Research|Natural Sciences (FNU) (09-064917) and the Lundbeck Foundation (R19-A2301) [to C.K.D.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Lee J.E., Cooper T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem.Soc. Trans. 2009;37:1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner C., Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J. Neurol. Neurosurg. Psychiatry. 2010;81:358–367. doi: 10.1136/jnnp.2008.158261. [DOI] [PubMed] [Google Scholar]
- 3.Udd B., Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- 4.Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T., et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 5.Botta A., Rinaldi F., Catalli C., Vergani L., Bonifazi E., Romeo V., Loro E., Viola A., Angelini C., Novelli G. The CTG repeat expansion size correlates with the splicing defects observed in muscles from myotonic dystrophy type 1 patients. J. Med. Genet. 2008;45:639–646. doi: 10.1136/jmg.2008.058909. [DOI] [PubMed] [Google Scholar]
- 6.Berul C.I., Maguire C.T., Aronovitz M.J., Greenwood J., Miller C., Gehrmann J., Housman D., Mendelsohn M.E., Reddy S. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Invest. 1999;103:R1–R7. doi: 10.1172/JCI5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jansen G., Groenen P.J., Bachner D., Jap P.H., Coerwinkel M., Oerlemans F., van den Broek W., Gohlsch B., Pette D., Plomp J.J., et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 1996;13:316–324. doi: 10.1038/ng0796-316. [DOI] [PubMed] [Google Scholar]
- 8.Reddy S., Smith D.B., Rich M.M., Leferovich J.M., Reilly P., Davis B.M., Tran K., Rayburn H., Bronson R., Cros D., et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996;13:325–335. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- 9.Dansithong W., Paul S., Comai L., Reddy S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J. Biol. Chem. 2005;280:5773–5780. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 10.Du H., Cline M.S., Osborne R.J., Tuttle D.L., Clark T.A., Donohue J.P., Hall M.P., Shiue L., Swanson M.S., Thornton C.A., et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ladd A.N., Charlet N., Cooper T.A. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol. 2001;21:1285–1296. doi: 10.1128/MCB.21.4.1285-1296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S., Thornton C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 13.Paul S., Dansithong W., Jog S.P., Holt I., Mittal S., Brook J.D., Morris G.E., Comai L., Reddy S. Expanded CUG repeats Dysregulate RNA splicing by altering the stoichiometry of the muscleblind 1 complex. J. Biol. Chem. 2011;286:38427–38438. doi: 10.1074/jbc.M111.255224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Philips A.V., Timchenko L.T., Cooper T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 15.Savkur R.S., Philips A.V., Cooper T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 16.Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S., et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones K., Jin B., Iakova P., Huichalaf C., Sarkar P., Schneider-Gold C., Schoser B., Meola G., Shyu A.B., Timchenko N., et al. RNA Foci, CUGBP1, and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Am. J. Pathol. 2011;179:2475–2489. doi: 10.1016/j.ajpath.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuyumcu-Martinez N.M., Wang G.S., Cooper T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahadevan M.S., Yadava R.S., Yu Q., Balijepalli S., Frenzel-McCardell C.D., Bourne T.D., Phillips L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006;38:1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward A.J., Rimer M., Killian J.M., Dowling J.J., Cooper T.A. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 2010;19:3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis B.M., McCurrach M.E., Taneja K.L., Singer R.H., Housman D.E. Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fardaei M., Larkin K., Brook J.D., Hamshere M.G. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001;29:2766–2771. doi: 10.1093/nar/29.13.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fardaei M., Rogers M.T., Thorpe H.M., Larkin K., Hamshere M.G., Harper P.S., Brook J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 24.Taneja K.L. Localization of trinucleotide repeat sequences in myotonic dystrophy cells using a single fluorochrome-labeled PNA probe. Biotechniques. 1998;24:472–476. doi: 10.2144/98243rr02. [DOI] [PubMed] [Google Scholar]
- 25.Taneja K.L., McCurrach M., Schalling M., Housman D., Singer R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dansithong W., Wolf C.M., Sarkar P., Paul S., Chiang A., Holt I., Morris G.E., Branco D., Sherwood M.C., Comai L., et al. Cytoplasmic CUG RNA foci are insufficient to elicit key DM1 features. PloS One. 2008;3 doi: 10.1371/journal.pone.0003968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laurent F.X., Sureau A., Klein A.F., Trouslard F., Gasnier E., Furling D., Marie J. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic acids research. 2012;40:3159–3171. doi: 10.1093/nar/gkr1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ravel-Chapuis A., Belanger G., Yadava R.S., Mahadevan M.S., DesGroseillers L., Cote J., Jasmin B.J. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J. Cell Biol. 2012;196:699–712. doi: 10.1083/jcb.201108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell R., Jarmoskaite I., Lambowitz A.M. Toward a molecular understanding of RNA remodeling by DEAD-box proteins. RNA Biol. 2013;10:44–55. doi: 10.4161/rna.22210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weston A., Sommerville J. Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res. 2006;34:3082–3094. doi: 10.1093/nar/gkl409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franks T.M., Lykke-Andersen J. The control of mRNA decapping and P-body formation. Mol. Cell. 2008;32:605–615. doi: 10.1016/j.molcel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minshall N., Kress M., Weil D., Standart N. Role of p54 RNA helicase activity and its C-terminal domain in translational repression, P-body localization and assembly. Mol. Biol. Cell. 2009;20:2464–2472. doi: 10.1091/mbc.E09-01-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu C.Y., Rana T.M. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4:e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fenger-Gron M., Fillman C., Norrild B., Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol. Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 35.Kedersha N., Anderson P. Regulation of translation by stress granules and processing bodies. Prog. Mol. Biol. Transl. Sci. 2009;90:155–185. doi: 10.1016/S1877-1173(09)90004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raoul C., Abbas-Terki T., Bensadoun J.C., Guillot S., Haase G., Szulc J., Henderson C.E., Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 37.Damgaard C.K., Lykke-Andersen J. Translational coregulation of 5’TOP mRNAs by TIA-1 and TIAR. Genes Dev. 2011;25:2057–2068. doi: 10.1101/gad.17355911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Margolis J.M., Schoser B.G., Moseley M.L., Day J.W., Ranum L.P. DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum. Mol. Genet. 2006;15:1808–1815. doi: 10.1093/hmg/ddl103. [DOI] [PubMed] [Google Scholar]
- 39.Damgaard C.K., Lykke-Andersen S., Kjems J. Stable Cell Lines with Splicing Reporters. In: Stamm S., Smith C. W. J., Lührmann R., editors. Alternative pre-mRNA Splicing: Theory and Protocols. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2012. pp. 408–414. [Google Scholar]
- 40.Ivanov P., Kedersha N., Anderson P. Stress puts TIA on TOP. Genes Dev. 2011;25:2119–2124. doi: 10.1101/gad.17838411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Damgaard C.K., Tange T.O., Kjems J. hnRNP A1 controls HIV-1 mRNA splicing through cooperative binding to intron and exon splicing silencers in the context of a conserved secondary structure. RNA. 2002;8:1401–1415. doi: 10.1017/s1355838202023075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amack J.D., Mahadevan M.S. The myotonic dystrophy expanded CUG repeat tract is necessary but not sufficient to disrupt C2C12 myoblast differentiation. Hum. Mol. Genet. 2001;10:1879–1887. doi: 10.1093/hmg/10.18.1879. [DOI] [PubMed] [Google Scholar]
- 43.Dansithong W., Jog S.P., Paul S., Mohammadzadeh R., Tring S., Kwok Y., Fry R.C., Marjoram P., Comai L., Reddy S. RNA steady-state defects in myotonic dystrophy are linked to nuclear exclusion of SHARP. EMBO Rep. 2011;12:735–742. doi: 10.1038/embor.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franks T.M., Lykke-Andersen J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 2007;21:719–735. doi: 10.1101/gad.1494707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrei M.A., Ingelfinger D., Heintzmann R., Achsel T., Rivera-Pomar R., Luhrmann R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA. 2005;11:717–727. doi: 10.1261/rna.2340405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liquori C.L., Ricker K., Moseley M.L., Jacobsen J.F., Kress W., Naylor S.L., Day J.W., Ranum L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 47.Larsen J., Pettersson O.J., Jakobsen M., Thomsen R., Pedersen C.B., Hertz J.M., Gregersen N., Corydon T.J., Jensen T.G. Myoblasts generated by lentiviral mediated MyoD transduction of myotonic dystrophy type 1 (DM1) fibroblasts can be used for assays of therapeutic molecules. BMC Res. Notes. 2011;4:490–498. doi: 10.1186/1756-0500-4-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dutta A., Zheng S., Jain D., Cameron C.E., Reese J.C. Intermolecular interactions within the abundant DEAD-box protein Dhh1 regulate its activity in vivo. J. Biol. Chem. 2011;286:27454–27470. doi: 10.1074/jbc.M111.220251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mallam A.L., Del Campo M., Gilman B., Sidote D.J., Lambowitz A.M. Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p. Nature. 2012;490:121–125. doi: 10.1038/nature11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coller J., Parker R. General translational repression by activators of mRNA decapping. Cell. 2005;122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coller J.M., Tucker M., Sheth U., Valencia-Sanchez M.A., Parker R. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA. 2001;7:1717–1727. doi: 10.1017/s135583820101994x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smillie D.A., Sommerville J. RNA helicase p54 (DDX6) is a shuttling protein involved in nuclear assembly of stored mRNP particles. J. Cell Sci. 2002;115:395–407. doi: 10.1242/jcs.115.2.395. [DOI] [PubMed] [Google Scholar]
- 53.Holt I., Mittal S., Furling D., Butler-Browne G.S., Brook J.D., Morris G.E. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cell. 2007;12:1035–1048. doi: 10.1111/j.1365-2443.2007.01112.x. [DOI] [PubMed] [Google Scholar]
- 54.Minshall N., Thom G., Standart N. A conserved role of a DEAD box helicase in mRNA masking. RNA. 2001;7:1728–1742. doi: 10.1017/s135583820101158x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.