

Abstract
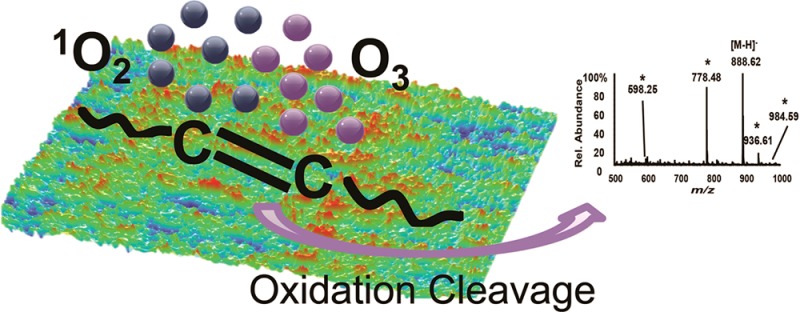
A simple, fast approach elucidated carbon–carbon double bond positions in unsaturated lipids. Lipids were deposited onto various surfaces and the products from their oxidation in ambient air were observed by electrospray ionization (ESI) mass spectrometry (MS). The most common oxidative products, aldehydes, were detected as transformations at the cleaved double bond positions. Ozonides and carboxylic acids were generated in certain lipids. Investigations of the conditions controlling the appearance of these products indicated that the surface oxidation depends on light and ambient air. Since the lipid oxidation was slower in a high concentration of ozone, singlet oxygen appeared to be a parallel oxidant for unsaturated lipids. The 3-hydroxyl group in the sphingoid base of sulfatides offered some protection from oxidation for the Δ4,5-double bond, slowing its oxidation rate relative to that of the isolated double bond in the N-linked fatty acyl chain. Direct sampling by thin-layer chromatography (TLC)-ESI-MS provides a powerful approach to elucidate detailed structural information on biological samples. Co-localization of the starting lipids and their oxidation products after TLC separation allowed assignment of the native unsaturation sites. Phosphatidylserine and N,N-dimethyl phosphatidylethanolamine isomers in a bovine brain total lipid extract were distinguished on the basis of their oxidation products. Meanwhile, the findings reported herein reveal a potential pitfall in the assignment of structures to lipids extracted from TLC plates because of artifactual oxidation after the plate development.
Lipids comprise a very diverse and complex group of molecules that are insoluble in water and soluble in organic solvents. The main biological functions of lipids include their central roles as reservoirs for energy storage, as structural components of cell membranes, and as important signaling molecules.1,2 The complex structural heterogeneity within lipid categories originates from variations in the numbers and lengths of fatty acyl chains, degrees of unsaturation, double bond positions, head groups, etc.
Lipid oxidation can be produced by a free radical chain reaction that is initiated either by removing an electron from an alkyl moiety or oxygen, or by changing the electron spin of the oxygen.3,4 Reactive oxygen species can be produced in the ambient environment. Singlet oxygen and ozone are the most well-known nonradical oxygen species. Singlet oxygen can be generated through various means and reacts readily with lipid double bonds. A very common, important route for generation of singlet oxygen is exposure of oxygen (air) to light in the presence of a photosensitizer.5 Metals can react with O2 and initiate lipid oxidation via formation of a metal–hydroperoxide catalyst complex.6 Irradiation of nontransition-metal oxide powders (e.g., silica gel, aluminum oxide, and magnesium oxide), in the presence of oxygen, results in the formation of singlet oxygen.7 Wynalda and Murphy have reported that low concentrations of environmental ozone react with phospholipids in lung surfactant.4 Ozonoloysis at unsaturated sites in lipids has been attributed to O3 at ambient levels (∼30–40 ppb).8 Unsaturated lipids can produce ozonides at carbon–carbon double bond positions by the Criegee mechanism.9 Therefore, lipid oxidation often compromises the interpretation and identification of lipid structures.
Since lipid structural details strongly affect their biological activities, precise determination of unsaturation sites is essential. For the identification, structural determination, and quantitation of lipids, different mass spectrometry (MS) ionization techniques are used: electron ionization (EI),10 fast atom bombardment (FAB),11 electrospray ionization (ESI),12 matrix-assisted laser desorption/ionization (MALDI),13 and desorption electrospray ionization (DESI).14 Recently, Mitchell et al. have reviewed MS methods to determine the positions of double bonds in lipids.15 Several techniques, for example, collision-induced dissociation (CID) tandem MS, covalent adduct chemical ionization, CID MS/MS of diols and ozonides generated by double bond oxidation, ozone electrospray ionization (ESI), and ozone-induced dissociation (OzID), provide such information. However, these techniques usually require multistep sample preparations (hydrolysis and derivatization), instrumental modification, or special equipment, and have limitations, in that most of these methods cannot assign the double bond positions with accuracy and efficiency. It has been reported that the location of the double bonds on the unsaturated fatty acyl groups can be assigned by a linear ion-trap multiple-stage mass spectrometric approach on the lithiated adduct of the unsaturated lipids. MS2 was sufficient for localization of the double bond in unsaturated fatty acids;16 however, MSn (n > 2) was required for the more complicated lipid classes, such as phospholipids,17 and triacylglycerols.18
The goal of the research reported herein is to provide a universal method to detect double bond positions in unsaturated lipids. This contribution describes a simple, new approach, surface oxidation under ambient air; the method provides an efficient, high throughput, convenient, and economical way to identify double bond positions in lipids; it requires neither sample derivatization nor any special MS instrument modification to detect unsaturation sites. In this study, lipid oxidation was determined by using liquid extraction surface analysis (LESA) coupled to an ESI quadrupole orthogonal time-of-flight (Q-o-TOF) MS, after unsaturated lipids were deposited onto several different surfaces and exposed to ambient air for 1 or 24 h. TLC-MS was applied to separate complex lipid mixtures and to elucidate the structures of the lipids and their oxidized products.
Experimental Section
Materials
Sulfatide (ST) standards (d18:1/C24:1 and d18:1/C24:0) and bovine brain total lipid extract were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). Octadecanoic acid (stearic acid, C18:0), 9Z-octadecenoic acid (oleic acid, C18:1 (9Z)), 9Z,12Z-octadecadienoic acid (linoleic acid, C18:2 (9Z,12Z)), tetracosanoic acid (lignoceric acid, C24:0), 15Z-tetracosenoic acid (nervonic acid, C24:1 (15Z)), and primuline were from Sigma Chemical Co (St. Louis, MO). Further details are in the Supporting Information.
HPTLC
For details, see Supporting Information.
Direct Sampling TLC-MS
Protocols we have developed for use of the LESA device (Advion BioSciences, Inc., Ithaca, NY) for lipid MS and MS/MS analyses are described elsewhere.19 For details, see Supporting Information.
Sample Preparation for Nanospray ESI-MS
Twenty picomoles of STs phosphatidylcholines and fatty acids in CHCl3/CH3OH (1:1, v/v) were deposited on aluminum- or glass-backed silica gel TLC plates, microscope glass slides, and aluminum foils. After exposure to the air for 0, 1, or 24 h, the samples were extracted using 10 μL of (CH3)2CHOH/CH3OH/H2O (9:1:1, v/v/v) and were analyzed using the nanoESI source of the QSTAR Pulsar i Q-o-TOF MS fitted with the TriVersa NanoMate.
Square pieces cut from aluminum foil (1 cm × 1 cm) were placed in a nitrogen-purged three-necked flask that had been half-filled with glass beads to provide a level surface. STs were applied to the substrate through a septum and dried with a nitrogen purge. At each time point, 10 μL of (CH3)2CHOH/CH3OH/H2O (9:1:1, v/v/v) was applied to the substrate to dissolve the lipid samples and the resulting solution was collected into an amber glass vial for analysis.
Twenty picomoles of the STs were deposited on pieces of aluminum foil (1 cm × 1 cm) and these were placed in a darkroom. STs were manually extracted by 10 μL of (CH3)2CHOH/CH3OH/H2O (9:1:1, v/v/v) and saved for analysis by nanoESI-MS at different time periods. Ozonide formation was confirmed by analyzing the samples after treatment under an ozone-saturated atmosphere. For this procedure, the ST samples on aluminum foil were placed in a 24-well plate and the plate was placed in an airtight container. After the container was purged with ozone-saturated air generated from a corona discharger (Model BD-20 V, Electro-Technic Products, Inc., Chicago, IL) for 10 min, the container was hermetically sealed to keep the samples in it under an ozone-saturated atmosphere for 24 h.
Ultra high-mass resolution spectra were acquired for nervonic acid with a 12-T SolariX (Bruker Daltonics Inc., Billerica, MA, USA) fitted with a modified Bruker nanoESI source, following deposition of the lipid onto aluminum foil and exposure to ozone for 0, 1, or 24 h. With this FTMS system, the mass accuracy was better than 1 ppm.
Characterization of Surface Roughness
The samples were characterized using white light interferometry, contact stylus interferometry and scanning electron microscopy. For details, see Supporting Information.
Results and Discussion
Observation of Lipid Surface Oxidation on TLC Plate
Consistent with the results reported by the Blanksby group using DESI MS8 and those we describe elsewhere,19 oxidation products were observed during direct-sampling TLC-ESI-MS of STs. Following deposition of 100 pmol of d18:1/C24:1 ST, the silica gel-coated TLC plate was developed in CHCl3/CH3OH/0.2% CaCl2 (55:45:10, v/v/v), then exposed to ambient air and the bands were analyzed by ESI-MS.
In addition to the expected [M – H]− at m/z 888.62, signals corresponding to larger species were observed at m/z 936.61 (+48 u) and 984.59 (+96 u). Peaks indicative of a degraded species appeared at m/z 778.48 (−110 u); this product apparently underwent a further loss of 180 u to generate the species observed at m/z 598.25. Assignments of these modified peaks were made on the basis of subsequent CID experiments (Supporting Information Figure S-1). The pathways are summarized in Figure 1c.
Figure 1.

Negative-ion mode TLC-ESI mass spectra; oxidation products are labeled with an asterisk.(*) (a) d18:1/C24:1 ST and (b) d18:1/C24:0 ST and their oxidation products. (c and d) Major oxidation pathways.
A similar ion series was observed for the reaction products of d18:1/C24:0 ST, which has one double bond on the sphingoid backbone chain and a saturated fatty acyl chain (Figure 1d). The [M – H]− was at m/z 890.64; the peak at m/z 938.62 indicated a component with an increase of 48 u and m/z 710.42 corresponded to [M – H]− for a species that arose by loss of 180 u from the one detected at m/z 890.64 (Figure 1b).
In CID MS/MS, all the above ions yielded ST-characteristic headgroup fragments at m/z 96.96, 241.00, and 259.01 (Supporting Information Figure S-1a). The ion at m/z 778.48 could be assigned as the oxidatively cleaved product bearing a terminal aldehyde at carbon-15 on the former fatty acyl chain of d18:1/C24:1 ST, and the ion at m/z 598.25 corresponded to the dialdehyde formed following the oxidative cleavage of the double bonds on both its fatty acyl chain and long chain base (LCB) (Supporting Information Figure S-1b). Evidence was found for oxidative cleavage of d18:1/C24:0 ST that has no fatty acyl double bond. Oxidation generated only one aldehyde group, at carbon-4 of its LCB, and the product was detected at m/z 710.42 (Supporting Information Figure S-1c). The ozonides were fairly stable (Figure 1c and d), consistent with previous reports.8,20 The [M – H]− ions observed at m/z 936.61 and 984.59, in the spectra of the ozonolysis products from d18:1/C24:1 ST, and m/z 938.62, from d18:1/C24:0 ST (Figure 1), could be assigned as ozonides formed on the double bond positions (Supporting Information Figure S-1d, e, and f) of these lipids. The signal at m/z 984.59 had low S/N in this TLC-MS experiment. CID decomposed all these ozonides to yield aldehyde and carboxylic acid fragments via initial homolytic cleavage of the peroxide bridge of the trioxolane followed by rearrangement.21 Ion signals at m/z 778.48 and 794.47 corresponded to the aldehyde and carboxylic acid products formed upon oxidation of the Δ15,16 double bond in the fatty acyl chain of d18:1/C24:1 ST (reaction at the lone double bond generated the ozonide at m/z 936.61). Observation of these two product ions confirmed that the ozonide was formed on the fatty acyl group but not along the LCB; otherwise, product ions at m/z 708.41 and 724.40 would also have been detected. Therefore, it became clear that the oxidation occurred at the double bond in the fatty acyl chain preferentially over the LCB. In contrast, in the CID MS/MS spectrum of m/z 938.62, the presence of product ions at m/z 710.42 and 726.41 indicated that an aldehyde and a carboxylic acid formed in the LCB at the Δ4,5 position of d18:1/C24:0 ST. Additionally, the presence of the hydroxyl group on the sphingoid backbone directed formation of other CID product ions. For instance, the precursor ion at m/z 938.62 provided the product ion at m/z 680.40, which results from secondary loss of 30 u (e.g., CH2O, formaldehyde) from the component observed at m/z 710.42. The m/z 680.40 peak thus represents the aldehyde formed with cleavage at the Δ4,5 site on the sphingoid backbone. Meanwhile, the precursor ion at m/z 984.59 corresponds to the fully ozonized d18:1/C24:1 ST; this generated a more complicated MS/MS spectrum. In addition to the cleavages at the trioxolane moiety (m/z 826.46, 842.46, 598.25, and 614.25), cleavage at the position one carbon further along the sphingosine chain formed an aldehyde at carbon-3, generating the ion at m/z 584.24 and its dehydration product detected at m/z 568.24. As noted above, the mass accuracy was within 15 ppm for all QStar experiments.
The discovery introduced above by direct sampling TLC-ESI MS could have many origins: the experimental processes of TLC, exposure during TLC in the air, sampling by the TLC-MS interface (LESA NanoMate), and nanoESI MS. Since signals were observed only at values corresponding to the [M – H]− ions predicted for the unmodified species when solutions of ST standards were directly introduced by nanospray into the MS (data not shown), it immediately seemed clear that nanoESI-MS itself is not the driver for the oxidation reaction.
To investigate the remaining factors that might be responsible for the lipid oxidation, we performed experiments on other surfaces: aluminum foil, glass slides, glass-backed TLC plates from two different manufacturers, and also aluminum-backed TLC plates without development. In the experiments whose results are shown in Figure 2, 100 pmol of d18:1/C24:1 and d18:1/C24:0 STs were deposited on these different surfaces and the products were analyzed using TLC-ESI-MS with a LESA TLC-MS interface. Analysis of a sulfatide (d18:1/C24:1) deposited on an aluminum-backed TLC plate showed different spectra over the analysis time (Figure 2a-1). The molecular ion at m/z 888.62 and ions at m/z 598.25, 778.48, 936.61, and 984.59 were observed after 1-h exposure to ambient air. After 24 h, all ions disappeared except for the ion at m/z 598.25. For d18:1/C24:0 ST, the spectrum from the 1-h treated sample showed ions at m/z 710.42 and 938.62. Similarly, after 24-h exposure, the abundance of the ion at m/z 710.42 increased with respect to that of the intact ST molecular ion (Figure 2a-2). STs deposited on the glass-backed TLC plate underwent changes similar to those on the aluminum-backed TLC plate (data not shown). The same pattern of results was obtained with a glass-backed TLC plate from a different manufacturer (data not shown). As shown in Figure 2b, both d18:1/C24:1 and d18:1/C24:0 ST were oxidized somewhat more slowly on aluminum foil than when deposited on an aluminum-backed TLC plate. In the spectrum of d18:1/C24:1 ST, the component detected at m/z 598.25 was attributed to loss of 180 u from the species with [M – H]−m/z 778.48. The relative abundance of the peak at m/z 598.25 was increased in comparison to that of the ion at m/z 778.48 in the spectrum of the sample on the TLC surface versus aluminum foil, for either 1 or 24 h (Figure 2b-1). Additionally, the oxidation products observed in the spectrum from the 1-h sample deposited on the TLC plate were similar to those found for the 24-h sample on aluminum foil. In the spectrum of the sample left for 24 h on the TLC plate, additional oxidation products were observed. For d18:1/C24:0 ST, the intensity ratio of the signal at m/z 710.42 to that at m/z 890.64 in the spectra collected at the same time point was much higher for samples deposited on TLC plates than for those that were deposited on aluminum foil (Figure 2b-2). The spectra shown in Figure 2c, indicate that the oxidation reactions on glass slides occurred more slowly than those on all other surfaces tested in this study. For d18:1/C24:0 ST, at least 3 days were required to observe the oxidation product at m/z 710.42 for samples deposited on glass slides. Therefore, these observations led to the conclusion that the order of oxidation reaction rates on the examined surfaces is silica gel TLC > aluminum foil ≫ glass slide.
Figure 2.

Negative-ion mode ESI-MS spectra acquired following deposition of d18:1/C24:1 ST (1) and d18:1/C24:0 ST (2) onto different surfaces, silica gel TLC plate (a), aluminum foil (b), and glass slide (c), and exposure to ambient air for 0, 1 h, and 1 day prior to analysis. Oxidation products are labeled with an asterisk (*).
The surface roughness was tested for these three materials. The average roughness, Ra, for silica gel TLC (∼606 nm) was slightly larger than that for aluminum foil (ca. 390 nm), and was much larger than that for the glass slide (∼3 nm) (Supporting Information Figure S-2). Rough surfaces provide increased contact area for the ambient air, and thus may be expected to cause oxidative cleavage reactions to take place more rapidly. Meanwhile, the mass spectra recorded for the samples treated on aluminum foil and glass slides were much cleaner than those obtained for the sample deposited on the TLC plate, and this improvement in spectral quality made it possible to detect the diozonide product of d18:1/C24:1 ST at m/z 984.59. On the basis of the spectra obtained from the samples treated on different surfaces, it should be noted that there was no evidence in the mass spectra to indicate that the ozonide had decomposed to the aldehyde. It is noteworthy that the spectra became more complicated after 1 day; new signals appeared, in addition to those that could be attributed to “aldehyde, carboxyl acid and ozonide”. This result warrants further investigation.
As discussed previously, d18:1/C24:1 ST generated an oxidation product that was detected at m/z 778.48 (from cleavage on the fatty acyl chain) and then produced an ion at m/z 598.25 (by cleavage of the double bonds in both the fatty acyl chain and the LCB). It is notable that there was no signal that would correspond to a fragment generated by cleavage solely on the LCB of d18:1/C24:1 ST and the disappearance of intact d18:1/C24:0 ST occurred much more slowly than d18:1/C24:1 ST, especially when the sulfatides were coated on glass slides (Figure 2c). These pieces of evidence suggest that the reaction rate for ST surface oxidation of the double bond on the fatty acyl chain is faster than the rate of oxidation on the LCB, at the site of unsaturation that has an adjacent hydroxyl group. The presence of the α-hydroxyl group evidently reduces the susceptibility of the double bond on the sphingoid backbone to oxidation.
The data presented above provide evidence that the oxidation occurs not only on the TLC plate, but also on other surfaces. To elucidate which element is critical for the oxidation, STs were deposited on aluminum foil, exposed under ambient air for different time periods and manually extracted. Extracted lipid samples were analyzed by nanoESI-MS performed with a TriVersa NanoMate and showed the same pattern of results as the data collected by LESA-MS for samples deposited on aluminum foil (data not shown). This suggests that the LESA NanoMate interface is not necessary for oxidation of an unsaturated lipid. The results do indicate that surface exposure to ambient air is critical for the lipid double bond oxidation.
Interestingly, after TLC development, the oxidation products of ST with shorter fatty acyl substituents or truncated LCBs were not observed in any position on the TLC plate below that of the intact STs with higher polarity. This result indicates that the STs were not oxidized prior to or during the TLC development. The TLC developing tank was saturated with the developing solvent vapor and this may have protected the unsaturated lipids embedded in the TLC gel from the ambient air. However, after development, during the time period utilized for drying, staining, and marking the spots of interest in the analytical TLC plate by pencil, the separated components were exposed to the ambient laboratory environment. During this time, the analytes must have undergone oxidation and formed oxidized products that were detected at the same migration position on the TLC plate where the relevant intact lipids were located. The TLC separation had been completed prior to oxidation and thus could not have separated the long chain ST from the truncated shorter chain STs. This phenomenon also suggests that the oxidation is caused by postdevelopment exposure of unsaturated lipids to ambient air.
Mechanism Study
Ambient ozone had been proposed as the reagent responsible for oxidation of unsaturated lipids and peptides that were air-dried prior to analysis by MALDI-MS, nanoassisted laser desorption/ionization mass spectrometry (NALDI-MS) and DESI-MS.8,22−24 Once we had determined that rate of the oxidation reaction was considerably faster on aluminum foil, it seemed easier to explore the oxidation mechanisms using aluminum foil rather than a glass slide. Aluminum foil also reduced the “chemical noise” background compared to TLC. Therefore, in subsequent experiments, we deposited 100 pmol of ST on aluminum foil under the “ozone” conditions (Supporting Information Figure S-3). After 24-h exposure to generated ozone, we detected only a small amount of ozonide (m/z 936.61) and no aldehyde (m/z 778.48) of d18:1/C24:1 ST by nanoESI-MS using the TriVersa NanoMate (Supporting Information Figure S-3a) for sampling. However, the MS spectrum recorded after 24-h exposure under ozone (Supporting Information Figure S-3b) showed no evidence for aldehyde (m/z 710.42) or ozonide (m/z 938.62) derived from the d18:1/C24:0 ST. The signal intensity from ozonide product of ST generated under the ozone environment was much lower than that recorded for the experiment performed under ambient air (as shown in Figure 2b) over the same time period. This result was consistent with the report that the amount of ozonides observed under ambient air was larger than that observed under the gas phase conditions of OzID.8 However, the results of our investigation suggest that the outcome does not depend on stabilization of the ozone adduct on the surface.23 It rather seems that the generated ozone somehow protects the unsaturated lipids from the ambient air on the surface. In addition, as we demonstrated with Figure 2, there was no evidence to indicate that the aldehyde was derived from the ozonide by decomposition during the air exposure process, though it appeared as a fragment of ozonide in the CID spectra. The aldehyde was generated earlier and ozonides were produced later in the course of lipid oxidation. Therefore, it seems that, in addition to ozone oxidation, there must be another oxidation mechanism that is deeply involved in the lipid double bond surface oxidation.
To elucidate the key role of ambient air in the observed lipid oxidation, gaseous nitrogen (N2) was used as protective environment (Supporting Information Figure S-4). The oxidation was totally inhibited by the protection of the sample by N2 after standard deposition of ST onto the aluminum foil. Even after 24 h under N2, neither d18:1/C24:1 nor d18:1/C24:0 ST generated any oxidative product. As a result, we could conclude that environmental air is required to oxidize the double bonds of lipids dried on the surfaces.
Since light has been considered to be involved in lipid oxidation by singlet oxygen in other cases, STs were deposited on aluminum foil and placed in the darkroom and spectra were collected over time. While kept in the dark, the substrates were protected from oxidation, despite there being sufficient airflow. No oxidation product was detected by nanoESI-MS with the TriVersa NanoMate after 24 h (Supporting Information Figure S-5). The data shown here provide evidence that oxidation of the unsaturated lipid surface is a light-sensitive reaction.
It has been strongly suggested that singlet oxygen (1O2) was formed by photosensitization in the atmosphere and may play a significant role as an oxidant in the air.25 Many researchers have pointed out that 1O2 is an important oxidizing agent present in ambient polluted air.26−28 Since the transition from the 1Δg state to the 3Eg– state is spin forbidden, the 1Δg O2 is considerably long-lived. It had been confirmed that the radiative lifetime of O2 (1Δg) is 45 min in the gas phase29 and 10–6 - 10–3 s in solution.30 This difference in the lifetime of singlet oxygen under the two conditions explains why oxidation of unsaturated lipids could only be observed on surfaces exposed to the air but not in solution. Singlet oxygen is significantly electrophilic, and reacts with unsaturated carbon–carbon bonds, neutral nucleophiles, and anions.31 Scheme 1 shows the proposed mechanism of the double bond oxidation by 1O2.32 The nucleophilic oxygen from the allylic hydroxyl group on carbon in the position α to the double bond enables the lipid to interact with the electrophilic 1O2, thereby directing the attack of this enophile.33 Resonance is not possible for the allylic hydroxyl group and the carbon–carbon double bond. Therefore, the hydroxyl group cannot participate in delocalization of π-electrons or function as an electron donor. In contrast, the hydroxyl oxygen is electron withdrawing by induction (−I) because the oxygen atom is relatively electronegative and is uncharged in that bonding arrangement. The “–I” effect makes the double bond less electron-rich and thus a poorer nucleophile than the carbon–carbon double bond without the allylic hydroxyl group. This consideration can explain why the double bond in the sphingoid backbone reacts more slowly than that in the fatty acyl chain.
Scheme 1. Proposed Mechanism for Carbon–Carbon Double Bond Oxidation by Singlet Oxygen.

Ozone is also a source of singlet oxygen because of photolysis of ozone in the Hartley region (3200 to 2000 Å).25 Singlet oxygen could act as a secondary oxidant by the reaction of ozone. The ethylene consumption rate constant of singlet oxygen is much greater than that of ozone.34 Consideration that the ozone concentration is higher than the concentration of singlet oxygen could explain the observation that the aldehyde generation starts earlier and ozonides are produced later in the course of lipid oxidation. The results obtained in Supporting Information Figure S-3, suggest that the presence of ozone somehow protected the unsaturated lipids from the ambient air on the surface. This result is consistent with the observation of Ellis et al.8 that ozonolysis was not observed when the samples were protected from airflow within a box containing a low concentration of ozone that was likely adsorbed by the surfaces of the box. Therefore, we propose that the reaction of singlet oxygen and ozone oxidation are two parallel reactions leading to double bond surface oxidation in unsaturated lipids.
Meanwhile, surface oxidation of phospholipids with poly unsaturated fatty acyl groups was investigated in this study. Phosphatidylcholine (16:0/18:1(9Z)) and phosphatidylcholine (16:0/18:2(9Z,12Z)) were compared after exposure to the ambient air (Supporting Information Figure S-6). Aldehyde formation was found in phosphatidylcholine (16:0/18:1(9Z)) at the isolated double-bond position (Supporting Information Figure S-6a) and two aldehyde products were detected in phosphatidylcholine (16:0/18:2(9Z,12Z)) at either of its double bond positions (Supporting Information Figure S-6b). With the presence of two aldehyde products, the existence of two carbon–carbon double bonds and their positions can be established.
Analysis of Free Fatty Acids and Bovine Brain Total Lipid Extract by TLC-ESI-MS
The direct sampling TLC-ESI-MS method for polar lipid analysis using the LESA TLC-MS interface recently developed by our group19 was applied to the analysis of free fatty acids and to a bovine brain total lipid extract. Herein, we present relevant mass spectra and discuss the identification of the double bond positions by analysis of TLC-MS data obtained for the oxidized products of the bovine brain lipid extract. Because of space limitations, the data obtained for the free fatty acids and discussion of these results are included in the Supporting Information.
The bovine brain total lipids were deposited on a normal phase silica gel TLC plate and their separation was primarily based on the differences in their polarity. After development, the TLC plate reserved for MS analysis was dried under ambient air. It was aligned with the primuline-stained TLC plate, marked with pencil to indicate the lipid locations, and cut to fit in the LESA holder. The total time for exposure to the air was about 0.5 h. Figure 3 shows LESA-TLC-ESI mass spectra obtained from TLC plate areas where the intensities of signals that correspond to phosphatidylserine (PS) and N,N-dimethyl phosphatidylethanolamine (PE-NMe2) candidates were the greatest. In Figure 3a, the most abundant peak (m/z 788.54) corresponded to the [M – H]− of PS (18:0/18:1), and could be assigned to this structure on the basis of its CID MS/MS spectrum. (Supporting Information Figure S-7a) The CID spectrum of the second most abundant signal (m/z 678.40) corresponded to the PS aldehyde that would have been formed by oxidation of the Δ9,10 double bond, in the PS that had a fatty acyl chain of 18:0, PS (18:0/9:1[O]) (Supporting Information Figure S-7b). Interestingly, an ion at m/z 591.37 that could be assigned as a PA aldehyde (18:0/9:1[O]) was found at the same retention factor (Rf) value; this assignment was confirmed by analysis of the corresponding PA standard (data not shown). Under exposure to the air, the PS lipid class reacted to form PS oxidation products and also PA oxidation products. The ion at m/z 706.43 corresponds to the expected aldehyde formed following the oxidation of the Δ11,12 double bond of the isomeric PS (18:0/18:1, Δ11,12) (Supporting Information Figure S-7d). Fatty acids 18:1 (Δ9,10) and 18:1, (Δ11,12) are common in nature and their presence in the mixture can explain the observations of PS (18:0/18:1, Δ9,10)17 and PS (18:0/18:1, Δ11,12). Other ions in the spectrum shown in Figure 3a corresponded to other less abundant PSs and their potential oxidation products.
Figure 3.

TLC-MS negative-ion mode spectra acquired following separation of the bovine brain total lipid extract. (a) PS (18:0/18:1) at m/z 788.54 and its oxidation products. (b) PE-NMe2 (16:0/18:1) m/z 744.55, PE-NMe2 (16:0/18:0) m/z 746.57, and PE-NMe2 (18:0/18:1) m/z 772.59, plus oxidation products and adducts. Oxidation products indicating the unsaturation Δ9,10 are labeled with (*) and those indicating the unsaturation Δ11,12 are labeled with a black square (■). The easily dissociated [M + C3H6O2 – H]− peaks at m/z 818 and 846 correspond to glyceryl esters or adducts.
Figure 3b shows the direct sampling LESA-TLC-MS spectrum from the PE-NMe2 candidate spot on a silica gel TLC plate. The ion at m/z 744.55 corresponds to PE-NMe2 (16:0/18:1), while m/z 746.57 corresponds to the molecular ion of PE-NMe2 (16:0/18:0). The structural assignments for both ions were confirmed by their CID spectra (Supporting Information Figure S-8a and b). The neutral loss of 71 Da, the signal at m/z 224.07 and other phosphate fragment ions are signatures that were used to identify the assignments of the PE-NMe2 molecular ion. Oxidation products were observed at m/z 634.41 and 662.44. The ion at m/z 634.41 in the CID MS/MS spectrum corresponded to PE-NMe2 aldehyde (16:0/9:1[O]) that was cleaved within the Δ9,10 double bond (Supporting Information Figure S-8c). The signal at m/z 662.44 was determined to be a mixture of the [M – H]− species from the PE-NMe2 aldehyde (16:0/11:1[O]) and PE-NMe2 aldehyde (18:0/9:1[O]) (Supporting Information Figure S-8d). The presence of both PE-NMe2 aldehydes (16:0/9:1[O]) and (16:0/11:1[O]) indicates that PE-NMe2 (16:0/18:1) exists as a mixture of isomers of PE-NMe2 (16:0/18:1, Δ9,10) and (16:0/18:1, Δ11,12). The peak at m/z 772.59 that could have provided evidence for the presence of PE-NMe2 (18:0/18:1) had lower S/N (Figure 3b); its signal intensity was not sufficient for an MS/MS experiment. It is significant that there were a few higher abundance peaks in the spectrum shown in Figure 3b, such as m/z 818.58, 846.62, 708.45, and 736.48. Their CID spectra showed a common 74-u neutral loss to yield fragments at m/z 744.55, 772.59, 634.41, and 662.44 respectively, which would correspond to expected values for a series of the PE-NMe2 lipids and their oxidized products. After the initial 74-u neutral loss from the molecular ions, the tandem MS spectra matched well for each precursor/primary fragment ion pair (m/z 818.58/744.55, m/z 708.45/634.41, m/z 736.48/662.44). The MS/MS spectrum of m/z 846.62 indicated it was a mixture of PE-NMe2 (18:0/18:1) and (16:0/20:1) with an additional 74 u (Supporting Information Figure S-8e). Combined with the oxidative product data, this result suggests that PE-NMe2 (18:0/18:1, Δ9,10), (16:0/20:1, Δ9,10) and (16:0/20:1, Δ11,12) were present in the sample as natural or artifactual species containing glyceryl esters on the phosphate group. The 74-u neutral loss corresponds to elimination of dehydroglycerol (C3H6O2).
It was notable that, at first, no oxidation products were observed for STs in the bovine brain total lipid extract by LESA-TLC-MS. However, when developed TLC plates were exposed to ambient air and light over longer time (more than 3 days), the corresponding aldehydes could be observed (data not shown). This observation might be explained by the presence of antioxidant components such as lipid-soluble vitamin E that coexists in the bovine brain. PEs that colocalized with STs on the TLC plate in this investigation showed phenomena similar to the STs, which supports the “position-related” protection theory.
A better TLC separation would provide a better opportunity for location of double bond positions on unsaturated lipids. Thus, 2-D TLC would be an ideal candidate to improve the separation of colocalized lipids.
Conclusions
The behaviors of various lipid classes were examined in this investigation. Surface oxidation under ambient air modified all categories of unsaturated lipids that were tested: fatty acids, GSLs, and phospholipids. The oxidation rates and products were found to be dependent on the types of double bonds. If an allylic hydroxyl group was present, its reaction rate was slower.
According to our study and the reports of others, surface oxidation in unsaturated lipids under ambient air occurs on a variety of surface materials. Moreover, we have found that the reaction rates differ, even for individual surface materials, based on their roughness. These data also indicate that both ozone oxidation and a second type of reaction occur. We propose a mechanism involving singlet oxygen, which is widely considered as an oxidant for lipid oxidation both in vivo and in vitro. When combined with the ozonolysis theory, our singlet oxygen mechanism can fully explain the phenomena observed in unsaturated lipids.
On the basis of our findings, extra care is needed in general lipid processing procedures. Liquid–liquid extraction is found to be safe for the unsaturated lipids becaue of the extremely low oxidation rates for singlet oxygen in solution. However, attention is needed for the lipids extracted from TLC plates, especially for studies that investigate oxidized lipids in biological samples. Artifactual oxidation may occur due to the ambient air exposure.
Surface oxidation under ambient air provides an efficient, convenient and economical way to identify double bond positions in unsaturated lipids. Surface oxidation does not require any special type of instrumentation to oxidize the unsaturated lipid. If the sample is reasonably pure, direct infusion supplemented by surface exposure on aluminum foil under ambient air for 1 h and liquid re-extraction are sufficient to elucidate the double bond positions within fatty acyl chains. For analysis of complex biological samples, TLC-MS is able to separate and analyze the components and their oxidation products, even those that are present at the same Rf value. In this investigation, LESA-TLC-ESI-MS was applied; TLC-MALDI-MS would also suitable. Therefore, our method could be used widely with almost any type of MS instrument to identify the location of double bonds in unsaturated lipids.
Acknowledgments
The authors are grateful to Dr. Xiaofeng Shi and Dr. Cheng Lin at Boston University School of Medicine for helpful discussion and Dr. James Weaver at Harvard University for SEM analysis. This investigation was supported by NIH grants P41 RR10888/GM104603 and S10 RR025082.
Supporting Information Available
Additional materials as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Ying Zhou: Astra Zeneca-US, Waltham, MA.
Author Present Address
⊥ Hyejung Park: Genzyme Corporation, Waltham, MA.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Y.Z. and H.P. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brown H. A.; Marnett L. J. Chem. Rev. 2011, 111, 5817–5820. [DOI] [PubMed] [Google Scholar]
- Gross R. W.; Jenkins C. M.; Yang J.; Mancuso D. J.; Han X. Prostaglandins Other Lipid Mediators 2005, 77, 52–64. [DOI] [PubMed] [Google Scholar]
- Porter N. A.; Caldwell S. E.; Mills K. A. Lipids 1995, 30, 277–290. [DOI] [PubMed] [Google Scholar]
- Wynalda K. M.; Murphy R. C. Chem. Res. Toxicol. 2010, 23, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley D. G.; Min D. B. Crit. Rev. Food Sci. Nutr. 1992, 31, 211–236. [DOI] [PubMed] [Google Scholar]
- Frankel E. N. Prog. Lipid Res. 1980, 19, 1–22. [DOI] [PubMed] [Google Scholar]
- Gohre K.; Miller G. C. J. Chem. Soc., Faraday Trans. I 1985, 81, 793–800. [Google Scholar]
- Ellis S. R.; Hughes J. R.; Mitchell T. W.; in het Panhuis M.; Blanksby S. J. Analyst 2012, 137, 1100–1110. [DOI] [PubMed] [Google Scholar]
- Criegee R. Angew. Chem., Int. Ed. Engl. 1975, 14, 745–752. [Google Scholar]
- Reich M.; Hannig C.; Al-Ahmad A.; Bolek R.; Kummerer K. J. Lipid Res. 2012, 53, 2226–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T.; Park Y. S.; Rho J. R.; Kim Y. H. Rapid Commun. Mass Spectrom. 2011, 25, 572–578. [DOI] [PubMed] [Google Scholar]
- Bang D. Y.; Lim S.; Moon M. H. J. Chromatogr., A 2012, 1240, 69–76. [DOI] [PubMed] [Google Scholar]
- Sparvero L. J.; Amoscato A. A.; Dixon C. E.; Long J. B.; Kochanek P. M.; Pitt B. R.; Bayir H.; Kagan V. E. Chem. Phys. Lipids 2012, 165, 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suni N. M.; Aalto H.; Kauppila T. J.; Kotiaho T.; Kostiainen R. J. Mass Spectrom. 2012, 47, 611–619. [DOI] [PubMed] [Google Scholar]
- Mitchell T. W.; Pham H.; Thomas M. C.; Blanksby S. J. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877, 2722–2735. [DOI] [PubMed] [Google Scholar]
- Hsu F. F.; Turk J. J. Am. Soc. Mass Spectrom. 2008, 19, 1673–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F. F.; Turk J. J. Am. Soc. Mass Spectrom. 2008, 19, 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F. F.; Turk J. J. Am. Soc. Mass Spectrom. 2010, 21, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.; Zhou Y.; Costello C. E. J. Lipid Res. 2014, 55, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. C.; Finlayson-Pitts B. J.; Willis W. V. Chem. Res. Toxicol. 1990, 3, 517–523. [DOI] [PubMed] [Google Scholar]
- Harrison K. A.; Murphy R. C. Anal. Chem. 1996, 68, 3224–3230. [DOI] [PubMed] [Google Scholar]
- Cohen S. L. Anal. Chem. 2006, 78, 4352–4362. [DOI] [PubMed] [Google Scholar]
- Pavlaskova K.; Strnadova M.; Strohalm M.; Havlicek V.; Sulc M.; Volny M. Anal. Chem. 2011, 83, 5661–5665. [DOI] [PubMed] [Google Scholar]
- Wisthaler A.; Weschler C. J. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6568–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummler R. H.; Bortner M. H.; Baurer T. Environ. Sci. Technol. 1970, 4, 1148–1150. [DOI] [PubMed] [Google Scholar]
- Kummler R. H.; Bortner M. H.; Baurer T. Environ. Sci. Technol. 1969, 3, 248–250. [DOI] [PubMed] [Google Scholar]
- Ogawa S.; Fukui S.; Hanasaki Y.; Asano K.; Uegaki H.; Fujita S.; Shimazaki R. Chemosphere 1991, 22, 1211–1225. [Google Scholar]
- Ogawa S.; Shimazaki R.; Soejima A.; Takamure E.; Hanasaki Y.; Fukui S. Chemosphere 1996, 32, 1823–1832. [Google Scholar]
- Arnold S. J.; Kubo M.; Ogryzlo E. A. Adv. Chem. Ser. 1968, 133–142. [Google Scholar]
- Merkel P. B.; Kearns D. R.; Nilsson R. J. Am. Soc. Mass Spectrom. 1972, 94, 7244–7253. [Google Scholar]
- DeRosa M. C.; Crutchley R. J. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar]
- Alberti M. N.; Orfanopoulos M. Chemistry 2010, 16, 9414–9421. [DOI] [PubMed] [Google Scholar]
- Griesbeck A. G.; Adam W.; Bartoschek A.; El-Idreesy T. T. Photochem. Photobiol. Sci. 2003, 2, 877–881. [DOI] [PubMed] [Google Scholar]
- Murray R. W.; L J. W.; Kaplan M. L. Ann. N.Y. Acad. Sci. 1970, 171, 121–129. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.