

Abstract
The majority of membrane proteins function as oligomers. However, it remains largely unclear how the oligomer stability of protein complexes correlates with their function. Understanding the relationship between oligomer stability and activity is essential to protein research and to virtually all cellular processes that depend on the function of protein complexes. Proteins make lasting or transient interactions as they perform their functions. Obligate oligomeric proteins exist and function exclusively at a specific oligomeric state. Although oligomerization is clearly critical for such proteins to function, a direct correlation between oligomer affinity and biological activity has not yet been reported. Here, we used an obligate trimeric membrane transporter protein, AcrB, as a model to investigate the correlation between its relative trimer affinity and efflux activity. AcrB is a component of the major multidrug efflux system in Escherichia coli. We created six AcrB constructs with mutations at the transmembrane intersubunit interface, and we determined their activities using both a drug susceptibility assay and an ethidium bromide accumulation assay. The relative trimer affinities of these mutants in detergent micelles were obtained using blue native polyacrylamide gel electrophoresis. A correlation between the relative trimer affinity and substrate efflux activity was observed, in which a threshold trimer stability was required to maintain efflux activity. The trimer affinity of the wild-type protein was approximately 3 kcal/mol more stable than the threshold value. Once the threshold was reached, an additional increase of stability in the range observed had no observable effect on protein activity.
The stability of water-soluble proteins and protein oligomers is tuned to match their biological function, the physiological environment, and the regulation mechanism.1−6 Usually, a protein is only marginally stable. This modest stability is thought to be critical for protein function, which usually requires a certain degree of structural flexibility and involves conformational changes. While steady progress has been made in recent years, our understanding of the stability–activity relationship still lags behind in the area of membrane protein research.7 Over the past 2 decades, much insight has been obtained on the causes and consequences of membrane protein oligomerization,7,8 yet a systematic study investigating the correlation between oligomer stability and in vivo activity of a multispan and multidomain helical membrane protein is still missing. Here, we used Escherichia coli AcrB as a model protein to investigate the relationship between its trimer stability and drug efflux activity.
AcrB belongs to the resistance-nodulation-cell-division (RND) family of efflux pumps, which are conserved in all Gram-negative bacteria.9−11 RND family efflux pumps play a major role in multidrug resistance in Gram-negative bacteria. AcrB is an obligate trimer that exists and functions exclusively in the trimeric form (Figure 1A).12 Trimeric AcrB associates with membrane fusion protein AcrA and outer membrane protein TolC to form a complex that spans the inner membrane, periplasmic space, and outer membrane.13−16 AcrB conducts the inward flow of protons across the inner membrane to drive conformational changes that facilitate substrate efflux. The structure of AcrB was first solved by X-ray crystallography in the asymmetric trimer form in 2006, which supports a conformational cycling model for drug transport.17,18 To date, more than 30 structures of AcrB have been deposited into the Protein Data Bank. However, crystal structures cannot provide insight into how its trimer affinity affects its drug efflux activity.
Figure 1.
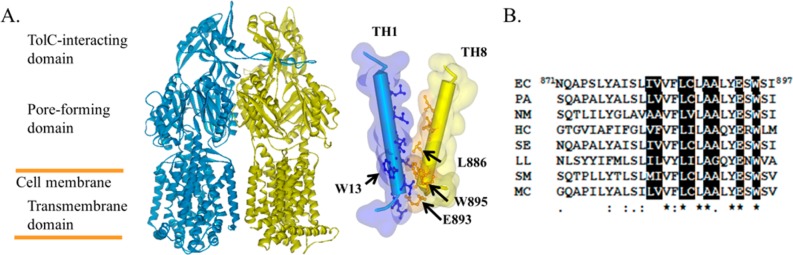
AcrB structure and sequence alignment. (A) Structure of AcrB trimer with the subunit at the back removed for clarity. Positions of the domains discussed in the text are labeled for the side view. Residues lining the interface between TH1 and TH8 are highlighted by ball-and-stick models. L886, E893, W895, and W13 are labeled. (B) Sequence alignment of residues in TH8. Residues at the interface are shown with white font in a black box. Numbers indicate the position of the starting and ending residues in the sequence of E. coli AcrB. Asterisks, colons, and periods indicate identical, conserved, and semiconserved residues, respectively. The sequences are as follows: EC, AcrB from E. coli; PA, MexB from Pseudomonas aeruginosa; NM, MtrD from Neisseria meningitidis; HC, HcanM9_00968 from Helicobacter canadensis; SE, acridine efflux pump from Salmonella enterica; LL, AcrB from Legionella longbeachae; SM, HAE1 from Tenotrophomonas maltophilia; and MC, AcrB from Moraxella catarrhalis RH4.
We have previously created several AcrB mutants with modifications on the periplasmic intersubunit interface that displayed different levels of drug efflux activity.19,20 These sites of mutation are located on a protruding loop critical for trimerization. A single amino acid substitution has been shown to almost completely dissociate the trimer structure (AcrBP223G). In this work, we introduced mutations at three sites on the transmembrane intersubunit interface, including both single, double, and triple replacements (Figure 1A), aiming at disrupting trimer association without affecting the structure of individual monomers. Structural characterization and stability measurement indicated that these mutations had little impact on monomer structure and stability, whereas they affected the trimer association affinity. We determined their relative trimer affinities and biologically relevant substrate efflux activities. When the efflux activity of the AcrB variants was plotted against their relative trimer stabilities, the requirement of a threshold stability for function was observed.
Materials and Methods
Protein Cloning, Expression, and Purification
Plasmid pQE70-AcrB was created during a previous study.21 Mutations were introduced into the acrB gene in plasmid pQE70-AcrB using the QuikChange site-directed mutagenesis kit following the manufacturer’s instructions (Agilent Technologies) and confirmed through DNA sequencing. The plasmid containing the acrB gene or its mutants was then transformed into E. coli strain BW25113ΔacrB for protein expression under basal conditions without induction. Briefly, a single colony from a freshly transformed plate was used to inoculate 3 mL of LB media supplemented with ampicillin and kanamycin and cultured overnight. The overnight culture was used to inoculate 600 mL of fresh LB media supplemented with ampicillin and kanamycin. The culture was incubated with shaking at 37 °C overnight. Cells were harvested the next morning by centrifugation. Protein was purified immediately without freezing the cell pellet.
Protein purification was conducted as described.21 Briefly, the cell pellet was suspended in a lysis buffer (30 mM NaPO4, 0.5 M NaCl, 0.5 mM phenylmethanesulfonylfluoride (PMSF), pH 7.5) and sonicated to break the cells. The cell lysate was centrifuged, and the pellet was suspended in an extraction buffer containing 30 mM NaPO4, 0.5 M NaCl, 1.5% (w/v) Triton, and 0.5 mM PMSF (pH 7.5). The supernatant, containing detergent-solubilized AcrB, was collected and subjected to metal-affinity chromatography. After elution, purified AcrB samples were dialyzed against a dialysis buffer (30 mM NaPO4, 0.1 M NaCl, 0.03% (w/v) DDM, 0.5 mM PMSF, pH 7.5). The expression level of each protein was determined using western blots as described previously.22 Band intensities were quantified using ImageJ.23
Structural Characterization Using Circular Dichroism (CD) and Fluorescence Spectroscopy
CD spectra of purified wild-type AcrB and its mutants were collected as described previously.24 SDS-induced unfolding was monitored using fluorescence spectroscopy as described previously.25 Briefly, unfolding of AcrB was initiated by titrating with a 0.25% (w/v) SDS solution. A fluorescence emission spectrum ranging from 300 to 400 nm was recorded at each titration point on a LS-55 fluorescence spectrometer (PerkinElmer, Inc., Waltham, MA) with an excitation wavelength of 280 nm. The maximum fluorescence intensity of each spectrum was normalized and plotted as a function of the SDS/DDM molar ratio.
| 1 |
A titration curve was fitted with a linear free energy equation to calculate the transition point of unfolding.
Drug Susceptibility Measurement and Ethidium Bromide (EtBr) Accumulation Assay
Drug susceptibility of different AcrB constructs was determined by measuring the minimum inhibitory concentration (MIC) of BW25113ΔacrB containing plasmid encoding each protein as described previously.24 The EtBr accumulation assay was performed following a published protocol.26 The slope of a plot of fluorescence intensity versus time in seconds was obtained as the accumulation rate in seconds–1. In both assays, BW25113ΔacrB containing plasmid pQE70-AcrB and empty vector pQE70 were used as positive and negative controls, respectively.
To compare data from different measurements, MIC values for the four substrates and EtBr accumulation rates were normalized against the controls to convert into residual percent activities. For example, the MIC of erythromycin (Ery) obtained for the strain containing wild-type AcrB (positive control) was 32-fold of the MIC obtained for the strain without AcrB (negative control). For a mutant (for example, AcrBL886G/E893G/W895G) with a MIC of 20 μg/mL, which is 8-fold of the MIC of the negative control, the residual activity is 25%. The EtBr accumulation rate was normalized similarly. For instance, the EtBr accumulation rate of BW25113ΔacrB was reduced by 14.7-fold with the expression of wild-type AcrB. The expression of AcrBL886G/E893G/W895G reduced the accumulation rate of BW25113ΔacrB by 1.6-fold, so the residual activity was calculated by the ratio of 1.6 and 14.7, which yielded 10.9%.
Method to Calculate Apparent Trimer Affinity of AcrB in Detergent Micelles
Freshly purified samples were analyzed using BN-PAGE as described previously.24 ImageJ was used to quantify the trimer and monomer bands, which was used to calculate the percentages of monomer and trimer in the sample.23 The concentration of AcrB was determined using Pierce BCA protein assay kit (Thermo Scientific Inc., TX).
Labeling with AlexaFluor350-Maleimide (AF350-MLM) and Fluorescence Resonance Energy Transfer (FRET) Measurement
Freshly purified CLAcrBW13C or CLAcrBW13C/P223G samples were incubated in a dialysis buffer in the presence of a 20-fold molar excess of AF350-MLM. The reaction mixture was incubated on ice for 1 h, and then Tris-Cl (pH 8.0) was added to a final concentration of 10 mM. Free dye was removed through dialysis. The molar ratio of labeling was estimated from the UV absorbance of the protein (extinction coefficient of 90 000 cm–1 M–1 at 280 nm) and AF350 (extinction coefficient of 19 000 cm–1 M–1 at 346 nm). For FRET measurement, the excitation wavelength was 280 nm. To correct for direct excitation of AF350 at 280 nm, emission spectra of a solution containing 1 μM of AF350-MLM and 1 mM DDT in the dialysis buffer were measured when excited at 280 and 346 nm, respectively. When excited at 346 nm, the emission peak intensity at 440 nm was measured to be 6.6-fold of the peak intensity when excited at 280 nm. For AF350-MLM-labeled AcrB samples, emission spectra were collected at excitation wavelengths of 280 and 346 nm. The peak intensity observed at 440 nm, when excited at 346 nm, was used to back calculate the contribution from the direct excitation at 280 nm. This reference trace was subtracted from the FRET spectra.
Results
AcrB Variants with Mutations at the Transmembrane Intersubunit Interface
Because the goal of this study is to investigate how trimer affinity correlates with AcrB efflux activity, we introduced mutations to disrupt protein–protein interactions at the intersubunit interface. To identify sites for mutation, we analyzed the intersubunit interface using the online server ProtorP27 and AcrB crystal structure 2DHH.18 When two AcrB subunits associate, approximately 47% of the overall accessible surface area (ASA) is contributed by the periplasmic loop-and-tunnel contacting site, and around 12% is contributed by interactions between transmembrane helices (TH). The interaction between the protruding loop from one subunit and residues that collectively form a tunnel around the loop from the other subunit is the major contributor to the binding interface in AcrB. Mutations in the loop or the tunnel that lead to changes of efflux activity have been shown to affect the stability of individual monomers.28 Here, we introduced mutations into the THs at the subunit interface to identify mutations that disrupted the intersubunit interactions between the THs to diminish AcrB trimer association without affecting the structure of individual monomers.
Although each AcrB subunit contains 12 THs, only a few residues from TH1 and TH8 are involved in intersubunit interaction in the crystal structure (Figure 1). On the basis of the analysis of the crystal structure as well as sequence alignment results (Figure 1B), we chose to mutate three invariable residues, Leu886, Glu893, and Trp895, in TH8 into Gly. To create partially active mutants with defects in TH1–TH8 packing, we trimmed the side chains of these three residues by replacing them with Gly. We have constructed single as well as double and triple mutants involving these residues.
AcrB Variants Displayed Various Residual Activities
To determine the effect of mutations on protein activity, we measured the activity of AcrB constructs using both drug susceptibility and EtBr accumulation assays. The drug susceptibility assay was performed as described previously.19 Briefly, E. coli strain BW25113ΔacrB was transformed with plasmids encoding the indicated AcrB constructs. The same strain containing plasmids encoding wild-type AcrB or the empty cloning vector pQE70 was used as positive and negative controls, respectively. MIC was measured under basal expression conditions. Four well-established AcrB substrates, Ery, novobiocin (Nov), rhodamine-6G (R6G), and tetraphenylphosphonium (TPP), were chosen to represent the diversity of AcrB substrates of different polarity, structure, and charged state. In addition, the presence or absence of AcrB causes a large MIC change for these drugs (ranging from 32- to 128-fold), which provides a large dynamic range to reveal differences between the residual levels of activity of different mutants (Table 1). We found that none of the single mutations had an observable effect on the MIC value of any substrate tested, whereas the maximum detrimental effect was observed for the triple mutant, which led to a 32-fold reduction of MIC for Nov and TPP.
Table 1. Monomer Stability of Purified AcrB Constructs and Their Activities in the BW25113ΔacrB Strain.
| MIC
(μg/mL) |
||||||
|---|---|---|---|---|---|---|
| protein | Ery | Nov | R6G | TPP | EtBr accumulation rate (s–1) | transition point ([SDS]/[DDM]) |
| no AcrBa | 2.5 | 5 | 5 | 5 | (1.4 ± 0.4) × 10–1 | |
| AcrB | 80 | 160 | 320 | 640 | (8.9 ± 5.0) × 10–3 | 1.6 ± 0.1 |
| AcrBL886G | 80 | 160 | 320 | 640 | (1.1 ± 0.4) × 10–2 | 1.5 ± 0.1 |
| AcrBE893G | 80 | 160 | 320 | 640 | (7.6 ± 3.0) × 10–3 | 1.5 ± 0.1 |
| AcrBW895G | 80 | 160 | 320 | 640 | (1.0 ± 0.5) × 10–2 | 1.5 ± 0.1 |
| AcrBL886G/E893G | 40 | 40 | 320 | 40 | (4.9 ± 0.6) × 10–2 | 1.4 ± 0.1 |
| AcrBE893G/W895G | 40 | 40 | 320 | 40 | (2.3 ± 0.3) × 10–2 | 1.5 ± 0.1 |
| AcrBL886G/E893G/W895G | 20 | 10 | 160 | 20 | (8.3 ± 0.8) × 10–2 | 1.5 ± 0.1 |
| AcrBC493A/C887A | 80 | 160 | 320 | 640 | ||
| AcrBW13C/C493A/C887A | 80 | 160 | 320 | 640 | ||
BW25113ΔacrB transformed with the empty vector pQE70 was used as the negative control for the activity assay.
Furthermore, we examined the substrate efflux rate of various AcrB constructs using an EtBr accumulation assay. Accumulation of EtBr by live E. coli cells was monitored by following the increase of fluorescence of EtBr.26 The accumulation rate was inversely correlated with the capacity of the efflux pumps to transport EtBr out of bacterial cells. EtBr is a substrate of AcrB, and the EtBr accumulation assay has been used in several studies to reveal the activity of AcrB.26,29,30 We measured the accumulation rate of BW25113ΔacrB containing a plasmid encoding the indicated AcrB constructs. The same strain containing a plasmid encoding wild-type AcrB or the empty cloning vector pQE70 was used as positive and negative controls, respectively. As shown in Table 1, removal of AcrB led to an approximately 15-fold increase in the accumulation rate. Overall, cells expressing a single mutant displayed a similar efflux rate as cells expressing wild-type AcrB, whereas cells expressing the double and triple mutants displayed significantly increased rates of accumulation.
Neither the MIC nor the EtBr accumulation assay directly measures the drug efflux rate. They reflect the combined effects of the drug entering the cell, mainly via diffusion, and exiting via active efflux and passive diffusion. We have also attempted to measure AcrB activity directly using the recently developed Nile Red efflux assay.31 We found that the difference between the positive and negative controls, BW25113ΔacrB containing wild-type AcrB or no AcrB, was not large enough and that the resolution was not high enough to resolve the differences in efflux activity among different mutants (data not shown).
Expression Levels of AcrB Mutants
We measured the relative expression level of all mutants used in this study. Plasmids encoding different AcrB constructs were transformed into BW25113ΔacrB for protein expression under basal conditions. Detergent-solubilized cell membrane samples obtained from freshly prepared log-phase cells were analyzed using SDS-PAGE and western blot analysis. The expression levels of all mutants were similar to the expression level of wild-type AcrB except for AcrBE893G, which appeared to have a significantly higher expression level (Figure 2A). Although it was not clear why the expression level of AcrBE893G was higher, these results indicate that the decrease in the observed drug efflux activity of AcrB mutants was not a result of decreased AcrB expression.
Figure 2.

Characterization of AcrB mutants. (A) Western blot analysis of the expression levels of AcrB constructs. Expression levels of AcrB mutants were normalized to the expression level of wild-type AcrB. Experiments were performed three times. The average value and standard deviation from three repeats are shown. A representative blot is shown. (B) CD spectra of wild-type AcrB (black) and AcrBL886G/E893G/W895G (gray).
Structure Characterization of AcrB Mutants
To examine if the decrease in activity was a result of variation on structures or stabilities of individual monomers, we purified all mutants and collected their far-ultraviolet (UV) circular dichroism (CD) spectra and fluorescence emission spectra. The CD spectra of all variants were similar to the spectrum of wild-type AcrB, indicating that these mutations had little effect on the overall protein structure. The spectra of the triple mutant and wild-type AcrB are shown in Figure 2B as examples. The spectra of the other five mutants are in the Supporting Information. Furthermore, we monitored SDS-induced denaturation by following the intrinsic fluorescence. We have previously shown that the unfolding of AcrB upon SDS titration can be followed by monitoring the decrease of the intrinsic fluorescence.25,28 Both trimer dissociation and monomer unfolding could potentially lead to a change of the intrinsic fluorescence emission. However, we have previously found that the presence of intersubunit disulfide bonds, which strengthen AcrB trimer stability by covalently linking the three subunits, has little effect on the transition point of AcrB unfolding monitored using fluorescence intensity.25 The fluorescence intensity change indicates the exposure of previously hidden aromatic residues, which results from the disruption of the hydrophobic core or dissociation of subunits. There are two potential reasons that may lead to this observation. First, trimer dissociation could be spectroscopically silent. Second, trimer dissociation could have occurred together with the global unfolding of individual subunits. We also found that the change of the fluorescence signal is largely contributed by aromatic residues in the soluble domain.25 The unfolding of the transmembrane domain may not generate a detectable fluorescence signal change, as the change of microenvironment for Trp residues in the transmembrane domain upon unfolding is not likely to be as dramatic as for Trp residues in the soluble domain.
Here, we used this method to probe the stability of each AcrB construct. The fluorescence intensity at 345 nm was normalizing as described in the Materials and Methods. The denaturation profiles of all mutants tested were very similar to the profile of the wide-type AcrB, with similar transition points. Figure 3A shows the unfolding profiles of wild-type AcrB and the triple mutant, AcrBL886G/E893G/W895G, as an example. The unfolding profiles of the other five mutants can be found in the Supporting Information. The transition point was determined by fitting the curve with a linear free energy equation (Table 1). There was no drastic difference among the transition points for all mutants tested, suggesting that the mutations that we introduced did not significantly disrupt the stability of AcrB tertiary structure or, more precisely, the unfolding of the periplasmic domain.
Figure 3.
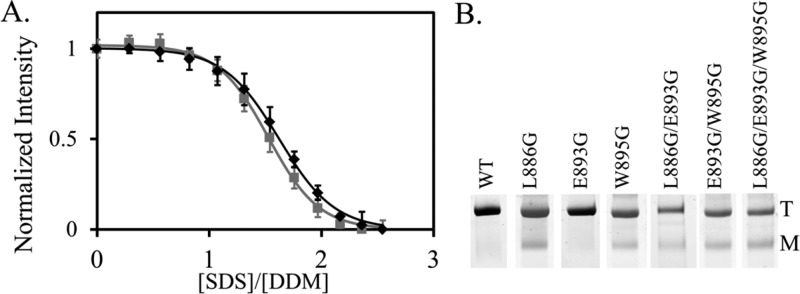
Unfolding and dissociation of AcrB mutants. (A) Representative SDS-induced unfolding profile of wild-type AcrB (black squares) and AcrBL886G/E893G/W895G (gray squares) monitored by fluorescence emission. The curves show the fitting using a single-exponential equation. (B) BN-PAGE analysis of purified AcrB constructs. Positions of monomer and trimer bands are marked by M and T, respectively. Each measurement was repeated at least three times. The average value and standard deviations are shown.
Determination of the Trimer Association Constant of Purified AcrB Variants
In the equilibration between AcrB monomer (M), dimer (D), and trimer (T)
The apparent trimer association constant, Kapp, is
| 2 |
in which MT, MD, and MM are the molar concentrations of trimer, dimer, and monomer, respectively. For membrane proteins, the actual volume that a protein can occupy is limited by the volume defined by detergent micelles.32,33 Therefore, a mole fraction concentration of protein versus detergent is a more proper expression of the association constant of AcrB trimer in detergent micelles
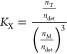 |
3 |
in which nT, nM, and ndet are moles of trimer, monomer, and detergent, respectively.32 Using this equation, we estimated and compared the relative trimer stability of AcrB mutants.
BN-PAGE was used to estimate the trimer to monomer ratio and thus their concentrations in the sample. BN-PAGE is a well-established method that has been used broadly in the characterization of membrane protein oligomers.34,35 All AcrB constructs migrated as a mixture of trimer and monomer. The positions of AcrB monomer and trimer in BN-PAGE have been previously established.19,24 No dimer band was observed in any AcrB construct, indicating that the portion of dimer was negligible and therefore neglected during calculation. The concentrations of AcrB monomer and trimer were obtained by multiplying the total AcrB concentration in the sample by the percent of AcrB existing as monomer or trimer, respectively. The trimer association constant, KX, was calculated using eq 3 from the concentrations of detergent and AcrB monomer and trimer (Table 2). Representative BN-PAGE gel images are shown in Figure 3B.
Table 2. Trimer Stability of AcrB Constructs.
| protein | KX | Kr | ΔΔGX (kcal/mol) |
|---|---|---|---|
| AcrB | (1.6 ± 0.8) × 109 | 1 | |
| AcrBE893G | (3.2 ± 1.4) × 109 | 1.9 | –0.4 |
| AcrBW895G | (2.2 ± 1.3) × 107 | 1.3 × 10–2 | 2.5 |
| AcrBL886G | (1.4 ± 0.5) × 107 | 8.9 × 10–3 | 2.8 |
| AcrBL886G/E893G | (1.0 ± 0.2) × 107 | 6.2 × 10–3 | 3.0 |
| AcrBE893G/W895G | (8.6 ± 1.6) × 106 | 5.3 × 10–3 | 3.1 |
| AcrBL886G/E893G/W895G | (1.0 ± 0.2) × 107 | 6.3 × 10–3 | 3.0 |
Correlation between AcrB Trimer Stability and Activity
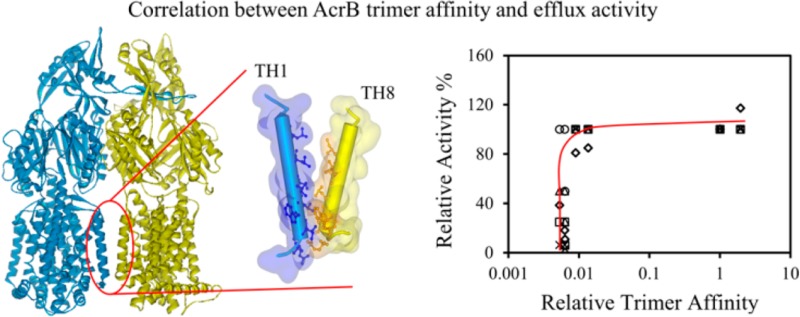
To quantitatively review the relation between the relative trimer stability of a mutant and its biological activity, we plotted the relative trimer affinity (Kr) of each mutant in detergent micelles versus the MIC (Figure 4A) and EtBr accumulation rate (Figure 4B) of wild-type AcrB and its variants. Relative trimer affinity, Kr, was obtained by designating the affinity of wild-type AcrB as 1 (Table 2). Data for all substrates showed the same trend. The correlation between the trimer stability and drug efflux activity was clearly not linear.
Figure 4.

Correlation between AcrB trimer affinity and substrate efflux activity. (A) MIC of four different AcrB substrates determined for BW25113ΔacrB containing a plasmid encoded wild-type AcrB or AcrB mutants plotted against their relative affinity. Substrates tested were Ery (triangles), TPP (crosses), R6G (circles), and Nov (squares). (B) EtBr accumulation rate of BW25113ΔacrB containing a plasmid encoded wild-type AcrB or AcrB mutants plotted against their relative affinity. Each measurement was repeated at least three times. The average value and standard deviations are shown. (C) Normalized MIC values and EtBr accumulation rate (diamonds) plotted against the relative trimer affinity. The same symbols were used for Ery, TPP, R6G, and Nov as those used in panel A.
To compare efflux activity measured using the MIC and EtBr accumulation assays, we normalized MIC values for four substrates and EtBr accumulation rates to obtain the residual percent activity. Various substrates are intrinsically different in their binding affinities and interactions with AcrB; therefore, it was not surprising that the fold change of the MIC and accumulation rate caused by the same mutation was not always the same. However, an overall trend could be observed in which a mutant with lower activity transported all substrates with lower efficiency (Table 1). These residual activities were plotted against the relative trimer stability for the seven AcrB constructs examined in this study (Figure 4C). The switch from no function to full function occurred in a very narrow stability range. A threshold trimer stability was required for efflux activity. This threshold value, in terms of relative affinity, is approximately 0.01, or 2 orders of magnitude lower than the affinity of the wild-type trimer.
Monitoring AcrB Trimer Dissociation/Unfolding Using FRET
FRET is one of the few techniques that are compatible with measurement in both detergent micelles and reconstituted lipid bilayers.36−39 To investigate the usefulness of such a method in the study of AcrB trimer stability, we chose to take advantage of the W13–W895 pair because these amino acids are close to each other at the intersubunit interface (Figure 1A). Trp could serve as a fluorescence donor to form a FRET pair with AF350.40,41 To enable site-specific labeling, we first replaced the two intrinsic Cys in AcrB by Ala to create a Cys-less AcrB (CLAcrB), and we then introduced a unique Cys to replace W13. We have confirmed that these mutations have no observable effect on AcrB activity, indicating that the structure of the AcrB trimer was not disrupted (Table 1). We have also constructed a quadruple mutation by introducing an additional P223G mutation. This mutation has been shown to drastically decrease trimer affinity and protein activity.19 The CLAcrBW13C and CLAcrBW13C/P223G pair was used to evaluate if the FRET method could reveal information about AcrB trimer stability, as the trimer association in CLAcrBW13C/P223G is expected to be weaker.
Freshly purified CLAcrBW13C and CLAcrBW13C/P223G were labeled under the same experimental conditions using AF350-MLM. We found that CLAcrBW13C/P223G was consistently labeled to a much higher level than CLAcrBW13C in all trials that we performed. The AF350 to AcrB molar ratios of labeled CLAcrBW13C and CLAcrBW13C/P223G were approximately 0.2 and 0.7, respectively. We speculate that this difference in labeling efficiency reflects the intrinsic accessibility of C13, as C13 in CLAcrBW13C should be less assessable because of the higher trimer affinity of the protein. To promote AcrB dissociation/unfolding, we titrated small aliquots of concentrated SDS into the samples (Figure 5A). The traces are color-coded to shift from red to blue with the increase in the SDS concentration. As discussed above, the intrinsic fluorescence of AcrB decreased drastically with the increase in the SDS concentration, presumably due to protein denaturation, and the observed change of fluorescence was mainly contributed by Trp residues in the soluble domain. Monitoring the change of intrinsic fluorescence could reflect the process of AcrB gradually losing the folded periplasmic domain, which could be coupled with trimer dissociation. The unfolding profile was very similar as the one we collected for wild-type AcrB, further confirming that the W13C mutation and the labeling did not have a large impact on the overall structure (Figure 5B).
Figure 5.
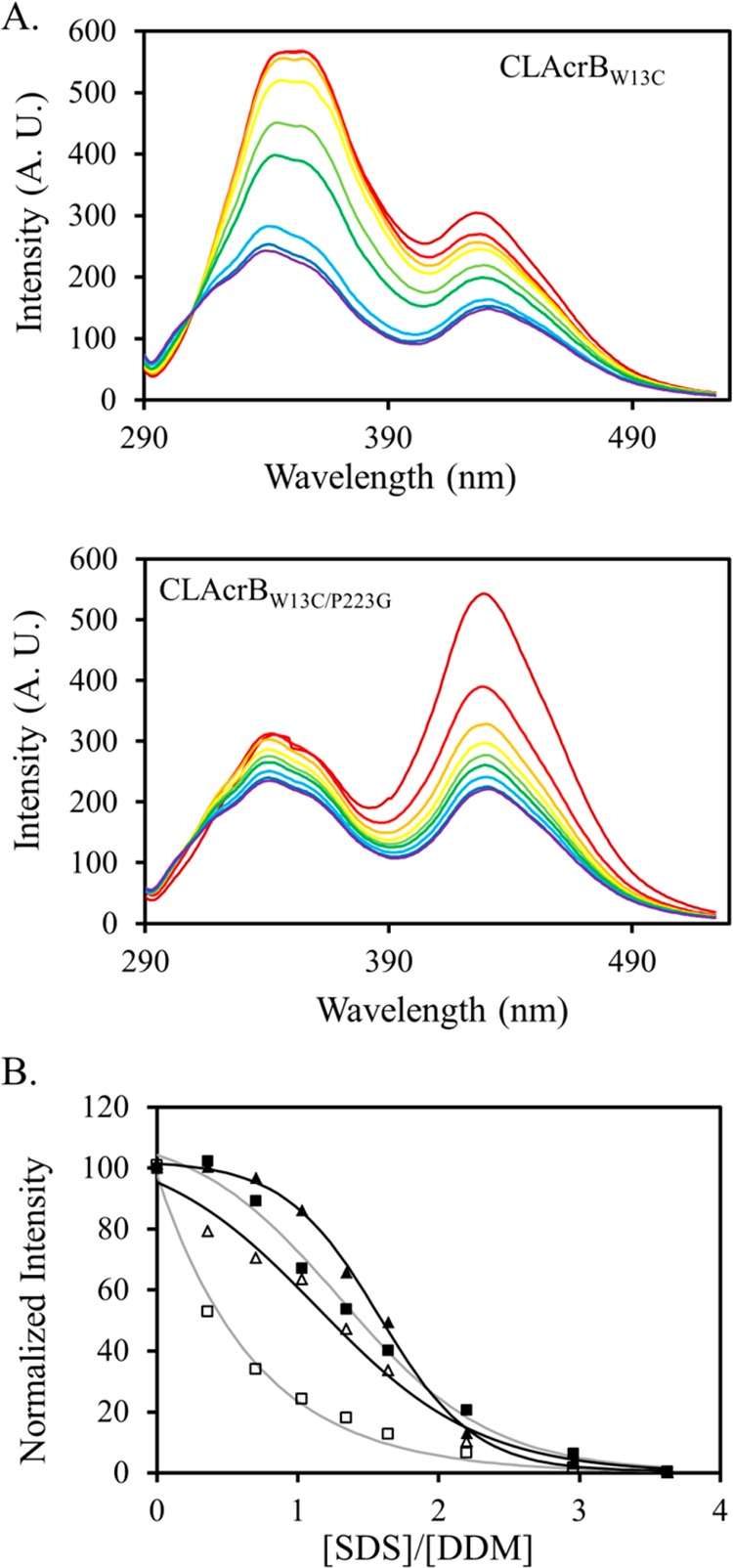
(A) Fluorescence emission of AF350–labeled CLAcrBW13C (top) and CLAcrBW13C/P223G (bottom) excited at 280 nm. The traces are color-coded to shift from red to blue with the increase in the SDS concentration. The two peaks at 345 and 430 nm correspond to the intrinsic fluorescence emission of AcrB and emission from AF350, respectively. Both decrease with the increase in the SDS concentration. (B) Normalized fluorescence intensity of CLAcrBW13C at 345 nm (filled triangles) and 430 nm (open triangles) as well as CLAcrBW13C/P223G at 345 nm (filled squares) and 430 nm (open squares). Although the structure of CLAcrBW13C/P223G began to unfold at a slightly lower SDS concentration than the structure of CLAcrBW13C, the decrease of the FRET efficiency occurred at a much lower SDS concentration for CLAcrBW13C/P223G. The lines are used to illustrate the trend of the data.
Using the change of the intrinsic fluorescence as a beacon of the process of AcrB dissociation/unfolding, we could evaluate the change in the FRET efficiency during the process (Figure 5B). Because CLAcrBW13C and CLAcrBW13C/P223G were intrinsically labeled to different levels, we normalized the AF350 signal changes using eq 1, with the peak intensity in the absence of SDS designated as Ymax and the peak intensity at the highest SDS concentration designated as Ymin. Although the complexity of the system ruled out a numerical interpretation of the trimer affinity, it was clear that the FRET efficiency decreased more dramatically with the increase in the SDS concentration for CLAcrBW13C/P223G.
Discussion
In this study, we obtained one of the first sets of data describing a direct correlation between oligomer association affinity and the activity of a multiple spanning helical membrane protein. First, we have to acknowledge the limitations of the method used in this study. Because the activity was measure in live cells, there are potentially factors that may interfere with the measurement. For example, the AcrAB–TolC complex is not the only drug efflux system in E. coli. However, it is the major one for the compounds used in this study, as revealed by the large difference in their efflux between the positive and negative controls. Furthermore, direct measurement of the oligomer stability of proteins in the cell membrane remains a challenging task. Therefore, we measured the trimer affinity of freshly purified samples in a mild detergent. It is generally believed that the oligomer affinities of membrane proteins are much higher in phospholipid bilayers than in detergent micelles. For example, in two studies, the association constants of model transmembrane helices were found to be 100-fold stronger in lipid bilayers than in detergent micelles.42,43 The relative trimer and monomer content in each mutant was estimated using BN-PAGE. BN-PAGE is a non-equilibrium method. During electrophoresis, the trimeric species and monomeric species are physically separated, which may cause more trimers to dissociate during the process and lead to a negative bias in the measured association constants.
The first important observation of this study is that the stability–activity correlation is not linear. The trimer stability of an AcrB mutant needs to reach a threshold value for the protein to display activity. A drastic increase in activity occurred in a narrow range of trimer stability. To discuss the affinity in thermodynamic energy terms, we calculated the free energy difference of trimer association (ΔΔGX) from the difference in the association constant (Kr) (Table 2). The association energy of the wild-type trimer, −11.9 kcal/mol, was approximately −3.0 kcal/mol higher than the threshold value.
Association energies have been determined for a few model membrane proteins and peptides in detergent micelles and phospholipids (Table 3). Glycophorin A transmembrane fragment (GpATM) was used as the model protein in three such studies, whereas fibroblast growth factor receptor 3 (FGFR3) and the transmembrane fragment of the M2 protein from influenza A virus were used in the other two studies. All model proteins form single transmembrane helices. Both GpATM and FGFR3 dimerize, whereas the M2 protein fragment forms a tetramer. The dimerization energy of GpATM in detergent micelles is 7 kcal/mol. The association constant for the tetrameric M2 protein fragment is 2 × 106 in detergent micelles, corresponding to an association free energy of 8.6 kcal/mol. The association free energy of the wild-type AcrB trimer was approximately 11.9 kcal/mol. This larger association energy could arise from the larger intersubunit interface in AcrB, as it is a much larger protein than the other model proteins.
Table 3. Oligomer Association Constant and Energy Reported in the Literature.
| system | method | KX | ΔGX | ref |
|---|---|---|---|---|
| glycophorin A transmembrane fragment (GpATM) in detergent micelles | analytical ultracentrifugation | –7 kcal/mol | (32), 49 | |
| transmembrane fragment of the M2 protein from influenza A virus in detergent micelles and phospholipid bilayer | thiol-disulfide equilibrium | 2 × 106 in detergent micelles 2 × 108 in lipid bilayer | (42) | |
| fibroblast growth factor receptor 3 (FGFR3) in phospholipid bilayer | FRET | –3 kcal/mol | (50) | |
| GpATM in vesicles derived from mammalian cell membranes | quantitative imaging FRET | –3.9 ± 0.2a kcal/mol | (51) | |
| GpATM in detergent micelles and phospholipid bilayer | steric trap | –7 kcal/mol in detergent micelles –12 kcal/mol in bilayer | (43) |
Standard state was defined as KD = 1/nm2.
On a final note, it is interesting that the oligomer association energies of soluble proteins are in a similar range. In a review of soluble protein quaternary structure stability, the free energy gain per subunit upon oligomerization was reported to be in the range of 2.1 to 6.3 kcal/mol.44 We are not attempting to directly compare the association free energy of membrane proteins with that of the soluble proteins. The experimental systems for soluble and membrane proteins are completely different: while soluble proteins can occupy the entire solution, membrane proteins can exist only in detergent micelles or lipid bilayers. Therefore, although the concentration of soluble protein is represented in units of moles per liter, the concentration of membrane protein needs to be discussed as mole fractions to detergents in the system. However, it is worth mentioning that for both soluble and membrane proteins the oligomerization free energy per subunit is quite modest, which might be necessary to maintain the flexibility of the protein structure and association/dissociation state to suit the functional need. As a matter of fact, a change in the quaternary state often affects a protein’s biological function or activity. Therefore, the stability of protein–protein interaction in such complexes is an important regulator of biological functions.1,2,45−48
Glossary
Abbreviations Used
- RND
resistance-nodulation-cell-division
- CD
circular dichroism
- SDS
sodium dodecyl sulfate
- MIC
minimum inhibitory concentration
- EtBr
ethidium bromide
- DDM
n-dodecyl β-d-maltoside
- ASA
accessible surface area
- TH
transmembrane helix
- Ery
erythromycin
- Nov
novobiocin
- R6G
rhodamine 6G
- TPP
tetraphenylphosphonium
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- BN-PAGE
blue native polyacrylamide gel electrophoresis
- AF350-MLM
Alexa Fluor 350-maleimide
- FRET
fluorescence resonance energy transfer
Supporting Information Available
CD spectra of AcrB mutants and SDS denaturation profiles of AcrB mutants. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the National Science Foundation (MCB 1158036, Y.W.) and National Institutes of Health (1R21AI103717).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Miller S.; Lesk A. M.; Janin J.; Chothia C. (1987) The accessible surface area and stability of oligomeric proteins. Nature 328, 834–836. [DOI] [PubMed] [Google Scholar]
- Blundell T. L.; Srinivasan N. (1996) Symmetry, stability, and dynamics of multidomain and multicomponent protein systems. Proc. Natl. Acad. Sci. U.S.A. 93, 14243–14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet B. K.; Baase W. A.; Kuroki R.; Matthews B. W. (1995) A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. U.S.A. 92, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. (1999) Structure and Mechanism in Protein Science, W. H. Freeman and Company, New York. [Google Scholar]
- Bloom J. D.; Labthavikul S. T.; Otey C. R.; Arnold F. H. (2006) Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U.S.A. 103, 5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields P. A. (2001) Protein function at thermal extremes: balancing stability and flexibility. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 129, 417–431. [DOI] [PubMed] [Google Scholar]
- Cymer F.; Schneider D. (2012) Oligomerization of polytopic α-helical membraneproteins: causes and consequences. Biol. Chem. 393, 1215–1230. [DOI] [PubMed] [Google Scholar]
- Hong H.; Joh N. H.; Bowie J. U.; Tamm L. K. (2009) Methods for measuring the thermodynamic stability of membrane proteins. Methods Enzymol. 455, 213–236. [DOI] [PubMed] [Google Scholar]
- Godoy P.; Molina-Henares A. J.; de la Torre J.; Duque E.; Ramos J. L. (2010) Characterization of the RND family of multidrug efflux pumps: in silico to in vivo confirmation of four functionally distinct subgroups. Microbiol. Biotechnol. 3, 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair J. M. A.; Piddock L. J. V. (2009) Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr. Opin. Microbiol. 12, 512–519. [DOI] [PubMed] [Google Scholar]
- Nikaido H.; Takatsuka Y. (2009) Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 1794, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka Y.; Nikaido H. (2009) Covalently linked trimer of the AcrB multidrug efflux pump provides support for the functional rotating mechanism. J. Bacteriol. 191, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons M. F.; Bokma E.; Koronakis E.; Hughes C.; Koronakis V. (2009) The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. U.S.A. 106, 7173–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova E. B.; Zgurskaya H. I. (2004) AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J. Biol. Chem. 279, 32116–32124. [DOI] [PubMed] [Google Scholar]
- Tamura N.; Murakami S.; Oyama Y.; Ishiguro M.; Yamaguchi A. (2005) Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochemistry 44, 11115–11121. [DOI] [PubMed] [Google Scholar]
- Tikhonova E. B.; Dastidar V.; Rybenkov V. V.; Zgurskaya H. I. (2009) Kinetic control of TolC recruitment by multidrug efflux complexes. Proc. Natl. Acad. Sci. U.S.A. 106, 16416–16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M. A.; Schiefner A.; Eicher T.; Verrey F.; Diederichs K.; Pos K. M. (2006) Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313, 1295–1298. [DOI] [PubMed] [Google Scholar]
- Murakami S.; Nakashima R.; Yamashita E.; Matsumoto T.; Yamaguchi A. (2006) Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443, 173–179. [DOI] [PubMed] [Google Scholar]
- Yu L. L.; Lu W.; Wei Y. N. (2011) AcrB trimer stability and efflux activity, insight from mutagenesis studies. PLoS One 6, e28390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J.; Yu L. L.; Wu M.; Wei Y. N. (2013) Dissecting the function of a protruding loop in AcrB trimerization. J. Biomol. Struct. Dyn. 31, 385–392. [DOI] [PubMed] [Google Scholar]
- Lu W.; Zhong M.; Wei Y. N. (2011) A reporter platform for the monitoring of in vivo conformational changes in AcrB. Protein Pept. Lett. 18, 863–871. [DOI] [PubMed] [Google Scholar]
- Lu W.; Chai Q.; Zhong M.; Yu L. L.; Fang J.; Wang T.; Li H. L.; Zhu H. N.; Wei Y. N. (2012) Assembling of AcrB trimer in cell membrane. J. Mol. Biol. 423, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramoff M. D.; Magelhaes P. J.; Ram S. J. (2004) Image processing with ImageJ. Biophoton. Int. 11, 36–42. [Google Scholar]
- Lu W.; Zhong M.; Wei Y. (2011) Folding of AcrB subunit precedes trimerization. J. Mol. Biol. 411, 264–274. [DOI] [PubMed] [Google Scholar]
- Ye C., Wang Z., Lu W., and Wei Y. (2014) Unfolding study of a trimeric membrane protein AcrB. Protein Sci., [Online early access], DOI: 10.1002/pro.2471, Published Online: April 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Z.; Poole K.; Nikaido H. (2003) Contributions of MexAB-OprM and an EmrE homolog to intrinsic resistance of Pseudomonas aeruginosa to aminoglycosides and dyes. Antimicrob. Agents Chemother. 47, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C.; Damerell D.; Jones S. (2009) ProtorP: a protein–protein interaction analysis server. Bioinformatics 25, 413–416. [DOI] [PubMed] [Google Scholar]
- Yu L.; Lu W.; Ye C.; Wang Z.; Zhong M.; Chai Q.; Sheetz M.; Wei Y. (2013) Role of a conserved residue R780 in Escherichia coli multidrug transporter AcrB. Biochemistry 52, 6790–6796. [DOI] [PubMed] [Google Scholar]
- Viveiros M.; Martins A.; Paixao L.; Rodrigues L.; Martins M.; Couto I.; Fahnrich E.; Kern W. V.; Amaral L. (2008) Demonstration of intrinsic efflux activity of Escherichia coli K-12 AG100 by an automated ethidium bromide method. Int. J. Antimicrob. Agents 31, 458–462. [DOI] [PubMed] [Google Scholar]
- Paixao L.; Rodrigues L.; Couto I.; Martins M.; Fernandes P.; de Carvalho C. C.; Monteiro G. A.; Sansonetty F.; Amaral L.; Viveiros M. (2009) Fluorometric determination of ethidium bromide efflux kinetics in Escherichia coli. J. Biol. Eng. 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert J. A.; Karamian B.; Nikaido H. (2010) Optimized Nile Red efflux assay of AcrAB-TolC multidrug efflux system shows competition between substrates. Antimicrob. Agents Chemother. 54, 3770–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming K. G. (2002) Standardizing the free energy change of transmembrane helix–helix interactions. J. Mol. Biol. 323, 563–571. [DOI] [PubMed] [Google Scholar]
- Sehgal P.; Mogensen J. E.; Otzen D. E. (2005) Using micellar mole fractions to assess membrane protein stability in mixed micelles. Biochim. Biophys. Acta 1716, 59–68. [DOI] [PubMed] [Google Scholar]
- Wittig I.; Braun H. P.; Schagger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428. [DOI] [PubMed] [Google Scholar]
- Heuberger E. H.; Veenhoff L. M.; Duurkens R. H.; Friesen R. H.; Poolman B. (2002) Oligomeric state of membrane transport proteins analyzed with blue native electrophoresis and analytical ultracentrifugation. J. Mol. Biol. 317, 591–600. [DOI] [PubMed] [Google Scholar]
- Nannepaga S. J.; Gawalapu R.; Velasquez D.; Renthal R. (2004) Estimation of helix–helix association free energy from partial unfolding of bacterioopsin. Biochemistry 43, 550–559. [DOI] [PubMed] [Google Scholar]
- Kriegsmann J.; Brehs M.; Klare J. P.; Engelhard M.; Fitter J. (2009) Sensory rhodopsin II/transducer complex formation in detergent and in lipid bilayers studied with FRET. Biochim. Biophys. Acta 1788, 522–531. [DOI] [PubMed] [Google Scholar]
- Reddy L.; Jones L.; Thomas D. (1999) Depolymerization of phospholamban in the presence of calcium pump: a fluorescence energy transfer study. Biochemistry 38, 3954–3962. [DOI] [PubMed] [Google Scholar]
- Adair B.; Engelman D. (1994) Glycophorin A helical transmembrane domains dimerize in phospholipid bilayers: a resonance energy transfer study. Biochemistry 33, 5539–5544. [DOI] [PubMed] [Google Scholar]
- Villamil Giraldo A. M.; Lopez Medus M.; Gonzalez Lebrero M.; Pagano R. S.; Labriola C. A.; Landolfo L.; Delfino J. M.; Parodi A. J.; Caramelo J. J. (2010) The structure of calreticulin C-terminal domain is modulated by physiological variations of calcium concentration. J. Biol. Chem. 285, 4544–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Seeger S. J. (2011) Multidonor deep-UV FRET study of protein–ligand binding and its potential to obtain structure information. Phys. Chem. B 115, 13643–13649. [DOI] [PubMed] [Google Scholar]
- Cristian L.; Lear J. D.; DeGrado W. F. (2003) Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc. Natl. Acad. Sci. U.S.A. 100, 14772–14777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Blois T. M.; Cao Z.; Bowie J. U. (2010) Method to measure strong protein-protein interactions in lipid bilayers using a steric trap. Proc. Natl. Acad. Sci. U.S.A. 107, 19802–19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz I. M.; Langerman N. R.; Darnall D. W. (1970) Quaternary structure of proteins. Annu. Rev. Biochem. 39, 25–62. [DOI] [PubMed] [Google Scholar]
- Ali M. H.; Imperiali B. (2005) Protein oligomerization: how and why. Biorg. Med. Chem. 13, 5013–5020. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S.; Olson A. J. (2000) Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 29, 105–153. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S. (1991) Inside a living cell. Trends Biochem. Sci. 16, 203–206. [DOI] [PubMed] [Google Scholar]
- Nooren I. M. A.; Thornton J. M. (2003) Diversity of protein–protein interactions. EMBO J. 22, 3486–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming K. G.; Ackerman A. L.; Engelman D. M. (1997) The effect of point mutations on the free energy of transmembrane alpha-helix dimerization. J. Mol. Biol. 272, 266–275. [DOI] [PubMed] [Google Scholar]
- You M.; Li E.; Wimley W. C.; Hristova K. (2005) FRET in liposomes: measurements of TM helix dimerization in the native bilayer environment. Anal. Biochem. 340, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Novicky L.; Merzlyakov M.; Hristov T.; Hristova K. (2010) Measuring the energetics of membrane protein dimerization in mammalian membranes. J. Am. Chem. Soc. 132, 3628–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.