

Background: Chemoattractant receptor Fpr2 interacts with host-derived agonist CRAMP and promotes dentritic cell maturation in immune responses.
Results: Deficiency in Fpr2 or CRAMP results in impaired maturation of dendritic cells in vitro and in vivo.
Conclusion: Fpr2 and its agonist CRAMP play a nonredundant role in DC maturation.
Significance: Fpr2 and its agonist CRAMP are potential targets for disease intervention.
Keywords: Cellular Immune Response, Dendritic Cell, G Protein-coupled Receptor (GPCR), Immunology, Inflammation, CRAMP, Fpr2, Maturation
Abstract
Mouse formylpeptide receptor 2 (Fpr2) is a homologue of the human G-protein coupled chemoattractant receptor FPR2, which interacts with pathogen and host-derived chemotactic agonists. Our previous studies revealed reduced allergic airway inflammation and immune responses in Fpr2-deficient (Fpr2−/−) mice in association with diminished dendritic cell (DC) recruitment into the airway and draining lymph nodes. These defects prompted us to investigate the potential changes in the differentiation and maturation of DCs caused by Fpr2 deficiency. Bone marrow monocytes from Fpr2−/− mouse mice incubated with GM-CSF and IL-4 in vitro showed normal expression of markers of immature DCs. However, upon stimulation with the TLR4 agonist LPS, Fpr2−/− mouse DCs failed to express normal levels of maturation markers with reduced production of IL-12 and diminished chemotaxis in response to the DC homing chemokine CCL21. Fpr2−/− DCs also failed to induce allogeneic T-cell proliferation in vitro, and their recruitment into the T-cell zones of the spleen was reduced after antigen immunization. The capacity of Fpr2 to sustain normal DC maturation was dependent on its interaction with an endogenous ligand CRAMP expressed by DCs, because neutralization of either Fpr2 or CRAMP inhibited DC maturation in response to LPS. We additionally observed that the presence of exogenous CRAMP in culture increased the sensitivity of WT mouse DCs to LPS stimulation. The importance of CRAMP for DC maturation was further demonstrated by the observations that DCs from CRAMP−/− mice expressed lower levels of costimulatory molecules and MHC II and exhibited poor chemotaxis in response to CCL21 after LPS stimulation. Our observations indicate a nonredundant role for Fpr2 and its agonist CRAMP in DC maturation in immune responses.
Introduction
Leukocyte trafficking and homing are mediated by G protein-coupled receptors (GPCRs)3 that recognize pathogen and host-derived chemoattractants (1, 2). Some chemoattractant GPCRs are involved in both innate and adaptive immune responses (3) and are essential for development, angiogenesis, and homing of antigen presenting dendritic cells (DCs) (4). Formylpeptide receptors (FPRs) belong to the GPCR family, which is increasingly recognized as important mediators in inflammatory and immune responses (5, 6). FPR subfamily consists of three members, FPR1, FPR2, and FPR3, in humans (5, 6). FPR1 is a high affinity receptor for the bacterial peptide formyl-methionyl-leucyl-phenylalanine (fMLF) and mediates fMLF-induced phagocyte chemotaxis and activation. FPR1 also mediates the leukocyte chemotactic activity of a neutrophil granule protein cathepsin G (7). In vivo, FPR1 plays a role in host defense against infection by Listeria monocytogenes as shown by evidence obtained in mice deficient in the FPR1 homologue, Fpr1 (8). FPR2 and its mouse counterpart Fpr2 are low affinity receptors for fMLF, but they interact with a number of endogenous chemotactic agonist peptides produced by the host in inflammation and immune responses (5, 6). FPR2 or Fpr2 has been reported to also recognize the lipid mediator lipoxin A4 and the N-terminal peptides of annexin I (AnxA1) that trigger anti-inflammatory responses (9, 10). FPR3 in human recognizes a chemotactic peptide fragment derived from Heme-binding protein that chemoattracts DCs (11). In mice, Fpr2 is likely a receptor that functions as both human FPR2 and FPR3 (8, 12).
Among endogenous chemoattractant ligands recognized by FPR2, LL-37 is a human cationic peptide derived from the cathelicidin hCAP-18 (13). In addition to its anti-bacteria and LPS binding activity, LL-37 is chemotactic for leukocytes through interaction with FPR2 (14). LL-37 has also been reported to promote endocytic capacity of DCs and the expression of costimulatory molecules. The mouse homologue of LL37 is CRAMP, which utilizes Fpr2 to induce leukocyte chemotaxis and activation (15). FPR2 and LL37, as well as their mouse counterparts, are proposed to play important roles in the initiation and progression of inflammatory and immune responses.
Our previous study showed severely reduced allergic airway inflammation in Fpr2−/− mice (16). Further investigation revealed that there is a significantly reduced recruitment of Ly6C+ inflammatory DCs into the bronchiolar area in the allergic inflammatory airway of Fpr2−/− or CRAMP−/− mice, suggesting that Fpr2 and its endogenous ligand CRAMP control DC trafficking (1).
However, it is unknown whether Fpr2 and CRAMP are also involved in DC maturation required for normal trafficking in disease states. In this study, we report that Fpr2 and CRAMP are important for the normal maturation of DCs and critical for DC recruitment in inflammatory and immune responses.
EXPERIMENTAL PROCEDURES
Animals
The generation of Fpr2−/− mice was previously described (16). To generate CRAMP−/− mice, CRAMP gene was retrieved from the mouse BAC clone RP23-77I19 into pLMJ235 vector containing the thymidine kinase gene. The targeting vector was then electroporated into C57BL/6 mouse ES cells (17). Recombinant ES cells were injected into blastocysts of albino C57BL/6 mice to generate CRAMP flox-neo mice, which were crossed to β-actin Cre mice on a C57BL/6 background. Heterozygous CRAMP+/− mice were mated to generate homozygous CRAMP−/− mice.4 Mice used in the experiments were 8–10 weeks old. They were allowed free access to standard laboratory chow and tap water. All animals were housed in an air-conditioned room with controlled temperature (22 ± 1 °C), humidity (65–70%), and day/night cycle (12 h light:12 h dark). Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals.
Reagents
FITC-, PE-PerCP-Cy5.5-conjugated, affinity-purified, rat or hamster IgG anti-mouse mAbs against CD16/32, CD11c, I-A/I-E, CD86/B7-2, CD80/B7-1, and CD40 as well as Armenian hamster IgG, rat IgG2b, and rat IgG2b were from eBioscience (San Diego, CA). Rabbit anti-mouse CRAMP Abs and rabbit anti Fpr2 (recognizing amino acids 208–280 in an internal region of Fpr2) were from Santa Cruz (Santa Cruz, CA). Anti-phosphorylated (p)-p38 MAPK (Thr180/Tyr182), anti-p38, anti-IκB, and anti-β-actin Abs for Western blotting were from Cell Signaling Technology (Beverly, MA). Cytokine ELISA kits were from eBioscience (San Diego, CA). GM-CSF and IL-4 were from PeproTech (Rocky Hill, NJ). LPS was from InvivoGen (San Diego, CA). Fpr2 agonist peptides MMK-1 and W-peptide (WKYMVm, W-pep) were synthesized at the Department of Biochemistry of Colorado State University (Fort Collins, CO) (18). Aβ42 peptide was from California Peptide Research (Napa, CA). Mouse CRAMP (cathelin-related antimicrobial peptide) (NH2-ISRLAGLLRKGGEKIGEKLKKIGQKIKNFFQKLVPQPE-OH) was synthesized by New England Peptide LLC (Gardner, MA). Mouse CD11c (N418) MicroBeads and anti-FITC MicroBeads were from Miltenyi Biotec Inc. (Auburn, CA).
Isolation of Mouse Bone Marrow Cells and Generation of BM-derived Dendritic Cells
BM cells were obtained by flushing femurs with PBS as described (1). Red cells were lysed with ACK Lysing Buffer (Cambrex Bio Science, MD). Immature BM-derived DCs (iDCs) were generated by culturing BM nucleated cells (106 cells/well/3 ml) with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) for 6 days (or indicated times). iDCs were stimulated with LPS (10 ng/ml or at the indicated concentrations) for 24 h to obtain mature BM-derived DCs (mDCs). For the activity of CRAMP on DC differentiation, BM nucleated cells from WT and Fpr2−/− mice were cultured in the presence or absence of CRAMP (50 μg/ml) with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) for 4 days. The cells were washed to remove CRAMP and cultured with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) for an additional 2 days and then were stimulated with LPS (100 ng/ml) for 24 h. The expression of costimulatory molecules was measured by FACS, or the cells were measured for chemotaxis induced by CCL21 at the indicated concentration.
RT-PCR
The expression of Fpr2 mRNA in DCs was examined by RT-PCR with primers as follows: sense, 5′-GTGTCCCCTGAATCTGGAAA-3′ and antisense, 5′-TAATTCAGGTGCTGTGGGTG-3′, which yield a 290-base pair (bp) product. Mouse β-actin primers were: sense, 5′-TGTGATGGTGGGAATGGGTGAG-3′ and antisense, 5′-TTTGATGTCACGCACGATTTCC-3′, which yield a 514-bp product. All PCR products were resolved by 1.5% agarose gel electrophoresis and visualized with ethidium bromide staining.
Chemotaxis Assays
Chemotaxis of DCs was measured with 48-well microchambers and polycarbonate filters (5-μm pore size) (NeuroProbe, Cabin John, MD) as described (1). The results are expressed as the means ± S.E. of the chemotaxis index, representing the fold increase in the number of migrated cells in response to chemoattractants over spontaneous cell migration (to control medium).
Flow Cytometry
DCs were preincubated in FACS buffer (PBS containing 1% FCS, 5 mm EDTA, and 0.1% NaN3) containing anti-CD16/32 mAb for 20 min at 4 °C to eliminate nonspecific binding of mAb to the FcγII/IIIR. Thereafter, cells were incubated with appropriate concentrations of mAbs for 30 min at 4 °C. For intracellular staining, the cells were then fixed and permeabilized with BD Cytofix/Cytoperm and resuspended in BD perm/wash buffer and stained with fluorochrome-conjugated anti-cytokine Abs or appropriate negative control Abs. The cells were also stained with rabbit anti-mouse CRAMP Ab followed by a biotinylated-conjugated anti-Ig secondary Ab and PE-conjugated streptavidin. To determine DC recruitment into the spleen after LPS injection, each mouse was injected intraperitoneally with 25 μg of LPS and 2.0 mg of OVA. Spleens were harvested 12 h later. After the lysis of red cells, splenocytes were stained with Abs (anti-mouse CD11c-FITC, CD4-PE, and CD8α-PerCP-Cy5.5 Abs) or anti-mouse CD11c-FITC and CCR7-PE and analyzed with FACS. A pool of three mice was used.
Fluorescence and Confocal Microscopy
DCs stimulated with CCL21 were spun on cover slides and stained with Phalloidin for F-actin and with DAPI for nuclei. DCs on slides were also stained with rabbit anti-mouse CRAMP Ab, followed by a biotinylated anti-Ig secondary Ab and PE-conjugated streptavidin. Samples were analyzed with fluorescence microscopy (Olympus IX 71) and confocal microscopy (Zeiss LSM510 NLO Meta).
Mixed Lymphocyte Reaction
BM-derived iDCs were treated with LPS (100 ng/ml) for 24 h to obtain mDCs with >90% viability. The cells were then treated with 50 ng/ml mitomycin C for 20 min at 37 °C. mDCs (stimulators) were washed and incubated with allogeneic naive CD3+ T-cells from Balb/C mouse spleen (responders) (2 × 105 cells/well) at the indicated ratios in 96-well U-bottom culture plates for 72 h in the presence of 1 μCi of 3H/well thymidine for the last 18 h. The cells were counted for β-emission in a TopCount scintillation counter (Packard Instrument, Downers Grove, IL).
Immunofluorescence and Immunohistology
Mice were injected intraperitoneally with 25 μg of LPS/2.0 mg of OVA/mouse. Spleens were harvested 12 h later. For immunofluorescence, frozen sections were stained with a hamster anti-mouse CD11c Ab followed by a biotinylated anti-Ig Ab (BD Biosciences) with streptavidin-PE and DAPI counter staining (InvitroGen).
ELISA
The supernatants from DCs (2 × 105 cells/ml) stimulated with LPS (1 μg/ml) for 24 h were measured for IL12/IL23 (total p40) by ELISA. Cytokine production by splenocytes from PGN−/OVA immunized mice was determined as described previously (16). Briefly, each mouse was injected intraperitoneally with 2 mg of OVA (Sigma) and 25 μg/mouse PGN (InvivoGen). Spleens were harvested 4 days later, and splenocytes were plated in triplicate in 12-well plates (1 × 106) in 1000 μl of RPMI 1640 supplemented with 10% FBS in the presence of 200 μg/ml OVA for 4 days. The supernatants were then assayed for IL-4, IL-10, and IL-13 by ELISA.
Statistical Analysis
All experiments were performed at least three times. Representative and reproducible results are shown. Statistical analysis was performed with Prism software (GraphPad Software, La Jolla, CA). The values are expressed as the means ± S.E. The significance of the differences between testing and control groups was assessed by Student's t test or one-way analysis of variance where appropriate. p < 0.05 was considered statistically significant.
RESULTS
The Expression of Fpr2 and CRAMP by DCs
We first confirmed the expression and function of Fpr2 in mouse DCs. Fpr2 mRNA was expressed by iDCs and mDCs from WT mice (Fig. 1A). These iDCs and mDCs migrated in response to the Fpr2 ligand Aβ42 (6) (Fig. 1, B and C). CD11c+ DCs isolated from mouse spleens also migrated in response to another Fpr2 ligand MMK-1 (Fig. 1D). In Fpr2−/− mice, neither iDCs nor mDCs expressed Fpr2 mRNA (Fig. 1A), and neither exhibited chemotaxis in response to Fpr2 agonist peptides (Fig. 1, B–D). In addition to Fpr2, iDCs from WT and Fpr2−/− mice expressed similar levels of the Fpr2 ligand CRAMP (Fig. 1, E and F).
FIGURE 1.

The expression of Fpr2 and CRAMP by mouse DCs. BM nucleated cells were isolated from WT and Fpr2−/− mice. The cells were cultured in the presence of GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) for 6 days as iDCs, which were subsequently stimulated with LPS (1 μg/ml) for 24 h as mDCs. A, the expression of Fpr2 mRNA in iDCs and mDCs was examined by RT-PCR. B, loss of Fpr2 agonist Aβ42-induced chemotaxis of iDCs from Fpr2−/− mice. C, loss of Aβ42-induced chemotaxis of mDCs from Fpr2−/− mice. Aβ42 was 75 μg/ml (B and C). D, failure of CD11c+ cells isolated from the spleen of Fpr2−/− mice to migrate in response to Fpr2 agonist MMK-1 (10−5 m). Chemotaxis results are expressed as the chemotaxis index (CI) representing fold increase in cell migration in response to stimulants over medium control (0). * indicates significantly increased migration in response to stimulants over spontaneous cell migration (to control medium, p < 0.05). All mice used were 8-week-old male littermates. Each experiment was repeated three times. E, the expression of CRAMP by iDCs. iDCs were spun on slides and stained with a rabbit anti-mouse CRAMP antibody followed by a biotinylated anti-rabbit IgG secondary antibody and PE-conjugated streptavidin. The cells were counterstained with DAPI for nuclei for analysis with confocal microscopy. Scale bars represent 10 μm. F, the fluorescence intensity of CRAMP+ iDCs. On each slide, 6–12 cells were examined. The results are expressed as the means ± S.E.
Defective in Vitro Maturation of DCs from Fpr2−/− Mice
To investigate the role of Fpr2 in DC maturation, we compared the expression of surface markers by BM-derived DCs from WT and Fpr2−/− mice. BM nucleated cells from Fpr2−/− and WT mice cultured in vitro with GM-CSF and IL-4 for 5 days showed no substantial difference in the expression levels of DC surface CD86 and MHC II molecules (Fig. 2, A, C, and D). However, the chemotaxis responses of iDCs from Fpr2−/− mice to the chemokines CCL3 and CCL4 (ligands for CCR5), as well as CCL20 (a ligand for CCR6), was reduced as compared with WT mouse iDCs (supplemental Fig. S2, A–C), suggesting that, despite the apparent normal expression of iDC markers, the Fpr2−/− mouse iDCs are defective in the function of certain chemokine receptors.
FIGURE 2.

Impaired maturation of DCs from Fpr2−/− mice. A, the expression of CD86 and MHC II by iDCs. B, the expression of CD86 and MHC II by mDCs after stimulation with LPS. C, MFI of CD86+ population in iDCs and mDCs. Results are expressed as the mean ± S.E. D, MFI of MHC II+ cell populations in iDCs and mDCs. The results are expressed as the means ± S.E. (n = 4 mice/group). Each experiment was repeated three times. *, p < 0.05 in C and D. E, reduced IL-12 producing DCs from Fpr2−/− mice. iDCs purified with CD11c+ MACS were stimulated with LPS (1 μg/ml) and GolgiStop (2 μm) for 4 h. The cells were stained with FITC-conjugated anti mouse CD11c and then fixed and permeabilized with BD Cytofix/Cytoperm, followed by intracellular staining with PE-conjugated anti mouse IL-12 (p40/p70) Ab and analysis with FACS. F, percentage of CD11c+ IL-12+ cells in CD11c+ population. The results are expressed as the means ± S.E. (n = 3 mice/group). **, p < 0.01. G, reduced production of IL-12/IL-23 (total p40) by Fpr2−/− mouse DCs. Supernatant from iDCs (2 × 106/ml) stimulated with LPS (1 μg/ml) for 24 h was measured for IL-12/IL23 (total p40) by ELISA. * indicates significantly reduced production of IL-12/IL-23 (total p40) by Fpr2−/− mouse DCs as compared with WT mouse DCs (p < 0.05) (n = 5 mice/group).
After iDCs were treated with LPS for 24 h to promote maturation, mDCs from Fpr2−/− mice showed a considerably lower level expression of the maturation markers CD86 and MHC II as compared with cells derived from WT littermates (Fig. 2, B-s-D). The expression of other DC maturation markers CD80 and CD40 was also reduced on mDCs from Fpr2−/− mice as compared with WT mouse mDCs (supplemental Fig. S1). The defective maturation of DCs from Fpr2−/− mice in vitro was accompanied by a reduced subpopulation of CD11c+ IL-12 (p40/p70)+ cells (Fig. 2, E and F) and the considerably lower production of IL-12/IL-23 (total p40) by Fpr2−/− iDCs after LPS stimulation as compared with cells from WT littermates (Fig. 2G).
It has been documented that in response to maturation stimulation, DCs alter their expression profile of chemokine receptors with down-regulation of CCR1, CCR5, and CCR6 but up-regulation of CCR7 (19). However, we found that both iDCs and LPS-stimulated mDCs from Fpr2−/− mice migrated poorly in response to the CCR7 ligand CCL21 (Fig. 3, A–C) as compared with WT mouse DCs. Consistent with defective CCR7-mediated cell migration, there was a marked reduction in CCL21-induced F-actin polymerization in mDCs from Fpr2−/− mice (Fig. 3, D and E). The defective function of CCR7 in mDCs from Fpr2−/− mice was associated with reduced CCR7 expression as compared with WT mouse mDCs (supplemental Fig. S3). In addition, stimulation of mDCs from Fpr2−/− mice with the CCR7 agonist CCL21 failed to induce p38 phosphorylation (supplemental Fig. S4A), albeit with comparable ERK1/2 activation (supplemental Fig. S4B) as compared with mDCs from WT mice. These results indicate that deficiency in Fpr2 impaired the normal signaling pathways mediated by the DC homing chemokine receptor CCR7.
FIGURE 3.

Reduced CCR7 and antigen-presenting cell function shown by Fpr2−/− mouse DCs in vitro. A, reduced chemotaxis of iDCs from Fpr2−/− mice in response to different concentrations of CCL21. B, reduced chemotaxis of mDCs from Fpr2−/− mice in response to CCL21 (100 ng/ml) at the indicated time points. C, reduced chemotaxis of mDCs from Fpr2−/− mice in response to different concentrations of CCL21. The results are expressed as the chemotaxis index (CI) representing fold increase in cell migration in response to CCL21 over the baseline migration (to medium). *, p < 0.05; **, p < 0.01, significantly reduced migration shown by DCs from Fpr2−/− mice as compared with WT littermate cells. Each experiment was repeated three times. D, CCL21-induced F-actin polymerization in mDCs. mDCs were stimulated with CCL21 (100 ng/ml) for indicated time points. The cells were then spun on cover slides and stained with phalloidin (red) for F-actin and DAPI (blue) for nuclei, followed by analysis with fluorescence confocal microscopy. Scale bar, 50 μm. E, the fluorescence intensity of F-actin+ cells. The results are expressed as the means ± S.E. (n = 6–12 cells/slide). The experiment was repeated two times. ***, p < 0.001. The mice used were 8–12-week-old male littermates. F, allogeneic mixed lymphocyte reaction stimulated by DCs. mDCs were treated with 50 ng/ml mitomycin C for 20 min at 37 °C. mDCs (stimulators) were then washed and incubated with allogeneic naive CD3+ T-cells from Balb/C mouse spleen (responders) at the indicated ratios in 96-well U-bottom culture plates for 72 h in the presence of 1 μCi of 3H/well thymidine for the last 18 h. The cells were then harvested and counted for β-emission in a TopCount scintillation counter. * indicates significantly reduced T-cell proliferation stimulated by mDCs from Fpr2−/− mice as compared with mDCs from WT littermates (p < 0.05). The mice used were 8-week-old females. G, the levels of IFN-γ in culture supernatants of mixed lymphocyte reaction determined by ELISA. The data shown represent the means ± S.E. * indicates significantly reduced level of IFN-γ produced by T-cells stimulated by DCs from Fpr2−/− mice as compared with the cells from WT littermates (p < 0.05) (n = 3 wells/group). The experiment was repeated three times (F and G).
Defective Allogeneic T-cell Stimulatory Capacity of Fpr2−/− Mouse mDCs
The impaired in vitro maturation of DCs from Fpr2−/− mice prompted us to measure their antigen-presenting cell capability. Using WT and Fpr2−/− B6 mouse mDCs as effectors and CD3+ splenocytes from BALB/c mice as responders, we found a markedly reduced ability of Fpr2−/− mouse mDCs to stimulate allogeneic T-cell proliferation as compared with WT mouse mDCs (Fig. 3F). In addition, the production of the effector cytokine IFN-γ was reduced in T-cell cultures in the presence of Fpr2−/− mouse DCs (Fig. 3G). Thus, the key function as antigen-presenting cells was defective in mDCs from Fpr2−/− mice.
Reduced DC Homing in Vivo in Fpr2−/− Mice
Having observed defective maturation of DCs from Fpr2−/− mice in vitro, we investigated whether these defects were associated with reduced DC homing and subsequent immune responses in vivo. Because mDCs from Fpr2−/− mice showed deficiency in CCR7 function, we examined whether mDC trafficking into the spleen of Fpr2−/− mice was impaired in OVA/LPS-induced immune response. Fewer CD11c+ cells (Fig. 4A), a lower number of CD11c+ CD4+ 8− Cells (Fig. 4B), and a lower ratio of CD11c+ CD4+ 8− DCs/CD11c+ CD4− 8+ DCs (Fig. 4C) and CD11c+ CCR7+ cells (Fig. 4D) were present in the spleen of Fpr2−/− mice after OVA/LPS immunization. Furthermore, after LPS/OVA immunization, CD11c+ cell accumulation in the T-cell zones of the spleen was reduced in Fpr2−/− mice as compared with WT mice (Fig. 4, E–G). These results indicate that the defective maturation of DCs from Fpr2−/− mice is associated with their reduced trafficking into lymphoid organs, resulting in impaired host immune responses. We further used OVA and a TLR2/6 agonist PGN to elicit a Th2 response in Fpr2−/− mice (20). Splenocytes from Fpr2−/− mice produced lower levels of IL-4, IL-10, and IL-13 as compared with the cells from WT mice (Fig. 5, A–C). These results were corroborated by earlier observation of defective OVA/LPS elicited Th1 responses in Fpr2−/− mice. Thus, both Th1 and Th2 immune responses were impaired in Fpr2−/− mice.
FIGURE 4.
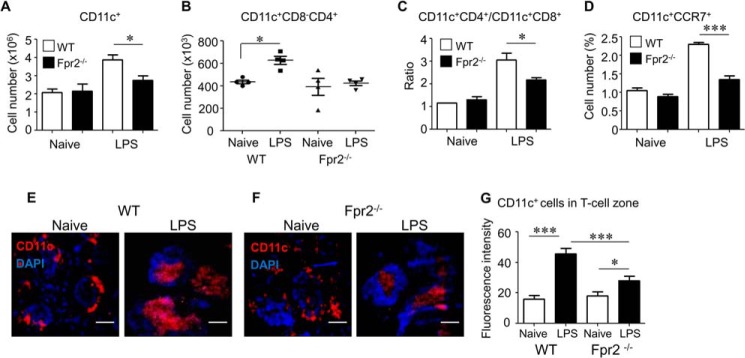
Reduction of DC recruitment into T-cell zones in the spleen in LPS/OVA immunized Fpr2−/− mice. A–D, DC subpopulations in the spleen. A, the percentage of CD11c+ DCs in the spleens. Each mouse was injected intraperitoneally with 25 μg of LPS and 2.0 mg of OVA. Spleens were harvested 12 h later. After the lysis of red cells, splenocytes were stained with three antibodies (anti-mouse CD11c-FITC, CD4-PE, and CD8α-PerCP-Cy5.5) and analyzed with FACS. Armenian hamster IgG-FITC, rat IgG2b-PE, and rat IgG2b-PerCP-Cy5.5 were used as isotype controls. The results are expressed as the means ± S.E. of CD11c+ cell percentage (n = 4 mice/group). B, CD11c+CD4+CD8α− DCs in the spleen. The mice were treated and splenocytes were stained as described for A and analyzed with FACS. The number of CD11c+CD4+CD8α− cell population was calculated. C, the ratio of CD11c+CD4+CD8α−/CD11c+CD4−CD8α+ cells in the spleen. The mice were treated, and splenocytes were stained as described for A and analyzed with FACS. The percentages of CD11c+CD4+CD8α− and CD11c+CD4−CD8α+ cell populations were calculated. The results are shown as the ratio of CD11c+CD4+CD8α− cells over CD11c+CD4−CD8α+ cells. * indicates significantly increased the ratio of CD11c+CD4+CD8α−/CD11c+CD4−CD8α+ cells in the spleen of immunized WT mice as compared with the spleen from Fpr2−/− mice (p < 0.05). D, CD11c+CCR7+cells in the spleen. The mice were treated and splenocytes were stained as described for A and analyzed with FACS. The results are shown as the percentage of CD11c+CCR7+ cells. * indicates significantly reduced percentage of CD11c+CCR7+ cells in the spleen of immunized Fpr2−/− mice as compared with the spleen of WT mice (p < 0.05) (n = 5 mice/group). The experiments were repeated three times (A–D). E and F, reduced recruitment of DCs in the spleen of Fpr2−/− mice after LPS/OVA stimulation. Each mouse was injected intraperitoneally with 25 μg of LPS and 2.0 mg of OVA. Spleens were harvested 12 h later. Frozen spleen sections were stained with a hamster anti-mouse CD11c antibody followed by a biotinylated anti-Ig antibody and streptavidin-PE and DAPI counter staining. Red, CD11c+ cells; blue, nuclei. Scale bars represent 100 μm. G, the fluorescence intensity of CD11c+ cells in the T-cell zone of the mouse spleen. The results are expressed as the means ± S.E. (n = 3 mice/group). 12–20 cells/slide were measured. *, p < 0.05; ***, p < 0.001. The mice used were 8–12-week-old male littermates.
FIGURE 5.

Reduced type 2 cytokine production by splenocytes and defective IκBα activation in DCs from Fpr2−/− mice. A–C, reduced type 2 cytokine production by splenocytes from Fpr2−/− mice. The mice were injected intraperitoneally with 2 mg of OVA and 25 μg of Staphylococcus aureus PGN. The spleens were harvested 4 days later, and splenocytes were plated in triplicate in 12-well plates (1 × 106/well) in 1000 μl of RPMI 1640 supplemented with 10% FBS in the presence of 200 μg/ml OVA for 4 days. The supernatants were then assayed for IL-4 (A), IL-13 (B), and IL-10 (C) by ELISA. The results are expressed as the means ± S.E. (n = 4 mice per group). * indicates significantly reduced Th2 cytokines in the supernatants of splenocytes from Fpr2−/− mice as compared with the cells from WT mice (p < 0.05). The mice used were 8-week-old females. D, reduced degradation of IκB-α in mDCs from Fpr2−/− mice. iDCs were stimulated with LPS (0.1 μg/ml) at the indicated time points (min). The cells were then harvested and lysed. Equal total proteins from same number of cells were electrophoresed and blotted. The protein bands were detected with anti-total IκB-α and β-actin antibodies.
Hyporesponsiveness of mDCs from Fpr2−/− Mice to Activation by LPS
To investigate the mechanisms of Fpr2 participation in DC maturation, we compared TLR4 expression by BM-derived DCs from WT and Fpr2−/− mice, because the level of TLR4 may affect the responsiveness of DCs to LPS-mediated maturation signal. BM nucleated cells from Fpr2−/− and WT mice cultured in vitro with GM-CSF and IL-4 for 6 days showed no significant difference in the expression levels of TLR4 (supplemental Fig. S5A). However, iDCs from WT mice showed rapid phosphorylation (5 min) of p38 MAPK to low levels of (0.1 μg/ml) LPS (supplemental Fig. S5B, left panel). In contrast, much higher levels of LPS (0.5 μg/ml) were required to stimulate a delayed phosphorylation of p38 (30 min) in iDCs from Fpr2−/− mice (supplemental Fig. S5B, right panel), albeit the cells from both mice showed comparable levels of ERK1/2 MAPK phosphorylation (supplemental Fig. S5B). In addition, there was a delayed degradation of IkB-α in Fpr2−/− mouse DCs after LPS stimulation as compared with WT mouse DCs (Fig. 5D). These results indicate that Fpr2 deficiency renders DCs hyporesponsive to the maturation stimulation by LPS.
Contribution of the Endogenous Fpr2 Agonist CRAMP to DC Differentiation
Because Fpr2 recognizes a host-derived agonists CRAMP, which is widely expressed in various cell types including DCs (21), we asked whether CRAMP may play a role in normal DC differentiation, because LL-37, a human homologue of CRAMP, has been shown to regulate DC differentiation (22). After LPS stimulation, the MFI level of CRAMP in DCs from WT mice was significantly reduced as compared with the level in iDCs (Fig. 6, A–C), suggesting that there is a dynamic change in the level of intracellular CRAMP during differentiation from iDCs to mDCs, presumably because of extracellular release. In contrast, mDCs from Fpr2−/− mice showed increased MFI as compared with iDCs (Fig. 6, A–C), suggesting that the Fpr2−/− mouse DCs did not efficiently release CRAMP in response to LPS stimulation.
FIGURE 6.

Fpr2-dependent priming activity of CRAMP on DC differentiation. A, CRAMP expression in iDCs and mDCs from WT and Fpr2−/− mice. B, CRAMP+ cell population (percent) in iDCs and mDCs. C, the MFI of CRAMP+ cells in iDCs and mDCs. The results are expressed as the means ± S.E. (n = 4 mice/group). Each experiment was repeated three times (A–C). D and E, CRAMP enhances CD86+ expression by WT mouse DCs. D, the frequency of CD11c+CD86+ cells analyzed by FACS. E, cumulative results for CD11c+CD86+ cell population. F and G, CRAMP fails to enhance CD86+expression by Fpr2−/− mouse DCs. F, the frequency of CD11c+CD86+ cell population measured by FACS. G, cumulative results for CD11c+CD86+ cells. The results are expressed as the means ± S.E. (n = 3 mice/group) (D–G). Each experiment was repeated three times (D and E). * indicates significantly increased CD11c+CD86+ cell population in CRAMP-primed DCs as compared with control DCs (p < 0.05).
To examine whether CRAMP was capable of regulating DC maturation, we tested the effect of exogenous and endogenous CRAMP in DC culture. WT mouse iDCs cultured with CRAMP (CRAMP-primed) increased CD11c+CD86+ population after stimulation with LPS as compared with DCs cultured in the absence of CRAMP (Fig. 6, D and E). The result was consistent with observations in mice deficient in the CRAMP receptor Fpr2 (Fpr2−/− mice) in which DCs contained a lower proportion of CD11c+CD86+ subpopulation (Fig. 6, F and G). WT mouse mDCs cultured with CRAMP showed a more potent chemotaxis in response to the DC homing chemokine CCL21 (Fig. 7A, left panel) and produced higher levels of IL-12/IL-23 than control mDCs (Fig. 7B, left panel). The effect of exogenous CRAMP on DC maturation was dependent on the receptor Fpr2, because CRAMP failed to show any stimulatory activity on DCs from Fpr2−/− mice (Fig. 7, A, right panel, and B, right panel). In addition, anti-CRAMP and anti-Fpr2 antibodies reduced the CD11c+CD86+ population in DCs from WT mice (Fig. 7, C and D). We further observed that in WT DCs cultured with CRAMP, LPS induced more rapid phosphorylation of IκB-α (5 min) (Fig. 7E). However, CRAMP should be removed 48 h before LPS stimulation, because CRAMP bounds LPS, thus attenuating its activity when present together in the culture (Fig. 7E). These results indicate that both exogenous and endogenous CRAMP plays a significant role in promoting the maturation of DCs via Fpr2.
FIGURE 7.

CRAMP enhances CCR7 function, IL-12 production, and IκB-α phosphorylation shown by WT mouse DCs. A, CRAMP enhances CCR7 function in DCs via Fpr2. CRAMP enhances CCR7 function in WT (left panel), but not Fpr2−/− (right panel), mouse DCs. CCL21 used was 1 ng/ml. The experiment was repeated three times. * indicates significantly increased chemotaxis of DCs cocultured with CRAMP (CRAMP-primed) as compared with DCs cultured in the absence of CRAMP (p < 0.05). B, CRAMP enhances IL-12 production by DCs via Fpr2. BM nucleated cells from WT and Fpr2−/− mice were cultured with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) in the absence or presence of CRAMP (50 μg/ml) for 4 days. The cells were washed to remove CRAMP and continued to be cultured with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) for an additional 2 days. The cells were purified and plated in 24-well cell culture clusters (5 × 106 cells/ml) and then were stimulated with LPS (100 ng/ml) for an additional 24 h. Cell-free supernatant was assayed for IL-12/IL-23 by ELISA. CRAMP enhances the production of IL-12/IL-23 by WT (left panel), but not Fpr2−/−, mouse DCs (right panel). The experiment was repeated three times. * indicates significantly increased IL-12 production by WT mouse DCs cocultured with CRAMP (CRAMP-primed) as compared with DCs cultured in the absence of CRAMP (p < 0.01). C and D, Abs to Fpr2 and CRAMP inhibited WT mouse DC differentiation. BM nucleated cells from WT mice were cultured in the presence or absence of anti-CRAMP or anti-Fpr2 Ab (20 μg/ml) for 6 days. The cells were washed and stimulated with LPS (100 ng/ml) for an additional 24 h. C, the frequency of CD11c+CD86+ cells was measured by FACS. D, cumulative results for CD11c+CD86+ cell population. The results are expressed as the means ± S.E. (n = 3 mice/group). The experiment was repeated three times. * indicates significantly reduced CD11c+CD86+ cell population in anti-CRAMP or anti-Fpr2 Ab treated DCs as compared with control DCs (p < 0.01). E, CRAMP enhances LPS-stimulated IκB-α phosphorylation in WT mouse DCs. BM nucleated cells from WT mice were cultured with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) in the presence or absence of CRAMP (50 μg/ml) for 4 days. The cells were washed to remove CRAMP and were cultured with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) for an additional 2 days. The cells were stimulated with LPS (100 ng/ml) or LPS in combination with CRAMP (50 μg/ml) at the indicated time points. The cells were then harvested and lysed for measurement of phosphorylated IκB-α and GAPDH by Western blotting. CI, chemotaxis index.
To examine whether other Fpr2 agonists might possess the same capacity as CRAMP to promote DC maturation, we examined the effect of AnxA1 (23). BM nucleated cells from WT mice were cultured in the presence of AnxA1 or anti-AnxA1 Ab with GM-CSF and IL-4 for 4 days. The cells were washed to remove AnxA1 and cultured with GM-CSF and IL-4 for an additional 2 days. The cells were then stimulated with LPS (100 ng/ml) for 24 h. We found that WT mouse iDCs cultured with AnxA1 increased CD11c+CD86+ and CD11+CD40+ populations after stimulation with LPS as compared with DCs cultured in the absence of AnxA1 (supplemental Fig. S6, A–C) and showing a more potent chemotactic response to CCL21 (supplemental Fig. S7A). In contrast, AnxA1 failed to show priming activity on DCs from Fpr2−/− mice (supplemental Fig. S7B) as measured by CCR7 function. These results indicate that Fpr2 was capable of interacting with multiendogenous agonists to prime DC maturation.
In an attempt to address whether CCR7 and Fpr2 might cooperate to induced DC migration, we examined the chemotaxis response of WT mouse DCs simultaneously to the ligands for these two GPCRs: CRAMP and CCL21. We found that CRAMP and CCL21 synergistically induced the migration of mDCs from WT mice (supplemental Fig. S8A), thus confirming the capacity of two GPCRs to cooperate, albeit with yet to be clarified biological significance in pathophysiological conditions.
Impaired DC Differentiation in CRAMP−/− Mice
Because CRAMP increases the sensitivity of iDCs to the maturation stimulant LPS via Fpr2, we hypothesized that CRAMP deficiency may also have a profound effect on DC differentiation. iDCs from CRAMP−/− showed no difference in the expression levels of surface costimulatory molecules as compared with the cells from WT mice (Fig. 8, A and B). In contrast, mDCs differentiated from CRAMP−/− mice showed a considerably lower level of expression of CD86, CD80, and MHC II as compared with the cells from WT mice (Fig. 8, C and D) after treatment with LPS. The addition of exogenous CRAMP restored the responses of iDCs from CRAMP−/− mice to LPS (Fig. 8, C and D). Thus, endogenous CRAMP is required for normal DC maturation. CRAMP−/− mouse DCs also showed reduced cell migration in response to CCL21 as compared with WT DCs (Fig. 8, E and F), albeit with a comparable level of CCR7 expression (supplemental Fig. S8, B and C). Taken together, our observations demonstrate the importance of CRAMP and Fpr2 interaction in DC maturation.
FIGURE 8.

Impaired maturation of DCs from CRAMP−/− mice. A and B, the expression of CD86, CD80, and MHC II by iDCs from CRAMP−/− mice. A, the frequency of CD11c+CD86+, CD11c+CD80+, and CD11c+MHC II+ cell populations was measured by FACS. B, cumulative results for CD11c+CD86+, CD11c+CD80+, and CD11c+MHC II+ cell populations. The results are expressed as the means ± S.E. (n = 3 mice/group). C and D, the expression of CD86, CD80, and MHC II by mDCs from CRAMP−/− and WT mice after stimulation with LPS before and after the addition of exogenous CRAMP. C, the frequency of CD11c+CD86+, CD11c+CD80+, and CD11c+MHC II+ cell populations was measured by FACS. D, cumulative results for CD11c+CD86+, CD11c+CD80+, and CD11c+MHC II+ cell populations. The results are expressed as the means ± S.E. n = 3 mice/group. E, reduced chemotaxis of iDCs from CRAMP−/− mice in response to CCL21 (1 ng/ml). F, reduced chemotaxis of mDCs from CRAMP−/− mice in response to CCL21 (100 ng/ml) at the indicated time points after LPS stimulation. The results are expressed as the chemotaxis index (CI) representing fold increase in cell migration in response to CCL21 over the baseline migration (to medium). * and ** indicate significantly reduced migration shown by DCs from CRAMP−/− mice as compared with DCs from WT mice (**, p < 0.01; ***, p < 0.001) (n = 3 mice/group). All experiments were repeated three times.
DISCUSSION
Leukocyte trafficking in inflammatory and immune response is believed to be mediated by chemokine GPCRs. However, these cells display a remarkable functional polyvalency and the ability to adapt to changes in the microenvironment, because of their vast cell surface receptor repertoire that continuously senses the signals in surrounding environment. Recently, studies have shown that multiple chemokine GPCRs are responsible for DC accumulation at the sites of inflammation and immune responses. For instance, the chemokine receptor CCR2 has not only been proven useful as a discriminative marker between inflammatory/classical monocytes (CCR2+Ly6Chigh) and resident/patrolling/nonclassical monocytes (CCR2−Ly6Clow) (24), but it is also actively participating in the recruitment of inflammatory monocytes to sites of infection, trauma, or tumor. However, the trafficking of inflammatory DCs/monocytes in disease states are complex, and receptors other than chemokine GPCRs have also been shown to play key roles in the relay of chemoattractant signals that direct DCs to the final destination of the immune responses. Among these nonchemokine GPCRs, Fpr2 has been shown to be essential for stepwise trafficking of inflammatory DCs in concert with chemokine receptors CCR2 and CCR7 to amplify immune responses (1). In this study, we further found that mouse DCs deficient in Fpr2 are hyporesponsive to LPS with impaired maturation and function, including reduction in the expression of MHC II and costimulatory molecules as well as severely diminished function of the DC homing receptor CCR7. As a consequence, Fpr2−/− DCs failed to stimulate allogeneic T-cell proliferation in vitro and reduced their accumulation in the spleen in both Th1 and Th2 immune responses (1).
FPRs, among numerous chemoattractant GPCRs, exhibit some unique features in host responses to pathogen and endogenous danger signals (5, 6). In human, three functional FPRs have been identified. FPR1 and FPR2 are both expressed by human monocytes and neutrophils, but not mature DCs. On the other hand, FPR3 was only detected on human monocytes and mature DCs and respond to a unique peptide ligand F2L (5, 11). In mouse, the identity of FPR3 analogue is not clear. Our study showed that mouse Fpr2 responded to F2L and neutrophils from Fpr2−/− mice completely lost responses to F2L. These findings in combination with our clear detection of Fpr2 on mature mouse DCs suggest that Fpr2 is likely an analogue for both human FPR2 and FPR3 (12).
In an effort to elucidate the mechanistic basis for the role of Fpr2 in promoting DC differentiation and maturation, we found that an endogenous Fpr2 agonist peptide, CRAMP, by interacting with Fpr2, is required for the functional maturation of DCs. CRAMP is the mouse orthologue of human LL37, which is an anti-microbial peptide contained in neutrophil granules and is also produced by normal epithelial cells (25) and cancer cells (26). In mice, CRAMP enhances OVA-induced immune responses including the production of Th1 and Th2 cytokines (15). Our study showed that CRAMP expressed in DCs promotes DC responses to maturation stimulants. The observations that DCs from Fpr2−/−and CRAMP−/−mice, as well as DCs from WT mice treated with anti-CRAMP or anti-Fpr2 antibodies, showed reduced maturation in response to LPS suggest that CRAMP released by DCs after LPS stimulation may interact with the receptor Fpr2 in an autocrine or paracrine loop in support of DC differentiation. This notion is supported by reduced intracellular CRAMP in DCs treated by LPS.
It is intriguing that under certain conditions, the human CRAMP analogue LL37 could inhibit the activation of DCs by TLR agonists (27). For instance, in the presence of LL37 throughout the culture period, the activation of iDCs by LPS was suppressed with diminished expression of DC markers and CCR7 (27). Also, there is evidence that CRAMP binds and neutralizes LPS. For instance, CRAMP inhibited the capacity of LPS to induce TNF-α expression by mouse macrophages (28) and suppressed the osteoclastogenesis in cocultures with LPS (29). We also found that to obtain optimal priming effect of CRAMP, CRAMP present in the culture should be removed 48 h before LPS stimulation. These results suggest that the effect of LL37/CRAMP on DC differentiation and function is well orchestrated in a time-dependent manner. Although the precise mechanisms for this fine tuning of DC differentiation by LL37/CRAMP require further investigation, one possibility is that LL37/CRAMP may also interact with other cellular receptors such as TLR or P2X7 with diverse signaling pathways and thus different biological consequences. In addition to CRAMP, another Fpr2 agonist AnxA1 has also been reported to play a role in normal DC function. AnxA1 is a member of calcium-dependent phospholipid binding proteins (30) and is expressed in a variety of cell types including iDCs and mDCs from mice (24). In mice deficient in AnxA1, when cultured in vitro, DCs showed diminished expression of maturation markers, decreased chemotaxis in response to chemokines, reduced production of inflammatory cytokines, hyporesponsiveness to LPS stimulation, and impaired capacity to stimulate allogeneic T-cell proliferation (23). AnxA1 was initially reported to activate the prototype FPR family member FPR1 in neutrophils and retain the cells in the blood vessel, thereby reducing their extravasation in response to other chemoattractants at the sites of inflammation (31, 32). Subsequently, AnxA1 and its N-terminal peptides were reported to exert anti-inflammatory effect via activation of human FPR2 or mouse Fpr2. These observations were substantiated by reduction of the anti-inflammatory activity of AnxA1 in Fpr2-deficient mice (31, 33). In our present study, AnxA1 was found to also prime mouse DC responses to LPS-mediated maturation signal. However, another synthetic Fpr2 agonist peptide MMK-1 did not show “priming” activity (data not shown) on mouse DC maturation, presumably because of its much shorter sequence, with a hypothetically simpler structure and different pattern of Fpr2 interaction as compared with CRAMP and AnxA1. It is therefore interesting to further clarify the interaction pattern of Fpr2 with its divergent agonists to better understand their role in pathophysiological conditions.
The biological function of FPR2/Fpr2 in vitro and in disease models has also been a subject of debate. In an earlier report (33), Fpr2 was described as a receptor transmitting inhibitory signals that dampen the host inflammatory responses. This was supported by results obtained by using a purported Fpr2 KO stain in which mice showed exacerbated pro-inflammatory responses to stimulation. However, such observations became inconclusive because of the later correction that the genotype of this Frp2 KO strain was incorrect, and the mice were deleted an additional Fpr2-like receptor (33), which presumably mediates anti-inflammatory responses. In contrast to this erroneous Fpr2 KO genotype, our Fpr2 KO mice clearly showed its capacity as one of the first line host defense molecules against pathological insults, thus mediating pro-inflammatory host responses (16, 34–36). It is therefore important to further delineate the function of Fpr analogues in mice and carefully interpret the results based on the precise mouse genotype and the context of the disease models.
It must be pointed out that despite a clear participation of Fpr2 and its agonist CRAMP in DC maturation and subsequent immune responses, the mechanistic basis underlying the impact of Fpr2 or CRAMP deficiency on molecular genetic changes in the cells remains to be determined. Our preliminary profiling of genes regulated by LPS in DCs revealed reduced expression of multiple genes by DCs from Fpr2−/− mice, including genes coding for TLR2, TLR4, and TLR7. Fpr2−/− mouse DCs also showed aberrant expression of genes coding for a number of transcription factors, cytokines, and chemokines and their receptors.5 Further study is ongoing to elucidate the precise mechanisms for the contribution of Fpr2 pathways to pathphysiological processes.
Supplementary Material
Acknowledgments
We thank Drs. J. J. Oppenheim for critically reviewing the manuscript and C. Lamb and S. Sheriff for secretarial assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant HHSN261200800001E. This work was also supported by the Intramural Research Program of the NCI, National Institutes of Health.

This article contains supplemental Figs. S1–S8.
T. Yoshimura, unpublished data.
K. Chen, data not shown.
- GPCR
- G protein-coupled receptor
- BM
- bone marrow
- CRAMP
- cathelin-related antimicrobial peptide
- DC
- dendritic cells
- FPR and Fpr
- formylpeptide receptor
- fMLF
- formyl-methionyl-leucyl-phenylalanine
- AnxA1
- annexin I
- iDC
- immature BM-derived DC
- mDC
- mature BM-derived DC
- Ab
- antibody
- MFI
- mean fluorescence intensity
- OVA
- ovalbumin
- PE
- phycoerythrin.
REFERENCES
- 1. Chen K., Liu M., Liu Y., Wang C., Yoshimura T., Gong W., Le Y., Tessarollo L., Wang J. M. (2013) Signal relay by CC chemokine receptor 2 (CCR2) and formylpeptide receptor 2 (Fpr2) in the recruitment of monocyte-derived dendritic cells in allergic airway inflammation. J. Biol. Chem. 288, 16262–16273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kristiansen K. (2004) Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Ther. 103, 21–80 [DOI] [PubMed] [Google Scholar]
- 3. Jin T., Xu X., Hereld D. (2008) Chemotaxis, chemokine receptors and human disease. Cytokine 44, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lukacs-Kornek V., Engel D., Tacke F., Kurts C. (2008) The role of chemokines and their receptors in dendritic cell biology. Front. Biosci. 13, 2238–2252 [DOI] [PubMed] [Google Scholar]
- 5. Ye R. D., Boulay F., Wang J. M., Dahlgren C., Gerard C., Parmentier M., Serhan C. N., Murphy P. M. (2009) International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 61, 119–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le Y., Murphy P. M., Wang J. M. (2002) Formyl-peptide receptors revisited. Trends Immunol. 23, 541–548 [DOI] [PubMed] [Google Scholar]
- 7. Sun R., Iribarren P., Zhang N., Zhou Y., Gong W., Cho E. H., Lockett S., Chertov O., Bednar F., Rogers T. J., Oppenheim J. J., Wang J. M. (2004) Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J. Immunol. 173, 428–436 [DOI] [PubMed] [Google Scholar]
- 8. Gao J. L., Lee E. J., Murphy P. M. (1999) Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J. Exp. Med. 189, 657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiang N., Fierro I. M., Gronert K., Serhan C. N. (2000) Activation of lipoxin A4 receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 191, 1197–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levy B. D., De Sanctis G. T., Devchand P. R., Kim E., Ackerman K., Schmidt B. A., Szczeklik W., Drazen J. M., Serhan C. N. (2002) Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat. Med. 8, 1018–1023 [DOI] [PubMed] [Google Scholar]
- 11. Migeotte I., Riboldi E., Franssen J. D., Grégoire F., Loison C., Wittamer V., Detheux M., Robberecht P., Costagliola S., Vassart G., Sozzani S., Parmentier M., Communi D. (2005) Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J. Exp. Med. 201, 83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Devosse T., Guillabert A., D'Haene N., Berton A., De Nadai P., Noel S., Brait M., Franssen J. D., Sozzani S., Salmon I., Parmentier M. (2009) Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J. Immunol. 182, 4974–4984 [DOI] [PubMed] [Google Scholar]
- 13. Tomasinsig L., Zanetti M. (2005) The cathelicidins: structure, function and evolution. Curr. Protein Pept. Sci. 6, 23–34 [DOI] [PubMed] [Google Scholar]
- 14. De Yang, Chen Q., Schmidt A. P., Anderson G. M., Wang J. M., Wooters J., Oppenheim J. J., Chertov O. (2000) LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 192, 1069–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurosaka K., Chen Q., Yarovinsky F., Oppenheim J. J., Yang D. (2005) Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J. Immunol. 174, 6257–6265 [DOI] [PubMed] [Google Scholar]
- 16. Chen K., Le Y., Liu Y., Gong W., Ying G., Huang J., Yoshimura T., Tessarollo L., Wang J. M. (2010) A critical role for the g protein-coupled receptor mFPR2 in airway inflammation and immune responses. J. Immunol. 184, 3331–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee E. C., Yu D., Martinez de Velasco J., Tessarollo L., Swing D. A., Court D. L., Jenkins N. A., Copeland N. G. (2001) A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56–65 [DOI] [PubMed] [Google Scholar]
- 18. Hu J. Y., Le Y., Gong W., Dunlop N. M., Gao J. L., Murphy P. M., Wang J. M. (2001) Synthetic peptide MMK-1 is a highly specific chemotactic agonist for leukocyte FPRL1. J. Leukocyte Biol. 70, 155–161 [PubMed] [Google Scholar]
- 19. Sallusto F., Schaerli P., Loetscher P., Schaniel C., Lenig D., Mackay C. R., Qin S., Lanzavecchia A. (1998) Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 28, 2760–2769 [DOI] [PubMed] [Google Scholar]
- 20. Dillon S., Agrawal A., Van Dyke T., Landreth G., McCauley L., Koh A., Maliszewski C., Akira S., Pulendran B. (2004) A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J. Immunol. 172, 4733–4743 [DOI] [PubMed] [Google Scholar]
- 21. Gallo R. L., Kim K. J., Bernfield M., Kozak C. A., Zanetti M., Merluzzi L., Gennaro R. (1997) Identification of CRAMP, a cathelin-related antimicrobial peptide expressed in the embryonic and adult mouse. J. Biol. Chem. 272, 13088–13093 [DOI] [PubMed] [Google Scholar]
- 22. Davidson D. J., Currie A. J., Reid G. S., Bowdish D. M., MacDonald K. L., Ma R. C., Hancock R. E., Speert D. P. (2004) The cationic antimicrobial peptide LL-37 modulates dendritic cell differentiation and dendritic cell-induced T cell polarization. J. Immunol. 172, 1146–1156 [DOI] [PubMed] [Google Scholar]
- 23. Huggins A., Paschalidis N., Flower R. J., Perretti M., D'Acquisto F. (2009) Annexin-1-deficient dendritic cells acquire a mature phenotype during differentiation. FASEB J. 23, 985–996 [DOI] [PubMed] [Google Scholar]
- 24. Geissmann F., Jung S., Littman D. R. (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82 [DOI] [PubMed] [Google Scholar]
- 25. Ménard S., Förster V., Lotz M., Gütle D., Duerr C. U., Gallo R. L., Henriques-Normark B., Pütsep K., Andersson M., Glocker E. O., Hornef M. W. (2008) Developmental switch of intestinal antimicrobial peptide expression. J. Exp. Med. 205, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Haussen J., Koczulla R., Shaykhiev R., Herr C., Pinkenburg O., Reimer D., Wiewrodt R., Biesterfeld S., Aigner A., Czubayko F., Bals R. (2008) The host defence peptide LL-37/hCAP-18 is a growth factor for lung cancer cells. Lung Cancer 59, 12–23 [DOI] [PubMed] [Google Scholar]
- 27. Kandler K., Shaykhiev R., Kleemann P., Klescz F., Lohoff M., Vogelmeier C., Bals R. (2006) The anti-microbial peptide LL-37 inhibits the activation of dendritic cells by TLR ligands. Int. Immunol. 18, 1729–1736 [DOI] [PubMed] [Google Scholar]
- 28. Nagaoka I., Hirota S., Niyonsaba F., Hirata M., Adachi Y., Tamura H., Heumann D. (2001) Cathelicidin family of antibacterial peptides CAP18 and CAP11 inhibit the expression of TNF-α by blocking the binding of LPS to CD14+ cells. J. Immunol. 167, 3329–3338 [DOI] [PubMed] [Google Scholar]
- 29. Horibe K., Nakamichi Y., Uehara S., Nakamura M., Koide M., Kobayashi Y., Takahashi N., Udagawa N. (2013) Roles of cathelicidin-related antimicrobial peptide in murine osteoclastogenesis. Immunology 140, 344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim L. H., Pervaiz S. (2007) Annexin 1: the new face of an old molecule. FASEB J. 21, 968–975 [DOI] [PubMed] [Google Scholar]
- 31. Gavins F. N., Yona S., Kamal A. M., Flower R. J., Perretti M. (2003) Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood 101, 4140–4147 [DOI] [PubMed] [Google Scholar]
- 32. Lim L. H., Solito E., Russo-Marie F., Flower R. J., Perretti M. (1998) Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc. Natl. Acad. Sci. U.S.A. 95, 14535–14539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dufton N., Hannon R., Brancaleone V., Dalli J., Patel H. B., Gray M., D'Acquisto F., Buckingham J. C., Perretti M., Flower R. J. (2010) Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 184, 2611–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu M., Chen K., Yoshimura T., Liu Y., Gong W., Wang A., Gao J. L., Murphy P. M., Wang J. M. (2012) Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y., Chen K., Wang C., Gong W., Yoshimura T., Liu M., Wang J. M. (2013) Cell surface receptor FPR2 promotes antitumor host defense by limiting M2 polarization of macrophages. Cancer Res. 73, 550–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen K., Liu M., Liu Y., Yoshimura T., Shen W., Le Y., Durum S., Gong W., Wang C., Gao J. L., Murphy P. M., Wang J. M. (2013) Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J. Clin. Invest. 123, 1694–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.