

Abstract
The pathophysiology of stroke is complex. Adaptive and maladaptive signalling occurs between multiple cell types in the brain. There is crosstalk between central and systemic responses. And there are overlapping pathways during initial injury and subsequent repair. These numerous feed-forward and feed-back interactions have made it difficult to translate experimental discoveries into clinical applications. An emerging hypothesis in biomedical research now suggests that contrary to a traditional model, translation may not be efficiently obtained without a rigorous understanding of mechanisms. Hence, to optimize diagnostics and therapeutics for stroke patients, it is necessary to identify and define causal mechanisms. Mirroring the multi-compartment interactions in stroke pathophysiology, bench-to-bedside, and bedside-back-to-bench advances in stroke may be best achieved with inter-disciplinary collaborations between basic research, neuroimaging, and broadly based clinical science. Causation can then be two-fold, ie, dissecting mechanisms and targets, as well as developing future scientists who can blur the boundaries between basic, translational, and clinical research. In systems theory, a critical goal is to distinguish causation from correlation. In stroke research, causation may perhaps be found through a collaborative search for mechanisms.
Keywords: brain injuries, cerebrovascular disorders, translational research
Translational Challenges
There have been numerous failures in clinical stroke trials. Why is it so difficult to translate basic science knowledge into clinical applications? Of course, for such a complex challenge, there are many reasons involved. However, during the past few years, it is increasingly recognized that 3 major issues may be worth discussing.
First, there is now an important movement to improve quality controls in animal model experiments.1,2 Meta-analyses of a wide spectrum of targets and drugs suggest that experimental design in some of the published literature may not be optimal.3 Is it possible that some of the false-positives were because of inadequate attention to key aspects of preclinical drug development, including proper randomization, blinding, and statistical powering of study design? Although this is surely not the only rate-limiting step in our translational process, more careful attention to these basic aspects of research design and execution should improve overall data quality not only in stroke, but also broadly across all of biomedical science.4
Potential limitations in study design may not be exclusive to the domain of experimental models. A second critical issue in stroke translation may pertain to the need to optimize clinical trial design as well. Because patient populations in stroke are heterogeneous, it is unlikely that any single target should be equally effective for all patients. Is it possible that a take-all-comers approach to power through all variations with large numbers and a single dichotomized end point may not necessarily be the most sensitive way to find effective therapeutics?5 Perhaps clinical trials may need to be restructured so that the emphasis is not on the sheer accumulation of large numbers per se but instead on the rigorous definition of target mechanisms and clinical biology in humans so that we can separate responders from nonresponders.
Beyond challenges in preclinical and clinical experimental design, the third and perhaps most important issue may be related to the relevance of mechanisms in translational research. The old standard model of translation was predicated on the principle of so long as it works, we do not need to know how and why. In retrospect, such an approach may not be useful because it is based on the assumption that one is lucky enough to have accidental discoveries. If one has a drug that works, then of course it is unnecessary to understand the how and why. But if an effective diagnostic or therapeutic does not yet exist, then relying on such a translational principle would be somewhat akin to just hoping for good luck. The power of serendipity should never be underestimated. But an emerging hypothesis in biomedical research now suggests that contrary to a traditional model, translation may not be efficiently obtained without a rigorous understanding of mechanisms.6,7 This may be especially true in a complex disease such as stroke.
Complex Mechanisms
Ischemic stroke is caused by a lack of blood flow. Hemorrhagic stroke is caused by a leak in blood vessels. For intracerebral hemorrhage, currently available therapeutic options comprise medical and surgical management of the hematoma. For the early stages of cerebral ischemia, recanalization with tissue-type plasminogen activator or mechanical devices may be efficacious in properly selected patients. However, treatment effects may sometimes be modest, and narrow time windows limit the number of patients who can be treated. Therefore, it is logical for ongoing research to focus on strategies that can amplify reperfusion8 or develop new biomarker methods for finding patients who are most responsive to therapy.9–12 But beyond these efforts to normalize blood flow or restore blood vessel integrity, it has been difficult to find effective treatments that target fundamental cell death processes in injured neurons.
The initial triggers in stroke may be deceptively simple. Loss of blood flow and energy supply or the traumatic stress of an expanding hematoma leads to rapid neuronal cell death in severely damaged core regions. Yet, belying these relatively straightforward early events, the subsequent pathophysiology is highly complex because surrounding penumbral areas continue to succumb. It is now clear that stroke-induced brain injury is not a purely neuronal disease. Multiple signals are induced in all neuronal, glial, and vascular cells.13,14 The penumbra decays over time not just because cell death programs are activated in susceptible neurons, but also because cell–cell signaling in the entire neurovascular unit becomes dysfunctional after stroke onset. Perturbations in astrocytic glutamate reuptake mechanisms may exacerbate excitotoxicity.15 Alterations in pericyte regulation may affect perfusion and blood–brain barrier function.16 And the blood vessel itself may not just be inert plumbing. Instead, the entire cerebrovascular network may act as a trophic organ embedded within brain itself.17,18 Thus, dysfunctional microvessels may lead to dysfunctional parenchyma even in the absence of immediate infarction. Furthermore, vascular signals in stroke may not be unique to the brain itself, and crosstalk between central and systemic responses is beginning to be revealed.19–22 The utility of targeting these pathways may begin to emerge because efforts are now underway to map vascular transcriptome and proteome signatures onto gene databases of human disease.23,24
Beyond the complexity of multiple signals in multiple cell types, another emerging concept in central nervous system disease postulates that there are no sharp boundaries between injury and repair.25 For stroke, this implies that the penumbra is not only actively dying, but may also be actively trying to recover.26 The same mediators that contribute to injury in the acute phase may surprisingly provide the substrates for endogenous repair and remodeling in the delayed phase.27,28 For example, overactivation of N-methyl-D-aspartate (NMDA) signaling leads to excitotoxic neuron death. But without appropriately regulated NMDA signaling, neuroplasticity cannot take place during recovery. It may not be possible to develop effective therapies without understanding these overlapping signals for injury and repair. As stroke mechanisms are further dissected for causality, might it be possible for future targets to decrease acute cell death and also simultaneously promote neurorecovery in the delayed phase after stroke?29–32
Finally, the entire network of cells and signals will be influenced by a whole host of modifying risk factors, including aging, hypertension, hyperlipidemia, diabetes mellitus, metabolic disease, and overall vascular inflammation. For example, in diabetic brains, upregulation of vascular proteases may degrade trophic signaling in neurons.33 Cardiovascular disease may augment negative feedback loops among the brain, heart, and diseased vessels.34,35 In the aged neurovascular niche, inflammatory microglia begin to suppress neurogenesis.36 In aging white matter, oligodendrocyte precursors may lose their endogenous abilities for repair.37 Defining causal mechanisms for stroke cannot be accomplished without the context of all these important stroke comorbidities.
Translation in stroke is difficult because the underlying mechanisms are highly complex. There are adaptive and maladaptive interactions between multiple cell types in the central nervous system, crosstalk between central and systemic responses, and overlapping cascades during initial injury and subsequent repair. Layered over all this interactive and recursive signaling is the influence of multiple factors that modify risk of disease, progression of injury, and response to treatments (Figure 1). Is it possible that our failure to translate may be due, in part, to the intense pressure to jump into clinical trials before the causality of complex mechanisms in stroke is fully elucidated?
Figure 1.
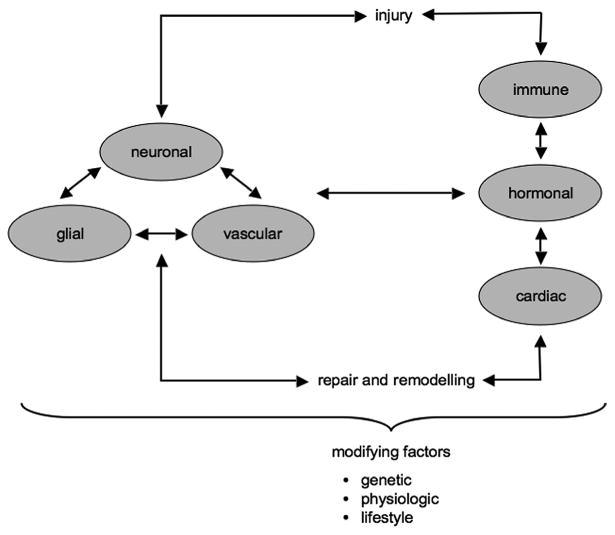
An extended neurovascular unit. Complexity of stroke mechanisms includes multiple recursive interactions among neuronal, glial, and vascular compartments, crosstalk between central nervous system and systemic responses, and overlapping pathways during initial injury and subsequent repair and remodeling. These cascades are further influenced by a wide spectrum of modifying factors that include aging, hypertension, hyperlipidemia, diabetes mellitus, obesity, metabolic disease, and overall inflammation because of genetic, physiological, and lifestyle factors.
Causation via Collaboration
Precisely because the science is so challenging, laboratories have become increasingly specialized. There are many advanced tools to explore causal mechanisms at the molecular, cellular, and systems levels. The convergence of inflammatory mechanisms in disease may be investigated using techniques of molecular evolution.38 Manipulation of neuronal signaling cascades can be performed with precise optogenetic methods.39 Cellular and animal models now span the range from Caenorhabditis elegans and drosophila to rats and mice.40 Transgenic nonhuman primates are being developed.41 In vivo imaging tools can map neurobiology in real time.42 Advances in molecular MRI may eventually allow the investigation of stroke pathophysiology in humans.43 These are all sophisticated and difficult tools. No one single laboratory can effectively use all these powerful technologies. Collaboration is necessary.
Mirroring the multicompartment interactions in stroke pathophysiology, bench-to-bedside and bedside-back-to-bench collaborations may perhaps be attempted with interdisciplinary networks that connect basic neurovascular research, neuroimaging, and broadly based clinical science (Figure 2). To find clinically effective solutions, causality in the underlying mechanisms of stroke must be defined and targeted. In systems theory, a key goal is to separate causation from correlation.44 In translational stroke research, it is likely that causation cannot be determined without extensive collaboration to define mechanisms.
Figure 2.

Representative network of collaborative publications among basic neuroscientists (white circles), clinician-scientists (black circles), and neuroimaging scientists (gray circles) based at Massachusetts General Hospital (drawn from author-name-and-year–based searches of the PubMed database). Various programs including P01 mechanisms funded by National Institute of Neurological Disorders and Stroke may augment opportunities for collaborations. The number of collaborative publications within the network after P01 initiation (2007–2013, right) are higher compared with the number of publications before the P01 program (2001–2006, left).
Opportunities
Translation in stroke research is challenging not because the fundamental biology is incorrect or irrelevant. Translation is difficult because the underlying mechanisms of stroke are highly complex. The risk of stroke, the transitions between injury and remodeling after stroke, and the overall response to potential treatments are mediated by recursive interactions between multiple cell types in central and systemic compartments, all of which are further influenced by an ever-widening array of risk factors. Because of this complexity, the dissection of mechanisms requires a broad approach that can only be achieved with interdisciplinary collaborations. It may be important to also recognize that the most effective collaborations are those that are encouraged but not designed. There is no master narrative that can be centrally directed. Committees and consortia tend to be less nimble. Top-down structures lead to homogeneity of ideas. In stroke research, as in any other intellectual endeavor, accumulated knowledge and the discovery of new knowledge may be a dispersed social phenomenon.45 Hence, to some degree, science will be personal and intuitive. The best opportunities for progress in stroke research will come in new ideas about new mechanisms from new people working together. By encouraging interdisciplinary collaborations that span the molecular gradients from cells and animals to systems and humans, we should strive to define causal mechanisms. These collaborations may also have a beneficial side effect. In pursuit of causation, we may also have the opportunity to train and inspire future scientists who will eventually blur the artificial boundaries between what is currently and incorrectly segregated as basic, clinical, and translational research.
Acknowledgments
Thanks to Kate Doan and Loc Pham for help with the network analysis schematic. I apologize to many colleagues whose work was not discussed because of space limitations. The majority of studies cited here were drawn from Massachusetts General Hospital colleagues because of proximity, not precedence.
Sources of Funding: This work supported by grants from National Institutes of Health, American Heart Association, and the Rappaport Foundation.
Footnotes
Disclosures: None.
References
- 1.Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR Guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res. 2013;4:279–285. doi: 10.1007/s12975-012-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macleod MR, Fisher M, O'Collins V, Sena ES, Dirnagl U, Bath PM, et al. Good laboratory practice: preventing introduction of bias at the bench. Stroke. 2009;40:e50–e52. doi: 10.1161/STROKEAHA.108.525386. [DOI] [PubMed] [Google Scholar]
- 3.O'Collins VE, Donnan GA, Macleod MR, Howells DW. Scope of preclinical testing versus quality control within experiments. Stroke. 2009;40:e497. doi: 10.1161/STROKEAHA.109.550335. [DOI] [PubMed] [Google Scholar]
- 4.Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bath PM, Lees KR, Schellinger PD, Altman H, Bland M, Hogg C, et al. European Stroke Organisation Outcomes Working Group Statistical analysis of the primary outcome in acute stroke trials. Stroke. 2012;43:1171–1178. doi: 10.1161/STROKEAHA.111.641456. [DOI] [PubMed] [Google Scholar]
- 6.Hobin JA, Galbraith RA. Engaging basic scientists in translational research. FASEB J. 2012;26:2227–2230. doi: 10.1096/fj.12-0601ufm. [DOI] [PubMed] [Google Scholar]
- 7.Zoghbi HY. The basics of translation. Science. 2013;339:250. doi: 10.1126/science.1234799. [DOI] [PubMed] [Google Scholar]
- 8.Fan X, Yu Z, Liu J, Liu N, Hajjar KA, Furie KL, et al. Annexin A2: a tissue plasminogen activator amplifier for thrombolytic stroke therapy. Stroke. 2010;41(10 suppl):S54–S58. doi: 10.1161/STROKEAHA.110.596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delgado Almandoz JE, Romero JM. Advanced CT imaging in the evaluation of hemorrhagic stroke. Neuroimaging Clin N Am. 2011;21:197–213. doi: 10.1016/j.nic.2011.01.001. ix. [DOI] [PubMed] [Google Scholar]
- 10.Fisher M, Albers GW. Advanced imaging to extend the therapeutic time window of acute ischemic stroke. Ann Neurol. 2013;73:4–9. doi: 10.1002/ana.23744. [DOI] [PubMed] [Google Scholar]
- 11.Heiss WD. The ischemic penumbra: correlates in imaging and implications for treatment of ischemic stroke. Cerebrovasc Dis. 2011;32:307–320. doi: 10.1159/000330462. [DOI] [PubMed] [Google Scholar]
- 12.Ning MM, Lopez M, Sarracino D, Cao J, Karchin M, McMullin D, et al. Pharmaco-proteomics opportunities for individualizing neurovascular treatment. Neurol Res. 2013;35:448–456. doi: 10.1179/1743132813Y.0000000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang JH, Badaut J, Tang J, Obenaus A, Hartman R, Pearce WJ. The vascular neural network–a new paradigm in stroke pathophysiology. Nat Rev Neurol. 2012;8:711–716. doi: 10.1038/nrneurol.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barreto G, White RE, Ouyang Y, Xu L, Giffard RG. Astrocytes: targets for neuroprotection in stroke. Cent Nerv Syst Agents Med Chem. 2011;11:164–173. doi: 10.2174/187152411796011303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- 17.Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, et al. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JA, Choi KS, Kim SY, Kim KW. Coordinated interaction of the vascular and nervous systems: from molecule- to cell-based approaches. Biochem Biophys Res Commun. 2003;311:247–253. doi: 10.1016/j.bbrc.2003.09.129. [DOI] [PubMed] [Google Scholar]
- 19.Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A. 2012;109:7505–7510. doi: 10.1073/pnas.1121146109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 22.Sharp FR, Jickling GC. Whole genome expression of cellular response to stroke. Stroke. 2013;44(6 suppl 1):S23–S25. doi: 10.1161/STROKEAHA.112.679357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo S, Zhou Y, Xing C, Lok J, Som AT, Ning M, et al. The vasculome of the mouse brain. PLoS One. 2012;7:e52665. doi: 10.1371/journal.pone.0052665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ning M, Sarracino DA, Kho AT, Guo S, Lee SR, Krastins B, et al. Proteomic temporal profile of human brain endothelium after oxidative stress. Stroke. 2011;42:37–43. doi: 10.1161/STROKEAHA.110.585703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lo EH. Degeneration and repair in central nervous system disease. Nat Med. 2010;16:1205–1209. doi: 10.1038/nm.2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- 27.Carmichael ST. Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol. 2006;59:735–742. doi: 10.1002/ana.20845. [DOI] [PubMed] [Google Scholar]
- 28.Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8:491–500. doi: 10.1016/S1474-4422(09)70061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hess DC, Fagan SC. Repurposing an old drug to improve the use and safety of tissue plasminogen activator for acute ischemic stroke: minocycline. Pharmacotherapy. 2010;30(7 pt 2):55S–61S. doi: 10.1592/phco.30.pt2.55S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lam TI, Bingham D, Chang TJ, Lee CC, Shi J, Wang D, et al. Beneficial effects of minocycline and botulinum toxin-induced constraint physical therapy following experimental traumatic brain injury. Neurorehabil Neural Repair. 2013;27:889–899. doi: 10.1177/1545968313491003. [DOI] [PubMed] [Google Scholar]
- 31.Pekcec A, Yigitkanli K, Jung JE, Pallast S, Xing C, Antipenko A, et al. Following experimental stroke, the recovering brain is vulnerable to lipoxygenase-dependent semaphorin signaling. FASEB J. 2013;27:437–445. doi: 10.1096/fj.12-206896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yigitkanli K, Pekcec A, Karatas H, Pallast S, Mandeville E, Joshi N, et al. Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann Neurol. 2013;73:129–135. doi: 10.1002/ana.23734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navaratna D, Fan X, Leung W, Lok J, Guo S, Xing C, et al. Cerebrovascular degradation of TRKB by MMP9 in the diabetic brain. J Clin Invest. 2013;123:3373–3377. doi: 10.1172/JCI65767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ning M, Lo EH, Ning PC, Xu SY, McMullin D, Demirjian Z, et al. The brain's heart—therapeutic opportunities for patent foramen ovale (PFO) and neurovascular disease. Pharmacol Ther. 2013;139:111–123. doi: 10.1016/j.pharmthera.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Impagnatiello F, et al. Aging-induced Nrf2-ARE pathway disruption in the subventricular zone drives neurogenic impairment in parkinsonian mice via PI3K-Wnt/β-catenin dysregulation. J Neurosci. 2013;33:1462–1485. doi: 10.1523/JNEUROSCI.3206-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyamoto N, Pham LD, Hayakawa K, Matsuzaki T, Seo JH, Magnain C, et al. Age-related decline in oligodendrogenesis retards white matter repair in mice. Stroke. 2013;44:2573–2578. doi: 10.1161/STROKEAHA.113.001530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Q, Zmasek CM, Godzik A. Domain architecture evolution of pattern-recognition receptors. Immunogenetics. 2010;62:263–272. doi: 10.1007/s00251-010-0428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Packer AM, Roska B, Häusser M. Targeting neurons and photons for optogenetics. Nat Neurosci. 2013;16:805–815. doi: 10.1038/nn.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Götz J, Ittner LM. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki E, Suemizu H, Shimada A, Hanazawa K, Oiwa R, Kamioka M, et al. Generation of transgenic non-human primates with germline transmission. Nature. 2009;459:523–527. doi: 10.1038/nature08090. [DOI] [PubMed] [Google Scholar]
- 42.Devor A, Sakadžić S, Srinivasan VJ, Yaseen MA, Nizar K, Saisan PA, et al. Frontiers in optical imaging of cerebral blood flow and metabolism. J Cereb Blood Flow Metab. 2012;32:1259–1276. doi: 10.1038/jcbfm.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deddens LH, Van Tilborg GA, Mulder WJ, De Vries HE, Dijkhuizen RM. Imaging neuroinflammation after stroke: current status of cellular and molecular MRI strategies. Cerebrovasc Dis. 2012;33:392–402. doi: 10.1159/000336116. [DOI] [PubMed] [Google Scholar]
- 44.Sugihara G, May R, Ye H, Hsieh CH, Deyle E, Fogarty M, et al. Detecting causality in complex ecosystems. Science. 2012;338:496–500. doi: 10.1126/science.1227079. [DOI] [PubMed] [Google Scholar]
- 45.Hayek FA. The use of knowledge in society. Am Econ Rev. 1945;35:519–530. [Google Scholar]