

Abstract
Vangl2 is one of the central proteins controlling the establishment of planar cell polarity in multiple tissues of different species. Previous studies suggest that the localization of the Vangl2 protein to specific intracellular microdomains is crucial for its function. However, the molecular mechanisms that control Vangl2 trafficking within a cell are largely unknown. Here, we identify Gipc1 (GAIP C-terminus interacting protein 1) as a new interactor for Vangl2, and we show that a myosin VI-Gipc1 protein complex can regulate Vangl2 traffic in heterologous cells. Furthermore, we show that in the cochlea of MyoVI mutant mice, Vangl2 presence at the membrane is increased, and that a disruption of Gipc1 function in hair cells leads to maturation defects, including defects in hair bundle orientation and integrity. Finally, stimulated emission depletion microscopy and overexpression of GFP-Vangl2 show an enrichment of Vangl2 on the supporting cell side, adjacent to the proximal membrane of hair cells. Altogether, these results indicate a broad role for Gipc1 in the development of both stereociliary bundles and cell polarization, and suggest that the strong asymmetry of Vangl2 observed in early postnatal cochlear epithelium is mostly a ‘tissue’ polarity readout.
Keywords: Polarity, Traffic, Actin, Cochlea, Van Gogh-like 2, Gipc1, Myosin VI, Planar polarity, Mouse, Rat
INTRODUCTION
In epithelia, two different, yet complementary, types of polarity regulate the orientation of individual cells. Apical-basal polarity refers to heterogeneity within a cell along the axis that extends between the basement membrane and the luminal surface with marked differences often observed between the luminal (apical) and basal and lateral (basal) surfaces. By contrast, planar cell polarity (PCP) refers to the uniform orientation of cells or cellular components within a horizontal plane of cells. The axis of orientation is therefore orthogonal to the axis of apical-basal polarity. In Drosophila, six ‘core genes’ were identified as a group of genes that function as a conserved signaling cassette for the specification of PCP: Van Gogh (Vang; also known as strabismus), flamingo (fmi; starry night), frizzled (fz), dishevelled (dsh), prickle (pk) and diego (Seifert and Mlodzik, 2007; Wang and Nathans, 2007; Zallen, 2007). In mammals, targeted or spontaneous mutations in homologs of these genes, such as cadherin EGF LAG seven-pass G-type receptor 1 (Celsr1), Van Gogh-like 2 (Vangl2), dishevelled1/2 (Dvl1/2) and Frizzled 3/6 (Fz3/6; Fzd3/6 – Mouse Genome Informatics), lead to defects in developmental processes that are thought to be mediated through PCP signaling, such as disruptions in closure of the neural tube and in the uniform orientation of stereociliary bundles in the inner ear (Montcouquiol et al., 2003; Curtin et al., 2003; Wang et al., 2005; Wang et al., 2006). The sensory epithelium of the mammalian cochlea, the organ of Corti (OC), comprises multiple cell types including mechanosensory hair cells (HCs) and non-sensory supporting cells (SCs). A bundle of modified microvilli, referred to as a stereociliary bundle, projects from the luminal surface of each HC, and all stereociliary bundles are orientated in the same direction, defining PCP within the epithelium. Defects in stereociliary bundle orientations have been used not only to demonstrate conservation of the PCP signaling pathway between vertebrates and invertebrates, but also to identify novel, mammalian-specific PCP components (Montcouquiol et al., 2003; Lu et al., 2004).
In both invertebrates and vertebrates, the establishment of PCP often correlates with the asymmetric localization of core planar polarity proteins to the proximal (Vang and Pk) and distal (Fz, Dvl and Diego) apicolateral membranes (Seifert and Mlodzik, 2007). However, the molecular mechanisms that dictate these asymmetric distributions are not clear (Tree et al., 2002; Bastock et al., 2003; Takeuchi et al., 2003; Torban et al., 2004; Das et al., 2004; Jenny et al., 2005). The mechanisms controlling PCP protein trafficking are therefore of interest, and the elegant and sensitive read-out of disruptions in PCP within the mammalian cochlea make this an ideal system in which to study these mechanisms.
MATERIALS AND METHODS
Plasmids constructs
Vangl2 full length was amplified from mouse cochlea and cloned into pEGFPC3 or pCLIG (Montcouquiol et al., 2006), or DsRed-monomer (Clontech). Site-directed mutagenesis was used to generate Vangl2 mutants lacking the last four amino acids (aa) (Vangl2Δ4), and the last 12 aa (Vangl2Δ12) (QuickChange, Stratagene). Rat myc-Gipc1 (pCM Vtag3C) and mouse synectin/GIPC (pEYFP-N1) cDNAs were obtained from R. Lefkowitz (Howard Hughes Medical Institute, Durham, NC, USA) and A. Horowitz (Dartmouth Medical School, Lebanon, NH, USA), respectively. Site-directed mutagenesis was used to induce mutations in the hydrophobic pocket of the PDZ domain (Gipc1PDZ1dead, LGL/AAA, aa 143-145). pSUPER.gfp/neo vector (sh-GFP) and pSUPER.gfp/neo Gipc1 (shGIPC1a-GFP, shGIPC1b-GFP) were obtained from R. Wenthold (NIDCD, NIH, Bethesda, USA). GFP-Myosin VI pEGFP-C1 was obtained from T. W. Hasson (UCSD, San Diego, USA).
Antibodies
The following primary antibodies were used: anti-Vangl2 (1:500; Montcouquiol et al., 2006), anti-Eea1 (1:500, BD Biosciences), anti-myc (1:1000, Covance, Ramona, CA, USA), anti-Gipc (M. Farquhar, UCSD, San Diego, USA, 1:800 and Proteus Biosciences, 1:400), anti-myosin VI (1:200, Proteus Biosciences), anti-β-catenin (1:1000, Chemicon International, Temecula, USA), anti-green fluorescent protein (GFP) (1:1000, Chemicon). Secondary antibodies were: Alexa Fluor-546/647 goat anti-rabbit and Alexa Fluor-546/647 goat anti-mouse (1:1000, Invitrogen), ATTO 647N anti-rabbit (1:400, Sigma), Star 635 anti-rabbit (Abberior) and Mega 520 anti-mouse (Sigma).
Yeast two-hybrid screening
The C-terminal portion of Vangl2 (aa 438-521) was used as a bait for the screening and was subcloned into pGBTK7 vector (Clontech) in-frame with the Gal4 DNA-binding domain (Vangl2 DNA-DBD). Yeast two-hybrid screening and assays were performed as described in the Clontech protocol (Matchmaker two-hybrid System). AH109 cells expressing GAL4-Vangl2 were combined with Y187 cells expressing an embryonic mouse cochlea cDNA library (Montcouquiol et al., 2006).
Construction and screening of PDZ-domain candidates
PDZ domains of interest [PDZ domain boundaries were obtained by cross-searching Interpro (V18), PFAM(V23) and SMART version 5.0 (http://smart.embl-heidelberg.de)] were cloned in a Gateway pDONR vector and then transferred into the AD vector pACT2GW by Gateway recombinational cloning and transfected into the haploid Y187 yeast strain. Interactions between each PDZ domain and the product of pGBTK7-Vangl2, pGBTK7-Vangl2Δ4 transfected into the haploid yeast strain AH109 were tested through mating of the two yeast strains.
Cell culture and transfections
HEK293T and COS-7 cells were obtained from American Type Culture Collection (ATCC) and were cultured in DMEM (Gibco BRL) supplemented with fetal bovine serum and antibiotics. Transfections were carried out using calcium phosphate co-precipitation (Sans et al., 2003). The cells were then cultured for an additional 24 hours before immunostaining. For endocytosis, 24 hours after transfection, the cells were incubated with 50 μg/ml Alexa Fluor-568-conjugated transferrin (Invitrogen) in DMEM with L-glutamine and bovine serum albumin, for 3 minutes at 37°C.
Cell lysis and subcellular fractionation
Twenty-four hours after transfection with 10 μg of indicated plasmids, HEK293T cells were harvested and suspended in cold 10 mM HEPES buffer. A sucrose gradient fractionation solution was added (320 mM sucrose, 20 mM HEPES, 5 mM EDTA), with protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA) (buffer A), and homogenized (Dounce homogenizer) to produce the homogenate (H). The homogenate was centrifuged (8 minutes at 1000 g), and the supernatant collected; the pellet was rinsed in buffer A, centrifuged again (7 minutes at 1000 g), and the final pellet was homogenized in buffer A (P1=nuclear pellet). The supernatant from the first centrifugation was centrifuged again twice (7 minutes at 1000 g then 20 minutes at 10,000 g). The pellet (P2=plasma membrane proteins) was homogenized in buffer A. The remaining supernatant was centrifuged (2 hours at 140,000 g) and the supernatant (S3=cytosol) and the pellet (P3=microsomes: trans-golgi, endoplasmic reticulum, vesicles, all remaining small membranes) were collected in buffer A. All the final pellets and lysates were resuspended in 2× SDS sample buffer. This experiment was carried out in triplicate.
Co-immunoprecipitation and immunoblotting
Detergent extracts from olfactory bulbs of newborn mice or cleared transfected cells were preincubated with specific antibodies to Vangl2, Gipc1 or GFP for 1 hour, then antibodies were immobilized on Protein A/G agarose beads (UltraLink resin, Thermo Scientific) overnight at 4°C. Washed beads were eluted with sample buffer and immunoprecipitated proteins were analyzed by SDS-PAGE and immunoblotting. Anti-Vangl2, anti-Gipc1, anti-MyoVI (1:500 each), anti-myc, anti-GFP (1:5000) and anti-Gad65/67 (1:500, Chemicon) antibodies were used for these experiments. Immunoreactive proteins were visualized using a chemiluminescence-based immunodetection of horseradish peroxidase (HRP) (Amersham).
Mutants
Fixed tissue of Snell's waltzers mutant mice were a generous gift of Karen Avraham (Tel Aviv University, Israel). For comparison analysis, cochleae from control and mutant animals were immunolabeled in the same tube.
Dissection and in vitro electroporations
Pregnant Sprague Dawley rats and CD1 mice were euthanized according to the European Communities Council Directives (86/609/EEC) and the French National Committee (87/848) recommendations. Embryonic day (E) 16.5 rat and E14.5 mice cochleae were dissected in cold HBSS (Invitrogen) as described previously (Montcouquiol et al., 2006).
In utero electroporation
For in utero electroporation, the PGK promoter of pSUPER.gfp/neo vector was replaced with the human elongation factor 1-α promoter (EF1-α; EEF1A1 – Human Gene Nomenclature Database). pSUPER-modified constructs were injected and electroporated into E11.5 mouse otocysts as described previously (Brigande et al., 2009). The organs of Corti were harvested seven days later for analysis by immunocytochemistry.
Microscopy
Immunocytochemistry on cochleae were carried out as described previously (Montcouquiol et al., 2008), except for images in Fig. 7A,B for which we used 10% trichloroacetic acid (TCA) for 1 hour at 4°C. We observed an overall stronger staining intensity with TCA fixation, coupled with a small disruption of the integrity of the tissue (Hayashi et al., 1999). For optical sectioning of whole mounts, image acquisition were obtained on an epifluorescence microscope system (AxioVision Z1; Carl Zeiss) fitted with an Apotome slide module with a 63× NA 1.4 Plan-Apochromat objective and a digital camera (AxioCam MRm; Carl Zeiss), or on a confocal microscope (TCS SP5; Leica). Imaging was carried out using a z-step from 0.21 to 0.4 μm. Stimulated emission depletion microscopy (STED) microscopy was performed with a TCS STED microscope (Leica), with a 63× 1.4 NA oil immersion and a 100× 1.4 NA oil immersion STED objective. Images were processed using Volocity software (PerkinElmer). Deconvolution was carried out with Huygens 4.2 using three iterations. For quantification of hair cell/bundle phenotype, we defined a PCP phenotype as a misorientated hair bundle within the plane of the epithelium but with a morphologically intact bundle.
Fig. 7.
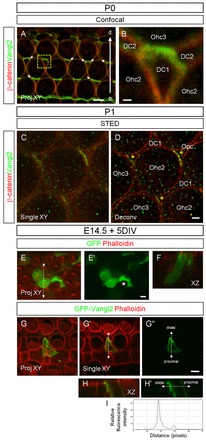
Vangl2 is enriched on the SC side at P0. (A ,B) Vangl2 (green) is asymmetrically accumulated at the junction between SC and HC (phalloidin, red). Arrows point to zones where SC membranes that are not in contact with a HC with strong Vangl2 enrichment. B shows a magnification of the boxed region in A. (C,D) STED microscopy (single plane view, XY) reveals numerous foci of Vangl2 expression and shows a strong enrichment of Vangl2 on the SC side. Asterisks indicate Vangl2 enrichment in SCs. D is the same image as in C after 3D deconvolution. (E-H′) Surface view projection (Proj XY), single plane view (single XY) and z-stack series (XZ) of the organ of Corti from rat cochlear cultures electroporated with GFP and GFP-Vangl2 constructs (left panels), and labeled with anti-GFP antibody (green) and phalloidin (red). (E-F) The empty vector expressing GFP alone fills the cytoplasm of transfected HCs and SCs (E′, asterisk). (G-H′) Full-length GFP-Vangl2 accumulates distally at the membrane of a SC. (H,H′) z-stack series along the dotted line in G′ and G″, respectively, showing the basolateral plasma membrane localization of GFP-Vangl2. (I) The corresponding line scan for GFP-Vangl2 expression along the dotted line in H′. Scale bars: in A,E-H′, 4 μm; in B, 0.5 μm; in C,D, 1 μm.
Colocalization analysis
Three 80×80 pixel regions were selected in the cytoplasm from each COS-7 cell. In each area, the value of colocalization was defined as the percentage ratio between the number of positive vesicles for GFP-Vangl2, GFP-Vangl2Δ4 and the number of myc-Gipc1-, Eea1- or transferrin-positive vesicles using Axiovision software (Zeiss). Measurements from three areas were averaged per cell. Means from different cells were averaged to obtain a final mean. Student's t-test was used in order to determine statistical significance.
RESULTS
Identification of Gipc1 as a novel Vangl2-interacting protein
To identify potential interacting partners of Vangl2, the C-terminal (C-ter) portion of Vangl2 was used as bait (Vangl2 C-ter, aa 438-521) in a yeast two-hybrid screen of an embryonic cochlear library (Fig. 1A). The bait included the last four amino acids of Vangl2 (-ETSV), which encode a PDZ-binding motif (PDZ-BM; Fig. 1A, orange). Thirteen of the 63 clones identified as interacting with Vangl2 encoded a portion of the protein Gipc1. Gipc1 is a cytoplasmic scaffold protein with an N-terminal (N-ter) proline-rich domain (PRD), a central type II class PDZ domain, and a C-ter acyl carrier protein (ACP) domain (Fig. 1B). We confirmed the interaction using a yeast two-hybrid assay with two of the thirteen clones, GIPC1C1 and GIPC1C15 (Fig. 1C). The deletion of the PDZ-BM (Vangl2Δ4), strongly reduced the interaction with Gipc1 in the yeast two-hybrid assay, but a further deletion in the C-ter of Vangl2 (Vangl2Δ12) was needed to eliminate the interaction (Fig. 1C). These results suggest that the Vangl2 PDZ-BM can bind Gipc1 but that, at most, eight aa upstream the PDZ-BM also participates in this interaction. The previously characterized interaction between Scribble1 (Scrib1) and Vangl2 was used as a positive control (Montcouquiol et al., 2006). Deletion assays demonstrated that the central PDZ domain of Gipc1 interacts with Vangl2, whereas the PRD and ACP domains do not (Fig. 1D). We also tested the interaction of our bait on a group of candidate molecules with PDZ domains. Results confirmed that Gipc1, along with Scrib1, Dlg or Magi are amongst the strongest interactors for the last 83 aa of Vangl2 (supplementary material Table S1). All of the positive interactions were lost after deletion of the last four or 12 aa of Vangl2. We confirmed an interaction between Vangl2 and Gipc1 using co-immunoprecipitation (Fig. 1E,F) and glutathione s-transferase (GST)-pull down assays (data not shown). This interaction depends on the integrity of PDZ-BM of Vangl2 and the PDZ domain of Gipc1.
Fig. 1.
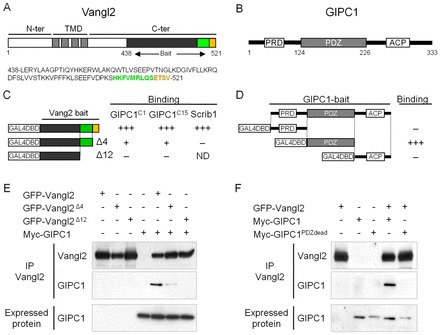
Gipc1 is a binding partner for Vangl2. (A) Vangl2 is a four transmembrane-domain (TMD) protein, with intracellular N-terminus (N-ter) and C-terminus (C-ter) ending with a PDZ binding motif (-ETSV, orange). The bait used to screen the embryonic cochlear library is indicated (aa 438-521). (B) Gipc1 is a cytosolic protein of 333 aa with a central PDZ domain, an N-ter proline-rich domain (PRD) and a C-ter acyl carrier protein domain (ACP). (C,D) Yeast-two-hybrid assay validating the interaction between Gipc1 and Vangl2. (C) The bait of Vangl2 positively interacts with two of the clones (GIPC1C1 and GIPC1C15) from the yeast-two-hybrid screen. Deletion of the PDZ-BM (last four aa, Δ4) leads to a strong reduction of the interaction, which is completely inhibited by deletion of the last 12 aa of the C-ter (Δ12). The interaction with Scrib1 was used as a positive control. (D) Only the PDZ domain of Gipc1 interacts with Vangl2. (E,F) Co-immunoprecipitation (co-IP) of GFP-Vangl2 with myc-Gipc1. (E) The interaction is strongly impaired by deletion of the PDZ-BM and abolished by removal of the last 12 aa of Vangl2. (F) The interaction is disrupted by mutation of the PDZ domain of Gipc1.
Gipc1 and Vangl2 are present in endocytic vesicles and at the membrane of COS-7 cells
Because Gipc1 is known to regulate the traffic of many transmembrane proteins through interaction with their C-ter (Hasson, 2003), we examined the localization of Vangl2 in COS-7 cells in the absence or presence of Gipc1. Single transfection of Vangl2 or Gipc1 revealed membrane and vesicular localization in COS-7 cells (Fig. 2A-D). When co-transfected, the two proteins colocalized in vesicles or in clusters, and in some domains of the plasma membrane (Fig. 2E-F, inset). Then, we examined the localization of Vangl2 in endocytic vesicles, using two different markers of endocytic compartments: early endosomal antigen 1 (Eea1), a classical marker of early endosomes, and transferrin (Fig. 2G). Vangl2 was present in 58.5±3% of Eea1-positive vesicles (Fig. 2H). When Vangl2 was co-transfected with Gipc1, we observed an 18% increase in Vangl2 localization in Eea1-positive vesicles. Removal of the PDZ-BM of Vangl2 led to a 33.3% reduction of this colocalization. The increase of Vangl2Δ4 presence in Eea1-positive vesicles when Gipc1 was co-expressed confirmed that the interaction persisted, at least partially, in the absence of the last four aa. Next, we used a short pulse of 3 minutes with Alexa 568-conjugated transferrin to label only the vesicles that are endocytosed by live cells. After this short pulse, 18.3±1% of Vangl2 vesicles colocalized with transferrin-positive vesicles, and co-transfection with Gipc1 significantly increased that colocalization to 38.5±2% (Fig. 2I). Similar to the Eea1 data, the removal of the PDZ-BM significantly reduced the localization of Vangl2 in endocytic vesicles, but a partial rescue was obtained by co-expression with Gipc1. Together, these data suggest that Vangl2 is endocytosed and that an interaction through its PDZ-BM with Gipc1 increases its localization in endocytic vesicles. Such mechanism could be efficient with a potent motor protein that would transport nascent endocytic vesicles away from the periphery of the cell along actin filaments. MyoVI (Myo6 – Mouse Genome Informatics) is a good candidate as it is an unconventional myosin that ‘walks’ along actin filaments towards the minus end of the filaments, and is also a well-characterized interactor of Gipc1 present in COS-7 cells (Hasson et al., 1997; Buss et al., 2001; Aschenbrenner et al., 2003; Buss and Kendrick-Jones, 2008). Co-expression of Vangl2, Gipc1 and MyoVI led to a striking re-localization of the three proteins in organelles located at the periphery of the cell, with very low levels of Vangl2 remaining at the plasma membrane (Fig. 2J-K, inset). These clusters require the presence of a functional Gipc1, as in its absence (Fig. 2L-M), or when its central PDZ domain was mutated (Fig. 2N-O, inset), we observed a strong reduction in the formation of clusters. COS-7 cells co-transfected with Vangl2, Gipc1 and MyoVI and treated for 3 minutes with fluorescent transferrin revealed a strong colocalization of the clusters with transferrin, suggesting that the clusters were, at least in part, of endocytic nature (Fig. 2P-Q), as suggested previously (Aschenbrenner et al., 2003). These results suggest that Gipc1 can associate with both MyoVI and Vangl2 simultaneously in clusters near the plasma membrane.
Fig. 2.
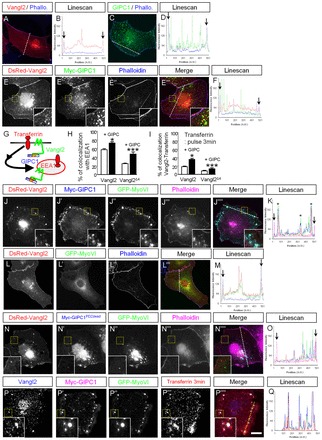
Vangl2 and Gipc1 colocalize in endosomal vesicles that are redistributed peripherally by MyoVI. (A-F) Immunofluorescence microscopy of COS-7 cells transiently transfected with DsRed-Vangl2, myc-Gipc1 or a combination of the constructs, and labeled with phalloidin (blue), with a line scan corresponding to the dashed line for each condition. The arrows on the line scans represent cell boundaries labeled by phalloidin (A-D). Single expression of DsRed-Vangl2 (red) or myc-Gipc1 (green) led to vesicular, membrane and perinuclear staining. (E-F) DsRed-Vangl2 and myc-Gipc1 colocalize in cytoplasmic vesicles, clusters and at the plasma membrane. (G) Schematic of the localization of transferrin and Eea1 in early endosomes containing Vangl2. (H) Sixty percent of GFP-Vangl2 vesicles colocalized with Eea1-labeled vesicles. This colocalization is increased in the presence of myc-Gipc1, and severely impaired by removal of the PDZ-BM of Vangl2 (Vangl2Δ4). (I) Similar profiles are observed when we measured colocalization of Vangl2-positive vesicles with fluorescent transferrin after a 3-minute treatment. (J-K) DsRed-Vangl2, GFP-MyoVI and myc-Gipc1 co-expression leads to an almost complete colocalization of the three proteins, with a re-localization of these clusters close to, but not at, the plasma membrane (magenta). Note on the line scan (K) that the peaks with high Vangl2 intensity (asterisks) do not correspond to the plasma membrane peaks (arrows). (L-M) DsRed-Vangl2 and GFP-MyoVI are expressed in the cytoplasm, in vesicle-like cytoplasmic punctae, weakly at the membrane and in the perinuclear region of the cell. (N-O) A mutation in the PDZ domain of Gipc1 (myc-Gipc1PDZdead) prevents the formation of the clusters and their re-localization. Note the presence of Vangl2 at the membrane (inset). (P-Q) A short treatment (3 minutes) with Alexa Fluor568-conjugated transferrin (red) reveals colocalization with the clusters. Insets show magnifications of boxed area within the same panel. *P≤0.05, **P≤0.01, ***P≤0.001. Error bars represent the standard error of the mean of triplicates. Scale bar: 15 μm.
Gipc1 and MyoVI form a protein complex that participates in Vangl2 removal from the plasma membrane
We confirmed that Vangl2, MyoVI and Gipc1 can be associated into a molecular complex, by co-immunoprecipitation (co-IP) (Fig. 3A,B). As anticipated, this complex was lost when the PDZ domain of Gipc1 was mutated (Fig. 3A,B). We also show the existence of a complex containing the three proteins in vivo, in mouse olfactory bulbs, by co-IP of Gipc1 with both endogenous Vangl2 and MyoVI (Fig. 3C). Because of the limitation in protein content, we could not perform a similar co-IP in cochlear tissue. Alternatively, we show that overexpression of a GFP-Gipc1 construct in HCs leads to the recruitment of endogenous Vangl2 in the GFP-Gipc1 clusters (Fig. 3D).
Fig. 3.
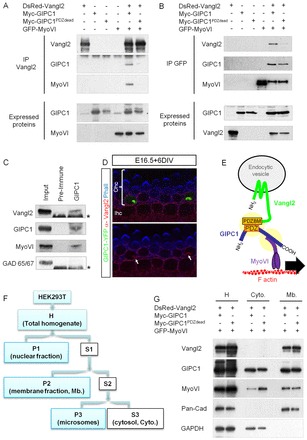
Gipc1 and MyoVI form a protein complex that participates in Vangl2 removal from the plasma membrane. (A,B) Co-IP of Gipc1 and MyoVI with Vangl2. HEK293T lysates were subjected to IP with an anti-Vangl2 antibody and probed with anti-myc (for Gipc1) and anti-GFP (for MyoVI), or with an anti-GFP antibody and probed with anti-Vangl2 and anti-myc (for Gipc1). The three proteins are present in a complex that is disrupted when the PDZ domain of Gipc1 is mutated (Gipc1PDZdead). (C) Endogenous Gipc1, Vangl2 and MyoVI co-immunoprecipitate in lysates of mouse olfactory bulbs, but not with Gad65/67. Asterisks denote IgG heavy chains. (D) Overexpression of Gipc1-YFP (green) in two Ohc1 cells leads to the formation of clusters containing endogenous Vangl2 (red). The image was taken just below the apical surface of the inner hair cell (Ihc) and outer hair cell (Ohc1); phalloidin labeling of the F-actin is in blue. (E) Vangl2 interacts with its PDZ-BM with the central PDZ domain of Gipc1, which can interact with MyoVI. The vesicular complex can be translocated along F-actin filaments by the motor function of MyoVI. (F) Schematic of the fractionation protocol. Fractions are as follows: H, total homogenate; S1, cell soma; P1, dense nuclei-associated membrane; S2, supernatant; P2, membrane fraction; S3, cytosolic; P3, microsomes. (G) Cellular fractionation assay. Antibodies against pan-cadherin and Gapdh show the segregation of the cytosolic (Cyto) and membrane (Mb) fractions from homogenate (H), and the antibody against myc reveals the presence of Gipc1 in the cytosolic fraction (with a weaker expression of the mutated form). Vangl2 is present in the membrane fraction, with higher levels when the PDZ of Gipc1 is mutated (Gipc1PDZdead).
To test the hypothesis that a protein complex consisting of Vangl2, Gipc1and MyoVI could move along the actin filament and away from the membrane (Fig. 3E), we transfected HEK293T cells and carried out subcellular fractionation (Fig. 3F). As a control for the fractionation procedure, some cells were transfected with a construct that expressed either wild-type (WT) Vangl2 or the Looptail form of Vangl2 (Vangl2Lp). As expected, Vangl2Lp levels in the membrane fraction were decreased by 58% compared with Vangl2 (data not shown) (Merte et al., 2010; Wansleeben et al., 2010). Next, similar fractionations were performed on cells transfected with either Vangl2/Gipc1/MyoVI or Vangl2/Gipc1-PDZdead/MyoVI. Our results show an increase of 186±39% in Vangl2 levels in the membrane fraction (normalized to pan-cadherin levels) when the PDZ domain of Gipc1 was mutated compared with WT Gipc1 (Fig. 3G). In addition, MyoVI levels were increased in the cytosolic fraction when Gipc1 was mutated, suggesting retention of MyoVI in the cytoplasm. These results are consistent with a process in which Gipc1 binds to Vangl2 at the plasma membrane, followed by a MyoVI-mediated removal of vesicles containing Vangl2 away from the plasma membrane, into the cytoplasm.
Gipc1 is localized in zones of intense traffic in cochlear hair cells
The subcellular localization of Gipc1 was analyzed in whole-mount preparations of postnatal day (P) 0 mouse cochleae. The lamina is derived from a mosaic of specialized cell types that includes a single row of inner hair cells (Ihcs), three rows of outer hair cells (Ohc1, 2 and 3), and several types of interdigitating non-sensory supporting cells (SCs) referred to as inner phalangeal cells (IPhs), inner and outer pillar cells (Ipcs, Opcs), and Deiter cells (DC1,2) (Fig. 4A). Two previously characterized Gipc1 antibodies were used to localize Gipc1 and gave a similar pattern to one another (De Vries et al., 1998; Dance et al., 2004). At P0, in the basal portion of the cochlear epithelium, Gipc1 is present in the cytoplasm of HCs and SCs as puncta or clusters (Fig. 4B,B′). In HCs, it is enriched around the cuticular plate of both Ihcs and Ohcs, in a region of intense traffic called the pericuticular necklace (Fig. 4B), where MyoVI also accumulates (Hasson et al., 1997; Kachar et al., 1997). We observed a preferential accumulation of clusters of Gipc1 on the proximal side of the HC, mainly restricted to an apical domain (Fig. 4B, white arrows and 4C,C′, asterisk), an observation confirmed by the expression of Gipc1-yellow fluorescent protein (YFP) in HCs (data not shown). The protein is also expressed in close apposition to the plasma membrane, in a more basolateral region (Fig. 4C,C′, curved bracket; supplementary material Fig. S1). At the apex of the cochlea, a region with a developmental delay from the base, this basolateral localization of Gipc1 is more pronounced (Fig. 4D-E′; supplementary material Fig. S1).
Fig. 4.
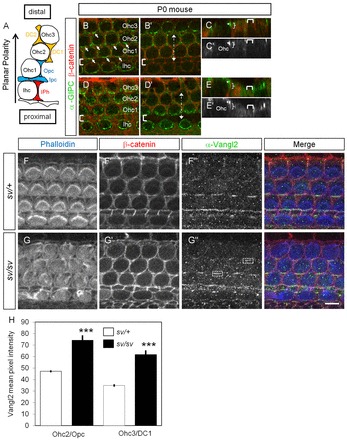
A MyoVI mutation in sv/sv mice leads to increased levels of Vangl2 at the plasma membrane. (A) Schematic of the various cell types composing the organ of Corti as indicated in the text. (B-E′) Surface view of a P0 rat cochlea in the basal (B-C′) and the apical (D-E′) regions, showing the localization of Gipc1 (green) and β-catenin (red). Gipc1 is expressed at the pericuticular necklace of Ihc and Ohc, with a preferential accumulation on the proximal side of the cells (Fig. 4B, arrows). (C,C′) xz view of the stack at the level indicated by the dotted line in B′. Gipc1 is apical (star) and more basolateral (bracket). (D-E′) In a more apical and less differentiated region of the same cochlea, Gipc1 localization extends more basolaterally (E,E′, bracket). Brackets in B,B′,D,D′ indicate the row of Ipcs. (F-G″) Surface views of sv/+ (F-F″) and sv/sv (G-G″) P1 mouse cochleae labeled with phalloidin (blue), β-catenin (red) and Vangl2 (green). (H) Quantification of the mean pixel intensity of Vangl2 at the junction between a HC and a SC (corresponding zones illustrated by boxes in G″) shows an increase in intensity at an Ohc2-Opc junction (mean=47.2 for sv/+, n=55; and mean=74.3, n=49 for sv/sv; P<0.001) and at an Ohc3-DC1 junction (mean=34.9 for sv/+, n=54; and mean=61.7, n=44 for sv/sv; P<0.001). Scale bar: 8 μm.
MyoVI participates in Vangl2 trafficking in HC
To test the hypothesis that MyoVI participates in Vangl2 endocytosis, the localization and levels of Vangl2 were examined in cochleae from Snell's waltzer (sv) mice, which arise as a result of an intragenic deletion leading to the absence of detectable MyoVI protein in homozygotes (Self et al., 1999). As described before, the structural integrity of the hair bundle was disrupted in these mutants (Fig. 4F,G). Asymmetric Vangl2 localization was still observed at HC-SC junctions in sv/sv mutants (Fig. 4G″). However, quantification of mean fluorescence intensity for the presence of Vangl2 at HC-SC junctions in WT and sv/sv cochlea (n=4 per genotype), indicated a significant increase in Vangl2 at HC-SC junctions in sv/sv mutants compared with sv/+ (Fig. 4H). These data suggest that a mutation in MyoVI affects Vangl2 levels at the plasma membrane.
Gipc1 downregulation leads to hair bundle phenotypes in vitro and in vivo
If Gipc1 and MyoVI are part of the same functional protein complex, we would expect that a decrease in Gipc1 would have similar HC phenotypes, as observed in MyoVI mutants (Avraham et al., 1995). To test this hypothesis, we used two previously described small hairpin RNAs (shRNAs) directed against Gipc1 (shGIPC1a-GFP and shGIPC1b-GFP) (Yi et al., 2007). We confirmed the efficacy of the two shRNA constructs in HEK293T, and although both constructs were able to reduce expression of transfected Gipc1, shGIPC1b-GFP was twice as effective (supplementary material Fig. S2A) (83±5% reduction in Gipc1 expression) than shGIPC1a-GFP (40±6%). Thus, shGIPC1b-GFP was used in subsequent studies. After testing that GFP alone did not have an impact on HC maturation (supplementary material Fig. S2B), we confirmed that shGIPC1b-GFP could downregulate endogenous Gipc1 in rat HCs (supplementary material Fig. S2C-C‴; n=28). Most HCs expressing shGIPC1b-GFP displayed stereociliary bundle abnormalities (supplementary material Fig. S2D-E′). The severity of the stereocilia phenotype was variable, probably as a result of variations in the efficacy of Gipc1 downregulation. In the most severely affected cells, the actin-rich stereocilia appeared to have completely lost structural integrity.
Because bundle polarity defects are ideally assessed in vivo, we evaluated the impact of Gipc1 downregulation by electroporation-mediated gene transfer in utero (Gubbels et al., 2008). We used shGIPC1b-GFP as its sequence is 100% identical between rat and mouse. Modified shRNA vectors (see Materials and methods) were microinjected into the otocysts of mice at E11.5 to induce an early downregulation of Gipc1 in epithelial progenitors of the inner ear. In samples transfected with the empty sh-GFP, GFP-positive cells were distributed along the length of the cochlea, including every cell type (Fig. 5A,A′), with intact and polarized stereociliary bundles (Fig. 5F, top pie chart). By comparison, the number of GFP-positive cells in embryos electroporated with shGIPC1b-GFP was markedly reduced, but examples of transfected cells that had developed as each of the different cell types within the organ of Corti were identified. Similar to observations in vitro, Gipc1 expression was downregulated in transfected HCs (supplementary material Fig. S2F-F″), and stereociliary bundle orientation and integrity was disrupted. The phenotype ranged from smaller and tilted bundles, consistent with a PCP phenotype (Fig. 5B,B″; supplementary material S2C,C′; Fig. 5F, n=11 HCs, average angle deviation=23±6°, 21% of total HCs), to a more extreme disruption of bundle integrity (Fig. 5C,C′D,D′,F, n=37 HCs, 71% of total HCs). Unequivocal results regarding the levels of Vangl2 at the plasma membrane of HC could not be obtained, largely owing to the disruption of a normal HC-SC junction; notably, the luminal surface area of HCs expressing the construct was reduced (Fig. 5B′,C′,D′ green stars; supplementary material Fig. S2F′). This dramatic phenotype suggests a role for Gipc1 in early HC development. In a few instances, the surface area of the Ohc transfected was not dramatically affected, and we were able to see Vangl2 labeling at the junctions (Fig. 5C″, green arrow). Finally, the levels of pericuticular MyoVI appeared to be reduced in transfected HC, suggesting a reduction of trafficking in these cells (Fig. 5B′, green star).
Fig. 5.
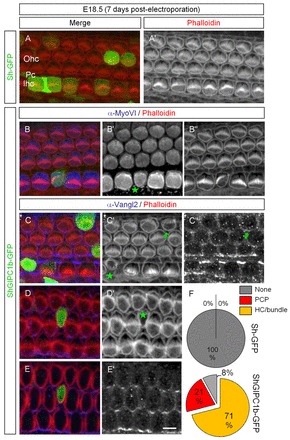
In utero downregulation of Gipc1 leads to PCP phenotype and HC maturation defects, including hair bundle integrity. (A,A′) Surface view of a mouse cochlea electroporated at E11.5 with sh-GFP (green) and labeled seven days later with phalloidin (red). HC development was not affected. (B-E′) The expression of shGIPC1b-GFP (green) leads to a PCP phenotype in HCs (B″). We observed a reduction of the apical surface area of the HC, and reduced pericuticular MyoVI expression (B′,C′,D′, green stars), and an impairment in hair bundle orientation and integrity (C′,D′). When downregulation of Gipc1 did not completely disrupt the HC-SC junction, Vangl2 expression is present (C″, green arrow), but when a strong HC phenotype is observed, there is a disruption of the HC-SC junction (E,E′). (F) Pie charts illustrating the variation in distribution of the HC phenotypes. Scale bar: 8 μm.
Gipc1 upregulation leads to hair bundle phenotypes in vitro
In Drosophila, it was shown recently that overexpression of dGIPC (Kermit – FlyBase) leads to PCP and multiple wing hair phenotypes (Toba et al., 1999; Djiane and Mlodzik, 2010). When electroporated with Gipc1-YFP, ten out of forty-two HCs in mouse cochlear epithelia had a misorientated hair bundle (Fig. 6A-A″,D; PCP phenotype), and six had a reduced apical surface or disrupted hair bundle (Fig. 6B-C″,D; HC/bundle phenotype). These results suggest that changes in levels of Gipc1 within HCs affect cytoskeleton dynamics. However, the upregulation of Gipc1 does not appear to produce a hair bundle/HC phenotype as severe as that induced by Gipc1 downregulation.
Fig. 6.
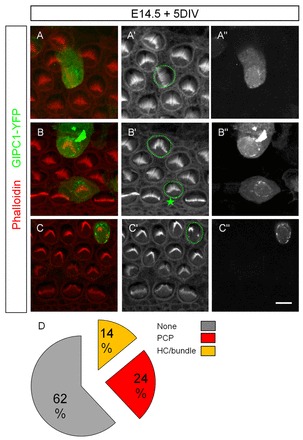
Upregulation of Gipc1 in vitro leads to a hair bundle phenotype. (A-D) The expression of the protein leads to a disruption of the orientation of the hair bundles in 23.8% of electroporated HCs (A-A″,D), a reduced apical surface area (B′, star; D) or a disrupted hair bundle phenotype (C″,D) in 14.2% of electroporated HCs. Scale bar: 8 μm.
Vangl2 is enriched at distal edges of supporting cells
In the light of our results, we re-examined the subcellular localization of Vangl2 within the cochlea, at HC-SC junction. As reported previously (Montcouquiol et al., 2006), Vangl2 is expressed on the basolateral membranes of SCs and HCs, and strongly accumulates in the apicolateral regions of cells at cell-cell junctions, a location where we also see Gipc1 expression (supplementary material Fig. S1). Using TCA fixation, we observed instances when the membrane of a SC was not in contact with the adjacent HC (Fig. 7A, arrows, and 7B). The intensity of Vangl2 labeling remained unchanged, suggesting that the enrichment was mostly on the SC side. Using STED microscopy, we show that Vangl2 is concentrated at foci on the SC side (Fig. 7C,D). Our data show that, at P1, Vangl2 is mostly enriched on the SC membrane. We confirmed this result in SC electroporated with a GFP-Vangl2 construct. In SCs with medium to weak expression of GFP-Vangl2, we observed an asymmetric localization of the protein with a greater accumulation on the distal side of the cell (Fig. 7G-I), consistent with what we observed by immunostaining (Fig. 7A-D).
DISCUSSION
Gipc1 regulates Vangl2 trafficking
Overall, our findings are compatible with a model in which Gipc1 can serve as a bridge between Vangl2 and MyoVI to participate in the trafficking of Vangl2-containing vesicles, including during endocytosis (supplementary material Fig. S3).
Gipc1 is a PDZ-domain-containing protein that can bind to proteins that have a conserved type I PDZ-BM at their carboxy termini. Gipc1 can then bind to MyoVI through its C-ter domain (Bunn et al., 1999), and MyoVI can then act as an actin-based motor to translocate these vesicles along the dense actin meshwork of the cuticular plate (Aschenbrenner et al., 2004). MyoVI is a minus-end motor protein, and its function is therefore compatible with moving vesicles away from the plasma membrane in polarized cells (Sweeney and Houdusse, 2007). The Gipc1-MyoVI complex could effectively reduce the presence of Vangl2 at the membrane, possibly in response to an unidentified endocytic signal. Consistent with this hypothesis is (1) the observation by STED microscopy that Vangl2 is expressed in both HCs and SCs but is enriched on the SC side of the HC-SC junctions at birth, (2) Gipc1 is expressed in both HCs and SCs but is enriched on the HC side of these junctions, and (3) MyoVI is expressed in HCs but not SCs. Because Gipc1 is expressed broadly in the cochlear epithelium, with accumulation of the proteins in HC progenitors as early as E15 (data not shown), Gipc1 could also regulate Vangl2 trafficking earlier in development. In other systems, MyoVI is also believed to anchor cytoplasmic organelles (Woolner and Bement, 2009), and we cannot exclude the possibility that the Gipc1-MyoVI complex could participate in the tethering of Vangl2 vesicles to the actin cytoskeleton, in a submembranous compartment.
The PCP effects we observed in our in vivo experiments were either mild or difficult to clearly assess because the majority of the cells affected had a distinct hair bundle morphology and/or HC phenotype. It is conceivable that a Gipc1-dependent disruption of Vangl2 trafficking might not translate into a strong PCP phenotype, or a clear change in Vangl2 levels at the membrane. Only mild PCP phenotypes have been reported in the inner ear of smurf ubiquitin ligase (Smurf1) and Sec24b, two regulators of PCP protein trafficking (Narimatsu et al., 2009; Merte et al., 2010; Wansleeben et al., 2010). The diversity of cellular phenotypes in cells with reduced levels of Gipc1 suggest additional roles for the protein (Bunn et al., 1999; Lanahan et al., 2010), and Gipc1 interactions are flexible and it is able to associate with a variety of proteins with divergent PDZ-BM (Abramow-Newerly et al., 2006). Members of the GIPC family are good candidates to regulate various functions, including traffic regulation of various proteins, through different binding partners and signaling pathways during the early stages of development of the HC.
Vangl2 is asymmetrically enriched on SC membranes in early postnatal cochlea
The results demonstrate an enrichment of Vangl2 on the SC side of the HC-SC junction. This enrichment in SC is consistent with the recent demonstration that asymmetric localization of Vangl2 is maintained in SCs in ototoxically injured avian inner ears when all HCs have been eliminated from the epithelium (Warchol and Montcouquiol, 2010). As we know that Vangl2 is expressed in both cell types (Montcouquiol et al., 2006), the interface between SCs and HCs could benefit from an intercellular negative-feedback loop to enrich Vangl2 on the SC side, but we cannot exclude directed targeting to increase Vangl2 asymmetry (see Fig. 7). The enrichment of Vangl2 on the SC fits with the reported localization of Frizzled 3 and 6 (Fzd3 and Fzd6) on the proximal side of HCs, i.e. opposite to the enriched domain of Vangl2 expression. By contrast, the reported expression of GFP-Dvl2 and GFP-Dvl3 on the distal side of HCs in transgenic animals (Wang et al., 2005; Wang et al., 2006; Etheridge et al., 2008) is more difficult to reconcile with a model similar to Drosophila. One would expect Fz and Dvl to both be on the proximal side of HCs because Dvl proteins are known effectors of Fz receptors (Vladar et al., 2009; Strutt and Strutt, 2009). However, Dvl proteins have also been shown to interact directly with Vangl2 C-ter in both invertebrates and vertebrates (Park and Moon, 2002; Bastock et al., 2003; Torban et al., 2004; Suriben et al., 2009). It is tempting to speculate that another Vangl2-dependent or -independent signaling cascade controls the hair bundle localization and orientation of individual HCs, perhaps before an observable Vangl2 asymmetric localization within the tissue. These apparent contradictions and our current limited knowledge of PCP molecular cascades emphasize the difficulty establishing a unifying model for PCP in mammals.
The asymmetry in SCs is also consistent with data in the vestibular system, in which Deans and colleagues showed that Pk2 is asymmetrically located at the HC/SC junction, and this distribution does not change at the line of reversal of hair bundle polarity within the vestibular maculae (striola) (Deans et al., 2007). This could be explained if Pk2 (Prickle2), a known interactor of Vangl2, is enriched on the SC side across the tissue. Together, these results suggest that the strong asymmetry of Vangl2 observed within the early postnatal cochlear epithelium is a planar ‘tissue’ polarity readout. In this context, the loss, or maintenance, of Vangl2 asymmetry could be indicative of a disruption of the transmission of a polarization signal across the tissue, owing to a disruption of the HC-SC junction, but not necessarily indicative of PCP disruption within individual HCs. This would explain why a large variety of genes have been linked to a loss of Vangl2 asymmetry combined with mild PCP phenotypes in the cochlear epithelium in the recent years.
Gipc1 is a regulator of hair bundle formation and maintenance
In the present study, expression of shRNA for Gipc1 in vivo and in vitro demonstrated a role for Gipc1 in several aspects of the differentiation of HCs. In particular, downregulation of Gipc1 led to reduction in the size of the apices of HCs, as well as the morphology of stereociliary bundles. Gipc1 and MyoVI are expressed in the cytoplasm of developing HCs as early as E13.5, well before the development of stereociliary bundles. They could function in early HC development, prior to the formation of the bundle. For example, Gipc1 could participate in the extensive membrane recycling that has been visualized at the apical HC domain during hair bundle formation and HC maturation (Souter et al., 1995; Forge and Richardson, 1993; Denman-Johnson and Forge, 1999). Consistent with this, dGIPC has recently been shown to modulate the actin cytoskeleton in Drosophila wing cells, at least in part through interactions with MyoVI (Djiane and Mlodzik, 2010). Although no obvious HC phenotypes have been observed in cochleae of Snell's waltzer mice before birth (Self et al., 1999), the survival of the homozygotes has led some authors to speculate that there might be a significant degree of functional redundancy among myosin proteins during mouse development and/or among other motor proteins that must be able to compensate for the loss of MyoVI. In humans, mutations in MYO6 cause hereditary hearing loss (DFNA22 and DFNB37 syndromes), which can be associated with hypertrophic cardiomyopathy (Ahmed et al., 2003; Melchionda et al., 2001).
In the course of this study, GIPC3, another member of the GIPC family was associated with progressive hearing loss in humans and mice (Charizopoulou et al., 2011). Interestingly, in the mutant mice, the identified mutation affected the PDZ domain of Gipc3, leading to early postnatal disruption of hair bundle structural integrity. In humans, one of the identified mutations leads to a potential truncated construct, preventing interaction with MyoVI. Also, a recent study correlated hearing impairment in four patients with a 359-kb deleted region in 19p13.12 harboring six genes, including GIPC1, leading the authors to suggest that haploinsufficiency of GIPC1 might contribute to hearing impairment through its interaction with MyoVI (Bonaglia et al., 2010). Altogether, these results strongly suggest that members of the GIPC family have essential roles in HC maturation and auditory function.
Supplementary Material
Acknowledgements
We thank the team of Bordeaux Imaging Center and Ulf Schwarz from Leica Microsystems, Germany, for their help in STED microscopy; the genotyping facility; and Helene Doat from the animal facility for technical assistance.
Footnotes
Funding
This research was supported by a French National Institute of Health and Medical Research (INSERM) AVENIR grant [R04208GS to M.M. and R04210GS to N.S.]; Conseil Regional d'Aquitaine [M.M. and N.S.]; La Fondation pour la Recherche Medicale [M.M., N.S., J.E.]; Agence Nationale de la Recherche [ANR-08-; MNPS-040-01 to M.M. and ANR-07-NEUR-031-01 to N.S.]; Aquitaine-INSERM Fellowship [A.P.G.]; International Reintegration Grants (IRG) Marie-Curie action [M.M.] and the European Commission Coordination Action and the Network of European Neuroscience Institutes ENINET [contract number LSHM-CT-2005-19063; N.S. and M.M.]; and the National Institute on Deafness and other Communication Disorders [DC R01 008595, DC R01 008595-03S2 (to J.B.) and P30 DC005983]. J.-P.B. is supported by Ligue Nationale Contre le Cancer [Label 2010 JPB], European Consortium for Anticancer Antibody Development (EUCAAD) (FP7 program), INCa and IBISa (Marseille Proteomic Platform).
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.074229/-/DC1
References
- Abramow-Newerly M., Roy A. A., Nunn C., Chidiac P. (2006). RGS proteins have a signalling complex: interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell Signal. 18, 579-591 [DOI] [PubMed] [Google Scholar]
- Ahmed Z. M., Morell R. J., Riazuddin S., Gropman A., Shaukat S., Ahmad M. M., Mohiddin S. A., Fananapazir L., Caruso R. C., Husnain T., et al. (2003). Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 72, 1315-1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschenbrenner L., Lee T., Hasson T. (2003). Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol. Biol. Cell 14, 2728-2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschenbrenner L., Naccache S. N., Hasson T. (2004). Uncoated endocytic vesicles require the unconventional myosin, Myo6, for rapid transport through actin barriers. Mol. Biol. Cell 15, 2253-2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham K. B., Hasson T., Steel K. P., Kingsley D. M., Russell L. B., Mooseker M. S., Copeland N. G., Jenkins N. A. (1995). The mouse Snell's waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat. Genet. 11, 369-375 [DOI] [PubMed] [Google Scholar]
- Bastock R., Strutt H., Strutt D. (2003). Strabismus is asymmetrically localised and binds to Prickle and Dishevelled during Drosophila planar polarity patterning. Development 130, 3007-3014 [DOI] [PubMed] [Google Scholar]
- Bonaglia M. C., Marelli S., Novara F., Commodaro S., Borgatti R., Minardo G., Memo L., Mangold E., Beri S., Zucca C., et al. (2010). Genotype-phenotype relationship in three cases with overlapping 19p13.12 microdeletions. Eur. J. Hum. Genet. 18, 1302-1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigande J. V., Gubbels S. P., Woessner D. W., Jungwirth J. J., Bresee C. S. (2009). Electroporation-mediated gene transfer to the developing mouse inner ear. Methods Mol. Biol. 493, 125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn R. C., Jensen M. A., Reed B. C. (1999). Protein interactions with the glucose transporter binding protein GLUT1CBP that provide a link between GLUT1 and the cytoskeleton. Mol. Biol. Cell 10, 819-832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F., Kendrick-Jones J. (2008). How are the cellular functions of myosin VI regulated within the cell? Biochem. Biophys. Res. Commun. 369, 165-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F., Arden S. D., Lindsay M., Luzio J. P., Kendrick-Jones J. (2001). Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J. 20, 3676-3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F., Spudich G., Kendrick-Jones J. (2004). MYOSIN VI: cellular functions and motor properties. Annu. Rev. Cell. Dev. Biol. 20, 649-676 [DOI] [PubMed] [Google Scholar]
- Charizopoulou N., Lelli A., Schraders M., Ray K., Hildebrand M. S., Ramesh A., Srisailapathy C. R., Oostrik J., Admiraal R. J., Neely H. R., et al. (2011). Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat. Commun. 2, 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin J. A., Quint E., Tsipouri V., Arkell R. M., Cattanach B., Copp A. J., Henderson D. J., Spurr N., Stanier P., Fisher E. M., Nolan P. M., Steel K. P., Brown S. D., Gray I. C., Murdoch J. N. (2003). Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 13, 1129-1133 [DOI] [PubMed] [Google Scholar]
- Dance A. L., Miller M., Seragaki S., Aryal P., White B., et al. (2004). Regulation of myosin-VI targeting to endocytic compartments. Traffic 5, 798-813 [DOI] [PubMed] [Google Scholar]
- Das G., Jenny A., Klein T. J., Eaton S., Mlodzik M. (2004). Diego interacts with Prickle and Strabismus/Van Gogh to localize planar cell polarity complexes. Development 131, 4467-4476 [DOI] [PubMed] [Google Scholar]
- De Vries L., Lou X., Zhao G., Zheng B., Farquhar M. (1998). G. GIPC, a PDZ domain containing protein, interacts specifically with the C terminus of RGS-GAIP. Proc. Natl. Acad. Sci. USA 95, 12340-12345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans M. R., Antic D., Suyama K., Scott M. P., Axelrod J. D., Goodrich L. V. (2007). Asymmetric distribution of prickle-like 2 reveals an early underlying polarization of vestibular sensory epithelia in the inner ear. J. Neurosci. 27, 3139-3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denman-Johnson K., Forge A. (1999). Establishment of hair bundle polarity and orientation in the developing vestibular system of the mouse. J. Neurocytol. 28, 821-835 [DOI] [PubMed] [Google Scholar]
- Djiane A., Mlodzik M. (2010). The Drosophila GIPC homologue can modulate myosin based processes and planar cell polarity but is not essential for development. PLoS ONE 5, e11228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheridge S. L., Ray S., Li S., Hamblet N. S., Lijam N., et al. (2008). Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 4, e1000259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forge A., Richardson G. (1993). Freeze fracture analysis of apical membranes in cochlear cultures: differences between basal and apical-coil outer hair cells and effects of neomycin. J. Neurocytol. 22, 854-867 [DOI] [PubMed] [Google Scholar]
- Gubbels S. P., Woessner D. W., Mitchell J. C., Ricci A. J., Brigande J. V. (2008). Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455, 537-541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson T. (2003). Myosin VI: two distinct roles in endocytosis. J. Cell Sci. 116, 3453-3461 [DOI] [PubMed] [Google Scholar]
- Hasson T., Gillespie P. G., Garcia J. A., MacDonald R. B., Zhao Y., Yee A. G., Mooseker M. S., Corey D. P. (1997). Unconventional myosins in inner-ear sensory epithelia. J. Cell Biol. 137, 1287-1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Yonemura S., Matsui T., Tsukita S. (1999). Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxylterminal threonine phosphorylated in cultured cells and tissues: application of a novel fixation protocol using trichloroacetic acid (TCA) as a fixative. J. Cell Sci. 112, 1149-1158 [DOI] [PubMed] [Google Scholar]
- Jenny A., Reynolds-Kenneally J., Das G., Burnett M., Mlodzik M. (2005). Diego and Prickle regulate Frizzled planar cell polarity signalling by competing for Dishevelled binding. Nat. Cell Biol. 7, 691-697 [DOI] [PubMed] [Google Scholar]
- Kachar B., Battaglia A., Fex J. (1997). Compartmentalized vesicular traffic around the hair cell cuticular plate. Hear. Res. 107, 102-112 [DOI] [PubMed] [Google Scholar]
- Lanahan A. A., Hermans K., Claes F., Kerley-Hamilton J. S., Zhuang Z. W., Giordano F. J., Carmeliet P., Simons M. (2010). VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev. Cell 18, 713-724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Borchers A. G., Jolicoeur C., Rayburn H., Baker J. C., Tessier-Lavigne M. (2004). PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 430, 93-98 [DOI] [PubMed] [Google Scholar]
- Melchionda S., Ahituv N., Bisceglia L., Sobe T., Glaser F., et al. (2001). MYO6, the human homologue of the gene responsible for deafness in Snell's waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 69, 635-640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merte J., Jensen D., Wright K., Sarsfield S., Wang Y., Schekman R., Ginty D. D. (2010). Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat. Cell Biol. 12, 41-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M., Rachel R. A., Lanford P. J., Copeland N. G., Jenkins N. A., Kelley M. W. (2003). Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 423, 173-177 [DOI] [PubMed] [Google Scholar]
- Montcouquiol M., Sans N., Huss D., Kach J., Dickman J. D., Forge A., Rachel R. A., Copeland N. G., Jenkins N. A., Bogani D., Murdoch J., Warchol M. E., Wenthold R. J., Kelley M. W. (2006). Asymmetric localization of Vangl2 and Fz3 indicate novel mechanisms for planar cell polarity in mammals. J. Neurosci. 26, 5265-5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M., Jones J. M., Sans N. (2008). Detection of planar polarity proteins in mammalian cochlea. Methods Mol. Biol. 468, 207-219 [DOI] [PubMed] [Google Scholar]
- Narimatsu M., Bose R., Pye M., Zhang L., Miller B., Ching P., Sakuma R., Luga V., Roncari L., Attisano L., Wrana J. L. (2009). Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell 137, 295-307 [DOI] [PubMed] [Google Scholar]
- Park M., Moon R. T. (2002). The planar cell-polarity gene stbm regulates cell behaviour and cell fate in vertebrate embryos. Nat. Cell Biol. 4, 20-25 [DOI] [PubMed] [Google Scholar]
- Sans N., Prybylowski K., Petralia R. S., Chang K., Wang Y. X., Racca C., Vicini S., Wenthold R. J. (2003). NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat. Cell Biol. 5, 520-530 [DOI] [PubMed] [Google Scholar]
- Seifert J. R., Mlodzik M. (2007). Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat. Rev. Genet. 8, 126-138 [DOI] [PubMed] [Google Scholar]
- Self T., Sobe T., Copeland N. G., Jenkins N. A., Avraham K. B., Steel K. P. (1999). Role of myosin VI in the differentiation of cochlear hair cells. Dev. Biol. 214, 331-341 [DOI] [PubMed] [Google Scholar]
- Souter M., Nevill G., Forge A. (1995). Postnatal development of membrane specialisations of gerbil outer hair cells. Hear. Res. 91, 43-62 [DOI] [PubMed] [Google Scholar]
- Strutt H., Strutt D. (2009). Asymmetric localisation of planar polarity proteins: mechanisms and consequences. Semin. Cell Dev. Biol. 20, 957-963 [DOI] [PubMed] [Google Scholar]
- Suriben R., Kivimäe S., Fisher D. A., Moon R. T., Cheyette B. N. (2009). Posterior malformations in Dact1 mutant mice arise through misregulated Vangl2 at the primitive streak. Nat. Genet. 41, 977-985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney H. L., Houdusse A. (2007). What can myosin VI do in cells? Curr. Opin. Cell Biol. 19, 57-66 [DOI] [PubMed] [Google Scholar]
- Takeuchi M., Nakabayashi J., Sakaguchi T., Yamamoto T. S., Takahashi H., Takeda H., Ueno N. (2003). The prickle related gene in vertebrates is essential for gastrulation cell movements. Curr. Biol. 13, 674-679 [DOI] [PubMed] [Google Scholar]
- Toba G., Ohsako T., Miyata N., Ohtsuka T., Seong K. H., Aigaki T. (1999). The gene search system. A method for efficient detection and rapid molecular identification of genes in Drosophila melanogaster. Genetics. 151, 725-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torban E., Wang H. J., Groulx N., Gros P. (2004). Independent mutations in mouse Vangl2 that cause neural tube defects in looptail mice impair interaction with members of the Dishevelled family. J. Biol. Chem. 279, 52703-52713 [DOI] [PubMed] [Google Scholar]
- Tree D. R., Shulman J. M., Rousset R., Scott M. P., Gubb D., Axelrod J. D. (2002). Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell 109, 371-381 [DOI] [PubMed] [Google Scholar]
- Vladar E. K., Antic D., Axelrod J. D. (2009). Planar cell polarity signaling: the developing cell's compass. Cold Spring Harb. Perspect. Biol. 1, a002964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Mark S., Zhang X., Qian D., Yoo S. J., Radde-Gallwitz K., Zhang Y., Lin X., Collazo A., Wynshaw-Boris A., Chen P. (2005). Regulation of polarized extension and planar cell polarity in the cochlea by the vertebrate PCP pathway. Nat. Genet. 37, 980-985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Nathans J. (2007). Tissue/planar cell polarity in vertebrates: new insights and new questions. Development 134, 647-658 [DOI] [PubMed] [Google Scholar]
- Wang Y., Guo N., Nathans J. (2006). The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 26, 2147-2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansleeben C., Feitsma H., Montcouquiol M., Kroon C., Cuppen E., Meijlink F. (2010). Planar cell polarity defects and defective Vangl2 trafficking in mutants for the COPII gene Sec24b. Development 137, 1067-1073 [DOI] [PubMed] [Google Scholar]
- Warchol M. E., Montcouquiol M. (2010). Maintained expression of the planar cell polarity molecule Vangl2 and reformation of hair cell orientation in the regenerating inner ear. J. Assoc. Res. Otolaryngol. 11, 395-406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolner S., Bement W. M. (2009). Unconventional myosins acting unconventionally. Trends Cell Biol. 19, 245-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z., Petralia R. S., Fu Z., Swanwick C. C., Wang Y. X., Prybylowski K., Sans N., Vicini S., Wenthold R. J. (2007). The role of the PDZ protein GIPC in regulating NMDA receptor trafficking J. Neurosci. 27, 11663-11675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen J. A. (2007). Planar polarity and tissue morphogenesis. Cell. 129, 1051-1063 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.